

Abstract
Introduction
Despite extensive prevention campaigns and scale-up of antiretroviral therapy, HIV incidence among young women in southern Africa remains high. While the development of an efficacious vaccine remains a challenge, the discovery of broadly neutralising monoclonal antibodies (mAbs) has created the opportunity to explore passive immunisation as a long-acting injectable HIV prevention strategy. The purpose of this trial is to provide safety, pharmacokinetic (PK) and functional activity data of VRC07-523LS and PGT121 when administered subcutaneously (SC) to young South African women. Going forward, the aim is to select the ideal dose and/or monoclonal antibody for co-formulation and testing with CAP256-VRC26.25LS, a potent monoclonal antibody against subtype C virus, in an efficacy trial.
Methods and analysis
CAPRISA 012A is a randomised, double blinded, placebo-controlled phase I trial to assess the safety and PK profile of two mAbs, VRC07-523LS and PGT121 administered SC to 35 young HIV negative women at low risk for HIV infection. Women will be randomised into seven groups of five participants each. In each group, women will be randomised (4:1) to the active intervention, VRC07-523LS and/or PGT121, or placebo. Participants will be followed up for 24 weeks after the administration of the last dose of study product with a total study duration of 72 weeks. Safety in the study will be assessed by the number and percentage of reactogenicity and adverse events experienced by participants and the relatedness to study product. The PK study design was based on preliminary PK data for VRC07-523LS and PGT121.
Ethics and dissemination
Ethical approval has been granted by the South African Health Products Regulatory Authority and by the University of KwaZulu-Natal Biomedical Research Ethics Committee. Results will be presented at international conferences and published in academic peer-reviewed journals. Trial results will be uploaded on the clinical trial registry.
Trial registration number
PACTR201808919297244; Pre-results.
Keywords: HIV prevention, monoclonal antibodies, VRC07-523LS, PGT121, South Africa
Strengths and limitations of this study.
This trial will provide new safety, pharmacokinetic and functional activity data for two monoclonal antibodies (mAbs), VRC07-523LS and PGT121, when administered subcutaneously alone or in combination to South African women.
The trial will inform the optimal dose and monoclonal antibody combination that will be selected for co-formulation and testing with the potent monoclonal antibody CAP256-VRC26.25LS, in an efficacy trial.
Data from this trial could inform the future development of an injectable HIV prevention method, with anticipated four-monthly or six-monthly dosing, that offers implementation and adherence advantages over available antiretroviral pre-exposure prophylaxis options.
While the use of mAbs are a promising HIV prevention strategy and high levels of protection have been demonstrated in animal studies, the efficacy in human clinical trials has not yet been established.
The sample size in this study is small (typical of phase I trials) and therefore all results and conclusions drawn must be prospectively validated. The potential clinical impact of these antibodies will depend on an efficacy signal established by phase IIb efficacy trials.
Introduction
Despite extensive prevention campaigns and scale-up of antiretroviral therapy (ART), South Africa remains an epicentre of the HIV pandemic.1 In southern and eastern Africa, the incidence of HIV among young women below 25 years remains high.1 2 While the HIV prevention landscape is changing rapidly, principally with the roll-out of pre-exposure prophylaxis (PrEP) and early ART (Treatment as Prevention), current HIV prevention programmes have had limited impact on reducing HIV incidence in young women.3–5 Clinical trials using daily oral tenofovir disoproxil fumarate alone and in combination with emtricitabine in African women demonstrated inconsistent results, most likely due to varying levels of medication adherence.6–8 New approaches that overcome these adherence challenges, are being tested. While the development of an efficacious vaccine remains a major challenge, the discovery of potent monoclonal antibodies (mAbs) has created the opportunity to explore passive immunisation as an HIV prevention strategy.
VRC07-523LS is a highly potent and broadly neutralising mAb that targets the HIV-1 CD4 binding site. It was developed by the Vaccine Research Center (VRC) at the National Institute of Health, USA. The antibody was engineered based on the VRC01 mAb, that was originally discovered in a subject infected with HIV-1, whose immune system controlled the virus without ART for more than 15 years.9 10 The neutralisation, potency and breadth of VRC01 was enhanced by next-generation sequencing and structure-guided design to create VRC07-523, which displayed fivefold to eightfold more potency than VRC01 and neutralised 96% of viruses tested.9 Thereafter, a lysine-serine (LS) mutation was designed to extend the half-life and increase concentrations in mucosal tissue. The LS mutation is in the Fc region and was introduced by site-directed mutagenesis to increase the binding affinity for the neonatal Fc-receptor, resulting in increased recirculation of functional IgG, thus increasing plasma half-life.9 Pharmacokinetic (PK) analyses in rhesus macaques demonstrated half-life values for VRC07-523LS ranging from 7 to 10 days, compared with 5 days for VRC07 and 5–6 days for VRC01. VRC07-523LS is approximately 10-fold more potent than VRC01 and active against 96% of diverse HIV-1 strains, including clade C.11 The VRC01 antibody is currently being evaluated in the HVTN 703/HPTN 081 phase IIb clinical trial (ClinicalTrials.gov Identifier: NCT02568215).
PGT121, is a recombinant human IgG1 mAb, isolated from an African donor in 2011, that targets the V3 glycan-dependent epitope region of the HIV envelope protein.12 PGT121 was developed by the Center for Virology and Vaccine Research at the Beth Israel Deaconess Medical Center and the International AIDS Vaccine Initiative. This mAb has a long heavy chain complementarity determining region that forms an antibody binding site with two functional surfaces and does not bind simply to the GPGR region of V3. Structural studies have shown that although PGT121 does not engage the CD4 binding site, it inhibits CD4 binding to gp120. PGT121 disrupts the Env-receptor engagement by an allosteric mechanism which interferes with CD4 binding and viral entry.13 Due to its mechanism of action, PGT121 has excellent potency and breadth.
In order to overcome the genetic diversity of HIV, combinations of mAbs targeting different epitopes on the viral envelope will likely be required.10 To identify the optimal combination of mAbs, Wagh and colleagues assessed the neutralising activity of 15 mAbs targeting four distinct epitopes of the envelope against a panel of 200 early/acute clade C HIV-1 Env-pseudoviruses and a mathematical model was developed to predict neutralisation by mAb combinations.14 15 The analysis revealed that the neutralisation profile of CAP256-VRC26.25LS (which targets the V2 loop) was particularly well suited as a complementary mAb with VRC07-523LS and PGT121. These two mAbs were found to be the best combinations in terms of neutralisation breadth and potency.
Results of preclinical and clinical studies have demonstrated promising results that support the clinical evaluation and development of mAbs for prevention.16–18 Currently there are two phase I trials assessing PGT121 and VRC07-523LS in the USA. These are separate trials that are investigating the safety and tolerability of each mAb used alone, but not in combination (ClinicalTrials.gov Identifier: NCT03015181 and NCT02960581). Preliminary data from these trials have demonstrated no safety concerns. Neither mAb has been investigated in an African population, particularly young African women, who are at high risk of HIV acquisition. The purpose of this protocol is to provide safety, and functional activity data from a phase I trial assessing VRC07-523LS and PGT121 when administered subcutaneously (SC), alone and in combination to HIV negative women in South Africa. Data from this trial will inform the optimal dose and mAb combination that will be selected for co-formulation and testing with the potent mAb, CAP256-VRC26.25LS, in a proof-of-concept trial.
Methods and analysis
(Standard Protocol Items: Recommendations for Interventional Trials reporting guidelines used.)
CAPRISA 012A is a randomised, double blinded, placebo-controlled phase I clinical trial.
Patient and public involvement
The CAPRISA community programme will inform, educate and mobilise the community to enhance community input into the research process. The local community research support groups play an active role as an interface between the researchers and community members serving as advocates for the community’s best interests and ensuring that the researchers are always aware of any concerns within the community about the research being conducted. The study concept is introduced to the CSRG and concerns are addressed, and feedback is given to the study team. The recruitment team will raise awareness of clinical trial opportunities and educate the community regarding eligibility, screening and enrolment. After enrolment into the study, study staff will make every reasonable effort to ensure retention by collecting adequate locator information for follow-up tracking, visit reminders and retention activities.
Study setting
The study will be conducted at the CAPRISA eThekwini Clinical Research Site in Durban, KwaZulu-Natal, South Africa.
Study population selection
The study will include 35 HIV negative women. Enrolment will be based on the following eligibility criteria (tables 1–3).
Table 1.
The distribution of study participants into individualised groups
| Group | Regimen | N | Dose (mg/kg) |
| 1 | VRC07-523LS/Placebo | 4/1 | 5 mg/kg SC one dose |
| 2 | VRC07-523LS/Placebo | 4/1 | 10 mg/kg SC one dose |
| 3 | VRC07-523LS/Placebo | 4/1 | 5 mg/kg SC with one repeat dose at 12 weeks |
| 4 | VRC07-523LS/Placebo | 4/1 | 10 mg/kg SC with one repeat dose at 24 weeks |
| 5 | PGT121/Placebo | 4/1 | 3 mg/kg SC one dose |
| 6 | PGT121/Placebo | 4/1 | 3 mg/kg SC with one repeat dose at 12 weeks |
| 7 | VRC07-523LS+PGT121/Placebo | 4/1 | 5 mg/kg SC+3 mg/kg SC one dose |
SC, subcutaneously
Table 2.
Probability of observing no events, one or more events and two or more events for a range of hypothetical true event rates
| True event rate (%) | Number of participants | No events | One or more events | Two or more events |
| 5 | 4 | 0.81 | 0.19 | 0.01 |
| 10 | 4 | 0.66 | 0.34 | 0.05 |
| 20 | 4 | 0.41 | 0.59 | 0.18 |
| 30 | 4 | 0.24 | 0.76 | 0.35 |
Table 3.
Probability of observing no events, at least one event or at least two events for a range of hypothetical true event rates
| True event rate (%) | Number of participants | No events | At least one event | At least two events |
| 1 | 4 | 0.96 | 0.04 | <0.01 |
| 8 | 0.92 | 0.08 | <0.01 | |
| 12 | 0.89 | 0.11 | 0.01 | |
| 16 | 0.85 | 0.15 | 0.01 | |
| 28 | 0.75 | 0.25 | 0.03 | |
| 5 | 4 | 0.81 | 0.19 | 0.01 |
| 8 | 0.66 | 0.34 | 0.06 | |
| 12 | 0.54 | 0.46 | 0.12 | |
| 16 | 0.44 | 0.56 | 0.19 | |
| 28 | 0.24 | 0.76 | 0.41 | |
| 10 | 4 | 0.66 | 0.34 | 0.05 |
| 8 | 0.43 | 0.57 | 0.19 | |
| 12 | 0.28 | 0.72 | 0.34 | |
| 16 | 0.19 | 0.81 | 0.49 | |
| 28 | 0.05 | 0.95 | 0.78 | |
| 20 | 4 | 0.41 | 0.59 | 0.18 |
| 8 | 0.17 | 0.83 | 0.50 | |
| 12 | 0.07 | 0.93 | 0.73 | |
| 16 | 0.03 | 0.97 | 0.86 | |
| 28 | <0.01 | >0.99 | 0.98 | |
| 30 | 4 | 0.24 | 0.76 | 0.35 |
| 8 | 0.06 | 0.94 | 0.74 | |
| 12 | 0.01 | 0.99 | 0.91 | |
| 16 | <0.01 | >0.99 | 0.97 | |
| 28 | <0.01 | >0.99 | >0.99 |
Inclusion criteria
18–40 years of age.
Female sex at birth.
Able and willing to complete the informed consent process.
Has understood the information provided, including the potential impact and/or risks linked to SC administration of the study product, and is willing to comply with protocol procedures.
Has access to the clinical research site and is available for the duration of the study.
Based on clinical assessment must be in good general health.
Assessed by site staff to be at low risk for HIV infection.
If of reproductive potential, has evidence of effective contraceptive use in the previous 21 days, and agrees to continued use during the study period.
Willing to have blood and genital samples collected, stored and used for research purposes.
Screening laboratory parameters
White blood cell count within institutional normal range.
Haemoglobin >10 g/dL.
Creatinine ≤upper limit of institutional normal range.
Alanine aminotransferase ≤upper limit of institutional normal range.
Negative for HIV infection by a Food and Drug administration (FDA)-approved method of detection in the last 30 days.
Negative β-Human chorionic gonadropin pregnancy test (urine or serum) within 21 days of enrolment.
Exclusion criteria
Any clinically significant acute or chronic medical condition that makes the participant unsuitable for participation in the study, or jeopardises the safety or rights of the volunteer.
If planning a pregnancy for the duration of the study, currently pregnant or breastfeeding.
Exceeding the weight of 90 kg (in order to restrict the amount of injections administered).
A history of alcohol or substance use judged to potentially interfere with participant study compliance.
Prior participation in an investigational HIV vaccine trial, except if proof of allocation to the placebo arm is available.
Administration of a mAb or polyclonal immunoglobulin within 28 days prior to enrolment.
Any history of anaphylaxis and related symptoms such as hives, respiratory difficulty and angioedema.
Evidence of autoimmune disease, or receiving immunosuppressive therapy.
Participants in this study may not take part in other concurrent research studies that would interfere with the objectives of this study.
Study schema
The 35 HIV negative participants at low risk for HIV infection will be randomised into seven groups of five participants each (table 1). In each group (n=5), four women will be randomly assigned to the active intervention, VRC07-523LS and/or PGT121 and one participant randomly assigned to placebo. The safety and PK profile of one and two doses of VRC07-523LS and/or PGT121 mAbs administered SC will be evaluated. As per table 1, VRC07-523LS will be administered alone at a dose of either 5 mg/kg or 10 mg/kg at one time point in Groups 1 and 2, respectively. VRC07-523LS will be administered alone at a dose of 5 mg/kg or 10 mg/kg at two time points in Group 3 (repeat dose at 12 weeks) and Group 4 (repeat dose at 24 weeks).
PGT121 will be administered alone at a dose of 3 mg/kg at one time point (Group 5) or at two time points in Group 6 (repeat dose at 12 weeks). VRC07-523LS and PGT121 will be administered in combination at one time point in Group 7. This mAb combination will not be administered as a single product containing two antibodies but rather as two separate injections, each containing a single mAb. Participants will be followed up for 24 weeks after the administration of the last dose of study product with a total study duration of 72 weeks.
Study objectives
Primary objective
To evaluate the safety of one and two doses of VRC07-523LS and/or PGT121 mAbs administered SC.
Secondary objectives
To characterise the PK profile of VRC07-523LS mAb (5 and 10 mg/kg) administered SC individually as a single dose or as two doses 12 and 24 weeks apart.
To characterise the PK profile of PGT121 mAb (3 mg/kg) administered SC individually as a single dose or as two doses 12 weeks apart.
To characterise the PK profile of VRC07-523LS and PGT121 mAbs administered in combination.
To assess the acceptability of VRC07-523LS and PGT121 mAbs SC injections.
To evaluate the concentrations and functional activity of VRC07-523LS and/or PGT121 mAb in plasma and genital samples following SC administration.
To determine whether SC administration of VRC07-523LS and/or PGT121 mAbs induces anti-mAbs.
Primary outcomes
Proportion of participants with mild, moderate and severe reactogenicity events within the first 3 days after SC administration.
Proportion of participants with mild, moderate and severe adverse events (AEs) up to 24 weeks after the last SC administration.
Proportion of participants with serious adverse events (SAEs) related to SC administration.
Secondary outcomes
Maximal concentration (Cmax), time of maximal concentration (Tmax), area under the concentration versus time curve (AUC), apparent clearance (CL/F) and terminal half-life (t1/2) of VRC07-523LS and PGT121 mAbs.
Proportion of participants who report that the SC injections are acceptable.
Concentration and function of mAbs in the systemic and genital tract compartments before and after SC mAb administration.
Changes in the concentration of serum anti-mAbs before and after SC mAb administration.
Sample size calculation
The analysis of the CAPRISA 012A trial will be primarily descriptive and a pragmatic approach was taken when choosing the sample size, ensuring enough participants to obtain safety data.
Currently, there is no safety data available to inform the true event rates that we might observe in the study. However, since the main objective of the study is to evaluate the safety of VRC07-523LS and PGT121 when administered SC, the ability of the study to detect SAEs was assessed for a range of hypothetical event rates. This was done by calculating the probability of detecting no SAE, at least one or two SAEs at a specified true event rate. These probabilities highlight the likelihood of the study to detect either rare or common AEs or SAEs as shown in tables 2 and 3. In addition, the 95% CI for the true event rates were calculated.
Among the four participants receiving active product in each of the seven groups, there is a 34% chance of observing at least one event if the true event rate is 10%. When the true event rate is twofold to threefold higher, this probability rises to 59% and 76%, respectively (table 2).
Since the phase I assessment of SC administration includes eight participants receiving PGT121, 16 participants receiving VRC07-523LS and 28 participants receiving active study product, the probability of observing no events, one or more events, and two or more events for a range of true event rates is provided in table 3. For example, among the eight participants receiving only PGT121 at enrolment there is an 8% chance of observing at least one event, if the true event is 1%, but 83% if the true event is 20%. However, if we combine all 16 women receiving only VRC07-523LS study product at enrolment these probabilities change to 15% and 97% if the true event rates are 1% and 20%, respectively. As expected, an increase in sample size, increases the likelihood of detecting rare events.
Study procedures
Informed consent
In accordance with South African Good Clinical Practice guidelines, informed consent is obtained from each study participant in English or isiZulu (the local African language) prior to screening and enrolment. Consent for pharmacogenetic studies as well as specimen storage is also obtained. Participants are provided with copies of their forms if they wish to receive them. For illiterate participants, an impartial witness is required for the entire informed consent process.
Screening and enrolment
Eligibility for the study is assessed in a stepwise manner at screening and enrolment. Potential participants will be invited to screen for the study and asked to provide informed consent for screening. Potential participants are checked for co-enrolment on the Biometric Co-Enrolment Prevention System and are asked to provide proof of contraception, that is, family planning card, as well as identity documents. Thereafter, they receive pretest counselling and two rapid HIV tests are performed. Post-test counselling is provided and if required, referral to one of several HIV/AIDS care programmes is facilitated. If both HIV test results are negative, the potential participant is asked to provide sociodemographic and behavioural information and undergoes a blood draw for screening bloods, as well as urine pregnancy testing. If potential participants remain eligible based on their screening blood results, they will undergo a complete physical examination. Screening blood tests include haematology, blood chemistry tests, liver function tests, serology, hepatitis B virus assays and serum and plasma storage. A genital specimen using the SoftCup collection device (SoftCup, EuroFemPro, Netherlands, or the SoftCup, Instead, San Diego, CA, USA) will also be obtained.19
Randomisation
Participants will be randomised according to the randomization schedule prepared by the blinded study statistician prior to study start. In this study, the pharmacist will remain unblinded. Sequentially numbered, sealed, opaque envelopes containing the intervention allocation, participant identification number and a treatment code (for use by the unblinded pharmacist only) will be provided to the study coordinator, to be opened once a participant has been deemed eligible and is ready to be enrolled into the study. Group 1 (n=5) will be randomised first. After administration of the first dose to the first participant, the study team will wait for 3 days before administering study product to the second participant in Group 1. After administration of the first dose to the second participant, the study team will wait a further 3 days before enrolling the remaining participants into Group 1. Thereafter, the next 25 participants will be randomly allocated to Groups 2–6. Enrolment of the final five participants into Group 7 will only occur once 12-week safety data has been reviewed for the first eight participants who received product.
Laboratory investigations
Serum, plasma and genital specimens will be taken at enrolment and designated follow-up visits and will be stored for PK analysis, detection of autoreactivity and assessments of markers of safety. The CAPRISA Research Laboratories will conduct specific laboratory testing required for this protocol. Sample processing and storage of specimens for possible future testing (blood and vaginal specimen) will also be undertaken. An accredited contract laboratory will perform all safety blood testing and provide a backup laboratory service when required.
Follow-up visits
At each scheduled study visit, enrolled participants in the intervention and control arm will also be provided with HIV risk reduction counselling, contraception counselling and contraceptive methods of choice. Participants will receive advice on how to contact study staff with additional questions about the study, how to request additional counselling, and how to report possible AEs. In addition to the regular follow-up requirements, genital specimen collection and scheduled safety blood draws as well as samples for PK analysis and storage will be completed.
Safety monitoring and AE reporting
Reactogenicity assessments
A baseline local and systemic reactogenicity assessment will be conducted prior to study product administration. Once the study product has been administered, participants will be directly observed in the clinic for a minimum of 1 hour, after which an early reactogenicity assessment will be performed. The participant will be seen at the clinic on the day of product administration (enrolment visit) as well as day 1 and day 3. Participants will be contacted telephonically on the evening of the enrolment visit and on Day two after product administration. In addition, all participants will keep a daily diary of local and systemic symptoms called a postinjection symptom log. This will aid to track and reconcile any local and systemic reactogenicity events that the participant experiences.
AEs and reporting requirements
Reporting of all AEs will occur during the period from the first study product administration through to the end of the study. After this period, only SAEs and AEs of special interest (potential immune-mediated diseases that include both autoimmune diseases and also other inflammatory and/or neurological disorders that may or may not have an autoimmune aetiology), will be recorded. Each AE will be graded for severity using The Division of AIDS Table for Grading the Severity of Adult and Paediatric Adverse Events, V.2.1, July 2017. Attribution categories used will be either ‘related’ (a reasonable possibility that the AE may be related to the study product) or ‘not related’ (not a reasonable possibility that the AE is related to the study product). All AEs will be captured in the database regardless of attribution. All participants reporting an AE will be followed clinically, until the AE resolves (returns to baseline or becomes non-gradable). Participants with unresolved AEs at study exit will be followed for up to 30 additional days and will be referred to a healthcare provider for further management, if required.
Data and safety monitoring
The trial will be audited by an external monitor prior to study start and during study conduct. Safety review decisions and the status of enrolment throughout the trial will be discussed as part of regular safety review meetings by a Protocol Safety Review Team (PSRT). In addition, a Data and Safety Monitoring Board (DSMB) consisting of three clinical trial specialists, a biostatistician and an ethicist, who are independent from the current study will review safety data during the trial. The study statisticians will prepare routine study progress reports which include reports of AEs experienced by study participants for review by the DSMB members. The members will meet in person and/or via teleconference during the study to conduct interim reviews of study progress, including rates of participant accrual, retention, completion of primary and secondary endpoint assessments, and clinical and laboratory AEs. Any deaths of study participants or other SAEs will be reviewed immediately by the PSRT prior to DSMB review. Following review of the data during the trial, the DSMB may recommend that the study proceed as designed, proceed with design modifications or be discontinued. Enrolment and administration of study product will be stopped, and a safety review conducted by the DSMB for any of the following criteria:
One or more participants experience an SAE that is related to the study product.
There is a participant death, regardless of relationship to the study product.
Two or more participants experience grade 3 AEs in the same category System Organ Class that are considered to be related to the study product.
Any grade 4 AE that is considered to be related to the study product (does not include subjective reactogenicity symptoms).
Statistical analyses
Baseline characteristics including demographics and laboratory measurements will be summarised using descriptive statistics by group and overall. Summaries of the number and percentage of participants experiencing any AE or reactogenicity will be analysed and presented along with 95% CIs. AEs and SAEs will be coded into Medical Dictionary for Regulatory Activities preferred terms. The number and percentages of participants experiencing each specific AE will be tabulated by severity and relationship to study product. For the calculations in these tables, each participant’s AE will be counted once under the maximum severity or strongest recorded causal relationship to study product. A complete listing of AEs for each participant will provide details including severity, relationship to study product, onset, duration and outcome.
Tolerability evaluation will be mostly descriptive and consist of solicited AEs that occur within 1 hour following study product administration and reasons for any withdrawal or discontinuation based on subject discomfort. This early assessment of tolerability of the mAbs will inform which parameters should be solicited or routinely assessed to further characterise the tolerability profile in a larger number of subjects. Analysis will be carried out using either SAS V.9.4 or higher or R.
Pharmacokinetic analyses
PK disposition of PGT121 and VRC07-523LS administered SC alone and in combination will be evaluated in this study. The PK study design was based on preliminary mAb PK data.16 Preliminary PK models have been constructed for VRC07-523LS and PGT121 and Monte Carlo simulation was performed to help design the sampling strategy and to predict overall distribution of mAb concentrations expected. The PK sampling design consists of three postdose samples collected during the first week at days 1, 3 and 7 in order to capture Cmax and Tmax following SC administration. A PK sample will also be collected at day 14. In the repeat dosing groups, PK samples are collected 7 days after the repeat dose; if the levels are different after the repeat dose, it will be important to know if it is due to changes in SC absorption or elimination.
Sample collections will continue for 24–40 weeks depending on study group allocation to capture the concentration versus time profile. Predicted concentrations following single dose VRC07-523LS for Groups 1 and 2 are shown in figure 1. VRC07-523LS concentrations are expected to be maintained >1 mcg/mL for more than 24 weeks following 5 or 10 mg/kg. With repeat administration, predicted VRC07-523LS concentrations following 5 mg/kg at 0 and 12 weeks (Group 3) are around 10 mcg/mL; even with an extended interval to 24 weeks they are predicted to be maintained >1 for more than 1 year with two doses (figure 2). Simulated PGT121 concentrations following single dose (Group 5) are expected to be lower than VRC07-523LS due to the smaller dose and shorter half-life. However, with repeat administration at 12 weeks (Group 6), PGT121 concentrations are expected to be maintained ≥1 mcg/mL as shown in figure 3.
Figure 1.
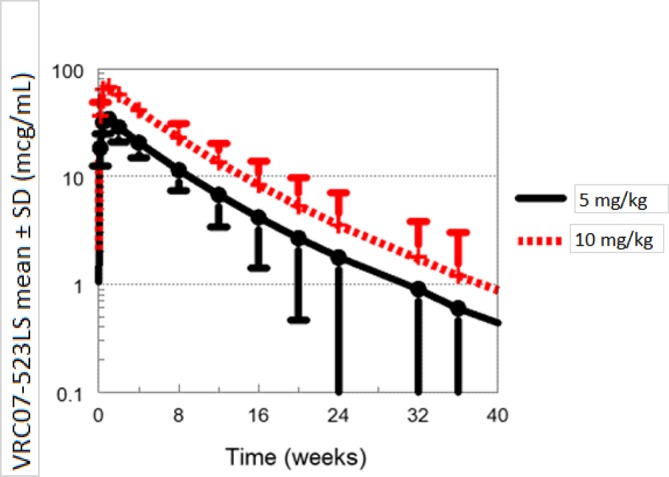
Predicted VRC07-523LS concentrations following single dose administration.
Figure 2.
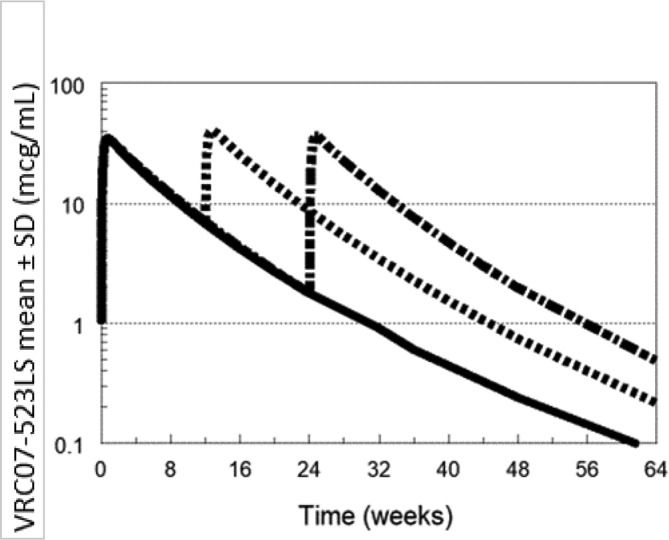
Predicted VRC07-523SL concentrations following repeat dose administration.
Figure 3.
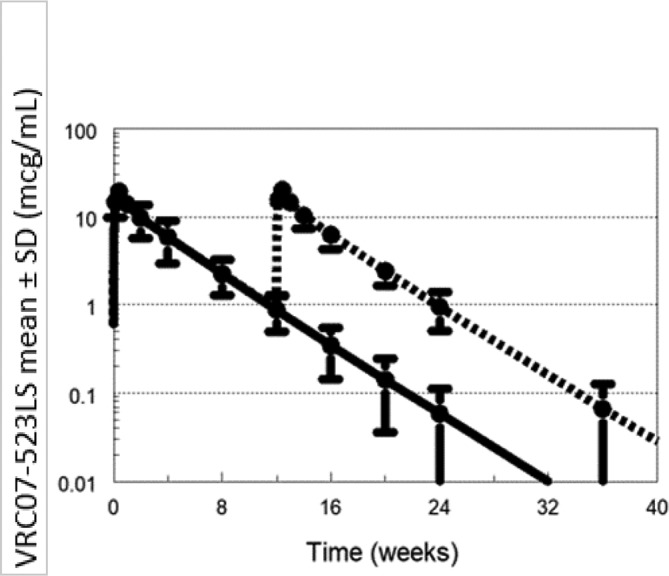
Simulated PGT121 concentrations following single dose administration and repeat dose administration.
PK analysis will be calculated using standard non-compartmental methods using the programme PKPlus. Additional compartmental population PK analysis will be performed using the computer program NONMEM. Cmax and Tmax will be derived directly from the observed data. AUC will be calculated using the trapezoidal method up to the last measured concentration (AUC0-Clast) and T1/2 from regression of the log-linear, terminal portion of the concentration versus time profile. If the final PK sample (Clast) has measurable mAb concentrations, the AUC after the final PK sample will be estimated as Clast/lz, where lz is the terminal slope of log-linear concentration versus time profile. The partial AUC over the first 12 and 24 weeks following the dose, AUC0-12WK and AUC0-24WK, will also be calculated for assessment of accumulation in the repeat dose arms.
A two-compartment model will be used for the compartmental analysis. Either first order or zero order with or without lag time will be used for absorption input. Apparent clearance (CL/F) and apparent volume of distribution (Vdss/F) will be estimated for each mAb across the study arms. Due to the small sample size for the study, a limited covariate assessment will be performed looking at potential effects of dose level, repeat dosing and combination dosing. Overall results will be reported by subgroup and overall after the first dose by mAb. Correlation between PK parameters and reported safety and pharmacodynamics outcomes will also be explored in order to examine exposure–effect relationships. The frequency and levels of anti-mAb antibodies will also be calculated and tabulated.
Data management
Data will be captured by study staff using standardised case report forms (CRFs). All source documents will be kept in the participants’ study files and medical charts at the clinical research site. CRFs will be faxed to the central CAPRISA Data Management Server using the iDataFax system (DF/Net Research, Seattle, USA). All data entry will undergo three stages of quality control including immediate source document review, internal quality audits and weekly quality reports generated by iDataFax. Queries arising during validation of the data will be recorded in quality control reports sent to the sites on a regular basis. The original CRFs and study related documents will be securely stored at the site, during the study and after study completion.
Ethics and dissemination
Ethical approval has been granted by the South African Health Products Regulatory Authority (SAHPRA) (Trial reference number: 20180522) and University of KwaZulu-Natal Biomedical Research Ethics Committee (Reference number: BFC108/18) for the study protocol (V.1.1, Dated 1 June 2018). Any future protocol modifications will be communicated to the relevant regulatory authorities. Eligible patients will be asked to provide written informed consent for study procedures and sample storage. All participants will receive a small financial compensation for their time, transport and inconvenience after each study visit in accordance with South African National Health Research Ethics Council Guidelines. Any breaches in confidentiality, study protocol or AEs attributable to this study will be reported to the above institutional review boards.
The study team will disseminate the trial results as broadly as possible. The research team will attend conferences periodically and present trial results to a multidisciplinary scientific community. The results from this research may also be disseminated through presentations at scientific institutions/meetings, and/or publication in scientific journals. All publications will be uploaded to the University of KwaZulu-Natal publication repository. After sharing the results with study participants, they will be presented to communities from which participants are drawn, following Good Participatory Practice guidelines. The results will also be shared with global and local policy makers. Summary results of the trial will be made publicly available through the clinical trial registry. Any datasets used for analysis in publications can be requested by investigators via an online request to the organisation. Measures will be taken to protect identifiable information in the datasets.
Trial status
The trial was registered on Pan African Clinical Trials Registry (PACTR 201808919297244) on www.pactr.org on 29 August 2018. Enrolment started in November 2018 and is predicted to be completed by June 2019.
Supplementary Material
Footnotes
Contributors: SAK conceived and designed the trial. SM and NG wrote the study protocol. EC will conduct pharmacokinetic (PK) simulations and analysis. NYZ performed sample size calculations and the statistical analysis strategy. CB, TG, DA, PM, NS, DHB, JM, JL and LM contributed to the planning and conduct of the trial. All authors contributed to the write up of the manuscript. All authors consented to final publication.
Funding: This study is being funded by the European and Developing Countries Clinical Trials Partnership (EDCTP Grant number: RIA2017S) and the South African Medical Research Council (SAMRC), Special Initiative on HIV Prevention Technology.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient consent for publication: Not required.
References
- 1. UNAIDS. UNAIDS data 2017. Geneva, Switzerland: Joint United Nations Programme on HIV/AIDS (UNAIDS), 2017. http://www.unaids.org/en/resources/documents/2017/2017_data_book (Accessed 28 Feb 2019). [Google Scholar]
- 2. de Oliveira T, Kharsany AB, Gräf T, et al. . Transmission networks and risk of HIV infection in KwaZulu-Natal, South Africa: a community-wide phylogenetic study. Lancet HIV 2017;4:e41–e50. 10.1016/S2352-3018(16)30186-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cohen MS, Chen YQ, McCauley M, et al. . Prevention of HIV-1 infection with early antiretroviral therapy. N Engl J Med 2011;365:493–505. 10.1056/NEJMoa1105243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baeten JM, Donnell D, Ndase P, et al. . Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N Engl J Med 2012;367:399–410. 10.1056/NEJMoa1108524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Van Damme L, Corneli A, Ahmed K, et al. . Preexposure Prophylaxis for HIV Infection among African Women. N Engl J Med Overseas Ed 2012;367:411–22. 10.1056/NEJMoa1202614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marrazzo JM, Ramjee G, Richardson BA, et al. . Tenofovir-based preexposure prophylaxis for HIV infection among African women. N Engl J Med 2015;372:509–18. 10.1056/NEJMoa1402269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Abdool Karim Q, Abdool Karim SS, Frohlich JA, et al. . Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science 2010;329:1168–74. 10.1126/science.1193748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fonner VA, Dalglish SL, Kennedy CE, et al. . Effectiveness and safety of oral HIV pre-exposure prophylaxis (PrEP) for all populations: A systematic review and meta-analysis. AIDS 2016;30:1973–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rudicell RS, Kwon YD, Ko SY, et al. . Enhanced potency of a broadly neutralizing HIV-1 antibody in vitro improves protection against lentiviral infection in vivo. J Virol 2014;88:12669–82. 10.1128/JVI.02213-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Walker LM, Huber M, Doores KJ, et al. . Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 2011;477:466–70. 10.1038/nature10373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gaudinski MR, Coates EE, Houser KV, et al. . Safety and pharmacokinetics of the Fc-modified HIV-1 human monoclonal antibody VRC01LS: A Phase 1 open-label clinical trial in healthy adults. PLoS Med 2018;15:e1002493 10.1371/journal.pmed.1002493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stephenson KE, Barouch DH. Broadly neutralizing antibodies for HIV eradication. Curr HIV/AIDS Rep 2016;13:31–7. 10.1007/s11904-016-0299-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Julien JP, Sok D, Khayat R, et al. . Broadly neutralizing antibody PGT121 allosterically modulates CD4 binding via recognition of the HIV-1 gp120 V3 base and multiple surrounding glycans. PLoS Pathog 2013;9:e1003342 10.1371/journal.ppat.1003342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wagh K, Bhattacharya T, Williamson C, et al. . Optimal Combinations of Broadly Neutralizing Antibodies for Prevention and Treatment of HIV-1 Clade C Infection. PLoS Pathog 2016;12:e1005520 10.1371/journal.ppat.1005520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morris L, Mkhize NN. Prospects for passive immunity to prevent HIV infection. PLoS Med 2017;14:e1002436 10.1371/journal.pmed.1002436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gaudinski MR, Houser KV, Chen G, et al. . A Phase I dose-escalation study of monoclonal antibody VRC07-523LS in healthy adults [Abstract: 1061]. Conference on Retroviruses and Opportunistic Infections; 2019 March 4-7, Boston, MA [Google Scholar]
- 17. Julg B, Barouch DH. Neutralizing antibodies for HIV-1 prevention. Curr Opin HIV AIDS 2019;14:318–24. 10.1097/COH.0000000000000556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sok D, Burton DR. Recent progress in broadly neutralizing antibodies to HIV (vol 19, pg 1179, 2018). Nature Immunology 2019;20:374–74. [DOI] [PubMed] [Google Scholar]
- 19. Archary D, Liebenberg LJ, Werner L, et al. . Randomized Cross-Sectional Study to Compare HIV-1 Specific Antibody and Cytokine Concentrations in Female Genital Secretions Obtained by Menstrual Cup and Cervicovaginal Lavage. PLoS One 2015;10:e0131906 10.1371/journal.pone.0131906 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.