

Supplemental Digital Content is available in the text.
Keywords: alternative splicing, mRNA decapping, myocardial contraction, myocardial infarction, phosphorylation
Abstract
Rationale:
RBPs (RNA binding proteins) play critical roles in the cell by regulating mRNA transport, splicing, editing, and stability. The RBP SRSF3 (serine/arginine-rich splicing factor 3) is essential for blastocyst formation and for proper liver development and function. However, its role in the heart has not been explored.
Objective:
To investigate the role of SRSF3 in cardiac function.
Methods and Results:
Cardiac SRSF3 expression was high at mid gestation and decreased during late embryonic development. Mice lacking SRSF3 in the embryonic heart showed impaired cardiomyocyte proliferation and died in utero. In the adult heart, SRSF3 expression was reduced after myocardial infarction, suggesting a possible role in cardiac homeostasis. To determine the role of this RBP in the adult heart, we used an inducible, cardiomyocyte-specific SRSF3 knockout mouse model. After SRSF3 depletion in cardiomyocytes, mice developed severe systolic dysfunction that resulted in death within 8 days. RNA-Seq analysis revealed downregulation of mRNAs encoding sarcomeric and calcium handling proteins. Cardiomyocyte-specific SRSF3 knockout mice also showed evidence of alternative splicing of mTOR (mammalian target of rapamycin) mRNA, generating a shorter protein isoform lacking catalytic activity. This was associated with decreased phosphorylation of 4E-BP1 (eIF4E-binding protein 1), a protein that binds to eIF4E (eukaryotic translation initiation factor 4E) and prevents mRNA decapping. Consequently, we found increased decapping of mRNAs encoding proteins involved in cardiac contraction. Decapping was partially reversed by mTOR activation.
Conclusions:
We show that cardiomyocyte-specific loss of SRSF3 expression results in decapping of critical mRNAs involved in cardiac contraction. The molecular mechanism underlying this effect likely involves the generation of a short mTOR isoform by alternative splicing, resulting in reduced 4E-BP1 phosphorylation. The identification of mRNA decapping as a mechanism of systolic heart failure may open the way to the development of urgently needed therapeutic tools.
Cardiovascular disease is the main cause of mortality worldwide, causing. 17.7 million deaths in 2015, corresponding to 31% of all deaths worldwide. Of these, 6.7 million were caused by myocardial infarction (MI), which is thus one of the main causes of cardiovascular disease deaths.1 Knowledge about the molecular mechanisms that regulate the progression from MI to heart failure is incomplete, precluding the development of new therapeutic approaches. In recent years, the development of massive mRNA sequencing technologies has identified gene expression patterns associated with the development of heart failure.2 However, understanding remains limited about post-transcriptional regulation and, more specifically, the role of RBPs (RNA binding proteins) in MI progression and in the development of heart failure.
In This Issue, see p 147
Meet the First Author, see p 148
RBPs contain RNA-binding domains that enable them to bind RNA molecules and regulate different processes associated with RNA metabolism, including constitutive and alternative splicing, nucleo-cytoplasmic transport, intracellular localization, translation, and degradation.3 Loss of function of specific RBPs is associated with clinical manifestations, including neuropathies, muscular atrophies, and cancer.3 In addition, recent studies show that some RBPs are crucial for proper cardiac function; loss of RBM20 (RNA-binding motif protein 20), TRBP (TAR RNA-binding protein), or RBFox2 (RNA-binding Fox-1 homolog 2) triggers the development and progression of dilated cardiomyopathy,4–7 whereas RBFox1 (RNA-binding Fox-1 homolog 1) deficiency leads to hypertrophic cardiomyopathy.8
SRSF (serine/arginine-rich splicing factors, also known as SR proteins) are a family of highly conserved RBPs containing one or 2 RNA recognition motifs in their N-terminal region, followed by an SR domain formed of at least 50 amino acids with a serine and arginine content above 40%.9 SR proteins were originally identified as splicing factors, but several studies have demonstrated that they are multifunctional proteins that take part in many mRNA regulation pathways.10 SR proteins are localized both in the nucleus and the cytoplasm and regulate both the transport and stability of mRNA.11 Deficiency of particular SR proteins has been linked to several disease states, including heart disease. SRSF1, SRSF2, and SRSF10 are essential for heart development and function. Loss of SRSF1 in the heart alters CaMKIIδ (calcium/calmodulin kinase IIdelta) splicing, which results in reduced phosphorylation of its target phospholamban, leading to calcium handling defects, hypercontraction, and death of the mice between 6 and 8 weeks of life.12 Cardiac-specific depletion of SRSF2 leads to excitation-contraction defects and nonlethal dilated cardiomyopathy, associated with a decrease in RyR2 (ryanodine receptor 2) expression.13 Ubiquitous deletion of SRSF10 induces cardiac developmental defects that result in embryonic death at E15.5.14 These defects are caused by alternative splicing of triadin, which results in reduced expression of both triadin and calsequestrin 2 and lead to calcium handling defects in cardiomyocytes.14
Like other members of the SR family, SRSF3 regulates splicing of several mRNAs,15–17 mRNA cytoplasmic transport,11 and mRNA localization.18 SRSF3 is essential for proper embryonic development. SRSF3-deficient embryos are unable to form blastocysts and die at the morula stage.19 Specific depletion of SRSF3 in the liver is not lethal, but mice present both embryonic and postnatal growth defects.20 As these mice get older, they develop hepatic carcinomas, mirroring the human disease, in which SRSF3 is also downregulated, in contrast to other cancer types.21 SRSF3 has also been reported to act as an oncogene in the liver by promoting cell proliferation.22 These studies establish the importance of SRSF3 for proper organ development and function. However, unlike other RBPs, the function of SRSF3 in the heart has not been reported previously.
In this study, we show that SRSF3 is essential for heart development in mice and that SRSF3 expression in the adult heart is decreased in the remote myocardium after MI. Induced loss of SRSF3 in adult cardiomyocytes results in widespread changes in alternative splicing and decapping of mRNAs encoding contraction proteins and its subsequent degradation, leading to severe systolic dysfunction and death. We also show that SRSF3 regulates mTOR (mammalian target of rapamycin) splicing, reducing the phosphorylation of 4E-BP1 (eIF4E-binding protein 1) required to prevent mRNA decapping.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
RNA-Seq data are available from GEO (GSE123002; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE123002). The entire iCOUNT script for the (individual-nucleotide resolution cross-linking and immunoprecipitation) iCLIP analysis is available on github: https://github.com/tomazc/iCount. Materials used in this work will be made available on request to the corresponding author.
Mice
The SRSF3 knockout mice were originally generated by Dr Peter J. Nielsen.19 The neomycin cassette was removed by breeding SRSF3-floxed mice with transgenic mice expressing CreERT2 under the polymerase II promoter (PolII-mER-Cre-mER). Treating these mice with low doses of tamoxifen generated an SRSF3-floxed mouse line that contained LoxP sites flanking SRSF3 exons 2 and 3. We performed 3 backcrosses with C57BL/6 to remove the Cre cassette and then crossed the resulting SRSF3-floxed mice with the cardiac-specific Cre lines, Nkx2.5-Cre and αMHC-Cre. Both crosses resulted in embryonic lethality, and lines were, therefore, maintained in heterozygosity for the SRSF3-floxed allele. In addition, we generated the cardiomyocyte-specific and inducible SRSF3 knockout mice by crossing SRSF3-floxed mice with αMHC-mER-Cre-mER mice. The resulting line was maintained in homozygosity. We used αMHC-mER-Cre-mER mice as negative controls. Genotyping was performed using the primers indicated in Online Table I.
Due to the effects of estrogens on the heart, only adult (2–3-month old) male mice were used for all experiments, to avoid potential effects of sex differences. Mice were euthanized by carbon dioxide asphyxiation. All procedures were approved by the ethics committees of the CNIC (Centro Nacional de Investigaciones Cardiovasculares) and the Regional Government of Madrid (PROEX 332-15). Animals were randomly assigned to groups. Researchers were blinded to the allocations.
Tamoxifen and Amino Acid Treatments
To activate the Cre recombinase in inducible knockout mice, SRSF3 knockout and control mice were treated with 3 doses of 1 mg 4-hydroxytamoxifen on alternate days (1 dose/d). To prepare tamoxifen doses, 50 mg of 4-hydroxytamoxifen (H6278, Sigma) was diluted in 500 µL 100% ethanol and stirred at 55°C for 1 hour, until completely dissolved. Preheated corn oil (4.5 mL) was then added, and the incubation continued with stirring at 55°C for a further hour. Once the tamoxifen and corn oil were fully dissolved, the solution was maintained at 4°C until use. Just before injection, the solution was warmed and maintained at 37°C for 10 minutes. A total of 100 µL were injected intraperitoneal per mouse per day. To activate mTOR, mice were injected intraperitoneal daily, for 7 days, starting on the day of the second tamoxifen injection, with 100 µL/g of a solution containing 6.0 g/L isoleucine, 12.0 g/L leucine, 7.2 g/L valine, and 6.04 g/L arginine in PBS (pH 7.4).23
Echocardiography
Transthoracic echocardiography was performed by blinded observers using a 40 MHz lineal transducer (Vevo 2100, VisualSonics). Mice were placed on a heating pad at 38.3°C and lightly anesthetized with isoflurane in 100% oxygen. The Isoflurane concentration was adjusted to maintain heart rate at 450 to 550 bpm.
Left ventricular (LV) systolic function was evaluated using the area/length method, with images acquired and recorded from 2-dimensional parasternal long axis views.24 Image analysis was performed by blinded observers using Vevo 2100 analysis software (VisualSonics).
Embryo Extraction, In Situ Hybridization, and Histology
Embryos were extracted from C57BL/6, αMHC-Cre, or SRSF3-floxed/αMHC-Cre mice at the stages indicated in the figures. In situ hybridization was performed on whole mount embryos (E9.5–E11.5) or 10 μm sections (E12.5–E14.5).25 The SRSF3 probe was designed to match the total length of the mRNA. Hematoxylin and eosin staining were performed on 5 μm sections according to standard protocols by the CNIC Histology Unit.
Gene Expression, Alternative Splicing, and Gene Ontology Analyses
Total RNA was extracted from left ventricles after 5 days of tamoxifen treatment using the RNeasy extraction kit (74104, Qiagen), including treatment with DNase. RNA sequencing was performed by the CNIC Genomics Unit. Libraries were prepared using polyA+ selection, and sequencing was performed using an Illumina-HiSeq 2500 platform, ensuring an average of 95 million paired-end reads per sample, with a minimum of 74 mol/L per sample. RNA-Seq analysis was performed by the CNIC Bioinformatics Unit. Fastq files containing the reads for each library were extracted and demultiplexed using the Casava v1.8.2 pipeline. Reads were preprocessed with a pipeline that used cutadapt v1.6 to remove Illumina adaptors, and FastQC to perform quality controls after each step. Gene expression was quantified using Kallisto v0.43.0,26 and the reference transcriptome was downloaded from Ensembl (Mus_musculus.GRCm38.cdna.all.fa). One hundred bootstraps were performed to account for quantification uncertainty. Sleuth27 with default parameters was then used to normalize the data and perform differential expression analysis at the gene level. Genes with an adjusted P value <0.05 and an absolute β >1 were considered to be differentially expressed for downstream enrichment analysis. Alternative splicing was analyzed using vast-tools to map reads against the mouse reference database and to perform differential splicing analysis.28,29 Gene ontology categories and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways were downloaded from the Enrichr website,28 and enrichment analysis of genes undergoing both differential expression or splicing was performed with one-sided Fisher tests and Benjamini-Hochberg False Discovery Rate correction using custom python scripts. RNA-Seq data were deposited in Gene Expression Omnibus (GSE123002).
Decapping Analysis
Decapped mRNA was quantified as previously described.30 Briefly, 4 μg of total RNA was treated with DNase and ligated with the rP5_RND primer (Online Table II), which binds specifically to 5′ ends lacking the cap structure and presenting a phosphate group. Afterwards, 100 ng were used for cDNA synthesis, and polymerase chain reaction (qRT-PCR) was performed using the primers indicated in Online Table II. Values were normalized to total mRNA expression.
Statistical Analysis
Data were analyzed for statistical significance using the unpaired Student t test for experiments with 2 conditions and 1 variable, 1-way ANOVA followed by the Bonferroni post-test for more than 2 conditions and 1 variable, and 2-way ANOVA followed by the Bonferroni post-test for analysis of multiple conditions and 2 variables, for normally distributed data, as indicated in each figure legend. The Mann-Whitney test was used for non-normally distributed data. Data were analyzed using GraphPad Prism 5.0, and changes were considered significant at P<0.05. Correction for multiple comparisons was used for all analyses with more than one variable.
Results
SRSF3 Expression in the Heart Decreases With Age
In situ hybridization in embryos from E9.5 to E14.5 revealed strong SRSF3 staining in the heart from E12.5 (Figure 1A). For a more detailed analysis of changes in SRSF3 expression during heart development and in adulthood, we isolated hearts from mice ranging from E12.5 to 6 months of age and quantified SRSF3 mRNA levels by qRT-PCR. SRSF3 expression was highest at E12.5 and decreased progressively during later embryonic stages and the postnatal period (Figure 1B). To identify the cardiac cell types expressing SRSF3 in the postnatal heart, we performed an immunofluorescence analysis of heart sections from P1, P7, and 1-month-old mice. Clear staining for SRSF3 was detected in cardiomyocyte nuclei, as well as in other cell types (Figure 1C).
Figure 1.
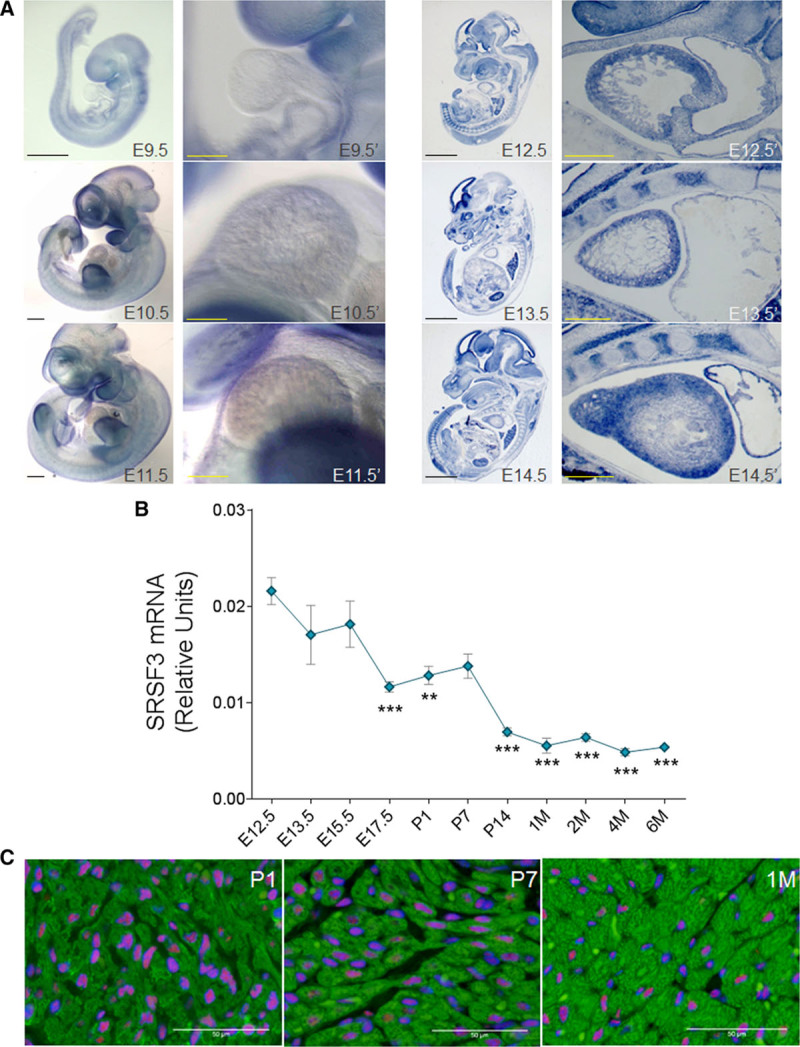
SRSF3 (serine/arginine splicing factor 3) expression in the heart decreases with age. A, In situ hybridization for SRSF3 performed at different embryonic stages, either in whole mount (Left set: E9.5, E10.5, and E11.5) or 10 μm sections (Right set: E12.5, E13.5, and E14.5). The region including the developing heart is shown at high magnification to the right of each whole-embryo view. B, qRT-PCR (polymerase chain reaction) analysis of SRSF3 mRNA expression (exons 3–5) normalized to Gapdh expression in hearts extracted from C57BL/6 mice at different time points. Data are shown as mean±SEM; n=3–4 mice per group. **P<0.01, ***P<0.001 vs E12.5, 1-way ANOVA followed by the Bonferroni multiple comparison test. C, Immunofluorescence analysis of hearts isolated from C57BL/6 mice at different ages using anti-SRSF3 (red), anti-TroponinT (green), and DAPI (blue). M, month; Bars, A, 1 mm (whole mount, full embryo); 0.5 mm (whole mount, heart); 2 mm (embryo section), 0.5 mm (heart section); C, 50 μm.
SRSF3 Downregulation Following MI Is Associated With Inclusion of Exon 4
To determine whether SRSF3 could be altered in heart disease, we measured its expression after MI in the region remote from the infarct (Figure 2A–2E). Both mRNA and protein SRSF3 expression was downregulated after MI (Figure 2B and 2C), suggesting a possible role in cardiac homeostasis. Published evidence shows that SRSF3 is regulated by the alternative splicing of exon 4, inclusion of which leads to degradation of the mRNA through nonsense-mediated decay (NMD; Figure 2A).31 qRT-PCR analysis revealed increased SRSF3 exon 4 inclusion in infarcted hearts, suggesting a role of this mechanism in the regulation of SRSF3 after MI (Figure 2D).
Figure 2.
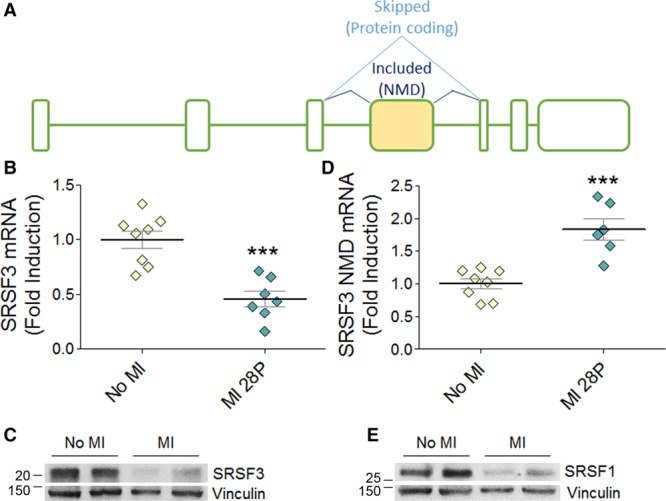
SRSF3 (serine/arginine splicing factor 3) is downregulated after myocardial infarction (MI) by alternative splicing. A, Schematic of the SRSF3 mRNA isoforms. The yellow box represents alternative exon 4. B and C, Analysis by qRT-PCR (polymerase chain reaction; B) and Western blot (C) of SRSF3 mRNA (exons 3–5 normalized to Gapdh) and protein expression in the remote myocardium of mouse hearts 28 days after induction of MI by left coronary artery ligation. MW markers are indicated. D, qRT-PCR analysis of the SRSF3 NMD (nonsense-mediated decay) isoform in the remote myocardium, normalized to total SRSF3 mRNA expression. E, Western blot analysis of SRSF1 protein expression in the remote myocardium. Data in B and D are mean±SEM, and symbols represent individual animals (n=7–8); *P<0.05 and ***P<0.001, Student t test.
SRSF1 has been shown to regulate SRSF3 expression by promoting exon 4 skipping.31 The post-MI increase in exon 4 inclusion thus prompted us to investigate whether SRSF1 expression was also affected. Western blot analysis showed decreased SRSF1 expression after MI (Figure 2E). These results suggest that the decrease in SRSF1 expression may favor the inclusion of SRSF3 exon 4, leading to SRSF3 downregulation after MI.
SRSF3 Cardiac Depletion During Embryonic Development Is Lethal
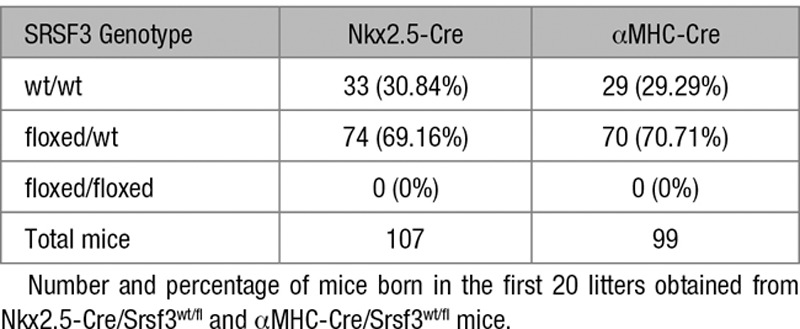
To investigate the role of SRSF3 in the heart, we developed several cardiac-specific knockout mouse models. We first crossed SRSF3-floxed mice with transgenic mice expressing Cre recombinase under the control of the Nkx2.5 promoter. No transgenic animals bearing both the Nkx2.5 promoter and the floxed SRSF3 allele were born (Table), suggesting that early SRSF3 depletion during heart development is embryonically lethal. To limit the recombination to cardiomyocytes and later developmental stages, we crossed SRSF3-floxed mice with mice expressing Cre recombinase under the αMHC promoter, which also was embryonically lethal (Table). Hematoxylin and eosin staining showed a reduction in myocardium thickness in embryonic hearts lacking SRSF3 expression (Figure 3A and 3B). Previous reports that show that SRSF3 promotes proliferation22 prompted us to investigate if cardiomyocyte proliferation was affected in these animals; we were unable to detect any cardiomyocytes with positive staining for the mitosis marker phospho-histone H3 in myocardium lacking SRSF3 expression (Figure 3C and 3D). These results suggest that SRSF3 expression in cardiomyocytes is essential for cell proliferation, proper heart development, and subsequent embryo survival.
Figure 3.
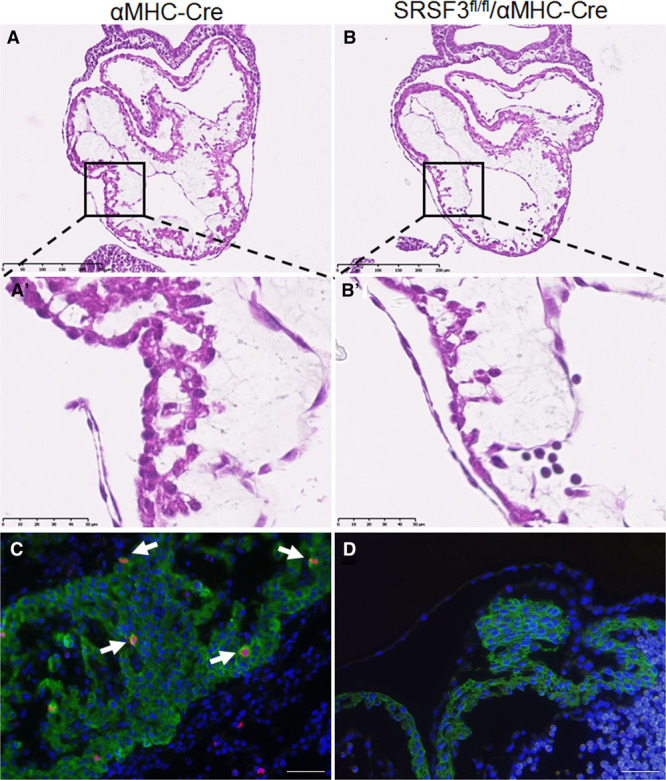
Loss of SRSF3 (serine/arginine splicing factor 3) in the developing heart results in embryonically lethal heart defects. A and B, Hematoxylin and eosin (H&E) staining in sections of αMHC-Cre embryos (A) and SRSF3-floxed/αMHC-Cre embryos (B) at E9.5. The boxed areas in A and B are shown at high magnification in A′ and B′, illustrating the cell deficit in the developing ventricular wall of SRSF3-floxed/αMHC-Cre embryos. Bar, 250 µm (A and B), 50 μm (A′ and B′). C and D, Immunofluorescence analysis in sections of αMHC-Cre embryos (C) and SRSF3-floxed/αMHC-Cre embryos (D) at E10.5, using anti-phospho-Histone H3 (red), anti-TroponinT (green), and Dapi (blue). Arrows indicate cardiomyocytes positive for phospho-histone H3; the developing SRSF3-floxed/αMHC-Cre heart is devoid of these cells. Bar, 20 μm.
Table.
Genotype of the Srsf3 Allele in the Offspring of Nkx2.5-Cre/Srsf3wt/fl and αMHC-Cre/Srsf3wt/fl Mice
Cardiac-Specific SRSF3 Knockout Mice Develop Severe and Fatal Contraction Defects
To investigate the role of SRSF3 in the adult heart, we developed a cardiac-specific tamoxifen-inducible knockout mouse line by crossing SRSF3-floxed and αMHC-mER-Cre-mER mice (SRSF3 knockout). SRSF3 depletion occurred rapidly, with a strong decline already observed on the first day after tamoxifen administration (Online Figure IA and IB). Immunofluorescence analysis confirmed that SRSF3 was depleted only in cardiomyocytes, although recombination was incomplete, and some cardiomyocytes were still positive for SRSF3 expression (Online Figure IC).
Unlike SRSF2 and SRSF1 cardiac-specific knockout mice, which either have a normal lifespan (SRSF2) or survive for several weeks after birth (SRSF1),12,13 no SRSF3 knockout mice survived beyond 8 days after SRSF3 depletion (Figure 4A). Echocardiography analysis of SRSF3 knockout mice 5 days after tamoxifen administration revealed a pronounced reduction in left ventricular ejection fraction and a strong increase in LV systolic volume (Figure 4B and 4C and Online Movie I), suggesting that these mice develop severe contraction defects that lead to death. No significant changes were observed in LV diastolic volume, likely due to the short lifespan after SRSF3 knockout (Figure 4D and Online Table III). Systolic dysfunction was accompanied by an increase in Nppb (natriuretic peptide B) and Acta1 gene expression in SRSF3 knockout hearts (Figure 4E and 4F). Together, these results demonstrate that the loss of SRSF3 in cardiomyocytes leads to severe cardiac dysfunction and premature death.
Figure 4.
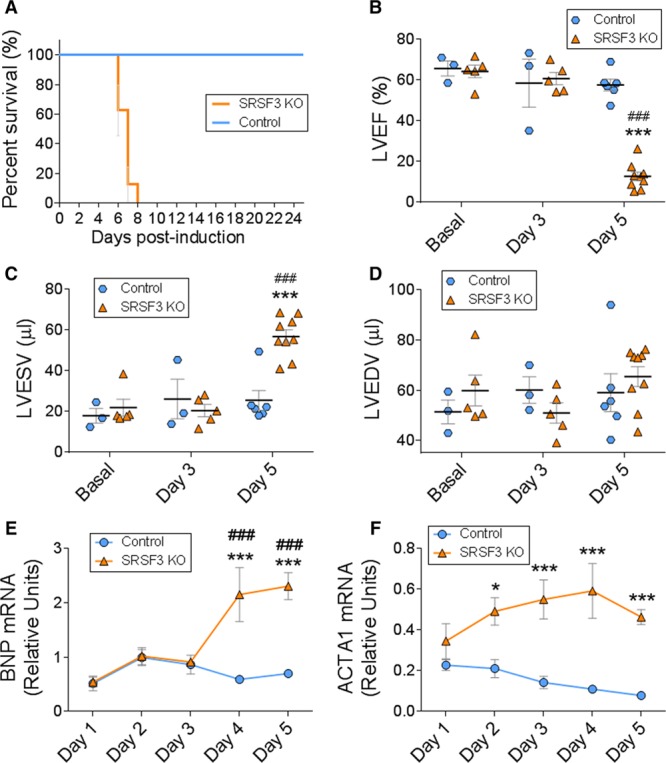
Cardiac-specific SRSF3 (serine/arginine splicing factor 3) knockout (KO) mice develop severe and fatal contraction defects. Deletion of SRSF3 in cardiomyocytes was induced in inducible SRSF3 KO mice by 3 hydroxytamoxifen injections on alternate days (post-induction days 0, 2, and 4). A, Survival curves for hydroxytamoxifen-injected control and SRSF3 KO mice. B–D, Echocardiography analysis of left ventricular ejection fraction (B), left ventricular end-systolic volume (C), and left ventricular end-diastolic volume (D); ***P<0.001 SRSF3 KO vs Control, ###P<0.001 vs Basal; 2-way ANOVA followed by the Bonferroni post-test. E and F, qRT-PCR (polymerase chain reaction) analysis of the cardiac expression of BNP (E) and Acta1 (F) mRNA over 5 days post-induction. Data are shown as mean±SEM. n=5–13 mice per group. Two-way ANOVA followed by Bonferroni post-test. *P<0.05, ***P<0.001 SRSF3 KO vs Control. ###P<0.001 vs day 1. ACTA1 indicates actin alpha 1; BNP, brain natriuretic peptide; LVEDV, left ventricular end diastolic volume; LVEF, left ventricular ejection fraction; and LVESV, left ventricular end systolic volume.
SRSF3 Depletion Results in Downregulation of Contraction-Related Genes
To investigate the mechanism underlying the contraction defects in SRSF3 knockout mice, we performed RNA-Seq in control and knockout hearts 5 days after tamoxifen administration. We found significant expression changes in several genes, both upregulated and downregulated (Online Table IV and Online Figure IIA). Gene ontology analysis showed a decrease in contraction-related genes and an increase in genes related to protein maturation and transport in the ER and to the immune response (Online Figure IIB and IIC). qRT-PCR analysis to confirm these results revealed that the decreased expression of sarcomeric genes was already evident 2 days after SRSF3 depletion (Figure 5A–5F). Protein levels of SERCA2A (sarcoplasmic/endoplasmic reticulum calcium ATPase 2a) and MYH (myosin heavy chain) were unchanged until day 5 after tamoxifen administration (Figure 5G), suggesting that cardiac function is preserved until the expression of contraction proteins decreases. To determine whether the downregulation of sarcomeric gene expression was due to general RNA degradation associated with apoptosis, we first measured the number of apoptotic cardiomyocytes in control and SRSF3 knockout mice 5 days after the last tamoxifen injection. We found a very low number of TUNEL+ (terminal deoxynucleotidyl transferase dUTP nick-end labeling) cardiomyocytes overall and no differences between both mouse lines (Online Figure IIIA). We observed a very mild decline in total RNA integrity 5 days after the last tamoxifen injection in knockout mice, which, in any case, takes place much later than the decrease of sarcomeric mRNA, which is already evident at day 2 (Online Figure IIIB, Figure 5A–5F). We found no differences in the apoptosis markers Raptor and Fas, and only a mild decrease in Bcl2 mRNA expression at late time points in SRSF3 knockout mice that was not accompanied by a decrease in Bclxl (Online Figure IIIC–IIIF). Therefore, the decrease in sarcomeric gene expression precedes early signs of apoptosis, which would start a few days later. Early stages of cell death would be in agreement with the induction of genes associated with the immune response observed in knockout mice 5 days after tamoxifen treatment (Online Figure IIB).
Figure 5.
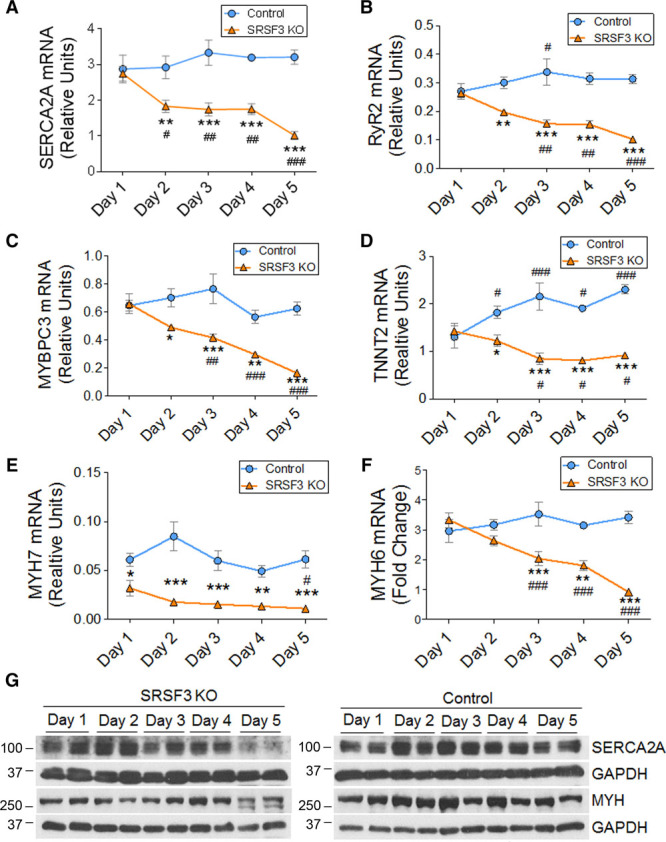
SRSF3 (serine/arginine splicing factor 3) depletion results in downregulation of contraction-related genes. A–F, qRT-PCR (polymerase chain reaction) analysis of the cardiac expression of SERCA2A (A), RyR2 (B), MYBPC3 (C), TNNT2 (D), MYH7 (E), and MYH6 (F) mRNA in control and SRSF3 KO mice over 5 days post tamoxifen induction. Data are shown as mean±SEM;. n=5–13 mice per group. *P<0.05, **P<0.01, and ***P<0.001 for SRSF3 knockout (KO) vs control; #P<0.05, ##P<0.01, ###P<0.001 vs day 1; 2-way ANOVA followed by the Bonferroni post-test. G, Western blot analysis of MYH and SERCA2A protein expression in the heart over 5 days post tamoxifen induction. MYBPC3 indicates myosin-binding protein C3; MYH6, myosin heavy chain 6; MYH7, myosin heavy chain 7; RYR2, ryanodine receptor 2; SERCA2A, sarcoplasmic/endoplasmic reticulum calcium ATPase 2a; and TNNT2, troponin T2.
SERCA2A Overexpression Partially Rescues Cardiac Contraction in SRSF3 Knockout Mice
Reduced expression of SERCA2A in cardiomyocytes is strongly associated with heart failure,32 and SERCA2A overexpression has been shown to improve cardiac function in several animal models.33,34 To determine whether the decreased expression of cardiac contraction proteins, specifically SERCA2A, is responsible for the severe systolic dysfunction in SRSF3 knockout mice, we overexpressed SERCA2A in control and knockout animals using serotype 9 adeno-associated viruses (AAV9-SERCA2A). AAV9-LUC, carrying the luciferase reporter gene, was used as a negative control. Viruses were injected into control and SRSF3 knockout mice 1 week before tamoxifen induction to ensure transgene overexpression in advance of the loss of SERCA2A expression. SERCA2A overexpression was confirmed by western blot 5 days after tamoxifen induction (Figure 6A).
Figure 6.
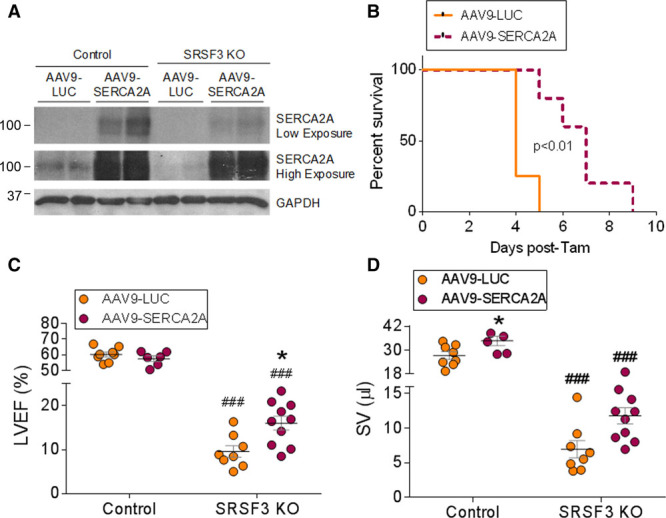
SERCA2A overexpression partially rescues systolic dysfunction in SRSF3 (serine/arginine splicing factor 3) knockout (KO) mice. A, Western blot analysis of SERCA2A protein expression in hearts of tamoxifen-induced control and SRSF3 KO mice administered AAV9 encoding luciferase (LUC) or SERCA2A (see Methods for details). B, Survival curve for SRSF3 KO mice treated either with AAV9-SERCA2A (dashed line; n=7) or AAV9-LUC as a control (continuous line; n=4). The x axis indicates time (days) after the last tamoxifen injection. C and D, Echocardiography analysis of left ventricular ejection fraction (C) and stroke volume (D) in mice treated as in A. *P<0.05 for AAV9-SERCA2A vs AAV9-LUC; ###P<0.001 for SRSF3 KO vs control; 2-way ANOVA followed by the Bonferroni post-test. AAV9 indicates adeno-associated virus 9; LVEF, left ventricular ejection fraction; SERCA2A, sarcoplasmic/endoplasmic reticulum calcium ATPase 2a; and SV, stroke volume.
We found that AAV9-SERCA2A significantly improved survival of SRSF3 knockout mice (Figure 6B). Echocardiography analysis of cardiac function showed a mild but significant improvement in left ventricular ejection fraction in mice overexpressing SERCA2A, and increased stroke volume (Figure 6C and 6D and Online Table V). Although this improvement was far from a full recovery, we conclude that the downregulation of SERCA2A is responsible, at least in part, for the contraction defects observed in SRSF3 knockout mice. Full recovery would likely require overexpression of additional genes involved in cardiac contraction whose expression was reduced in the knockout mice.
SRSF3 Binds to the Untranslated Region of Contraction-Related mRNAs
To gain further insight into the mechanisms underlying the contraction defects in SRSF3 knockout mice, we looked for direct SRSF3 target mRNAs in cardiac tissue. For this, we performed iCLIP on mouse neonatal cardiomyocytes transfected with modified RNA encoding an SRSF3-GFP (green fluorescent protein) chimera, and analyzed the distribution of iCLIP binding sites along different regions of the genes detected to be expressed in the previous RNA-Seq analysis (Online Table VI). SRSF3 showed a tendency to bind exons, particularly in coding regions (Online Figure IVA). Genes with SRSF3-enriched exons were associated with post-transcriptional processing and insulin signaling, whereas SRSF3-bound untranslated regions (UTRs) were present in genes associated with muscle contraction (Online Figure IVB and IVC). These analyses identified SRSF3 binding to several contraction-related mRNAs, including MYBPC3 (myosin-binding protein C), TNNT2 (troponin T2), and TNNC1 (troponin C1; Online Table VI). The SRSF3 consensus motif derived from iCLIP peaks (significant X-links) was identical to the bona fide SRSF3 motif (CNYC) and was independent of the bound transcript region (whole transcript versus UTR; Online Figure VID and VIE).
Based on these results, we decided to study whether SRSF3 could increase the stability of an mRNA by binding to its 3′-UTR. To test this hypothesis, we used a stability assay based on a protein-RNA binding system present in the MS2 virus (Online Figure VA).35 P19 cells were transfected with MS2-CP or MS2-CP-SRSF3 and Rluc8 (containing 8 repeats of the MS2-CP binding loop in the 3′UTR region of R-Luc) or Rluc0 (containing no loops). Cells were treated with Actinomycin D, and R-Luc expression and enzymatic activity were measured at different time points after treatment. Expression of the MS2-CP-SRSF3 chimera had no effect either on luciferase mRNA stability (Online Figure VB and VC) or on protein activity (Online Figure VD and VE). These results suggest that the downregulation of contraction-related genes after SRSF3 depletion is independent of the binding of this RBP to the 3′-UTR in the target mRNAs.
SRSF3 Depletion Leads to a Late Induction of NMD
SRSF3 has been shown to promote the inclusion of alternative exons that lead to the appearance of premature stop codons, triggering mRNA degradation by NMD.36 Therefore, we explored whether NMD was responsible for the degradation of cardiac contraction-related mRNAs in SRSF3 knockout hearts. qRT-PCR analysis of SRSF3 knockout mouse hearts showed induced expression of the NMD genes SMG1, SMG8, SMG9, SMG6, and RBM8A (Online Figure VIA–VIE). In agreement with these results, we also observed an increased proportion of the NMD isoform of SERCA2A (Online Figure VIF). However, most of these changes took place 4 or 5 days after tamoxifen treatment. Thus, although SRSF3 depletion in the heart induces NMD, it is unlikely that this mechanism mediates the early downregulation of contraction-related mRNAs already observed at day 2.
SRSF3 Regulates mTOR Alternative Splicing
Given that SRSF3 regulates alternative splicing,15–17 we looked for alternative splicing changes in our RNA-Seq data. We identified changes in different types of alternative splicing events, ranging from alternative usage of splice donor and acceptors to intron retention (Online Figure VIIA and Online Table VII). Cassette exons were the event type showing the highest proportion (about 7%) of significant changes. There were more than twice as many exons with decreased inclusion rates than exons with increased inclusion, suggesting that the loss of SRSF3 mainly results in exon skipping. Skipped exons were associated with cytoskeletal organization and transcription, whereas retained introns affected genes associated with the regulation of transcription and translation, which could eventually contribute to the decreased expression of contraction-related genes (Online Figure VIIB and VIIC).
To determine whether the observed splicing changes were directly mediated by SRSF3, we identified SRSF3 iCLIP binding sites (significant X-links) on regulated exons and flanking regions that were potentially involved in splicing regulation (Online Figure VIIIA–VIIID and Online Tables VIII and IX). We found that SRSF3 bound preferentially to skipped exons, suggesting that the lack of direct SRSF3 binding results in exclusion of these exons. In contrast, for included exons, we observed increased SRSF3 binding in the flanking introns and exons, suggesting that SRSF3 acts here as an inhibitor of exon inclusion, for example, by promoting skipping (Online Figure VIIIA and VIIIB). For skipped introns, SRSF3 binding sites were over-represented in the intronic region and its flanking exons, indicating that SRSF3 promotes the retention of these introns. On the contrary, introns showing increased retention (inclusion) in knockout mice did not show a significant association with SRSF3 binding sites, suggesting that they may be an indirect consequence of SRSF3 deletion (Online Figure VIIIC and VIIID).
Among other changes, we found a significant change in alternative splicing of mTOR mRNA, which was bound to SRSF3 (Online Tables VI–IX). More specifically, we found that retention of mTOR intron 5 in SRSF3 knockout mice led to a decrease in the full-length mTOR isoform and in turn favored the expression of a much shorter isoform containing only the first 5 exons of the gene. This short isoform lacks the mTOR kinase domain (Figure 7A). We next checked whether this alternative splicing change was already present from day 2 after tamoxifen administration, when mRNAs related to contraction begin to decrease. Western blot analysis of SRSF3 knockout hearts confirmed loss of the long isoform and gain of the short isoform at day 2 after the induction of recombination (Figure 7B). This was associated with decreased phosphorylation of the mTOR targets AKT and 4E-BP1 (Figure 7B).37,38
Figure 7.
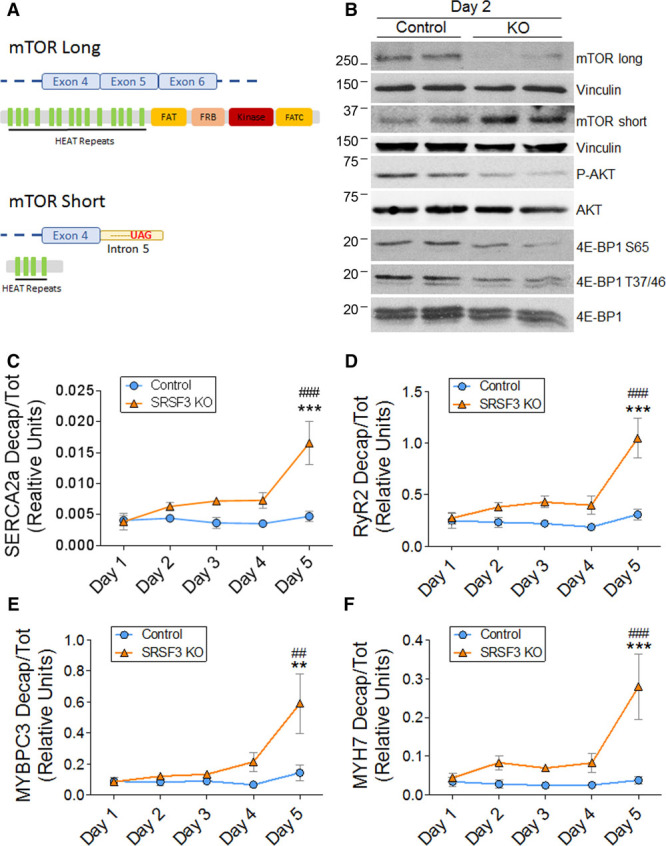
SRSF3 (serine/arginine splicing factor 3) regulates mTOR (mammalian target of rapamycin) splicing, and its depletion induces decapping of contraction-related mRNAs. A, mTOR long and short isoforms. B, Western blot analysis of mTOR splicing isoforms and the phosphorylation states of AKT and 4E-BP1 in hearts of control and SRSF3 knockout (KO) mice on day 2 post tamoxifen induction. C–F, qRT-PCR (polymerase chain reaction) analysis of SERCA2A (C), RyR2 (D), MYBPC3 (E), and MYH7 (F) decapped mRNAs in hearts of tamoxifen-induced control and SRSF3 KO mice. Data are normalized to total mRNA expression and are shown as mean±SEM; n=5–9 mice per group. **P<0.01 and ***P<0.001 for SRSF3 KO vs Control; ##P<0.01 and ###P<0.001 vs day 1; 2-way ANOVA followed by the Bonferroni post-test. 4E-BP1 indicates eIF4E-binding protein 1; eIF4E, eukaryotic translation initiation factor 4E; MYBPC3, myosin-binding protein C3; MYH7, myosin heavy chain 7; RYR2, ryanodine receptor 2; and SERCA2A, sarcoplasmic/endoplasmic reticulum calcium ATPase 2a.
SRSF3 Depletion Induces Decapping of Contraction-Related mRNAs
The phosphorylation state of 4E-BP1 determines its interaction with eIF4E (eukaryotic translation initiation factor 4E); hypo-phosphorylation leads to a stronger interaction, inhibiting eIF4E and preventing its binding to mRNA cap structures.38 Thus, a potential consequence of reduced 4E-BP1 phosphorylation is increased mRNA decapping due to increased accessibility of the mRNA cap. Decapping of an mRNA induces its degradation,39 and analysis of the decapping status of contraction-related mRNAs in SRSF3 knockout hearts revealed an increase in the proportion of decapped SERCA2A, RyR2, MYBPC3, and MYH7 mRNAs. Although the largest increase was observed at day 5 post tamoxifen administration, increased decapping could be detected as early as day 2 (Figure 7C–7F). These results indicate that the downregulation of contraction-related genes observed after cardiac SRSF3 depletion might be due to an increase in mRNA decapping activity.
To determine whether loss of mTOR activity was responsible for the increased decapping of these genes, we overactivated mTOR in SRSF3 knockout mice by treating them with amino acid injections starting on the second day of tamoxifen administration and for a total of 7 days. As shown in Online Figure IX, mTOR activation reduced decapping of some sarcomeric genes but did not rescue systolic function overall. Together, these results suggest that the loss of mTOR activity is partially responsible for the decapping of at least some sarcomeric genes. However, it is likely that additional mechanisms controlled by SRSF3 are dysregulated in the knockout mice and contribute to their pathological phenotype.
Discussion
In this study, we describe for the first time the role of SRSF3 in the embryonic and adult heart. The expression pattern of SRSF3 in the heart correlates with cell proliferation, with the highest expression levels found in the developing heart. This finding is in line with the suggestion that it functions as a proto-oncogene by promoting cell cycle progression and tumorigenesis.22 These results are also consistent with the role that SRSF3 plays during early embryo formation and with the embryonic lethality we observed in constitutive cardiac-specific SRSF3 knockout mice, which died in utero, likely due to deficient cardiomyocyte proliferation.
A key finding of our study is that loss of SRSF3 in adult cardiomyocytes leads to decapping and degradation of mRNAs encoding proteins involved in cardiac contractility, resulting in severe systolic dysfunction and death. mRNA decapping has mostly been viewed as a process for RNA recycling once it is no longer useful.39,40 However, recent studies have begun to link mRNA decapping to disease progression. Specifically, the expression of the decapping protein DCP1A (decapping mRNA 1A) has been associated with melanoma progression,41 and the tumor suppressor PNRC1 (proline-rich nuclear receptor coactivator 1) has been shown to act by recruiting the decapping complex, thus hampering ribosomal RNA maturation.42 Given that SRSF3 expression is associated with tumor progression,22,43 it would be interesting to assess if mRNA decapping is affected in cancer types in which SRSF3 is essential for progression; this would provide new insights into tumor progression mechanisms and possible therapeutic targets.
The role of mRNA decapping in heart homeostasis and disease is virtually unknown. Our study provides the first evidence of the importance of decapping deregulation in the adult heart. Overactivation of this process can have fatal consequences for cardiac function, as we observed on SRSF3 depletion. Post-transcriptional regulation plays an important role in heart disease, and several tools have been developed in recent years to modulate this process, most of them based on noncoding RNAs.44 A recent report showed that an inhibitor of the mRNA decapping scavenger enzyme can improve spinal muscular atrophy,45 suggesting that this mRNA degradation mechanism could become an effective therapeutic target.
The finding that direct binding of SRSF3 to 3′-UTRs has no effect on mRNA stability suggested that SRSF3 likely regulates mRNA degradation indirectly. The function of SRSF3 as a splicing regulator is widely known, and our results confirm this role in the heart. Binding to mTOR mRNA and regulation of its alternative splicing was previously described for the RBP Sam68 during adipogenesis; however, that study did not detect the endogenous short mTOR protein isoform.46,47 Our results thus provide the first evidence for the protein expression of the endogenous mTOR short isoform, and ours is also the first study to describe this alternative splicing change in heart tissue. Interestingly, a previous iCLIP study also identified mTOR as a direct SRSF3 target in P19 cells.36 Although this finding was not explored further, taken together with our report, it suggests that SRSF3 may regulate mTOR splicing in several tissues. Moreover, SRSF3 binds a further 210 genes in both P19 cells and cardiomyocytes. These genes are involved in post-transcriptional mRNA processing and are enriched in nucleic acid binding proteins, suggesting that SRSF3 may regulate these processes in several cell types. In line with our results, loss of mTOR activity has been related to a decrease in 5′-capped mRNA,48 and mTOR activity in the heart has been reported to be essential for proper cardiac function and survival.49 However, the phenotype of mTOR knockout mice differs from that of SRSF3 knockout mice. On cardiac mTOR depletion, mice develop dilated cardiomyopathy and survive for several weeks after tamoxifen treatment,49 whereas in our analysis cardiac SRSF3 depletion led to acute systolic dysfunction and animal death in <8 days after treatment. It is thus likely that SRSF3 knockout animals do not develop dilated cardiomyopathy because there is insufficient time before they die. The phenotype of SRSF3 knockout mice is more aggressive than that of mTOR knockout mice, and hearts lacking SRSF3 expression also undergo many other changes. It is thus possible that the observed phenotype involves contributions from additional molecular processes in which SRSF3 participates, not necessarily through alternative splicing. Alternatively, the mTOR short isoform could act as a dominant negative protein, precluding mTOR proper function and promoting decapping. We also cannot exclude the possibility that the increased decapping and mRNA degradation effects of mTOR deficiency may involve contributions from targets other than 4E-BP1. Furthermore, SRSF3 is involved in other biological processes that could potentially contribute to the severe phenotype observed in knockout mice. SRSF3 controls mRNA nuclear export and translation through a number of different mechanisms, which has an impact on different biological processes, ranging from microglia activation to tumor cell apoptosis.11,18,50 Although apoptosis and inflammation appear in SRSF3 knockout mice are unlikely to contribute to systolic dysfunction; they may aggravate the animals’ condition at late time points.
In conclusion, we have identified SRSF3 as an essential factor for proper heart contraction and animal survival. Loss of SRSF3 in the heart leads to a critical downregulation of contraction-related genes, most probably caused by induction of the decapping process. We have also shown that SRSF3 regulates mTOR alternative splicing and presented the first report of the endogenous mTOR short isoform protein. This alternative splicing change leads to a dephosphorylation of 4E-BP1 that would explain the induction of mRNA decapping (Figure 8). Given that SRSF3 expression is downregulated after MI, we propose both SRSF3 and mRNA decapping as key regulators of cardiac function, paving the way to new therapeutic approaches to treat heart disease.
Figure 8.
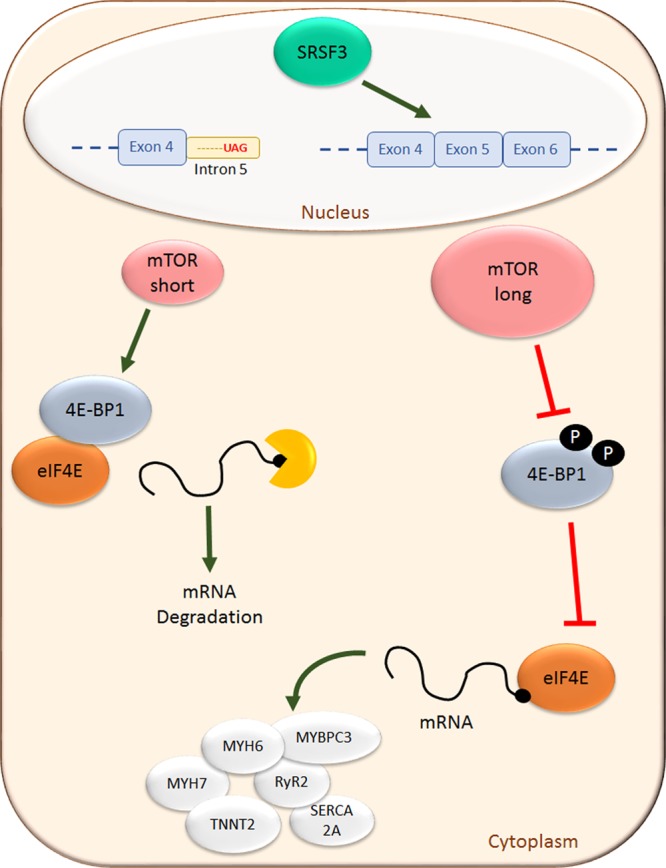
Summary of SRSF3 (serine/arginine splicing factor 3) signaling in cardiomyocytes. SRSF3 regulates mTOR (mammalian target of rapamycin) splicing, promoting synthesis of the long isoform. This isoform phosphorylates 4E-BP1, preventing it from inhibiting interaction of eIF4E with the mRNA caps and the subsequent mRNA translation. In the absence of SRSF3, the short mTOR (mammalian target of rapamycin) isoform prevails, 4E-BP1 phosphorylation is reduced, and mRNA decapping increases, leading to its mRNA degradation. 4E-BP1 indicates eIF4E-binding protein 1; eIF4E, eukaryotic translation initiation factor 4E; MYBPC3, myosin-binding protein C3; MYH6, myosin heavy chain 6; MYH7, myosin heavy chain 7; RYR2, ryanodine receptor 2; and SERCA2A, sarcoplasmic/endoplasmic reticulum calcium ATPase 2a.
Acknowledgments
We are grateful to Igor de los Mozos and François McNicoll for assistance with the individual-nucleotide resolution cross-linking and immunoprecipitation experiments and analyses. We thank the CNIC (Centro Nacional de Investigaciones Cardiovasculares) animal facility staff members for mouse work, the Histology Unit for hematoxylin and eosin staining, and the Genomics and Bioinformatics Units for RNA-Seq and analysis.
Sources of Funding
This study was supported by grants from the European Union (CardioNeT-ITN-289600 and CardioNext-ITN-608027 to E. Lara-Pezzi), from the Spanish Ministerio de Economía y Competitividad (RTI2018-096961-B-I00, SAF2015-65722-R, and SAF2012-31451 to E. Lara-Pezzi; BIO2015-67580-P and PGC2018-097019-B-I00 to J. Vázquez), the Spanish Carlos III Institute of Health (CPII14/00027 to E. Lara-Pezzi, RD12/0042/066 to P. García-Pavía and E. Lara-Pezzi, and RD12/0042/0056, PRB2-IPT13/0001-ISCIII-SGEFI/FEDER, ProteoRed to J. Vázquez), the Madrid Regional Government (2010-BMD-2321 Fibroteam to E. Lara-Pezzi). This study was also supported by the Plan Estatal de I+D+I 2013–2016—European Regional Development Fund (ERDF) A way of making Europe, Spain. The CNIC is supported by the Ministerio de Ciencia, Innovación y Universidades (MCNU) and the Pro CNIC Foundation, and is a Severo Ochoa Center of Excellence (SEV-2015-0505).
Disclosures
None.
Supplementary Material
{kind=link}
Nonstandard Abbreviations and Acronyms
- iCLIP
- individual-nucleotide resolution cross-linking and immunoprecipitation
- LV
- left ventricular
- MI
- myocardial infarction
- mTOR
- mammalian target of rapamycin
- NMD
- nonsense-mediated decay
- RBP
- RNA Binding Protein
- SRSF3
- serine/arginine splicing factor 3
- UTR
- untranslated region
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCRESAHA.118.314515.
Novelty and Significance
What Is Known?
RNA binding proteins regulate mRNA localization, stability, splicing, and activity.
SRSF3 (serine/arginine splicing factor 3) plays a role in blastocyst formation and in liver function.
What New Information Does This Article Contribute?
The expression of SRF3 in the adult heart is essential for proper cardiac contraction and animal survival.
Loss of SRSF3 results in expression of a smaller mTOR (mammalian target of rapamycin) isoform, which has no enzymatic capacity.
SRSF3 prevents mRNA decapping and loss of SRSF3 results in increased mRNA decapping and degradation of mRNAs encoding sarcomeric proteins, which leads to systolic dysfunction.
RNA binding proteins play diverse roles in the cell, including mRNA transport, regulation of mRNA stability, and post-transcriptional mRNA modifications, among others. SRSF3 is an RNA binding protein that plays a role in embryonic development and in the liver. Its role in the heart was unknown. Using mice that lack SRSF3 specifically in cardiomyocytes, we found that SRSF3 is necessary for embryonic cardiomyocyte proliferation and heart development. In the adult heart, loss of SRSF3 causes severe cardiac contraction defects that lead to heart failure. We show that the loss of SRSF3 leads to decapping and degradation of mRNAs, specifically those encoding sarcomeric proteins. This is the main mechanism underlying cardiac dysfunction in our mice. We also show that the absence of SRSF3 causes aberrant splicing of mTOR, producing a shorter isoform with reduced enzymatic activity, which is known to be necessary to prevent decapping. Based on these findings, it is tempting to speculate that mRNA decapping may have an important role in heart disease and that therapies aiming at modulating decapping may have a positive impact on cardiac contraction.
References
- 1.WHO. Cardiovascular Diseases (CVDs). 2017.
- 2.Barth AS, Kumordzie A, Frangakis C, Margulies KB, Cappola TP, Tomaselli GF. Reciprocal transcriptional regulation of metabolic and signaling pathways correlates with disease severity in heart failure. Circ Cardiovasc Genet. 2011;4:475–483. doi: 10.1161/CIRCGENETICS.110.957571. doi: 10.1161/CIRCGENETICS.110.957571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corbett AH. Post-transcriptional regulation of gene expression and human disease. Curr Opin Cell Biol. 2018;52:96–104. doi: 10.1016/j.ceb.2018.02.011. doi: 10.1016/j.ceb.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo W, Schafer S, Greaser ML, et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med. 2012;18:766–773. doi: 10.1038/nm.2693. doi: 10.1038/nm.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Bruin RG, Rabelink TJ, van Zonneveld AJ, van der Veer EP. Emerging roles for RNA-binding proteins as effectors and regulators of cardiovascular disease. Eur Heart J. 2017;38:1380–1388. doi: 10.1093/eurheartj/ehw567. doi: 10.1093/eurheartj/ehw567. [DOI] [PubMed] [Google Scholar]
- 6.Ding J, Chen J, Wang Y, Kataoka M, Ma L, Zhou P, Hu X, Lin Z, Nie M, Deng ZL, Pu WT, Wang DZ. Trbp regulates heart function through microRNA-mediated Sox6 repression. Nat Genet. 2015;47:776–783. doi: 10.1038/ng.3324. doi: 10.1038/ng.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei C, Qiu J, Zhou Y, Xue Y, Hu J, Ouyang K, Banerjee I, Zhang C, Chen B, Li H, Chen J, Song LS, Fu XD. Repression of the central splicing regulator RBFox2 is functionally linked to pressure overload-induced heart failure. Cell Rep. 2015;10:1521–1533. doi: 10.1016/j.celrep.2015.02.013. doi: 10.1016/j.celrep.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao C, Ren S, Lee JH, Qiu J, Chapski DJ, Rau CD, Zhou Y, Abdellatif M, Nakano A, Vondriska TM, Xiao X, Fu XD, Chen JN, Wang Y. RBFox1-mediated RNA splicing regulates cardiac hypertrophy and heart failure. J Clin Invest. 2016;126:195–206. doi: 10.1172/JCI84015. doi: 10.1172/JCI84015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manley JL, Krainer AR. A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins). Genes Dev. 2010;24:1073–1074. doi: 10.1101/gad.1934910. doi: 10.1101/gad.1934910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Änkö ML. Regulation of gene expression programmes by serine-arginine rich splicing factors. Semin Cell Dev Biol. 2014;32:11–21. doi: 10.1016/j.semcdb.2014.03.011. doi: 10.1016/j.semcdb.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 11.Müller-McNicoll M, Botti V, de Jesus Domingues AM, Brandl H, Schwich OD, Steiner MC, Curk T, Poser I, Zarnack K, Neugebauer KM. SR proteins are NXF1 adaptors that link alternative RNA processing to mRNA export. Genes Dev. 2016;30:553–566. doi: 10.1101/gad.276477.115. doi: 10.1101/gad.276477.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu X, Yang D, Ding JH, et al. ASF/SF2-regulated CaMKIIdelta alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell. 2005;120:59–72. doi: 10.1016/j.cell.2004.11.036. doi: 10.1016/j.cell.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 13.Ding JH, Xu X, Yang D, Chu PH, Dalton ND, Ye Z, Yeakley JM, Cheng H, Xiao RP, Ross J, Chen J, Fu XD. Dilated cardiomyopathy caused by tissue-specific ablation of SC35 in the heart. EMBO J. 2004;23:885–896. doi: 10.1038/sj.emboj.7600054. doi: 10.1038/sj.emboj.7600054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng Y, Valley MT, Lazar J, Yang AL, Bronson RT, Firestein S, Coetzee WA, Manley JL. SRp38 regulates alternative splicing and is required for Ca(2+) handling in the embryonic heart. Dev Cell. 2009;16:528–538. doi: 10.1016/j.devcel.2009.02.009. doi: 10.1016/j.devcel.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong J, Garner B, Halliday GM, Kwok JB. Srp20 regulates TrkB pre-mRNA splicing to generate TrkB-Shc transcripts with implications for Alzheimer’s disease. J Neurochem. 2012;123:159–171. doi: 10.1111/j.1471-4159.2012.07873.x. doi: 10.1111/j.1471-4159.2012.07873.x. [DOI] [PubMed] [Google Scholar]
- 16.Sen S, Talukdar I, Webster NJ. SRp20 and CUG-BP1 modulate insulin receptor exon 11 alternative splicing. Mol Cell Biol. 2009;29:871–880. doi: 10.1128/MCB.01709-08. doi: 10.1128/MCB.01709-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonçalves V, Matos P, Jordan P. Antagonistic SR proteins regulate alternative splicing of tumor-related Rac1b downstream of the PI3-kinase and Wnt pathways. Hum Mol Genet. 2009;18:3696–3707. doi: 10.1093/hmg/ddp317. doi: 10.1093/hmg/ddp317. [DOI] [PubMed] [Google Scholar]
- 18.Kim J, Park RY, Chen JK, Kim J, Jeong S, Ohn T. Splicing factor SRSF3 represses the translation of programmed cell death 4 mRNA by associating with the 5’-UTR region. Cell Death Differ. 2014;21:481–490. doi: 10.1038/cdd.2013.171. doi: 10.1038/cdd.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jumaa H, Wei G, Nielsen PJ. Blastocyst formation is blocked in mouse embryos lacking the splicing factor SRp20. Curr Biol. 1999;9:899–902. doi: 10.1016/s0960-9822(99)80394-7. [DOI] [PubMed] [Google Scholar]
- 20.Sen S, Jumaa H, Webster NJ. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat Commun. 2013;4:1336. doi: 10.1038/ncomms2342. doi: 10.1038/ncomms2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sen S, Langiewicz M, Jumaa H, Webster NJG. Deletion of splicing factor SRSF3 in hepatocytes predisposes to hepatocellular carcinoma in mice. Hepatology. 2015;61:171–183. doi: 10.1002/hep.27380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jia R, Li C, McCoy JP, Deng CX, Zheng ZM. SRp20 is a proto-oncogene critical for cell proliferation and tumor induction and maintenance. Int J Biol Sci. 2010;6:806–826. doi: 10.7150/ijbs.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.González-Terán B, López JA, Rodríguez E, Leiva L, Martínez-Martínez S, Bernal JA, Jiménez-Borreguero LJ, Redondo JM, Vázquez J, Sabio G. p38g and d promote heart hypertrophy by targeting the mTOR-inhibitory protein DEPTOR for degradation. Nat Commun. 2016;7:10477. doi: 10.1038/ncomms10477. doi: 10.1038/ncomms10477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Padrón-Barthe L, Villalba-Orero M, Gómez-Salinero JM, Acín-Pérez R, Cogliati S, López-Olañeta M, Ortiz-Sánchez P, Bonzón-Kulichenko E, Vázquez J, García-Pavía P, Rosenthal N, Enríquez JA, Lara-Pezzi E. Activation of serine one-carbon metabolism by calcineurin Aβ1 reduces myocardial hypertrophy and improves ventricular function. J Am Coll Cardiol. 2018;71:654–667. doi: 10.1016/j.jacc.2017.11.067. doi: 10.1016/j.jacc.2017.11.067. [DOI] [PubMed] [Google Scholar]
- 25.Nus M, Martínez-Poveda B, MacGrogan D, Chevre R, D’Amato G, Sbroggio M, Rodríguez C, Martínez-González J, Andrés V, Hidalgo A, de la Pompa JL. Endothelial Jag1-RBPJ signalling promotes inflammatory leucocyte recruitment and atherosclerosis. Cardiovasc Res. 2016;112:568–580. doi: 10.1093/cvr/cvw193. doi: 10.1093/cvr/cvw193. [DOI] [PubMed] [Google Scholar]
- 26.Bray NL, Pimentel H, Melsted P, Pachter L. Erratum: near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:888. doi: 10.1038/nbt0816-888d. doi: 10.1038/nbt0816-888d. [DOI] [PubMed] [Google Scholar]
- 27.Pimentel H, Bray NL, Puente S, Melsted P, Pachter L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods. 2017;14:687–690. doi: 10.1038/nmeth.4324. doi: 10.1038/nmeth.4324. [DOI] [PubMed] [Google Scholar]
- 28.Irimia M, Weatheritt RJ, Ellis JD, et al. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell. 2014;159:1511–1523. doi: 10.1016/j.cell.2014.11.035. doi: 10.1016/j.cell.2014.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tapial J, Ha KCH, Sterne-Weiler T, et al. An atlas of alternative splicing profiles and functional associations reveals new regulatory programs and genes that simultaneously express multiple major isoforms. Genome Res. 2017;27:1759–1768. doi: 10.1101/gr.220962.117. doi: 10.1101/gr.220962.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pelechano V, Wei W, Steinmetz LM. Genome-wide quantification of 5’-phosphorylated mRNA degradation intermediates for analysis of ribosome dynamics. Nat Protoc. 2016;11:359–376. doi: 10.1038/nprot.2016.026. doi: 10.1038/nprot.2016.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jumaa H, Nielsen PJ. The splicing factor SRp20 modifies splicing of its own mRNA and ASF/SF2 antagonizes this regulation. EMBO J. 1997;16:5077–5085. doi: 10.1093/emboj/16.16.5077. doi: 10.1093/emboj/16.16.5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawase Y, Hajjar RJ. The cardiac sarcoplasmic/endoplasmic reticulum calcium ATPase: a potent target for cardiovascular diseases. Nat Clin Pract Cardiovasc Med. 2008;5:554–565. doi: 10.1038/ncpcardio1301. doi: 10.1038/ncpcardio1301. [DOI] [PubMed] [Google Scholar]
- 33.Byrne MJ, Power JM, Preovolos A, Mariani JA, Hajjar RJ, Kaye DM. Recirculating cardiac delivery of AAV2/1SERCA2a improves myocardial function in an experimental model of heart failure in large animals. Gene Ther. 2008;15:1550–1557. doi: 10.1038/gt.2008.120. doi: 10.1038/gt.2008.120. [DOI] [PubMed] [Google Scholar]
- 34.del Monte F, Williams E, Lebeche D, Schmidt U, Rosenzweig A, Gwathmey JK, Lewandowski ED, Hajjar RJ. Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase in a rat model of heart failure. Circulation. 2001;104:1424–1429. doi: 10.1161/hc3601.095574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rambout X, Detiffe C, Bruyr J, et al. The transcription factor ERG recruits CCR4-NOT to control mRNA decay and mitotic progression. Nat Struct Mol Biol. 2016;23:663–672. doi: 10.1038/nsmb.3243. doi: 10.1038/nsmb.3243. [DOI] [PubMed] [Google Scholar]
- 36.Änkö ML, Müller-McNicoll M, Brandl H, Curk T, Gorup C, Henry I, Ule J, Neugebauer KM. The RNA-binding landscapes of two SR proteins reveal unique functions and binding to diverse RNA classes. Genome Biol. 2012;13:R17. doi: 10.1186/gb-2012-13-3-r17. doi: 10.1186/gb-2012-13-3-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 38.Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, Sonenberg N. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 1999;13:1422–1437. doi: 10.1101/gad.13.11.1422. doi: 10.1101/gad.13.11.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Houseley J, Tollervey D. The many pathways of RNA degradation. Cell. 2009;136:763–776. doi: 10.1016/j.cell.2009.01.019. doi: 10.1016/j.cell.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Kiledjian M. Regulation of mRNA decapping. Wiley Interdiscip Rev RNA. 2010;1:253–265. doi: 10.1002/wrna.15. doi: 10.1002/wrna.15. [DOI] [PubMed] [Google Scholar]
- 41.Tang Y, Xie C, Zhang Y, Qin Y, Zhang W. Overexpression of mRNA-decapping enzyme 1a predicts disease-specific survival in malignant melanoma. Melanoma Res. 2018;28:30–36. doi: 10.1097/CMR.0000000000000406. doi: 10.1097/CMR.0000000000000406. [DOI] [PubMed] [Google Scholar]
- 42.Gaviraghi M, Vivori C, Pareja Sanchez Y, Invernizzi F, Cattaneo A, Santoliquido BM, Frenquelli M, Segalla S, Bachi A, Doglioni C, Pelechano V, Cittaro D, Tonon G. Tumor suppressor PNRC1 blocks rRNA maturation by recruiting the decapping complex to the nucleolus. EMBO J. 2018;37:e99179. doi: 10.15252/embj.201899179. doi: 10.15252/embj.201899179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang YL, Hsu YJ, Chen Y, Wang YW, Huang SM. Theophylline exhibits anti-cancer activity via suppressing SRSF3 in cervical and breast cancer cell lines. Oncotarget. 2017;8:101461–101474. doi: 10.18632/oncotarget.21464. doi: 10.18632/oncotarget.21464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laina A, Gatsiou A, Georgiopoulos G, Stamatelopoulos K, Stellos K. RNA therapeutics in cardiovascular precision medicine. Front Physiol. 2018;9:953. doi: 10.3389/fphys.2018.00953. doi: 10.3389/fphys.2018.00953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gopalsamy A, Narayanan A, Liu S, et al. Design of potent mRNA Decapping Scavenger Enzyme (DcpS) inhibitors with improved physicochemical properties to investigate the mechanism of therapeutic benefit in Spinal Muscular Atrophy (SMA). J Med Chem. 2017;60:3094–3108. doi: 10.1021/acs.jmedchem.7b00124. doi: 10.1021/acs.jmedchem.7b00124. [DOI] [PubMed] [Google Scholar]
- 46.Huot MÉ, Vogel G, Zabarauskas A, Ngo CT, Coulombe-Huntington J, Majewski J, Richard S. The Sam68 STAR RNA-binding protein regulates mTOR alternative splicing during adipogenesis. Mol Cell. 2012;46:187–199. doi: 10.1016/j.molcel.2012.02.007. doi: 10.1016/j.molcel.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 47.Wang H, Chen Y, Lu XA, Liu G, Fu Y, Luo Y. Endostatin prevents dietary-induced obesity by inhibiting adipogenesis and angiogenesis. Diabetes. 2015;64:2442–2456. doi: 10.2337/db14-0528. doi: 10.2337/db14-0528. [DOI] [PubMed] [Google Scholar]
- 48.Rahman H, Qasim M, Oellerich M, Asif AR. Crosstalk between Edc4 and mammalian target of rapamycin complex 1 (mTORC1) signaling in mRNA decapping. Int J Mol Sci. 2014;15:23179–23195. doi: 10.3390/ijms151223179. doi: 10.3390/ijms151223179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang D, Contu R, Latronico MV, et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest. 2010;120:2805–2816. doi: 10.1172/JCI43008. doi: 10.1172/JCI43008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boutej H, Rahimian R, Thammisetty SS, Béland LC, Lalancette-Hébert M, Kriz J. Diverging mRNA and protein networks in activated microglia reveal SRSF3 suppresses translation of highly upregulated innate immune transcripts. Cell Rep. 2017;21:3220–3233. doi: 10.1016/j.celrep.2017.11.058. doi: 10.1016/j.celrep.2017.11.058. [DOI] [PubMed] [Google Scholar]