

Abstract
Recent thymic emigrants (RTEs) that fail post-positive selection maturation are also targeted by complement proteins. T cells likely acquire complement-resistance during maturation in the thymus, a complement-privileged organ. To test this, thymocytes and fresh serum were separately obtained and incubated together in vitro to assess complement deposition. Complement binding decreased with development and maturation. Complement binding decreased from the double positive (DP) thymocyte to the single positive (SP) stage, and within SP thymocytes, complement binding gradually decreased with increasing intra-thymic maturation. Binding of the central complement protein, C3, to WT immature thymocytes required the lectin but not the classical pathway. Specifically, MBL2 but not MBL1 was required, demonstrating a unique function for MBL2. Previous studies demonstrated that the loss of NKAP, a transcriptional regulator of T cell maturation, caused peripheral T cell lymphopenia and enhanced complement susceptibility. To determine whether complement causes NKAP-deficient T cell disappearance, both the lectin and classical were genetically ablated. This blocked C3 deposition on NKAP-deficient T cells but failed to restore normal cellularity, indicating that complement contributes to clearance but is not the primary cause of peripheral T cell lymphopenia. Rather, the accumulation of lipid peroxides in NKAP-deficient T cells was observed. Lipid-peroxidation is a salient feature of ferroptosis, an iron-dependent non-apoptotic cell death. Thus, WT thymocytes naturally acquire the ability to protect themselves from complement targeting by MBL2 with maturation. However, NKAP deficient immature peripheral T cells remain scarce in complement-deficient mice likely due to ferroptosis.
Keywords: Complement, MBL2, T cell maturation, NKAP, ferroptosis
Introduction:
The complement system is an integral component of innate immunity and consists of soluble protein fragments and complexes found in the circulation (1–4). Although initially thought to be predominantly important for host defense by aiding in antibody-mediated clearance of pathogens, it is now well-appreciated that functions of the complement system extend to tissue homeostasis, immune response modulation, and elimination of unwanted or dying cells (5, 6). Complement activation relies on interplay between multiple soluble proteins, and receptors via three main mechanisms – the classical, alternative, and lectin pathways (2). These pathways converge at the C3 (complement component 3) convertase, which catalyzes formation of effector molecules, opsonins and anaphylatoxins by enzymatic fragmentation of C3. This leads to outcomes such as phagocytosis of microbial targets, modulation of immune cell activation, and clearance of host antigens generated via apoptotic or necrotic processes (2, 4). Classical complement pathway can be initiated by antibody-dependent, or antibody-independent C1q-mediated mechanisms. On the other hand, lectin pathway activation depends on the collectin family of molecules including mannose-binding lectins (MBL) and ficolins. These molecules recognize and bind specific glycosylation patterns such as mannose residues, which are predominantly found on bacterial cell walls (7, 8). Lastly, the alternative pathway is spontaneously activated leading to membrane attack complex (MAC) formation in the absence of regulatory molecules (4). Multiple factors determine the activity of complement proteins including inhibitory molecules, glycosylation patterns on the cell surface, and accessibility of tissues to complement proteins (9).
The thymus is a complement-privileged organ, as the deletion of a complement inhibitor encoding gene, complement receptor 1-related (Crry) gene, leads to peripheral T cell lymphopenia mediated by complement attack while thymic cellularity is maintained; there is no evidence of complement deposition on thymocytes when analyzed directly ex vivo (10). Glycosylation patterns change as thymocytes progress through development in the thymus (11). There is a gradual increase in cell surface sialylation due to increasing expression of sialic acid transferases (12–15). Increasing sialylation masks exposed mannose residues on developing thymocytes (11, 16). Sialylation is required to prevent complement activation as demonstrated by enzymatic stripping of sialic acid by neuraminidase resulting in complement activation and cell death (17). Taken together, these observations suggest that T cells gain resistance to complement prior to egress into blood, which contains complement proteins. Previously, we have shown that the transcriptional regulator NKAP is required for T cell maturation (15, 18). NKAP-deficient thymocytes fail to upregulate α−2,8 sialyltransferases (15, 19). Peripheral T cells lacking NKAP are opsonized by complement proteins and eliminated at the recent thymic emigrant (RTE) stage, further indicating that complement resistance is gained intrathymically as part of T cell maturation (15).
In this study, we directly tested whether resistance to complement is gained during thymic T cell maturation. Susceptibility of thymocytes to complement as a function of maturation and contribution of complement proteins in mediating disappearance of NKAP-deficient T cells was investigated. To circumvent restricted access of the thymus to complement (10), freshly harvested WT thymocytes were incubated with freshly isolated serum in vitro and assessed for complement binding. C3 and C4 deposition on thymocytes was inversely proportional to development and maturation. Deposition required the lectin pathway while the classical pathway was dispensable. Specifically, MBL2 was required for C3 and C4 deposition while MBL1 was not. Finally, ablation of both the classical and lectin pathways (C1q KO MBL1 MBL2 double KO) was needed to prevent C3 deposition on NKAP-deficient mature naïve T cells (MNTs) in the periphery, but failed to restore normal naïve T cell percentages and absolute numbers in CD4-cre NKAP cKO mice, suggesting another mode of cell death as the primary cause of T cell lymphopenia. Increased lipid peroxidation in NKAP-deficient cells indicated ferroptosis, a form of regulated cell death driven by reactive oxygen species (ROS) derived from iron metabolism. Overall, this study is the first to provide evidence that thymocytes gain resistance to the lectin pathway of complement deposition as a function of increasing thymic T cell maturation, and that the death of NKAP-deficient T cells is driven by ferroptosis, followed by complement-mediated clearance.
Materials and Methods:
Mice:
C57BL6, C1q knock out (KO), C3 KO, MBL1/MBL2 dKO mice were obtained from the Jackson Laboratory. NKAP fl mice (28) were interbred with MBL1/MBL2 dKO mice or CD4-cre NKAP cKO mice (19) to create MBL1/MBL2 dKO CD4-cre NKAP cKO mice. CD4-cre NKAP cKO mice were crossed to C1q KO mice to generate C1q KO CD4-cre NKAP cKO mice. C1q KO CD4-cre NKAP cKO were interbred with MBL1/MBL2 dKO CD4-cre-NKAP cKO mice to generate C1q KO MBL½ dKO CD4-cre NKAP cKO mice. MBL1/MBL2 dKO mice were outbred to generate MBL1 KO and MBL2 KO mice. Mice between 6–10 weeks of age were used for all the experiments.
Flow Cytometry:
For complement deposition experiments, freshly harvested wildtype (6–10 week old) thymocytes were incubated in freshly isolated sera in GVB++ buffer (Complement Technology) at a 1:5 (serum:GVB++) ratio for one hour at room temperature. Serum was isolated by bleeding mice (6–10 week old) retro-orbitally, incubating blood at room temperature to allow coagulation followed by centrifugation at 8000 rpm. Cells were washed with FACS buffer and stained with fluorescent-conjugated cell surface antibodies against CD4 (1:500 dilution, clone RM4–5 or GK1.5), CD8 (1:500 clone 53–6.7 or 2.43), CD24 (1:1000, clone M1/69) and CCR7 (1:100, clone 4B12,) (Biolegend, eBioscience and Tonbo) and biotinylated antibodies for complement C1q (1:100), complement C3 (1:200) and complement C4 (1:500) (Cedarlane). Surface stain of CCR7 was performed for 30 minutes at 37° C followed by washing and staining of other surface antigens on ice for 30 minutes and washing to remove unbound antibodies. Secondary stain was performed with PE (1:1000) or APC (1:1000) conjugated streptavidin (Biolegend and eBioscience) and unbound antibodies were washed. All experiments included fixable viability dye (Tonbo) for analysis of live cells. Stained cells were analyzed using an Attune NxT flow cytometer (Thermofisher). Data was analyzed using FlowJo (Tree Star) 9.8 or 10. For analysis of all experiments, doublets were excluded based on forward and side scatter, and dead cells positive for fixable viability dye were also excluded. For mannose inhibition of C3 deposition, freshly isolated WT sera was first treated for 5 minutes with mannose (Sigma-Aldrich) at various concentrations and incubated with WT thymocytes as described above. A large range of dilutions of mannose was used to determine whether low levels of mannose could impose a block on C3 deposition. These concentrations did not alter osmotic potential of cells as determined using forward and side scatter of total thymocytes.
Analysis of cell stress and death:
Freshly harvested splenocytes were cell surface stained on ice followed by incubation with fluorescent-conjugated Annexin-V in Annexin-V binding buffer for 10 minutes at room temperature (Biolegend) immediately or following a one-hour incubation at 37˚ C in RPMI 1640 media (Corning) supplemented with 10% fetal bovine serum and 1 μM BODIPY-C11 (ThermoFisher). For examination of Liperfluo (Dojindo Molecular technologies) positivity, freshly harvested splenocytes were incubated with 10 μM LiperFluo (resuspended in 1% DMSO) in HBSS for 30 minutes at 37˚ C. Cells were then washed and surface stained. To examine the impact of ferroptosis inhibitors on lipid peroxidation, total splenocytes were incubated with 100 μM alpha-tocopherol (Millipore Sigma) and/or 10 μM Ferrostatin-1 (Millipore Sigma) for 15 minutes followed by staining with 1 μM BODIPY-C11 for 1 hour. Cells were then washed and surface stained. For intracellular protein detection, cells were fixed and permeabilized (Tonbo TNB-0607-KIT) and stained with fluorescent-conjugated antibodies against Bcl2 and Cleaved Caspase-3 (Biolegend and Cell Signaling). For determining intracellular GPX4 expression, cells were surface stained, fixed and permeabilized (BD 558049 and 558050), and stained with primary antibody against GPX4 (Abcam) followed by anti-rabbit secondary antibody (SouthernBiotech). For cellular dyes, 1 μM YO-PRO1 Iodide (ThermoFisher) was added and splenocytes were stained on ice for 30 minutes followed by cell surface staining. MitoTracker, MitoSOX, TMRM, CellROX, and ThiolTracker dyes (all from ThermoFisher) were added at 50 nM, 5 μM, 100 nM, 5 μM, and 10 μM, respectively. Data was analyzed as above using flow cytometry methods.
Western blot analysis of complement proteins:
Mouse sera were diluted 500-fold in sample buffer and boiled before loading on gels. Blotting was performed overnight at 4 degrees with the following antibodies: 0.4 μg/ml rabbit anti-C3 (Abcam ab200999), 0.1 μg/ml goat anti-MBL1 (R&D Systems AF2077), 0.1 μg/ml goat anti-MBL2 (R&D Systems AF2208), and 0.4 μg/ml rabbit anti-Transferrin (Abcam ab82411). Proteins were then detected by incubation for 1 hour at room temperature with horseradish peroxidase coupled secondary antibodies at 0.1 μg/ml, using either goat anti-rabbit IgG (Southern Biotech 4050) or donkey anti-goat IgG (Southern Biotech 6420). All antibodies were diluted in tris buffered saline with 3% bovine serum albumin. Bands were detected on film with ECL Pro reagent (Perkin Elmer).
Statistical Analysis:
All statistical analysis involved enumeration of absolute cell counts or MFI, calculation of standard error of the mean, and statistical significance using one-way ANOVA (GraphPad Prisim software) for multiple comparisons.
Results:
Susceptibility to complement binding decreases as post-positive selection maturation increases.
NKAP-deficient T cells have a defect in post-positive selection maturation, as peripheral T cells lacking NKAP are both phenotypically and functionally immature (15, 18). Furthermore, NKAP-deficient T cells are opsonized by complement proteins in the periphery and eliminated at the recent thymic emigrant (RTE) stage suggesting that complement resistance is gained as part of T cell maturation. In contrast, complement factors do not bind to NKAP-deficient thymocytes when directly assessed ex vivo (15). This suggests that the thymus is a complement privileged organ with restricted access to complement proteins. To circumvent restricted access of the thymus to complement (10) and to directly assess whether complement susceptibility of thymocytes decreases with maturation, freshly harvested WT thymocytes were incubated with serum in vitro and assessed for complement binding. The binding of C1q, C4 and C3 to double positive (DP), CD8 single positive (SP), CD4 SP, CD4 semi-mature (SM), CD4 mature 1 (M1) and CD4 mature 2 (M2) WT thymocytes was assessed by incubating with serum from WT mice in vitro (Figure 1). Deposition of C1q, C3 and C4 decreased as T cell development and maturation progressed. In particular, there was almost no complement deposition on M2 thymocytes, the most mature SP cells that are licensed to egress. Thus, thymocytes become resistant to complement prior to egress from the thymus.
Figure 1. Susceptibility to complement binding decreases during thymic development and maturation.
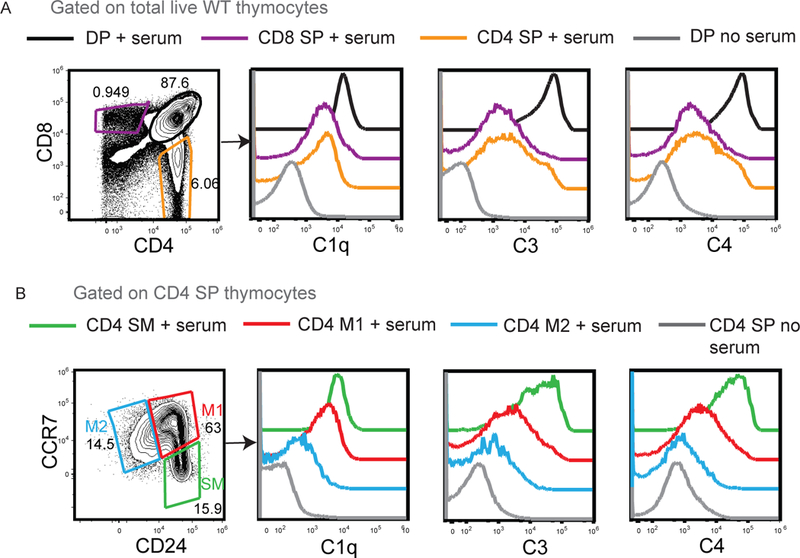
(A) Examination of C1q, C3 and C4 binding on double positive (DP), CD4 single positive (SP), CD8 SP thymocytes after incubation with WT serum. DP thymocytes incubated without serum was used as a negative control. (B) Examination of C1q, C3 and C4 complement binding on CD4 SP thymocytes divided into CD24+CCR7− SM, CD24+CCR7+ M1 and CD24−CCR7+ M2 populations. CD4 SP thymocytes incubated without serum was used as a negative control. Data is representative of 4 WT mice in total from 4 independent experiments.
The lectin pathway is necessary and sufficient for C3 and C4 deposition.
Relative contributions of the lectin and the classical complement pathways to C3 deposition on thymocytes were examined. These two pathways converge at C4 (Figure 2A). The C3 convertase, C4bC2a, formed via activation of either the lectin or the classical pathway enables C3 deposition. Sera from C1q knock out (KO), MBL1/MBL2 double KO (dKO) and C3 KO mice were examined for their ability to deposit C1q, C3 and C4 on thymocytes. Absence of C3, MBL1 and MBL2 in sera was confirmed by western blot (Supplement Figure 1). Sera were isolated from WT, C1q KO, MBL1/MBL2 dKO as well as C3 KO mice, and individually incubated with WT thymocytes (Figure 2B–E, gating strategy in Supplementary Figure 2). Compared to WT thymocytes incubated with sera from WT mice, WT thymocytes incubated with sera from MBL1/MBL2 dKO mice lacked C3 and C4 deposition while C1q deposition was intact. WT thymocytes incubated with sera from C3 KO mice was used as a negative control (Figure 2B). The extent of C3 binding on WT thymocytes incubated with sera from C3 KO mice was comparable to WT thymocytes incubated with sera from MBL1/MBL2 dKO mice ruling out the alternative pathway and demonstrating that the lectin pathway is necessary for complement deposition on thymocytes (Figure 2B, D). Loss of C1q from sera did not prevent C3 and C4 deposition on WT thymocytes (Figure 2C) demonstrating that the classical pathway is not required. Further, pre-incubation of sera that are WT mice with mannose abrogated C3 deposition, indicating that soluble mannose binds mannose-binding molecules in sera were responsible for C3 deposition (Figure 2F).
Figure 2. The lectin pathway is required for C3 deposition.
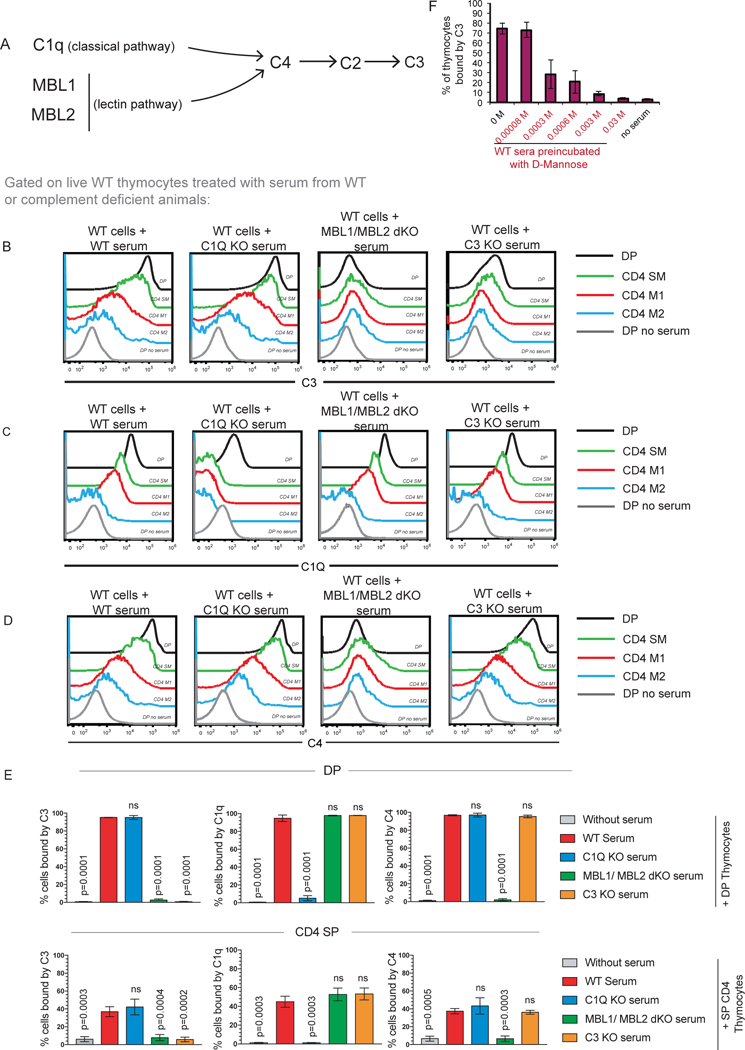
(A) Simplified schema of the classical and lectin pathways of complement activation; both pathways converge at C4 deposition. (B, C, D) Examination of C3, C1q, and C4 binding to wildtype (WT) DP, CD4 SM, CD4 M1, CD4 M2 cells after incubation with either WT, C1Q KO, MBL1 MBL2 dKO or C3 KO sera. Data is representative of 4 WT mice in total from 4 independent experiments. (E) Quantification of percent DP (black bars) and CD4 SP (orange bars) thymocytes bound by C3, C1Q and C4. Data is averaged from 4 mice in total from 4 independent experiments and error bars represent standard error of mean (SEM). P values were calculated by one-way ANOVA. (F) Pre-incubation of sera with varying concentrations of D-mannose blocks C3 deposition on thymocytes. Data was averaged from two independent experiments with 3 mice in total and error bars indicate SEM.
MBL2 is required for C3 and C4 deposition and MBL1 is dispensable.
Mice express two highly homologous MBL genes (MBL1 and MBL2), which are thought to be redundant in function (20). Sera from either MBL1 KO or MBL2 KO mice were incubated with WT thymocytes (Figure 3, gating strategy in Supplementary Figure 3). Sera from MBL1 KO mice led to similar levels of C3, C1q and C4 deposition on WT thymocytes as WT sera (Figure 3A–C). On the other hand, WT thymocytes incubated with sera from MBL2 KO mice exhibited negligible C3 and C4 deposition, similar to WT thymocytes incubated with C3 KO sera, while C1q binding was unaffected (Figure 3A–D). Thus, although MBL1 and MBL2 are thought to be redundant (20), MBL2 is required for C3 deposition on thymocytes while MBL1 is not.
Figure 3. MBL2 but not MBL1 is required for C3 deposition.
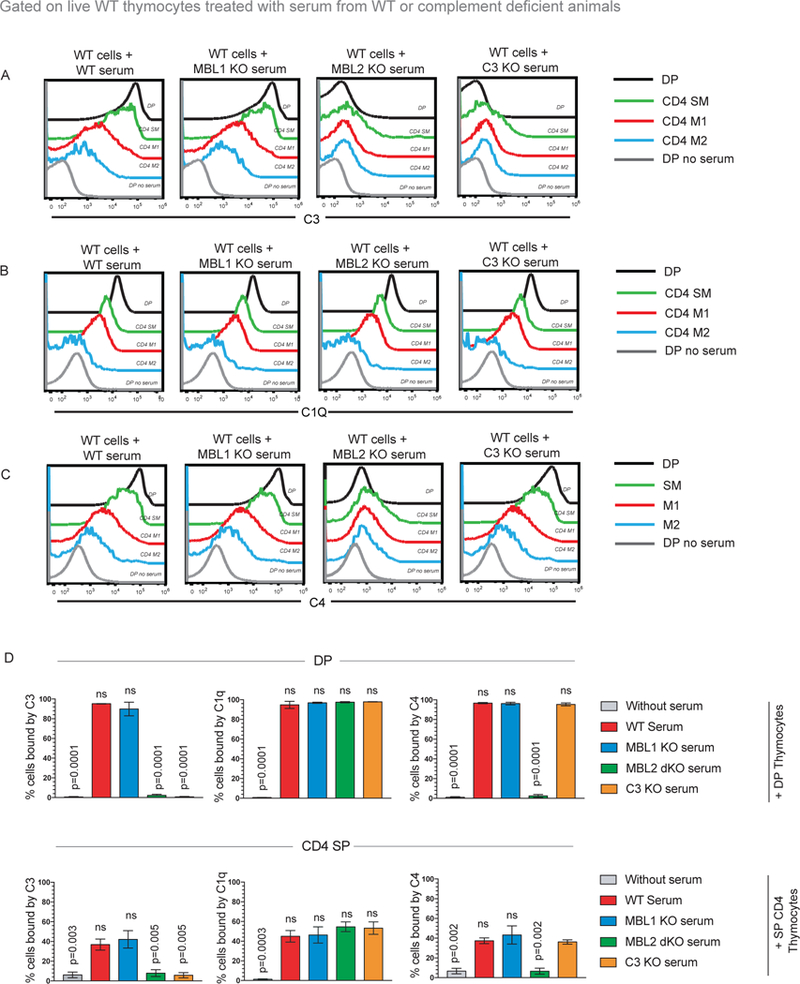
(A, B, C) Examination of C3, C1q or C4 binding on wildtype (WT) DP, CD4 SM, CD4 M1, CD4 M2 stages incubated with WT, C1Q KO, MBL1 KO, MBL2 KO or C3 KO sera. Data is representative of at least 4 independent experiments with at least 4 mice in total. (D) Quantification of percentage of DP and CD4 SP thymocytes bound by C3, C1Q or C4 in the presence of WT, C1Q KO, MBL1 KO, MBL2 KO or C3 KO sera. Data is averaged from 4 mice in total from 4 independent experiments and error bars represent (SEM). P values were calculated by one-way ANOVA comparing percent bound by C3, C1q and C4 between WT and complement KO sera in either DP or CD4 SP thymocytes.
Disruption of the lectin and classical pathways prevents C3 deposition, but does not rescue survival of NKAP-deficient naïve T cells.
The transcriptional regulator NKAP is critical for T cell maturation (15, 18). The majority of NKAP-deficient naïve peripheral T cells are recent thymic emigrants (RTEs). Very few NKAP-deficient mature naïve T cells (MNTs) remain and exhibit significant deposition of complement proteins C1q, C3 and C4 on their cell surface. Deletion of the classical pathway by crossing C1q KO and CD4-cre NKAP cKO mice did not restore the number or percent of naïve CD4 T cells to normal (Fig 4A, 4B) or prevent deposition of C3 on CD4+CD62L+CD44−CD45RB+ MNTs (Figure 4C, gating strategy shown in Supplement Figure 4). Given the observation that MBL2 was required for complement deposition on immature thymocytes incubated with serum, we next examined whether deficiency of the lectin pathway could rescue naïve T cell numbers in CD4-cre NKAP cKO mice by preventing C3 deposition on MNTs. MBL1/MBL2 dKO mice were crossed to CD4-cre NKAP cKO mice to ablate the lectin pathway. In the absence of both MBL proteins, C3 deposition still occurred on NKAP-deficient MNTs, in stark contrast to the observations made with developing thymocytes (Figure 4C). The elimination of both classical and lectin pathways by crossing CD4-cre NKAP cKO mice to C1q KO MBL1/MBL2 dKO mice was required to prevent C3 deposition on NKAP-deficient MNTs (Figure 4C). Despite prevention of C3 binding, there were no enhancement of naïve peripheral NKAP-deficient T cell proportions or numbers (Figure 4A, 4B). In addition, peripheral T cell numbers and proportions were not rescued when C3 KO mice were crossed to CD4-cre NKAP cKO mice. Thus, complement proteins likely act as a clearance mechanism of NKAP-deficient peripheral T cells rather than the primary cause of cell death.
Figure 4. C3 deposition on NKAP-deficient naïve T cells requires both the lectin and classical pathways.
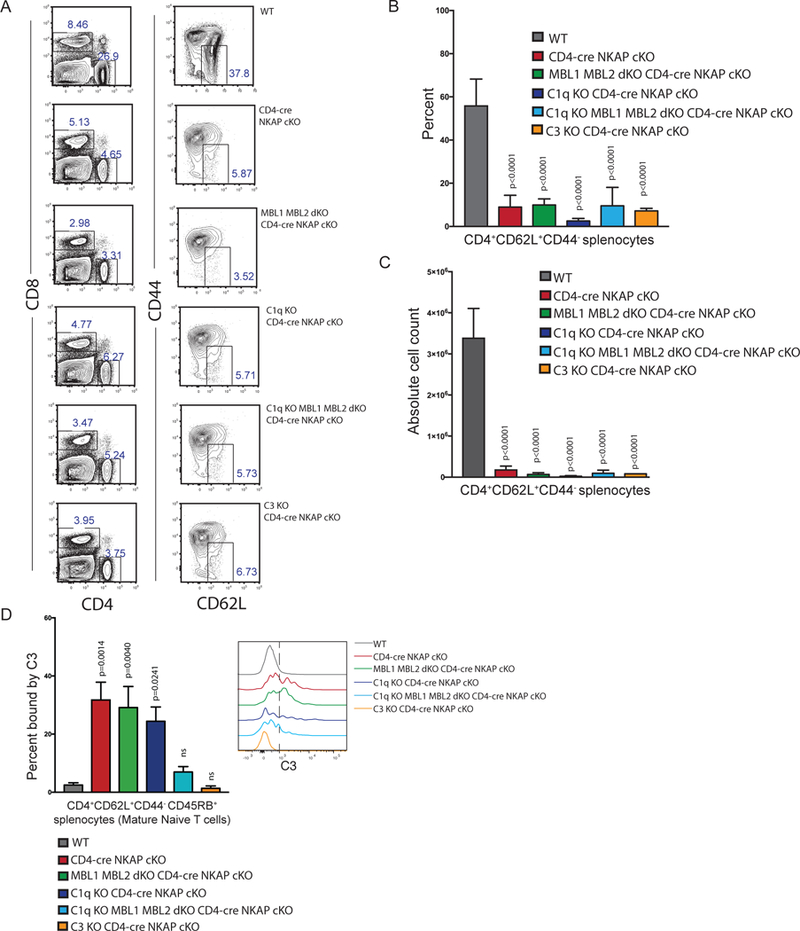
(A) Representative frequencies of CD4+ and CD4+CD62L+CD44− splenocytes in WT, CD4-cre NKAP cKO, MBL1 MBL2 dKO CD4-cre NKAP cKO, C1q KO CD4-cre NKAP cKO, C1q KO MBL1 MBL2 dKO CD4-cre NKAP cKO, and C3 KO CD4-cre NKAP cKO mice. (B, C) Enumeration of absolute cell counts and frequencies of CD4+CD62L+CD44− splenocytes in WT, CD4-cre NKAP cKO, MBL1 MBL2 dKO CD4-cre NKAP cKO, C1q KO CD4-cre NKAP cKO, C1q KO MBL1 MBL2 dKO CD4-cre NKAP cKO, and C3 KO CD4-cre NKAP cKO mice. (D) Enumeration of frequencies of CD4+CD62L+CD44−CD45RB+ mature naïve splenocytes bound by C3 from WT, CD4-cre NKAP cKO, MBL1 MBL2 dKO CD4-cre NKAP cKO, C1q KO CD4-cre NKAP cKO, C1q KO MBL1 MBL2 dKO CD4-cre NKAP cKO, and C3 KO CD4-cre NKAP cKO mice. All data are representative of at least 3 independent experiments with at least 6 mice per genotype in total. P-values were calculated by one-way ANOVA with multiple comparisons. Error bars for all figures represent SEM.
NKAP-deficient naïve T cells have increased lipid peroxidation, a defining feature of ferroptosis.
In order to determine the underlying cause for peripheral T cell deficiency in CD4-cre NKAP cKO mice, additional mechanisms of cell death and mitochondrial health were examined in CD4+CD62L+CD44− naïve T cells from WT, CD4-cre NKAP cKO and C1q KO MBL1/MBL2 dKO CD4-cre NKAP cKO mice (Figure 5). Levels of Bcl2 expression and cleaved caspase-3 were equivalent in CD4+CD62L+CD44− naïve T cells from mice of all three genotypes (Figure 5A). Binding of YO-PRO-1, a large dye that indicates presence of pores in the cell membrane (21), was also unchanged for all three genotypes (Figure 5B). Parameters of mitochondrial and cellular health, including presence of mitochondrial superoxide (MitoSOX), intact mitochondrial membrane potential (TMRM), cellular oxidative stress (CellROX) and mitochondrial content (MitoTracker) were examined. However, there were no notable differences between naïve T cells from WT, CD4-cre NKAP cKO, or C1q KO MBL1/MBL2 dKO CD4-cre NKAP cKO mice (Figure 5C). Although there is no difference in percentage of apoptotic (Annexin V+) or dead (FVD+) cells within freshly harvested and stained naïve T cells from CD4-cre NKAP cKO mice compared to WT mice (18), after 1 hour in culture at 37° C, live naïve T cells from CD4-cre NKAP cKO and C1q KO MBL½ KO CD4-cre NKAP cKO mice were slightly decreased in frequency, but this was not significant (Figure 5D). Finally, ferroptosis, a form of regulated cell-death caused by dysregulated iron metabolism was analyzed. Iron-overload results in lipid peroxidation, the defining characteristic of ferroptosis (22–27). Live naïve T cells from CD4-cre NKAP cKO and C1q KO MBL1/MBL2 dKO CD4-cre NKAP cKO mice had significantly increased lipid peroxidation as measured using C11-BODIPY 581/591, a lipophilic fluorescent molecule that shifts fluorescence when oxidized after reaction with peroxyl radicals (Figure 5E). Similarly, Liperfluo, another lipophilic ROS sensor indicated increased hydroxyl-peroxides present in NKAP-deficient T cells from complement-sufficient and deficient backgrounds (Figure 5F). To examine whether lipid peroxidation was reversible, WT, CD4-cre NKAP cKO and C1q KO MBL1/MBL2 dKO CD4-cre NKAP cKO naïve CD4 T cells were treated with alpha-tocopherol (αToc), the most abundant form of vitamin E and a lipid soluble antioxidant (26–28). Naïve CD4 T cells from CD4-cre NKAP cKO and C1q KO MBL1/MBL2 dKO CD4-cre NKAP cKO mice showed a significant decrease in oxidation of C11-BODIPY compared to untreated cells indicating a reduction in lipid peroxidation (Figure 5G). Next, the ferroptosis inhibitor Ferrostatin-1 was used to assess mitigation of lipid peroxidation (28). Similar to treatment with αToc, Ferrostatin-1 treated naïve CD4 T cells from CD4-cre NKAP cKO and C1q KO MBL1/MBL2 dKO CD4-cre NKAP cKO mice showed a significant decrease in oxidation of C11-BODIPY compared to untreated cells indicating decreased lipid peroxidation. Treatment of naïve CD4 T cells from WT, CD4-cre NKAP cKO and C1q KO MBL1/MBL2 dKO CD4-cre NKAP cKO with αToc and Ferrostatin-1 further reduced accumulation of lipid peroxides indicating synergistic anti-oxidant effects of both inhibitors of ferroptosis. Previously, αToc and Ferrostatin-1 were shown to reverse ferroptosis stemming from genetic ablation of GPX4 in CD4 T cells (27). Glutathione peroxidase 4 (GPX4) limits ferroptosis and T cell specific GPX4 deletion does not hinder T cell development but results in increased lipid peroxidation and peripheral T cell lymphopenia as a result of ferroptosis (27). However, GPX4 expression and intracellular glutathione levels, a substrate of GPX4, did not differ between naïve T cells from WT, CD4-cre NKAP cKO and C1q KO MBL1/MBL2 dKO CD4-cre NKAP cKO mice, suggesting ferroptosis independent of GPX4 (Figure 5H).
Figure 5. NKAP-deficient naïve T cells have increased lipid peroxidation.
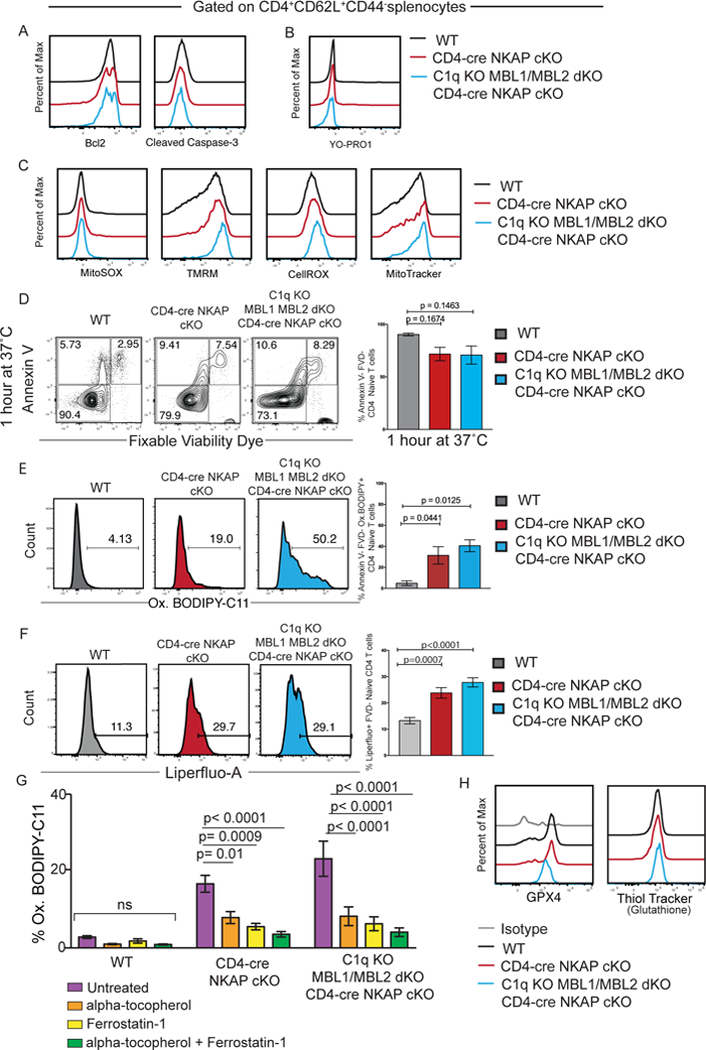
(A) Bcl2 and Cleaved Caspase-3 expression. (B) Levels of YO-PRO1. (C) Levels of MitoSOX, TMRM, CellROX, and MitoTracker. (D) Examination of frequencies of Annexin-V−FVD− (live), Annexin-V+FVD− (dying), and Annexin-V+FVD+ (dead) CD4+CD62L+CD44− splenocytes from WT, CD4-cre NKAP cKO, and C1q KO MBL1 MBL2 dKO CD4-cre NKAP cKO mice after 1 hour in culture. (E) Frequencies of oxidized BODIPY C-11 (Ox.BODIPY) CD4+CD62L+CD44−Annexin-V−FVD− splenocytes (non-apoptotic and non-necrotic naïve T cells) after 1 hour in culture. (F) Frequencies of Liperfluo positive CD4+CD62L+CD44− splenocytes after 30 minutes in culture. (G) Frequencies of oxidized BODIPY C-11 (Ox.BODIPY) CD4+CD62L+CD44−FVD− splenocytes treated without and with ferroptosis inhibitors alpha-tocopherol (Vitamin E), Ferrostatin-1 and a combination of alpha-tocopherol and Ferrostatin-1. (H) GXP4 expression and levels of glutathione. Flow cytometry plots are representative of at least 3 independent experiments with at least 3 mice. Bar graphs show the mean of 3 independent experiments with at least 3 mice per group in total. P-values were calculated by one-way ANOVA with multiple comparisons. Error bars represent SEM.
Discussion:
Our data demonstrates that murine thymocytes gradually gain resistance to complement proteins as C3, C4 and C1q deposition decreases with increasing T cell maturation (Figure 1). Cumulatively, these results provide evidence that complement resistance is achieved during maturation in the complement-privileged thymus and prior to T cell egress into blood, which harbors complement proteins. Mechanistically, the lectin pathway is required for C3 and C4 deposition on thymocytes while the classical pathway is dispensable suggesting that exposed mannose residues of developing T cells underlies complement susceptibility (Figure 2). The lectin pathway consists of two key initiating molecules, MBL1 and MBL2. It is known that serum MBL binds to immature BALB/c thymocytes and this binding decreases during development (29), but whether MBL protein binding is the primary determinant of C3 and C4 deposition has not been previously demonstrated. Further, C3 and C4 binding are dependent on MBL2 but not MBL1 (Figures 3). Although murine MBL1 and MBL2 are structurally homologous (20), their ability to recognize or bind to specific glycosylation patterns on thymocytes’ surface may vary as indicated by our data. Murine and primate MBL1 and MBL2 are thought to be products of a gene duplication event (20). While the murine MBL1 and MBL2 genes encode functional proteins that are thought to be redundant, in humans only MBL2 encodes a protein product and MBL1 is silenced (20). Whether MBL2 may interact with human thymocytes is unknown. On the other hand, the observation that MBL1 cannot substitute MBL2 in mice for C3 deposition on thymocytes is a novel finding and challenges the existing paradigm (20) that MBL1 and MBL2 are likely functionally interchangeable and redundant. The exclusion, of complement proteins from the thymus is likely important to ensure proper T cell development and maturation, as immature thymocytes are extremely susceptible to complement-mediated attack. However, it remains unclear how the thymus achieves complement exclusion, and it is unknown why the immature thymocyte surface remains largely desialylated until the single positive stage (11, 15, 16).
Our data also raises the question of functional significance of complement susceptibility of immature T cells that egress into the periphery. One possible explanation is that complement proteins selectively target T cells that fail maturation. NKAP-deficient T cells exhibit a maturation block characterized by elimination in the periphery at the RTE and MNT stages, and opsonization by complement proteins (15, 18). It has previously been shown that stripping of sialic acids using neuraminidase renders erythrocytes susceptible to complement attack and consequent death, providing direct evidence of the importance of sialylation in complement protection (30). NKAP deficient T cells fail to upregulate sialic acid transferases, suggesting faulty glycosylation as the initiator of complement deposition (15). Previously, to determine whether absence of C3 could restore long-term survival of NKAP-deficient T cells, Rag GFP WT and Rag GFP CD4-cre NKAP cKO derived bone marrow were transferred into irradiated C3 KO or WT recipients to assess kinetics of maturation in C3 deficient and sufficient backgrounds, respectively. Some NKAP deficient RTEs (GFP+) were able to transition to the MNT (GFP−) stage as higher frequencies of NKAP deficient MNTs (GFP−) were found in the spleen of C3 KO recipients compared to WT recipients. NKAP deficient RTEs also expressed increased levels of Qa2 and CD55 in C3 KO recipients compared to WT recipients suggesting survival was prolonged (15). However, NKAP deficient RTEs were present at a higher frequency compared to WT RTEs in C3 KO recipients suggesting that maturation was not completely restored and transition to MNT stage was incomplete. Unforeseen effects of bone marrow transplantion on maturation or possible contribution of C3 production by hematopoietic cells could potentially halt transition of NKAP-deficient RTEs to MNTs. Crossing CD4-cre NKAP cKO mice to C3 KO mice failed to restore peripheral T cell lymphopenia, and therefore prompted us to examine additional mechanisms that could explain NKAP-deficient T cell paucity.
By crossing CD4-cre NKAP cKO mice to C1q KO mice, MBL½ dKO mice or C1qKO MBL½ dKO mice, we investigated the relative contributions of the lectin and classical pathways to C3 deposition on peripheral NKAP-deficient T cells. NKAP deficient T cells were not restored to normal frequency or numbers in any of these crosses (Figures 4, 5). In contrast to thymocytes incubated with serum in vitro, ablation of the lectin pathway is not sufficient to prevent C3 deposition on NKAP-deficient T cells in vivo. Rather, loss of both the classical and lectin pathway was required. Thus, combined deficiencies of C1q, MBL1 and MBL2 prevented C3 deposition but failed to rescue naïve T cell persistence. This suggested that another mechanism of cell death must be the primary cause of CD4-cre NKAP cKO RTE and MNT clearance. However, the possibility of substitution of thrombin as a C5 convertase (31), or the recognition of the desialylated cell surface and engulfment by resident macrophages (10) as mechanism of NKAP deficient T cell clearance cannot be ruled out.
In this study, we used a number of assays to assess the metabolic health of NKAP-deficient T cells, along with alternative modes of cell death, including ferroptosis. While parameters of mitochondrial health and apoptosis were comparable between CD4-cre NKAP cKO, C1q KO MBL1 MBL2 dKO CD4-cre NKAP cKO and WT naïve T cells, levels of lipid peroxidation were significantly increased as assessed by two different fluorescently labeled lipophilic dyes. The use of BODIPY-C11 is a well-established indicator of ferroptosis, and Liperfluo, which becomes fluorescent due to the presence of hydroxyl-peroxides confirmed that NKAP-deficient T cells have significantly increased levels of lipid peroxidation (22–26, 29). Lipid peroxidation could be reversed by the ferroptosis inhibitors αToc and Ferrostatin-1 providing further evidence that NKAP-deficiency leads to oxidative stress causing enhanced lipid peroxidation, an essential feature of ferroptosis. However, the levels of neither GPX4 nor glutathione (GSH), known inhibitors of ferroptosis (27, 32), were altered in NKAP-deficient cells. Although levels of lipid peroxidation can increase due to decreased GPX4 expression or decreased levels of its cofactor GSH, multiple alternative genetic and environmental factors independent of GPX4 and GSH, although not overt regulators of ferroptosis, can also influence sensitivity to ferroptosis (25). For example, DP thymocytes express high levels of the cysteine/glutamate transporter, or system Xc- (SLC7A11), but this decreases to low expression in SP thymocytes and peripheral T cells (Immgen database). System Xc- imports cystine into cells, which is then reduced to cysteine, and acts in conjunction with glutamate to produce glutathione, the cofactor for GPX4 important for maintaining low levels of lipid peroxides and prevention of ferroptosis (25). A low level of system Xc- alone in peripheral T cells is not enough to induce ferroptosis in peripheral WT T cells, but it may synergize with NKAP deficiency to favor a ferroptotic fate. Although GPX4 deletion causes peripheral T cells to undergo ferroptosis, rate limited enzymatic activity of GPX4, or limited GSH availability could feasibly be overwhelmed by other genes or environments that increase susceptibility to ferroptosis, including but not limited to the regulation of mitochondrial or cellular free iron abundance by iron transporters, or abundance of lipid peroxides by arachidonate lipoxygenases (ALOXs) (22, 24–25). Taken together, ferroptosis is a highly complex process involving amino acid, glutathione and lipid metabolic pathways that are affected by several genes and environments (26). We propose that NKAP deficiency may prompt sensitivity to ferroptosis independent of the glutathione/GPX4 pathway, as our data suggests.
While it is known that complement proteins mediate clearance of apoptotic and necrotic cells (3), it has not been shown whether they also mediate clearance of cells undergoing ferroptosis. As NKAP deficient T cells in complement-deficient (combined classical and lectin pathways) mice undergo ferroptosis similar to complement-sufficient mice, it is likely that complement-mediated clearance is secondary to ferroptosis. While a causal link between ferroptosis and complement activation has not been established, both pathways have been implicated in disease settings such as cardiac and renal ischemic injuries as well as neurodegenerative disorders, suggesting co-occurrence (7, 24, 33). Interestingly, a recent study demonstrated heme-mediated complement activation on endothelial cells, raising the possibility that alterations in iron metabolism may precipitate complement attack (34).
Overall, this study presents an intriguing link between glycosylation, complement activation and ferroptosis. Our data provides evidence that T cells gain resistance to complement as a function of maturation in the thymus, a complement-privileged organ, and that complement binding to immature thymocytes is completely dependent on MBL2, After egress, WT thymocytes are protected from complement by their heavily sialylated cell surface. Previous studies by our lab demonstrated that NKAP-deficient T cells, which are less sialylated and immature after egress, are opsonized by complement in the periphery. Our present data expands on this observation and concludes that this is due to the underlying cell death process of ferroptosis, with complement aiding T cell clearance. The functional significance of MBL2’s potential interaction with developing T cells, and the mechanism by which NKAP increases susceptibility to ferroptosis will be investigated in the future.
Supplementary Material
Key Points:
In vivo, T cell maturation entails increasing resistance to complement proteins.
In vitro, MBL2 is required for complement deposition on immature thymocytes.
NKAP-deficient T cells that fail maturation die by ferroptosis, not by complement.
Acknowledgements:
We thank Derek Glaser for assisting with western blots for confirmation of complement knockout models and Michael Lehrke for critical reading of the manuscript.
This work was supported by NIH R01 AI083279 to V.S.S., Mayo Graduate School funds to B.D. and P.J.B., and the Mayo Clinic Wilbur T. and Grace C. Pobanz Predoctoral Research Fellowship awarded to P.J.B.
Abbreviations used
- C3
complement factor 3
- C4
complement factor 4
- C1q
complement factor 1q
- MBL
Mannose Binding Lectin
- DP
Double Positive
- SP
Single Positive
- RTE
Recent Thymic Emigrant
- MNT
Mature Naïve T cell
Footnotes
The authors declare no competing financial interests.
References
- 1.Delves PJ, and Roitt IM. 2000. The Immune System: First of Two Parts. N. Engl. J. Med 343: 37–49. [DOI] [PubMed] [Google Scholar]
- 2.Ricklin D, Reis ES, and Lambris JD. 2016. Complement in disease: a defence system turning offensive. Nat. Rev. Nephrol 12: 383–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sarma JV, and Ward PA. 2011. The complement system. Cell Tissue Res 343: 227–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elvington M, Liszewski MK, and Atkinson JP. 2016. Evolution of the complement system: from defense of the single cell to guardian of the intravascular space. Immunol. Rev 274: 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stephan AH, Barres BA, and Stevens B. 2012. The Complement System: An Unexpected Role in Synaptic Pruning During Development and Disease. Annu. Rev. Neurosci 35: 369–89. [DOI] [PubMed] [Google Scholar]
- 6.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SWM, and Barres BA. 2007. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 131: 1164–1178. [DOI] [PubMed] [Google Scholar]
- 7.La Bonte LR, Dokken B, Davis-Gorman G, Stahl GL, and McDonagh PF. 2009. The mannose-binding lectin pathway is a significant contributor to reperfusion injury in the type 2 diabetic heart. Diabetes Vasc. Dis. Res 6: 172–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iadecola C, Anrather J, Nat, Med, Ezekowitz RA, Sastry K, Bailly P, Warner A, 4 Eisen DP, and Ogden CA. 2011. Mannose-binding lectin—the forgotten molecule? Nat. Med 17: 796–808. [DOI] [PubMed] [Google Scholar]
- 9.Ritchie GE, Moffatt BE, Sim RB, Morgan BP, Dwek RA, and Rudd PM. 2002. Glycosylation and the complement system. Chem. Rev 102: 305–319. [DOI] [PubMed] [Google Scholar]
- 10.Miwa T, Zhou L, Kimura Y, Kim D, Bhandoola A, and Song W-C. 2009. Complement-dependent T-cell lymphopenia caused by thymocyte deletion of the membrane complement regulator Crry. Blood 113: 2684–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daniels MA, Hogquist KA, and Jameson SC. 2002. Sweet “n” sour: The impact of differential glycosylation on T cell responses. Nat. Immunol 3: 903–910. [DOI] [PubMed] [Google Scholar]
- 12.Hsu FC, Shapiro MJ, Dash B, Chen CC, Constans MM, Chung JY, Romero Arocha SR, Belmonte PJ, Chen MW, McWilliams DC, and Shapiro VS. 2016. An Essential Role for the Transcription Factor Runx1 in T Cell Maturation. Sci. Rep 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Despont JP, Abel CA, and Grey HM. 1975. Sialic acids and sialyltransferases in murine lymphoid cells: Indicators of T cell maturation. Cell. Immunol 17: 487–494. [DOI] [PubMed] [Google Scholar]
- 14.Hsu F, Belmonte PJ, Constans MM, Chen MW, McWilliams DC, Hiebert SW, and Shapiro VS. 2015. Histone Deacetylase 3 Is Required for T Cell Maturation. J. Immunol 195: 1578–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu F, Shapiro MJ, Chen MW, McWilliams DC, Seaburg LM, Tangen SN, and Shapiro VS. 2014. Immature recent thymic emigrants are eliminated by complement. J. Immunol 193: 6005–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clark MC, and Baum LG. 2012. T cells modulate glycans on CD43 and CD45 during development and activation, signal regulation, and survival. Ann. N. Y. Acad. Sci 1253: 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lambre CR, Thibon M, Le Maho S, and Di Bella G. 1983. Auto-antibody dependent activation of the autologous classical complement pathway by guinea-pig red cells treated with influenza virus or neuraminidase: in vitro and in vivo study. Immunology 49: 311–319. [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu F-C, Pajerowski AG, Nelson-Holte M, Sundsbak R, and Shapiro VS. 2011. NKAP is required for T cell maturation and acquisition of functional competency. J. Exp. Med 208: 1291–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dash B, Shapiro MJ, Chung JY, Romero Arocha S, and Shapiro VS. 2018. Treg-specific deletion of NKAP results in severe, systemic autoimmunity due to peripheral loss of Tregs. J. Autoimmun 89: 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seyfarth J, Garred P, and Madsen HO. The “involution” of mannose-binding lectin . [DOI] [PubMed]
- 21.Fujisawa S, Romin Y, Barlas A, Petrovic LM, Mesruh T Ning F Ke X, Garcia AR, Monette S, Klimstra DS, Erinjeri JP, Solomon SB, Manova-Todorova K, Sofocleous CT, Fujisawa S, Romin Y, Barlas ÁA, Turkekul ÁM, Fan ÁN, Xu ÁK, Manova-Todorova ÁK, Petrovic LM, Garcia AR, Erinjeri ÁJP, Solomon ÁSB, Monette S, and Klimstra DS. 2014. Evaluation of YO-PRO-1 as an early marker of apoptosis following radiofrequency ablation of colon cancer liver metastases. Cytotechnology 66: 259–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miyajima H, Kono S, Takahashi Y, and Sugimoto M. 2002. Increased Lipid Peroxidation and Mitochondrial Dysfunction in Aceruloplasminemia Brains. Blood Cells, Mol. Dis 29: 433–438. [DOI] [PubMed] [Google Scholar]
- 23.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, and Tang D. 2016. Ferroptosis: process and function. Cell Death Differ 23: 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu Y, Song J, Wang Y, Wang X, Culmsee C, and Zhu C. 2019. The Potential Role of Ferroptosis in Neonatal Brain Injury. Front. Neurosci 13: 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, and Stockwell BR. 2012. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149: 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Overholtzer M, Woerpel KA, Torti FM, Zhang DD, Ran Q, Salnikow K, Jiang X, Linkermann A, Fulda S, V Torti S, Hatzios SK, Rosenfeld CS, Tang D, Park J, Murphy ME, Kagan VE, Gascón S, Oyagi A, Toyokuni S, Dixon SJ, Noel K, and Pagnussat GC. 2017. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171: 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, and Kopf M. 2015. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med 212: 555–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, and Pratt DA. 2017. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci 3: 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uemura K, Yokota Y, Kozutsumi Y, and Kawasaki T. 1996. A unique CD45 glycoform recognized by the serum mannan-binding protein in immature thymocytes. J. Biol. Chem 271: 4581–4584. [DOI] [PubMed] [Google Scholar]
- 30.Durocher JR, Payne RC, and Conrad ME. 1975. Role of Sialic Acid in Erythrocyte Survival. Blood 45: 11–20. [PubMed] [Google Scholar]
- 31.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, and Ward PA. 2006. Generation of C5a in the absence of C3: a new complement activation pathway. Nat. Med 12: 682–687. [DOI] [PubMed] [Google Scholar]
- 32.Finkel T, and Holbrook NJ. 2000. Oxidants, oxidative stress and the biology of ageing. Nature 408: 239–247. [DOI] [PubMed] [Google Scholar]
- 33.Rocha-Ferreira E, and Hristova M. 2015. Antimicrobial peptides and complement in neonatal hypoxia-ischemia induced brain damage. Front. Immunol 6: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merle NS, Paule R, Leon J, Daugan M, Robe-Rybkine T, Poillerat V, Torset C, Frémeaux-Bacchi V, Dimitrov JD, and Roumenina LT. 2019. P-selectin drives complement attack on endothelium during intravascular hemolysis in TLR-4/heme-dependent manner. Proc. Natl. Acad. Sci. U. S. A 1–6. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.