

Abstract
Preterm birth is a major contributor to neonatal mortality and morbidity, and infection is a major risk factor. Chorioamnionitis, inflammation of the placenta and fetal membranes (FMs), is commonly observed in preterm birth and is characterized by neutrophil infiltration. However, interactions between FMs and neutrophils remain incompletely understood. The objectives of this study were to determine how FMs, with or without bacterial lipopolysaccharide (LPS) stimulation, affect neutrophil recruitment, activation, and the formation of neutrophil extracellular traps (NETs); and to elucidate the signaling mechanisms involved. Using a combination of in vitro, ex vivo, and in vivo approaches, we show that human resting FMs can directly recruit neutrophils and induce them to produce pro-inflammatory factors. Furthermore, neutrophils release vital NETs in response to FM-derived factors. LPS-stimulated FMs further augmented neutrophil recruitment, inflammatory cytokine/chemokine secretion, and vital NET release; and also induced ROS production and degranulation. We demonstrate a role for FM-derived TNF-α in mediating these effects through activation of neutrophil p38 MAPK. We propose that, during infection, while neutrophil recruitment and activation may neutralize pathogens, vital NET formation and prolonged neutrophil viability; and in combination with degranulation, ROS production and inflammatory chemokine/cytokine production, may contribute to tissue injury at the maternal-fetal interface.
Keywords: Inflammation, fetal membrane, pregnancy, preterm birth, neutrophils, NETs
INTRODUCTION
Preterm birth is a common pregnancy complication affecting over 10% of live births globally (1). Despite advances in neonatal care, preterm birth remains a major contributor of neonatal mortality and morbidity; and is associated with long-term complications for the offspring (2, 3). Although the causes of spontaneous preterm birth are incompletely understood, infection is an important risk factor (4). Chorioamnionitis, which is inflammation of the placenta and fetal membranes (FM), is seen in 40–70% of preterm births and commonly occurs in the presence of an infection (5, 6). Some proinflammatory cytokines that have been associated with preterm birth include IL-1β, IL-6 and TNF-α (7, 8). Chorioamnionitis is characterized by neutrophil infiltration to the FMs (9).
Neutrophils are short-lived immune cells that are rapidly recruited to sites of infection, following chemoattractants such as IL-8. They neutralize pathogens by phagocytosis; degranulation of antimicrobial enzymes, such as myeloperoxidase (MPO) and elastase; and the release of neutrophil extracellular traps (NETs) (10, 11). NETs are networks of extracellular chromatin decorated with histones and antimicrobial enzymes, like MPO and elastase, which entrap and neutralize microbes (10, 12). NET release has been reported in response to bacterial, viral, fungal and parasitic infections but their mechanisms of release differ greatly depending on the stimulus (12–19). Classic NET inducers, such as phorbol myristate acetate (PMA) and calcium ionophores, induce suicidal NETosis, where neutrophils die after the release of NETs (20), while Staphylococcus aureus has been reported to induce the release of vital NETs, whereby neutrophils retain their viability and function after NET extrusion (14).
While infection is a well-recognized trigger for preterm birth, and neutrophils are commonly observed in FMs from these deliveries, little is known about the mechanisms of neutrophil function at the FMs. In pregnant mice administered either high dose bacterial lipopolysaccharide (LPS) or E. coli, neutrophil depletion does not prevent preterm birth, suggesting that these immune cells do not contribute to the initiation of preterm birth (21, 22). These studies do, however, show that neutrophils contribute to utero-placental inflammation (21). Thus, it is important to understand the mechanisms governing neutrophil function at the FMs, particularly since chorioamnionitis, even in the absence of preterm birth, can seriously impact fetal heath and development (23, 24).
We previously reported that human FMs are very sensitive to low levels of bacterial LPS, a common factor in all gram-negative bacteria. Following exposure to low dose LPS, human FM explants secrete many chemotactic and pro-inflammatory factors that could potentially recruit neutrophils and other immune cells to the maternal-fetal interface (25, 26). However, to our knowledge, no studies have investigated the interaction between the human chorioamnion and neutrophils, and how this affects neutrophil function. Since neutrophil recruitment and accumulation is a key feature of chorioamnionitis, this study aimed to investigate the effects of human FMs, after exposure to LPS, on neutrophil recruitment, activation, and NET formation; and to determine the signaling mechanisms involved. Herein, we show that human FMs can directly recruit neutrophils and activate them to produce pro-inflammatory factors and to release vital NETs. LPS-stimulated FMs further augmented neutrophil recruitment, inflammatory cytokine/chemokine secretion, and vital NET release; and also induced ROS production and degranulation. Neutrophil recruitment and NET formation were confirmed in vivo in pregnant mice after exposure to LPS. Furthermore, we demonstrate a role for FM-derived TNF-α in mediating these effects through activation of the neutrophil MAPK signaling pathway.
MATERIALS AND METHODS
Study approvals
Human tissue collection was approved by the Yale University’s Human Research Protection Program (#0607001625). All animal studies were conducted in accordance with NIH guide for the Care and Use of Laboratory Animals and approved by Yale University’s Institutional Animal Care & Use Committee (#2016–11589).
Collection of human fetal membrane conditioned media (FM-CM)
Human FMs were collected from planned uncomplicated Caesarean deliveries between 37–41 weeks of gestation without labor or known infection/inflammation. FM explants, with the amnion and chorion intact, were prepared as previously described (25, 26). The next day, FM explants were treated with or without 1ng/ml LPS isolated from Escherichia coli O111:B4 (Sigma-Aldrich, St Louis, MO) in serum-free OptiMEM (Life Technologies, Grand Island, NY). We selected this dose of LPS as we previously reported that 1ng/mL was sufficient to induce a robust cytokine response by human FMs (25) and wanted a concentration that would not directly affect neutrophil function. After 24 hrs, cell-free FM conditioned media (CM) was collected under sterile conditions, spun at 2,000rpm for 10 mins and stored at −80°C. Conditioned media from three separate FMs were pooled together to reduce the influence of patient variability. In total, FMs from 42 women were used for collecting CM for this study. Prior to treating neutrophils, 2% fetal bovine serum (FBS; Gemini Bio-Products, West Sacramento, CA) was added into the pooled FM-CM.
Isolation of human neutrophils
Neutrophils were isolated from the peripheral blood of healthy, non-pregnant women and used immediately. Briefly, blood was collected by venepuncture into heparin-coated vacutainers (BD Biosciences, San Jose, CA) and red blood cells were sedimented using dextran (Sigma-Aldrich). Neutrophils were isolated by density centrifugation over a Histopaque 1077/1119 gradient (Sigma-Aldrich). Neutrophils were washed with Hank’s balanced salt solution without calcium or magnesium (Life Technologies) and hypotonic red blood cell lysis was performed. The isolated neutrophil fraction was >95% pure as determined by CD16b staining (#302005, Biolegend, San Diego, CA, 1:100 dilution) by flow cytometric analysis (FACs Calibur, BD Biosciences).
Neutrophil migration
Neutrophil migration was quantified using a two chamber transwell migration assay, as previously described (27). Briefly, 2×105 neutrophils were seeded into transwells with 3μm pores (top chamber) that are placed in a lower chamber (24 well plate) containing either: no treatment (NT; OptiMEM alone); untreated FM-CM (FM); LPS-stimulated FM-CM (FM+LPS); or as a control for the FM+LPS, LPS alone (1ng/ml). After 1 hr at 37°C, the percentage of neutrophils that migrated to the lower chamber was quantified using the QCM 24-well Colorimetric Cell Migration assay (Chemicon International, Temecula, CA) and optical densities read at 560 nm (27). For each experiment, data is presented relative to a 100% migration control consisting of neutrophils directly placed in the 24-well plate without any transwells.
Treatment of neutrophils with FM-CM
Neutrophils were treated with OptiMEM media as a no treatment (NT) control, untreated FM-CM (FM); LPS-stimulated FM-CM (FM+LPS); or as a control for the FM+LPS, LPS alone (1ng/ml) for 1 hr before changing the medium to fresh RPMI media with 2% FBS for 4 hrs at 37°C. Cell-free culture supernatants were collected and stored at −80°C. In some experiments, FM-CM was first incubated with 1μg/ml of either a neutralizing anti-TNF-α mAb (#MAB610, R&D systems, Minneapolis, MN) or an isotype-matched control IgG (R&D systems) for 1 hr at 20°C before adding to neutrophils. In other experiments, prior to exposure to FM-CM, neutrophils were pre-treated for 30 mins at 37°C with either: the NADPH oxidase inhibitor, Diphenyleneiodonium (DPI, 10μM, Sigma-Aldrich); the p38 MAP kinase inhibitor, SB203580 (1μM, Selleckchem, Houston, TX); the ERK inhibitor, SCH772984 (10nM, Selleckchem); or the JNK inhibitor, SP600125 (5μM, Selleckchem). The doses of inhibitors used have previously been reported to be effective in neutrophils (20, 28–30). The inhibitors were kept in the culture system during the whole treatment process through the media changes.
Neutrophil cytokine secretion and degranulation
Neutrophil culture supernatants were measured for IL-8/CXCL8 (ENZO Life Sciences, Farmingdale, NY) and TNF-α (R&D systems) by ELISA. The following cytokines/chemokines were quantified by multiplex analysis (Bio-Rad): G-CSF/CSF3, GM-CSF/CSF2, GRO-α/CXCL1, IL-1β, IL-6, IL-8, IL-10, IL-12, IL-17, IFN-γ, IP-10/CXCL-10, MCP-1/CCL2, MIP-1α/CCL3, MIP-1β/CCL4, RANTES/CCL5, and TNF-α (25, 26). Neutrophil degranulation was quantified by measuring the levels of myeloperoxidase (MPO) in the culture supernatants by ELISA (R&D systems).
Neutrophil extracellular trap (NET) release
Neutrophils were treated with OptiMEM media as a no treatment (NT) control, untreated FM-CM (FM); LPS-stimulated FM-CM (FM+LPS); or LPS alone (1ng/ml) for 5 mins to 3 hrs at 37°C. As a positive control in some cases, treatment with phorbol myristate acetate (PMA, 100nM) was included. NET release was quantified by measuring dsDNA release into culture supernatants using the Quant-iT PicoGreen dsDNA assay (Thermo Fisher Scientific, Waltham, MA), and fluorescence measured at 485/535nm using the Tecan Infinite M1000 Pro microplate reader (Thermo Fisher Scientific). NET release was also visualized by immunofluorescence as follows. Extracellular DNA was stained using Sytox Green (167nM, Thermo Fisher Scientific) or Hoechst (1μg/ml, Life technologies). Cells were then fixed in 4% paraformaldehyde (PFA) before staining with antibodies against citrullinated histone 3 (#ab5103, 1:100 dilution, Abcam, Cambridge, UK) and/or neutrophil elastase (#ab21595, 1:100 dilution, Abcam). Primary antibody binding was detected using the Alexa Fluor 564-labelled anti-rabbit antibody (1:500 dilution, Thermo Fisher Scientific). Cells were viewed using the Observer Z1 inverted fluorescence microscope (Zeiss, Thornwood, NY). Images were captured by the Volocity software (PerkinElmer, Waltham, MA) and merged on ImageJ (31).
Neutrophil viability
After treatments for 1 hr as described above, short-term neutrophil viability was investigated by Propidium iodide (PI) staining for dead cells followed by flow cytometric analysis (FACs Calibur, BD Biosciences). An additional positive control for cell death was prepared by treating neutrophils with 4% PFA. Long-term (24 hr) neutrophil viability was investigated using the CellTiter 96™ viability assay (Promega, Madison, WI) as per the manufacturer’s instructions. Briefly, neutrophils were treated for 24 hrs prior to adding the CellTiter substrate, MTS tetrazolium, for 4 hrs at 37°C. Optical densities at 490nm were recorded. All samples were assayed in triplicate and cell viability was presented as a fold change relative to the no treatment (NT) control.
Neutrophil phagocytosis assay
After treatments for 1 hr, pHRodo™ Red E. coli BioParticles (Thermo Fisher Scientific) that were opsonized with 10% human serum were added. Neutrophils were incubated with the beads for 2 hrs at 37°C before fixation with 4% PFA and flow cytometric analysis (FACs Calibur, BD Biosciences). The phagocytosis index was calculated by number of beads phagocytosed after treatment/number of beads phagocytosed by the NT control.
Neutrophil reactive oxygen species (ROS) production
Neutrophil superoxide production was quantified using luminol chemiluminescence (32, 33). Briefly, on fibronectin-coated white 96-well plates, neutrophils were incubated with luminol (300mM, Sigma-Aldrich) and horseradish peroxidase (40U/ml, Sigma) for 5 mins at 37°C. Then, treatments were added and chemiluminescence was immediately measured on an EnVision 2104 Multilabel plate reader (PerkinElmer, CT). Readings were made every second for 2 mins. Peak luminescence values were recorded.
Western blot
Neutrophils were treated for 30 mins at 37°C after which cells were lysed for protein and Western blot analysis was performed as previously described (34). Membranes were probed with the following primary antibodies from Cell Signaling Technology (Danvers, MA): phosphorylated (P) p38 MAPK (#9211S; 1:10,000 dilution); total (T) p38 MAPK (#9212; 1:30,000 dilution); P-ERK (#9101; 1:1000 dilution), T-ERK (#4695S; 1:2000 dilution); P-JNK (#9251; 1:1000 dilution); T-JNK (#9252A; 1:1000 dilution); P-p65 (#3033S; 1:10,000 dilution); or T-p65 (#8242S; 1:5000 dilution). Images were recorded and semi-quantitative densitometry was performed using the Gel Logic 100 (Eastman Kodak, Rochester, NY) and Carestream software (Carestream Molecular Imaging, CT). Levels of phosphorylated protein were normalized against the total amount of that specific protein.
Detection of NET formation in vivo
C57BL/6 mice were time-mated (The Jackson Lab, Bar Harbor, ME) and at gestational day 15.5, 20μg LPS or PBS was administered intraperitoneally. 6 hrs after injection, mice were sacrificed and the placentae and FMs were dissected together and fixed in 4% PFA. This dose of LPS administered ip to pregnant mice is able to increase FM expression of proinflammatory cytokines when compared to PBS controls (IL-1β: 2.4 fold; IL-6: 1.3 fold; KC (IL-8) 1.6 fold) (25 and unpublished data). 5μm tissue sections were analyzed by immunofluorescence using the following primary antibodies: Ly6G (#551459, BD Biosciences; 1:200 dilution); MPO (#AF3667, R&D systems; 1:200 dilution); citrullinated histone 3 (#ab5103, Abcam; 1:100 dilution). Primary antibody binding was detected using the appropriate Alexa Fluor-conjugated secondary antibodies (Thermo Fisher Scientific, 1:200 dilution), sections were counterstained with DAPI and mounted using Vectashield Antifade Mounting Medium. Sections were visualized using a Leica SP5 confocal microscope and images recorded using LAS AF software (Leica, Wetzlar, Germany).
Statistical analysis
Each experiment was performed at least three times using neutrophils from different women. The number of independent experiments that data were pooled from are indicated in the figure legends as “n=“. All data are reported as mean ± standard error of the mean (SEM) of pooled experiments. Statistical significance was set at p<0.05 and determined using Prism Software (Graphpad, Inc; La Jolla, CA). For normally distributed data, significance was determined using either one-way analysis of variance (ANOVA) for multiple comparisons or a t-test. For data not normally distributed, significance was determined using a non-parametric multiple comparison test or the Wilcoxon matched-pairs signed rank test.
RESULTS
Unstimulated FM-CM induce neutrophil migration and activation, and this is further augmented by LPS-stimulated FM-CM
In order to determine whether human FMs can recruit neutrophils and whether this is augmented during an infection, neutrophils were exposed to conditioned media (CM) from unstimulated or low dose LPS-stimulated FMs. The composition of this FM-CM has been previously characterised, showing that LPS exposure increased FM production of many chemokines and cytokines, including IL-6, IL-8 and TNF-α (25, 26). Unstimulated FM-CM significantly induced a 1.6±0.1 fold increase in neutrophil migration compared to the no treatment control (NT, OptiMEM media treated neutrophils). LPS-stimulated FM-CM further and significantly augmented this by 1.3±0.1 fold when compared to unstimulated FM-CM; and 2.0±0.2 fold when compared to the NT control (Figure 1A). To control for the LPS in the stimulated FM-CM, neutrophils were treated with low-dose LPS alone, which did not significantly affect neutrophil migration (Figure 1A).
Figure 1. LPS-stimulated FM-CM induce neutrophil migration and activation.
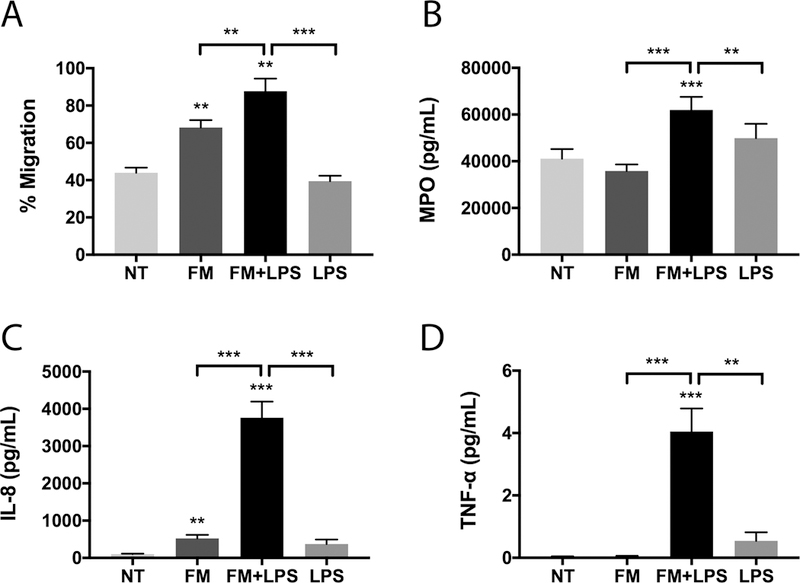
(A) Neutrophil migration towards no treatment (NT); unstimulated FM-CM (FM); LPS-stimulated FM-CM (FM+LPS); or LPS alone (n=8). (B-D) Neutrophils were exposed to NT; FM; FM+LPS; or LPS, after which activation was measured as: (B) degranulation of MPO (n=12); (C) secretion of IL-8 (n=12); and (D) secretion of TNF-α (n=12). *p<0.05, **p<0.01, ***p<0.001 compared to NT unless otherwise indicated.
In order to determine whether FM-CM also induced neutrophil activation, neutrophil degranulation and cytokine/chemokine secretion were measured. Unstimulated FM-CM did not significantly affect neutrophil myeloperoxidase (MPO) degranulation, while LPS-stimulated FM-CM significantly induced neutrophil degranulation by 1.6±0.1 fold compared with the NT control (Figure 1B). LPS alone had no effect on neutrophil MPO degranulation (Figure 1B). By ELISA, we demonstrated that unstimulated FM-CM significantly increased neutrophil IL-8/CXCL8 secretion by 5.7±0.9 fold compared with the NT control. LPS-stimulated FM-CM further and significantly augmented neutrophil IL-8 secretion by 8.6±1.0 fold when compared to unstimulated FM-CM, and by 42.5±6.0 fold when compared to the NT control (Figure 1C). In contrast, only LPS-stimulated, but not unstimulated, FM-CM induced neutrophil secretion of TNF-α by 118.4±21.6 fold relative to the NT control (Figure 1D). LPS alone had no effect on neutrophil IL-8 or TNF-α secretion (Figure 1C & D). Multiplex analysis validated these findings and demonstrated that unstimulated FM-CM also induced neutrophils to secrete increased levels of G-CSF/CSF3, IL-6, IL-17, IP-10/CXCL10 and MCP-1/CCL2 compared to the NT control (Figure 2). The secretion of all of the above factors were further and significantly augmented in the presence of LPS-stimulated FM-CM (Figure 2). In addition, LPS-stimulated FM-CM significantly induced neutrophil secretion of GM-CSF/CSF2, GRO-α/CXCL1, IL-1β, IL-10, IFN-γ, MIP-1α/CCL3, MIP-1β/CCL4 and RANTES/CCL5 (Figure 2). For the majority of our cytokine studies moving forward, we focused on IL-8 secretion since it is a major chemoattractant and marker of neutrophil activation (35); and its secretion by neutrophils was altered by both unstimulated and LPS-stimulated FM-CM.
Figure 2. Neutrophil cytokine secretion profile in response to FM-CM.
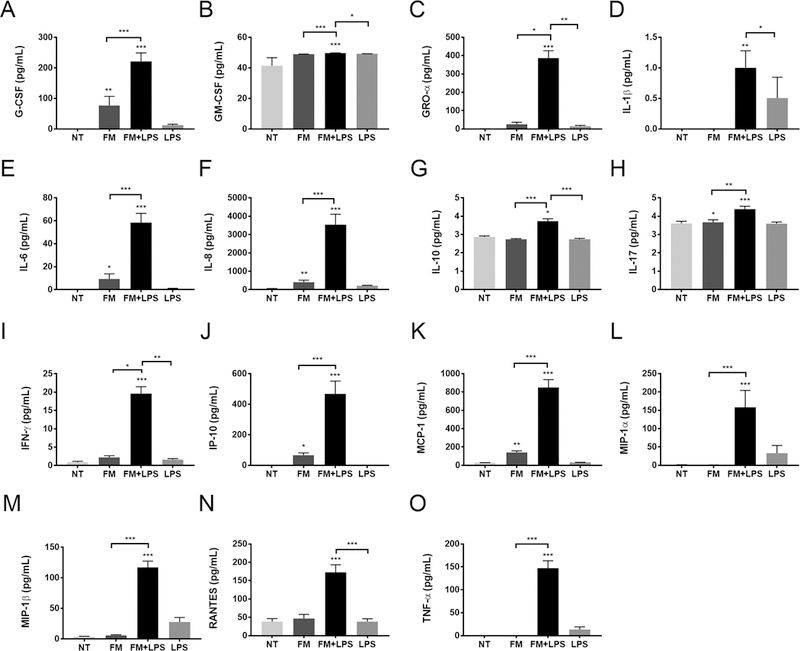
Neutrophils were exposed to no treatment (NT); unstimulated FM-CM (FM); LPS-stimulated FM-CM (FM+LPS); or LPS alone (n=11), after which supernatants were measured by multiplex analysis for (A) G-CSF; (B) GM-CSF; (C) GRO-α; (D) IL-1β; (E) IL-6; (F) IL-8; (G) IL-10; (H) IL-17; (I) IFNγ; (J) IP-10; (K) MCP-1; (L) MIP-1α; (M) MIP-1β; (N) RANTES; and (O) TNF-α. *p<0.05, **p<0.01, ***p<0.001 compared to NT unless otherwise indicated.
Unstimulated FM-CM induce NET release and this is further augmented by LPS-stimulated FM-CM
Having shown that FM-CM can recruit and activate neutrophils, we next investigated whether FMs could induce NET release. As shown in Figure 3, both unstimulated FM-CM and LPS-stimulated FM-CM induced the release of NETs as determined by (A and B) visualisation, and (C) quantification of extracellular DNA, while NT or LPS control conditions did not (Figure 3). The extracellular DNA extruded by neutrophils in response to both unstimulated and LPS-stimulated FM-CM co-localized with the NET markers, citrullinated histone 3 and neutrophil elastase, confirming NET formation (Figure 3A). Unstimulated FM-CM induced a 1.3±0.1 fold increase in DNA release compared to the NT control (Figure 3C). LPS-stimulated FM-CM further and significantly augmented DNA release by 1.3±0.1 fold when compared to unstimulated FM-CM, and by 1.6±0.1 fold when compared to the NT control (Figure 3C).
Figure 3. Neutrophils release NETs in response to FM-CM.
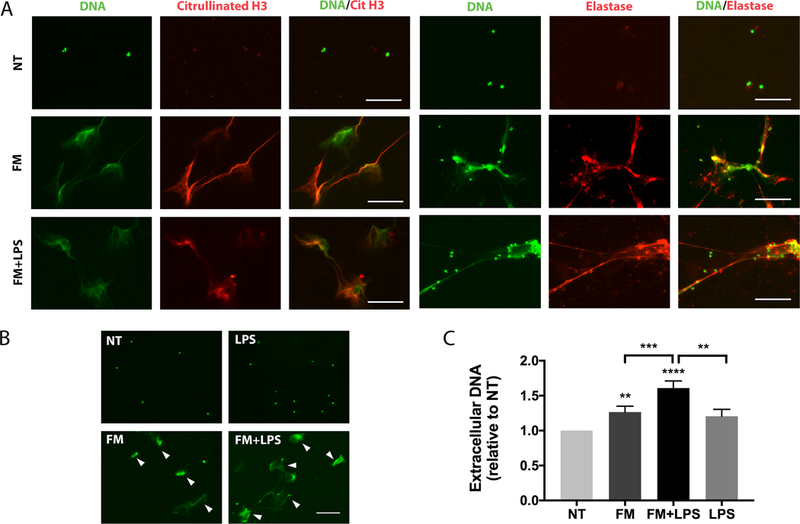
Neutrophils were exposed to no treatment (NT); unstimulated FM-CM (FM); LPS-stimulated FM-CM (FM+LPS); or LPS alone, after which: (A) Extracellular DNA (green) of released NETs colocalized with citrullinated histone 3 (red) or neutrophil elastase (red). Images are representative of three independent experiments. Scale bar= 100μm. (B) NETs were visualized using Sytox Green (arrowed). Images are representative of five independent experiments. (C) Extracellular DNA was quantified using PicoGreen (n=13). *p<0.05, **p<0.01, ***p<0.001 compared to NT unless otherwise indicated.
Both unstimulated and LPS-stimulated FM-CM induce vital NET formation
Neutrophils can form NETs through a suicidal or vital pathway which have different downstream consequences for neutrophil function. In order to determine which pathway was induced by FM-CM, the kinetics of NET release, neutrophil viability and function were assessed. FM-CM induced rapid NET release within 5 mins of stimulation, whereas PMA, a classic inducer of suicidal NETosis, induced visible NETs only after 30 mins (Figure 4A). 97.5±0.4% of neutrophils exposed to CM from resting FMs (FM) and 96.8±0.4% of neutrophils exposed to CM from LPS-stimulated FMs (FM+LPS) for 1 hr remained propidium iodide (PI)-negative which was not significantly different from untreated neutrophils (Figure 4B). Conversely, the exposure of neutrophils to PMA significantly increased the number of PI-positive neutrophils (Figure 4B). Furthermore, exposure of neutrophils to both unstimulated and LPS-stimulated FM CM significantly increased their long-term viability over 24 hrs when compared with untreated neutrophils, while PMA significantly reduced neutrophil viability (Figure 4C). Finally, neutrophils that have been exposed to both unstimulated and LPS-stimulated FM CM for 1 hr to induce NET formation retained their phagocytic ability compared to untreated neutrophils, while treatment with PMA significantly reduced neutrophil phagocytic capability (Figure 4D).
Figure 4. Neutrophils release NETs rapidly in response to FM-CM without compromising viability.
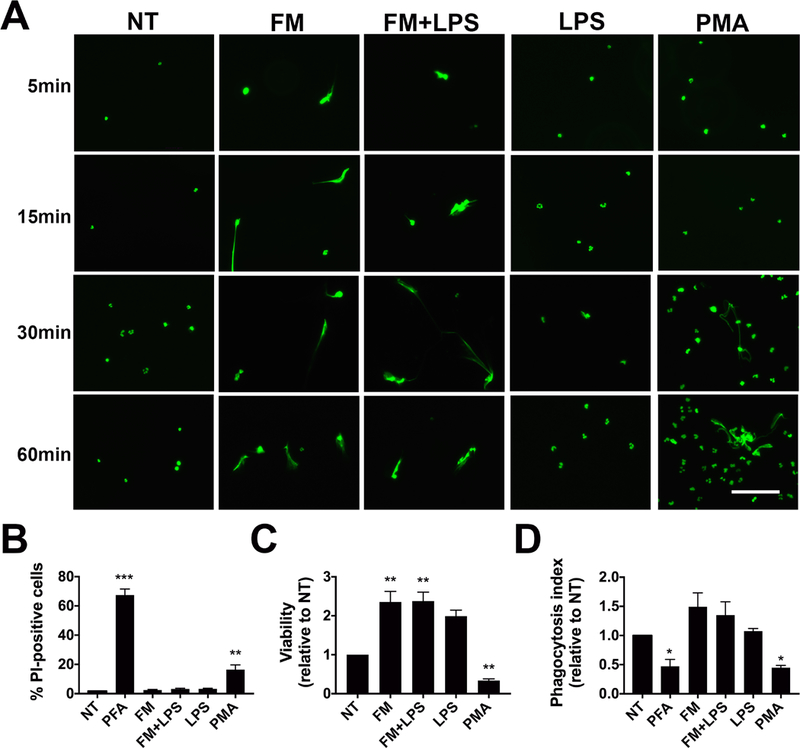
Neutrophils were exposed to no treatment (NT); PMA (100nM); LPS (1ng/ml); unstimulated FM-CM (FM); or LPS-stimulated FM-CM (FM+LPS) after which: (A) NETs were visualized using Sytox Green. Images are representative of three independent experiments. Scale bar= 100μm. (B) Neutrophil viability after treatment for 1 hr was quantified by propidium iodide (PI) staining (n=8). (C) Long-term neutrophil viability after treatment for 24 hrs was quantified by the Cell Titre Assay (n=10). (D) The phagocytosis index of treated neutrophils were measured using fluorescent E. coli bioparticles and normalized to NT (n=6). Neutrophils were treated with 4% PFA as a dead cell control. *p<0.05, **p<0.01, ***p<0.001 compared to NT unless otherwise indicated.
LPS-stimulated FM-CM induce neutrophil ROS production which contributes to downstream IL-8 secretion and NET release
Reactive oxygen species (ROS) production is an important signaling mechanism in neutrophils (36). LPS-stimulated FM-CM significantly increased ROS production by 2.9±0.6-fold when compared to the unstimulated FM-CM. There was no significant increase in ROS by the unstimulated FM-CM when compared to the NT control (Figure 5A). LPS alone has no effect on neutrophil ROS levels (Figure 5A). In order to investigate whether NADPH oxidase activation and subsequent ROS production mediates some of the neutrophil activity observed in response to LPS-stimulated FM-CM, diphenyleneiodonium (DPI), an inhibitor of NADPH oxidase was employed. DPI significantly reduced the ability of LPS-stimulated FM-CM to increase neutrophil IL-8 secretion by 9.0±2.3% (Figure 5B). The only other cytokine induced by LPS-stimulated FM-CM that was inhibited by DPI was IL-17 (data not shown). While DPI did not affect neutrophil MPO degranulation (Figure 5C), it did significantly inhibit NET release in response to LPS-stimulated FM-CM by 22.0±5.4% (Figure 5D). In the presence of DPI, the amount of NET release in response to LPS-stimulated FM-CM was not significantly different to that from the NT control (Figure 5D).
Figure 5. LPS-stimulated FM-CM induce neutrophil ROS production which contributes to downstream IL-8 secretion and NET release.
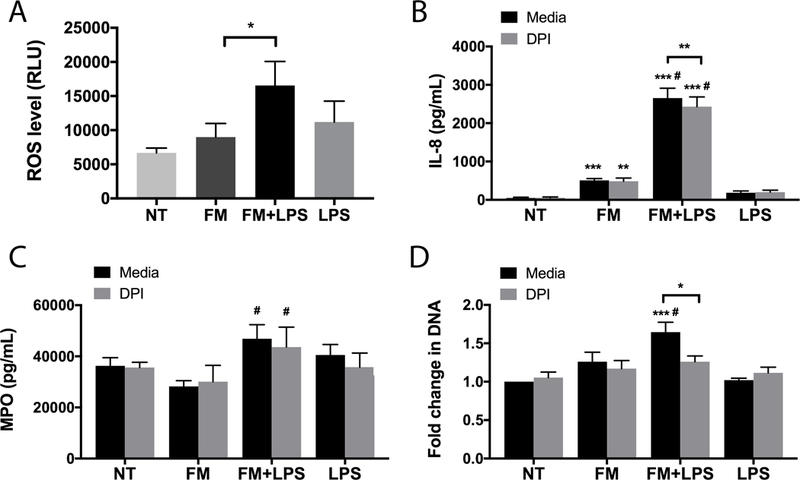
Neutrophils were exposed to no treatment (NT); unstimulated FM-CM (FM); LPS-stimulated FM-CM (FM+LPS); or LPS alone in the presence of media or the ROS inhibitor, diphenyleneiodonium (DPI). (A) Barchart shows ROS production by neutrophils under media conditions in relative light units (RLU, n=6). (B-D) In the presence of either media or DPI (n=7), barcharts show: (B) IL-8 secretion; (C) MPO degranulation; and (D) NET-associated extracellular DNA relative to the NT. *p<0.05, **p<0.01, ***p<0.001 compared to NT (under media or DPI conditions) unless otherwise indicated. #p<0.05 compared to FM (under media or DPI conditions).
LPS-stimulated FM-CM activates the neutrophil MAP kinase pathway
Since neutrophil activation in response to FM-CM was only partially mediated by ROS, we examined the intracellular pathways activated in neutrophils by FM-CM by investigating two central inflammatory pathways: NFκB and MAP kinase (MAPK). Neutrophils under NT control conditions expressed high basal levels of phosphorylated p65 (P-p65), a central mediator of the NFκB pathway. The levels of P-p65 were not significantly altered in neutrophils exposed to either unstimulated or LPS-stimulated FM-CM (Figure 6A). In contrast, LPS-stimulated FM-CM significantly increased the levels of P-p38 MAPK (Figure 6B); P-ERK (Figure 6C); and P-JNK (Figure 6D) by 26.7±4.9-fold; 147.5±67.8-fold; and 54.9±30.8-fold, respectively, when compared to the NT control; and by 13.0±2.844-fold; 28.7±9.0-fold; and 11.2±4.2-fold; respectively, when compared to unstimulated FM-CM.
Figure 6. LPS-stimulated FM-CM activate MAP kinase signaling pathways in neutrophils.
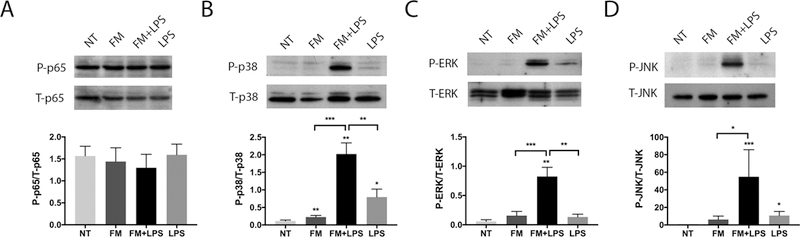
Neutrophils were exposed to no treatment (NT); unstimulated FM-CM (FM); LPS-stimulated FM-CM (FM+LPS); or LPS alone. After 30mins, proteins were collected and analysed by Western blot for phosphorylated (P) and total (T): (A) p65 NFκB; (B) p38 MAPK; (C) ERK; and (D) JNK (n=3). Blots are representative of three independent experiments and barcharts show levels of expression as determined by densitometry. *p<0.05, **p<0.01, ***p<0.001 compared to NT unless otherwise indicated.
Inhibition of the p38 MAPK and ERK, but not JNK, signaling pathways reduced the neutrophil response to LPS-stimulated FM-CM
MAPK signalling can be mediated by p38, ERK or JNK. In order to investigate which kinases were activated by LPS-stimulated FM-CM to induce neutrophil activation and NET production, specific inhibitors were employed. As shown in Figure 7A, SB203580, a specific p38 MAPK inhibitor, significantly reduced the ability of LPS-stimulated FM-CM to increase neutrophil (i) IL-8 secretion by 37.3±4.5%; (ii) MPO release by 27.3±6.2%; (iii) and NET release by 12.9±3.8%. As shown in Figure 7B, SCH772984, a specific ERK inhibitor, similarly reduced the ability of LPS-stimulated FM-CM to increase neutrophil (i) IL-8 secretion by 22.7±7.1%; (ii) MPO release by 24.8±6.9%; and (iii) NET release by 16.6±3.1%. In contrast, SP600125, a specific JNK inhibitor, did not affect neutrophil (i) IL-8 secretion; (ii) MPO degranulation; or (iii) NET release in response to LPS-stimulated FM-CM (Figure 7C). Multiplex analysis also revealed that inhibition of p38 MAPK significantly reduced the ability of LPS-stimulated FM-CM to increase neutrophil secretion of IL-6, IL-17, RANTES and TNF-α (Supplemental Figure 1). In contrast, inhibition of ERK (Supplemental Figure 2) or JNK (Supplemental Figure 3) only significantly inhibited neutrophil secretion of RANTES in response to LPS-stimulated FM-CM.
Figure 7. Neutrophil activation and NET release in response to LPS-stimulated FM-CM is mediated by p38 MAPK and ERK.
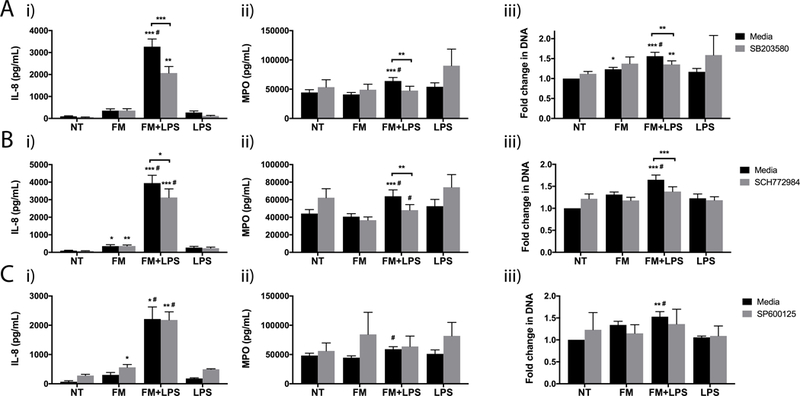
Neutrophils were exposed to no treatment (NT); LPS alone; unstimulated FM-CM (FM); or LPS-stimulated FM-CM (FM+LPS) in the presence of media or (A) the p38 MAPK inhibitor, SB203580 (n=11); (B) the ERK inhibitor, SCH772984 (n=9); or the JNK inhibitor, SP600125 (n=5). Barcharts show: (i) IL-8 secretion; (ii) MPO degranulation; and (iii) NET-associated extracellular DNA relative to the NT. *p<0.05, **p<0.01, ***p<0.001 compared to NT (under media or inhibitor conditions) unless otherwise indicated. #p<0.05 compared to FM (under media or inhibitor conditions).
FM-derived TNF-α induces neutrophil MAPK signaling and subsequent neutrophil activation and NET release.
TNF-α is a well-recognized pro-inflammatory cytokine that is associated with chorioamnionitis and preterm birth (37, 38). We previously reported that low dose LPS triggers FMs to secrete elevated levels of TNF-α (25, 26). Thus, we investigated whether TNF-α in the FM-CM could be responsible for activating neutrophils at the chorioamnion. To test this, a TNF-α neutralizing antibody was added to the FM-CM. Compared to the isotype control IgG, the presence of the anti-TNF-α antibody significantly reduced the ability of LPS-stimulated FM-CM to induce neutrophil expression of P-p38 MAPK by 61.0±6.0% (Figure 8A i & ii); P-ERK by 46.6±4.5% (Figure 8A i & iii); and P-JNK by 61.9±4.1% (Figure 8A i & iv). Furthermore, compared to control IgG, the presence of the anti-TNF-α antibody significantly reduced the ability of LPS-stimulated FM-CM to induce neutrophil IL-8 secretion by 35.3±3.8% (Figure 8B); MPO degranulation by 26.2±7.7% (Figure 8C); and NET release by 12.7±4.0% (Figure 8D). Multiplex analysis also revealed that the neutralization of FM-derived TNF-α significantly inhibited neutrophil secretion of GRO-α and TNF-α (Supplemental Figure 4).
Figure 8. FM-derived TNF-α activates neutrophil MAP kinase signaling and subsequent neutrophil activation and NET release.
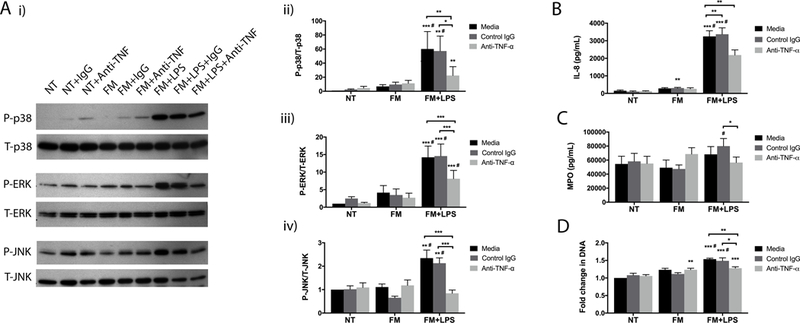
Neutrophils were exposed to no treatment (NT); unstimulated FM-CM (FM); or LPS-stimulated FM-CM (FM+LPS) in the presence of either media, an isotype control IgG (IgG), or a neutralizing anti-TNF-α mAb (Anti-TNFα). (A) Proteins were analysed by Western blot for phosphorylated (P) and total (T): p38 MAPK; ERK; and JNK (n=5). (i) Blots are representative of 5 experiments. (ii-iv) Barcharts show levels of expression as determined by densitometry. Supernatants were analysed for (B) IL-8 secretion (n=8); (C) MPO degranulation (n=8); and (D) NET-associated extracellular DNA relative to the NT (n=8). *p<0.05, **p<0.01, ***p<0.001 compared to NT (under media, control IgG, or anti-TNFα conditions) unless otherwise indicated. #p<0.05 compared to FM (under media, control IgG, or anti-TNFα conditions).
Placental neutrophil recruitment and NET release in response to LPS in vivo
In order to validate our in vitro findings, low dose LPS or PBS (control) was administered to pregnant mice to induce FM inflammation (25) (unpublished data) and immunofluorescence performed on placental and their associated FM tissues for the markers of neutrophils and NETs: MPO and citrullinated histone 3. By immunofluorescence and morphological analysis, neutrophils and NETs were identified in the placentae of both control mice (Figure 9A & B) and mice injected with LPS (Figure 9C & D). Quantification of immunofluorescence at 10x magnification revealed significantly more neutrophils recruited to the placental-FM unit of mice injected with LPS (10.5±0.8 neutrophils per mm2 of placental tissue, n=6) compared to control mice (6.7±0.8 neutrophils per mm2 of placental tissue, n=3, Figure 9E).
Figure 9. Placental neutrophil recruitment and NET release in vivo in response to LPS.
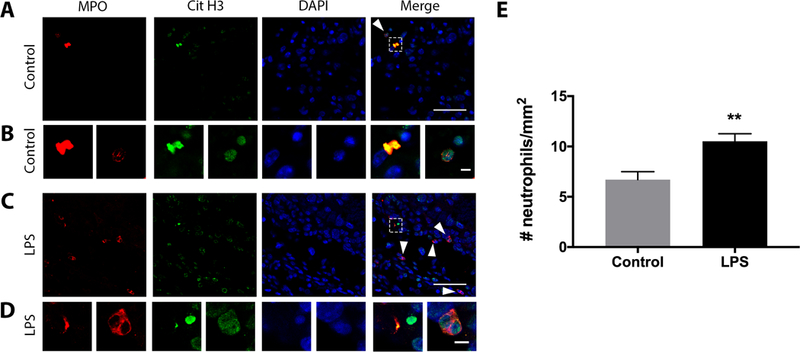
LPS or PBS (Control) was administered to pregnant mice on gestational day 15. 6 hrs later, placentae and associated FMs were dissected, fixed, and double immunofluorescence was performed. Neutrophils (arrowhead) and NETs (boxed) that were positive for MPO (red) and citrullinated histone 3 (Cit H3, green) were identified in the placentae of control mice (A & B, n=3) and mice exposed to LPS (C & D, n=6). Scale bar= 20μm (A & C), 5μm (C & D). Images are representative of 3 control and 6 LPS-injected mice. (E) Neutrophil recruitment to the placenta was quantified by double-staining at 10x magnification. **p<0.01 compared to control.
DISCUSSION
Preterm birth remains a major healthcare burden worldwide. Despite accounting for over 70% of all neonatal deaths and being associated with neurodevelopmental issues, and increased risk of metabolic and cardiovascular disease later in life, the triggers and pathogenesis of spontaneous preterm birth remain unclear (2). Neutrophil infiltration of the FMs is a central hallmark of chorioamnionitis that is commonly observed in preterm birth; however, the functions of these immune cells at the maternal-fetal interface, and whether the FMs themselves govern neutrophil recruitment and activation, have yet to be fully demonstrated. While clinical and experimental studies have shown correlations between bacterial infection, inflammation and preterm birth (39–41), the identified microbes associated with preterm birth are often common to the genital tract or placenta (42), and no one causative bacterium has been identified (43). Thus, this study used bacterial LPS to broadly model a gram-negative bacterial infection at the FMs. Using a combination of ex vivo, in vitro and in vivo approaches, we demonstrated a direct interaction between human FMs and neutrophils, demonstrating that FM-derived factors can directly recruit and activate neutrophils, as well as induce their release of vital NETs that do not compromise neutrophil viability or their downstream functions.
Somewhat surprisingly, we showed that conditioned media from resting human FM explants that had been previously characterized (25, 26) was able to recruit neutrophils; induce limited secretion of a restricted inflammatory profile (IL-8, IL-6, G-CSF, IP-10, MCP-1); and induce vital NET release. Compared to resting FMs, factors derived from LPS-exposed FM explants further increased neutrophil migration; the robust secretion of a wide range of inflammatory cytokines and chemokines; and vital NET release. Additionally, LPS-exposed FM explants also released factors that triggered neutrophil ROS production and degranulation. The dose of LPS used in this study was low to induce FM inflammation without directly activating neutrophils (25, 26). We specifically investigated the effects of FMs on neutrophil cytokine/chemokine secretion, degranulation and NET release as these are mechanisms that are involved in the resolution of an infection, yet may also inflict collateral tissue injury in the chorioamnion. Increased neutrophil recruitment and NET release at the maternal-fetal interface in response to LPS was also validated in a pregnant mouse model. Mechanistically, the increase in neutrophil IL-8 secretion, MPO degranulation and NET release in response to LPS-stimulated FMs was partially mediated by FM-derived TNF-α causing the activation of the p38 MAPK and ERK signaling pathways in neutrophils. Increased neutrophil IL-8 and NET release were also ROS-dependent.
Several proinflammatory cytokines have been implicated in the pathogenesis of preterm birth, including IL-1β, IL-6, IL-8 and TNF-α (37). In this study, we have shown for the first time that neutrophils can release these factors in response to conditioned media from LPS-stimulated FM explants. Additionally, in response to conditioned media from LPS-stimulated FM explants, neutrophils also released a number of other inflammatory cytokines and chemokines (IL-17, IFNγ, G-CSF, GRO-α, IP-10, MCP-1, MIP-1α, MIP-1β, RANTES) as well as anti-inflammatory IL-10. A local increase in these pro-inflammatory factors may further promote the infiltration of maternal neutrophils and other immune cells, as well as local prostaglandin production, leading to an increased risk of preterm birth (44, 45). Furthermore, IL-8 has also been shown to induce NET release by neutrophils in a paracrine manner (46).
Mechanistically, neutrophil secretion of IL-8 and IL-17 in response to LPS-stimulated FMs was ROS-dependent while the secretion of IL-6, IL-8, IL-17, RANTES and TNF-α was partially mediated by activation of the p38 MAPK pathway. In contrast, inhibition of the ERK signaling reduced neutrophil IL-8 and RANTES secretion, while inhibition of JNK signaling only reduced neutrophil RANTES secretion in response to LPS-stimulated FM-CM.
While inhibition of p38 MAPK and NADPH oxidase activity each significantly reduced neutrophil IL-8 secretion in response to LPS-stimulated FM-CM, no inhibitor completely abrogated the IL-8 response. This indicates that these identified signaling mechanisms are not the only pathways activated in neutrophils by FM-derived factors. PI3K/mTOR signaling, for example, may also be involved (47). Alternatively, compensatory mechanisms may be triggered in neutrophils when single signaling molecules are blocked, such that there is no overall change in the global secreted cytokine levels. Neutralization of FM-derived TNF-α also did not completely reverse neutrophil cytokine/chemokine secretion induced by LPS-stimulated FM-CM. Indeed, we have previously reported that in addition to TNF-α, LPS-stimulated FM explants also release increased levels of IL-1β and other pro-inflammatory cytokines (25, 26). Intra-amniotic administration of IL-1β to pregnant Rhesus Macaques has been reported to recruit neutrophils to the chorioamnion where they expressed IL-8 and TNF-α (48). Thus, FM-derived IL-1β may be another key factor in activating neutrophil cytokine/chemokine production at the maternal-fetal interface.
In contrast to cytokine/chemokine secretion, inhibition of p38 MAPK and ERK pathways almost completely reversed neutrophil MPO degranulation; and inhibition of ROS signaling almost completely reversed neutrophil NET release in response to conditioned media from LPS-stimulated FMs. Here, a role for FM-derived TNF-α was also found: TNF-α appeared to be a major driver of neutrophil degranulation, while it was only partially responsible for NET production. This supports the notion that different neutrophil activation processes are occurring at least partially independently of each other through distinct intracellular signaling mechanisms.
In contrast to the LPS-stimulated FM-CM, neutrophil IL-8 secretion and NET release in response to unstimulated FM-CM was found to be independent of TNF-α, ROS, or MAPK signaling. This suggests that neutrophil activation by resting FMs is mediated by a distinct signaling pathway(s) and by a factor other than TNFα. Indeed, unstimulated FMs have a distinct cytokine/chemokine secretion profile compared to LPS-stimulated FMs; unstimulated FMs secrete low, if any, TNFα (25). Since NET release can also be induced by IL-1β, IL-8 and IL-17 (8, 49), it is possible that one of these factors may be responsible for neutrophil activation in response to resting FMs. Indeed, we previously reported that resting FMs secrete appreciable levels of IL-8 and IL-17 (25).
Classic “suicidal” NET release in response to stimuli such as PMA and calcium ionophores induce neutrophils to undergo cell death and optimal NETs are observed after 3–4 hrs of treatment (20). In contrast to this, NET release in response to FM conditioned media occurred rapidly, after only 5 mins of exposure. This is consistent with the timeline of “vital” NET release by neutrophils in response to Staphylococcus aureus (14). To further explore this idea, we demonstrated that NET release by neutrophils in response to FM conditioned media did not compromise their short- or long- term viability, nor their ability to phagocytose. Indeed, long-term viability of neutrophils was enhanced in the presence of FM conditioned media. These observations were the case for conditioned media from both resting FMs and LPS-stimulated FMs. These strong lines of evidence support that FM conditioned media induces neutrophils to release NETs through a vital pathway, by budding and exocytosis of DNA instead of membrane lysis as in suicidal NETosis (50, 51). That FMs can prolong neutrophil survival is supported by an in vivo study of the choriodecidua of Rhesus Macaques (48). Nevertheless, the mechanisms of vital NET release in response to FM conditioned media were not completely distinct from that of suicidal NETosis as we also showed that NET release induced by FM conditioned media was dependent on ROS signaling and the activation of the p38 MAPK and ERK pathways, which are also activated in the case of PMA-induced suicidal NETosis (12, 20, 52, 53). In summary, FM-derived factors induce neutrophils to undergo vital NET release that does not cause neutrophil demise. This might explain why in chorioamnionitis, neutrophils can accumulate in the FM tissue, rather than being cleared. Thus, in the presence of an infection, while continued neutrophil recruitment and NET release may act to neutralize pathogens; neutrophil vital NET formation prolongs viability and function, and in combination with degranulation, ROS production and inflammatory cytokine/chemokine secretion, this may instead result in tissue injury and contribute to preterm birth.
While a previous study reported that systemic depletion of maternal neutrophils did not prevent immediate preterm birth induced by intrauterine administration of high dose LPS to pregnant mice at gestational day 17 (21), it is possible that recruited neutrophils may still be pathological by causing tissue damage progressively in a more physiological model of prolonged low-grade infection. Indeed, in the aforementioned study, neutrophil depletion reduced uterine IL-1β and TNF-α levels in response to LPS (21). Since resting FMs also recruited neutrophils and induced vital NET release, we speculate that in normal pregnancy, neutrophils may have a surveillance role at the FMs to protect the tissue against pathological infections. Indeed, we observed that neutrophils and NETs are found in low numbers in normal murine placentae at gestational day 15.5. Administration of low dose LPS to pregnant mice augmented neutrophil influx to the placental-FM unit and NET release. This further validates the clinical observation where NETs have been reported in FMs from women with acute chorioamnionitis who underwent spontaneous term or preterm labor (54). While we co-localized myeloperoxidase and citrullinated histone 3 to the NETs in vivo, these markers are also present in macrophage-derived extracellular traps (METs), thus it is possible that the ETs observed in vivo could also be derived from resident placental macrophages (55). Recently, neutrophil infiltration to the choriodecidua and NET release has been reported in a non-human primate model (19) as well as a mouse model of an ascending vaginal infection with Group B Streptococcus (13), however, our current study is the first to report NET release in murine placentae in response to a single bacterial factor, LPS. Furthermore, from our in vitro studies, it appears that human FM-derived factors induce neutrophils to undergo vital NET release which may have different roles to NETs formed via the classic suicidal pathway.
In summary, this study demonstrated a direct interaction between human FMs and neutrophils, and showed that FM-derived factors, released in response to bacterial LPS, can directly recruit and activate neutrophils, inducing them to secrete inflammatory cytokines/chemokines, degranulate and release vital NETs. Induction of neutrophil activation and NET release was mediated, in part, by FM-derived TNF-α leading to the activation of neutrophil MAPK signaling pathways, as well as by ROS production. Increased neutrophil activation and vital NET release may contribute to the inflammation and tissue injury observed at the maternal-fetal interface in women with chorioamnionitis.
Supplementary Material
KEY POINTS.
Resting FMs induce low levels of neutrophil cytokine secretion and vital NET release.
LPS-stimulated FMs augment neutrophil activation and vital NET release via TNF-α.
NET release induced by LPS-stimulated FMs was ROS- and p38 MAP kinase-dependent.
ACKNOWLEDGEMENTS
The authors would like to thank the staff of Labor and Delivery, Yale-New Haven Hospital and the Yale University Reproductive Sciences Biobank for blood and tissue collection.
This study was supported by a grant from the NIAID, NIH (R01AI121183, to VMA).
REFERENCES
- 1.Goldenberg RL, Culhane JF, Iams JD, and Romero R. 2008. Epidemiology and causes of preterm birth. Lancet 371: 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hack M, and Fanaroff AA. 2000. Outcomes of children of extremely low birthweight and gestational age in the 1990s. Semin. Neonatol 5: 89–106. [DOI] [PubMed] [Google Scholar]
- 3.Clark EA, and Varner M. 2011. Impact of preterm PROM and its complications on long-term infant outcomes. Clin. Obstet. Gynecol 54: 358–369. [DOI] [PubMed] [Google Scholar]
- 4.McElrath TF, Hecht JL, Dammann O, Boggess K, Onderdonk A, Markenson G, Harper M, Delpapa E, Allred EN, Leviton A, and Investigators ES. 2008. Pregnancy disorders that lead to delivery before the 28th week of gestation: an epidemiologic approach to classification. Am. J. Epidemiol 168: 980–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andrews WW, Goldenberg RL, Faye-Petersen O, Cliver S, Goepfert AR, and Hauth JC. 2006. The Alabama Preterm Birth study: polymorphonuclear and mononuclear cell placental infiltrations, other markers of inflammation, and outcomes in 23- to 32-week preterm newborn infants. Am. J. Obstet. Gynecol 195: 803–808. [DOI] [PubMed] [Google Scholar]
- 6.Tita AT, and Andrews WW. 2010. Diagnosis and management of clinical chorioamnionitis. Clin. Perinatol 37: 339–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Romero R, Mazor M, Sepulveda W, Avila C, Copeland D, and Williams J. 1992. Tumor necrosis factor in preterm and term labor. Am. J. Obstet. Gynecol 166: 1576–1587. [DOI] [PubMed] [Google Scholar]
- 8.Garley M, Jablonska E, Surazynski A, Grubczak K, Ratajczak-Wrona W, Iwaniuk A, Dabrovska D, Palka JA, and Moniuszko M. 2017. Cytokine Network & NETs. Folia Biol. (Praha) 63: 182–189. [DOI] [PubMed] [Google Scholar]
- 9.Menon R, Taylor RN, and Fortunato SJ. 2010. Chorioamnionitis--a complex pathophysiologic syndrome. Placenta 31: 113–120. [DOI] [PubMed] [Google Scholar]
- 10.Kolaczkowska E, and Kubes P. 2013. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol 13: 159–175. [DOI] [PubMed] [Google Scholar]
- 11.Nathan C 2006. Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol 6: 173–182. [DOI] [PubMed] [Google Scholar]
- 12.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, and Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science 303: 1532–1535. [DOI] [PubMed] [Google Scholar]
- 13.Kothary V, Doster RS, Rogers LM, Kirk LA, Boyd KL, Romano-Keeler J, Haley KP, Manning SD, Aronoff DM, and Gaddy JA. 2017. Group B Streptococcus Induces Neutrophil Recruitment to Gestational Tissues and Elaboration of Extracellular Traps and Nutritional Immunity. Front Cell Infect Microbiol 7: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, Robbins SM, Green FH, Surette MG, Sugai M, Bowden MG, Hussain M, Zhang K, and Kubes P. 2010. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol 185: 7413–7425. [DOI] [PubMed] [Google Scholar]
- 15.Rochael NC, Guimaraes-Costa AB, Nascimento MT, DeSouza-Vieira TS, Oliveira MP, Garcia e Souza LF, Oliveira MF, and Saraiva EM. 2015. Classical ROS-dependent and early/rapid ROS-independent release of Neutrophil Extracellular Traps triggered by Leishmania parasites. Sci. Rep 5: 18302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, and Chow VT. 2011. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol 179: 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, Uehata T, Iwasaki H, Omori H, Yamaoka S, Yamamoto N, and Akira S. 2012. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 12: 109–116. [DOI] [PubMed] [Google Scholar]
- 18.Byrd AS, O’Brien XM, Johnson CM, Lavigne LM, and Reichner JS. 2013. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J. Immunol 190: 4136–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boldenow E, Gendrin C, Ngo L, Bierle C, Vornhagen J, Coleman M, Merillat S, Armistead B, Whidbey C, Alishetti V, Santana-Ufret V, Ogle J, Gough M, Srinouanprachanh S, MacDonald JW, Bammler TK, Bansal A, Liggitt HD, Rajagopal L, and Waldorf KMA. 2016. Group B streptococcus circumvents neutrophils and neutrophil extracellular traps during amniotic cavity invasion and preterm labor. Science immunology 1: eaah4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, and Zychlinsky A. 2007. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol 176: 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rinaldi SF, Catalano RD, Wade J, Rossi AG, and Norman JE. 2014. Decidual neutrophil infiltration is not required for preterm birth in a mouse model of infection-induced preterm labor. J. Immunol 192: 2315–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Filipovich Y, Agrawal V, Crawford SE, Fitchev P, Qu X, Klein J, and Hirsch E. 2015. Depletion of polymorphonuclear leukocytes has no effect on preterm delivery in a mouse model of Escherichia coli-induced labor. Am. J. Obstet. Gynecol 213: 697.e691–697.e610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burd I, Brown A, Gonzalez JM, Chai J, and Elovitz MA. 2011. A mouse model of term chorioamnionitis: unraveling causes of adverse neurological outcomes. Reprod. Sci 18: 900–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elovitz MA, Brown AG, Breen K, Anton L, Maubert M, and Burd I. 2011. Intrauterine inflammation, insufficient to induce parturition, still evokes fetal and neonatal brain injury. Int. J. Dev. Neurosci 29: 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cross SN, Potter JA, Aldo P, Kwon JY, Pitruzzello M, Tong M, Guller S, Rothlin CV, Mor G, and Abrahams VM. 2017. Viral Infection Sensitizes Human Fetal Membranes to Bacterial Lipopolysaccharide by MERTK Inhibition and Inflammasome Activation. J. Immunol 199: 2885–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoang M, Potter JA, Gysler SM, Han CS, Guller S, Norwitz ER, and Abrahams VM. 2014. Human fetal membranes generate distinct cytokine profiles in response to bacterial Toll-like receptor and nod-like receptor agonists. Biol. Reprod 90: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mulla MJ, Myrtolli K, Brosens JJ, Chamley LW, Kwak-Kim JY, Paidas MJ, and Abrahams VM. 2010. Antiphospholipid antibodies limit trophoblast migration by reducing IL-6 production and STAT3 activity. Am. J. Reprod. Immunol 63: 339–348. [DOI] [PubMed] [Google Scholar]
- 28.Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, and Lee JC. 1995. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett 364: 229–233. [DOI] [PubMed] [Google Scholar]
- 29.Arndt PG, Young SK, Lieber JG, Fessler MB, Nick JA, and Worthen GS. 2005. Inhibition of c-Jun N-terminal kinase limits lipopolysaccharide-induced pulmonary neutrophil influx. Am. J. Respir. Crit. Care Med 171: 978–986. [DOI] [PubMed] [Google Scholar]
- 30.Zhang ER, Liu S, Wu LF, Altschuler SJ, and Cobb MH. 2016. Chemoattractant concentration-dependent tuning of ERK signaling dynamics in migrating neutrophils. Sci Signal 9: ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider CA, Rasband WS, and Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lundqvist H, and Dahlgren C. 1996. Isoluminol-enhanced chemiluminescence: a sensitive method to study the release of superoxide anion from human neutrophils. Free Radic. Biol. Med 20: 785–792. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, Tang W, Zhang H, Niu X, Xu Y, Zhang J, Gao K, Pan W, Boggon TJ, Toomre D, Min W, and Wu D. 2013. A network of interactions enables CCM3 and STK24 to coordinate UNC13D-driven vesicle exocytosis in neutrophils. Dev. Cell 27: 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mulla MJ, Salmon JE, Chamley LW, Brosens JJ, Boeras CM, Kavathas PB, and Abrahams VM. 2013. A role for uric acid and the Nalp3 inflammasome in antiphospholipid antibody-induced IL-1beta production by human first trimester trophoblast. PLoS One 8: e65237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuhns DB, Young HA, Gallin EK, and Gallin JI. 1998. Ca2+-dependent production and release of IL-8 in human neutrophils. J. Immunol 161: 4332–4339. [PubMed] [Google Scholar]
- 36.Mitra S, and Abraham E. 2006. Participation of superoxide in neutrophil activation and cytokine production. Biochim. Biophys. Acta 1762: 732–741. [DOI] [PubMed] [Google Scholar]
- 37.Saji F, Samejima Y, Kamiura S, Sawai K, Shimoya K, and Kimura T. 2000. Cytokine production in chorioamnionitis. J. Reprod. Immunol 47: 185–196. [DOI] [PubMed] [Google Scholar]
- 38.Burdet J, Sacerdoti F, Cella M, Franchi AM, and Ibarra C. 2013. Role of TNF-alpha in the mechanisms responsible for preterm delivery induced by Stx2 in rats. Br. J. Pharmacol 168: 946–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kemp MW, Saito M, Newnham JP, Nitsos I, Okamura K, and Kallapur SG. 2010. Preterm birth, infection, and inflammation advances from the study of animal models. Reprod. Sci 17: 619–628. [DOI] [PubMed] [Google Scholar]
- 40.Elovitz MA, and Mrinalini C. 2004. Animal models of preterm birth. Trends Endocrinol. Metab 15: 479–487. [DOI] [PubMed] [Google Scholar]
- 41.Cardenas I, Mulla MJ, Myrtolli K, Sfakianaki AK, Norwitz ER, Tadesse S, Guller S, and Abrahams VM. 2011. Nod1 activation by bacterial iE-DAP induces maternal-fetal inflammation and preterm labor. J. Immunol 187: 980–986. [DOI] [PubMed] [Google Scholar]
- 42.Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, and Versalovic J. 2014. The placenta harbors a unique microbiome. Sci. Transl. Med 6: 237ra265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ganu RS, Ma J, and Aagaard KM. 2013. The role of microbial communities in parturition: is there evidence of association with preterm birth and perinatal morbidity and mortality? Am. J. Perinatol 30: 613–624. [DOI] [PubMed] [Google Scholar]
- 44.Romero R, Manogue KR, Mitchell MD, Wu YK, Oyarzun E, Hobbins JC, and Cerami A. 1989. Infection and labor. IV. Cachectin-tumor necrosis factor in the amniotic fluid of women with intraamniotic infection and preterm labor. Am. J. Obstet. Gynecol 161: 336–341. [DOI] [PubMed] [Google Scholar]
- 45.Casey ML, Cox SM, Beutler B, Milewich L, and MacDonald PC. 1989. Cachectin/tumor necrosis factor-alpha formation in human decidua. Potential role of cytokines in infection-induced preterm labor. J. Clin. Invest 83: 430–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gupta AK, Hasler P, Holzgreve W, Gebhardt S, and Hahn S. 2005. Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum. Immunol 66: 1146–1154. [DOI] [PubMed] [Google Scholar]
- 47.Weichhart T, and Saemann MD. 2008. The PI3K/Akt/mTOR pathway in innate immune cells: emerging therapeutic applications. Ann. Rheum. Dis 67 Suppl 3: iii 70–74. [DOI] [PubMed] [Google Scholar]
- 48.Presicce P, Park CW, Senthamaraikannan P, Bhattacharyya S, Jackson C, Kong F, Rueda CM, DeFranco E, Miller LA, Hildeman DA, Salomonis N, Chougnet CA, Jobe AH, and Kallapur SG. 2018. IL-1 signaling mediates intrauterine inflammation and chorio-decidua neutrophil recruitment and activation. JCI Insight 22;3(6). pii: 98306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, Barthwal MK, and Dikshit M. 2012. Cytokines induced neutrophil extracellular traps formation: implication for the inflammatory disease condition. PLoS One 7: e48111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yipp BG, and Kubes P. 2013. NETosis: how vital is it? Blood 122: 2784–2794. [DOI] [PubMed] [Google Scholar]
- 51.Delgado-Rizo V, Martinez-Guzman MA, Iniguez-Gutierrez L, Garcia-Orozco A, Alvarado-Navarro A, and Fafutis-Morris M. 2017. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front. Immunol 8: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keshari RS, Verma A, Barthwal MK, and Dikshit M. 2013. Reactive oxygen species-induced activation of ERK and p38 MAPK mediates PMA-induced NETs release from human neutrophils. J. Cell. Biochem 114: 532–540. [DOI] [PubMed] [Google Scholar]
- 53.Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, and Waldmann H. 2011. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol 7: 75–77. [DOI] [PubMed] [Google Scholar]
- 54.Gomez-Lopez N, Romero R, Leng Y, Garcia-Flores V, Xu Y, Miller D, and Hassan SS. 2017. Neutrophil extracellular traps in acute chorioamnionitis: A mechanism of host defense. Am. J. Reprod. Immunol 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doster RS, Sutton JA, Rogers LM, Aronoff DM, and Gaddy JA. 2018. Streptococcus agalactiae Induces Placental Macrophages To Release Extracellular Traps Loaded with Tissue Remodeling Enzymes via an Oxidative Burst-Dependent Mechanism. MBio; 9(6). pii: e02084–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.