

Abstract
Neurodegenerative stimuli are often associated with perturbation of the axon initial segment (AIS), but it remains unclear whether AIS disruption is causative for neurodegeneration or is a downstream step in disease progression. Here we demonstrate that either of two separate, genetically parallel pathways that disrupt the AIS induce axonal degeneration and loss of neurons in the central brain of Drosophila. Expression of a portion of the C-terminal tail of the Ank2-L isoform of Ankyrin severely shortens the AIS in Drosophila mushroom body (MB) neurons, and this shortening occurs through a mechanism that is genetically separate from the previously described Cdk5α-dependent pathway of AIS regulation. Further, either manipulation triggers morphological degeneration of MB axons and is accompanied by neuron loss. Taken together, our results are consistent with the hypothesis that disruption of the AIS is causally related to degeneration of fly central brain neurons, and we suggest that similar mechanisms may contribute to neurodegeneration in mammals.
Keywords: Axon Initial Segment, Axonal degeneration, Neurodegeneration, Ankyrin 2, Cdk5, Drosophila
INTRODUCTION
The axon initial segment (AIS) is a key neuronal domain that lies between the somatodendritic and axonal compartments. The AIS is a gatekeeper for intracellular transport that maintains neuronal polarity [1], and it is essential for proper neuronal excitability as it is the site of action potential (AP) initiation and a critical target for AP modulation [2,3]. The AIS performs these tasks by virtue of a unique composition of membrane proteins, including a distinctive set of voltage-gated ion channels and submembranous cytoskeletal proteins [4–7]. The structure of the AIS in vertebrate neurons depends on the presence of a “master organizer”, Ankyrin G (ankG), a giant ankyrin isoform that recruits and anchors the specialized set of proteins that performs its distinctive functions [8–10].
Aberrant AIS regulation and function have been linked to a variety of diseases. Changes in sequence or expression levels of proteins localized to the AIS are involved in epilepsy [11], bipolar disorder [12], schizophrenia [13], and autism spectrum disorder [14]. Perturbation of the AIS has also been linked to neurodegeneration under a variety of contexts. The AIS contributes to proper tau localization, and breakdown of its barrier function leads to tau missorting [15], while expression of Alzheimer’s disease (AD)-related tau mutants was found to decrease AIS-associated proteins and shorten the overall length of the AIS [16]. Sanchez-Mut et al identified SPTBN4 (encoding non-erythrocyte β-spectrin 4) in a DNA methylation screen of genes related to AD; β-spectrin works in coordination with ankG to localize proteins to the AIS [17]. Further, in a mouse model of AD, it was found that AIS length was reduced near Aβ plaques [18]. Aβ plaques are also associated with microglia recruitment [18], which is itself associated with AIS structural plasticity [19]. While recent literature suggests that various provocations, including pathology, can trigger structural plasticity of the AIS, the underlying mechanisms remain enigmatic.
Our lab demonstrated previously that some Drosophila neurons include a domain that exhibits the characteristic hallmarks of the mammalian AIS, providing a simpler model in which to study the molecular pathways contributing to AIS biogenesis and regulation [20]. First, there is selective accumulation of an anchoring protein within this subcellular domain. Drosophila only has two ankyrin genes: Ank1, which is expressed ubiquitously, and the neuron-specific Ank2 [21,22]. While Ank1 is expressed in all cells, it is present at elevated levels in the AIS of MB neurons [20]. Second, the Drosophila AIS contain a unique combination of voltage-gated potassium channels. Specifically, Elk, Shaw (Kv3), and Shal (Kv4) channels were enriched in the AIS, while dORK-C2 (Ork1), Shaker (Kv1.3), and EKO (Kv1.3) were selectively excluded. Lastly, there is an altered F-actin cytoskeleton within the AIS. In the somatodendritic and axonal regions, actin is highly expressed and ubiquitous; within the AIS, actin levels are reduced significantly and appear to have a distinctively patterned distribution [20]. An AIS-like domain has also been observed in multipolar dendritic arborization neurons of flies. Jegla et al identified a diffusion barrier localized to the proximal axon of ddaE neurons that coincided with the enrichment of Shal and Elk potassium channels [23]. Furthermore, they found that a fragment of Ank2-L was targeted to the proximal axon in the same area. As Ank2-L is one of two Ank2 isoforms that have giant exons that share structural similarities with AnkG [24,25], this suggests that Ank2-L is an analog, and possibly an ortholog, of mammalian AnkG.
Similar to the mammalian AIS, the AIS in flies is also linked to neurodegenerative stimuli. Deletion of Cdk5α (also called D-p35), encoding the activating subunit for cyclin dependent kinase 5 (Cdk5), has been shown to induce multiple degenerative phenotypes in Drosophila central brain neurons, including impaired autophagy, swelling of proximal axons, histologically-evident tissue loss (i.e. formation of ‘vacuoles’ in the brain), and age-dependent loss of neurons [26,27]. Cdk5α-null flies also exhibit an AIS that is severely shortened, and indeed nearly absent: though a barrier remains that separates the somatodendritic and axonal compartments, the domain is so short that the characteristic pattern of accumulation of molecular markers cannot be detected [20]. Notably, the position of the missing AIS correlates with the location where axonal swellings form in affected neurons, and where histological tissue loss is observed [26]. However, it remains unclear if disruption of the AIS merely correlates with neurodegeneration, or plays a causative role.
We show here that overexpression of a portion of the carboxyl domain of the Ank2-L isoform results in a severely shortened AIS in Drosophila central brain neurons, reminiscent of that observed in Cdk5a-null. Ank2-L4-mediated modulation of the AIS, however, occurs through a pathway that is genetically parallel to the previously identified Cdk5α-dependent mechanism. Strikingly, dysregulation of either pathway results in morphological degeneration of axons, and is associated with neuron loss. Therefore, an orthogonal molecular manipulation that shortens the AIS, acting by a genetically independent pathway, also causes axonal degeneration, just as we observed with altered Cdk5α. This provides evidence supporting the hypothesis that disruption of the AIS is apt to be causal for degeneration in fly MB neurons, and may contribute to neurodegeneration in mammalian disease.
METHODS
Fly stocks and genetics
Ank2 constructs (UAS-VENUS-Ank2-S, UAS-VENUS-Ank2-L4, UAS-VENUS-Ank2-L8) were graciously provided by Jan Pielage (University of Kaiserslautern, Kaiserslautern, Germany). Ank2-RNAi lines and the Ank22001 stock were procured from the Bloomington Drosophila Stock Center (BDSC). To enhance efficacy, some RNAi constructs were also co-expressed with UAS- Dcr2, or crosses were carried out at 29°C to enhance GAL4 expression and raise the level of Ank2-RNAi. Additional stocks for assessing the AIS included UAS-Act-RFP (BDSC) and UAS-Syt-HA (provided by Andreas Prokop, University of Manchester, Manchester, UK). Cdk5α overexpression conditions (UAS-Cdk5α, also called UAS-p35) have been previously described [28,27,29,30,20,26]. In all experiments, MB-specific expression was accomplished with the gamma-neuron specific GAL4 driver 201Y-GAL4.
For the lethality screen, virgins heterozygous for the ELAV-GAL4 pan-neuronal driver and the curly wing balancer (CyO) were crossed with males expressing either UAS-Ank2-RNAi or UAS-VENUS-Ank2-L4, and the ratio of balancer to non-balancer offspring was quantified.
For MARCM experiments, the following stocks were acquired from BDSC: P{ry[+t7.2]=hsFLP}1, y[1] w[*] P{w[+mC]=UAS-mCD8::GFP.L}Ptp4E[LL4]; P{w[+mC]=tubP-GAL80}LL9 P{w[+mW.hs]=FRT(w[hs])}2A/TM3, Sb[1]; and P{w[+mW.hs]=GawB}OK107 ey[OK107] (Bloomington # 44404). FRT2A and FRT40A were graciously provided by Dr. Chi-hon Lee (Institute of Cellular and Organismic Biology, Taipei, Taiwan). UAS-Cdk5α clones were generated using hs-Flp122,UAS-mCD8-GFP; FRT2A,TubP- GAL80/TM3,Sb; OK107 female flies crossed to UAS-Cdk5α/CyO;FRT2A/TM6B males. MARCM clones of UAS-VENUS-Ank2-L4 were obtained using hs-Flp122,UAS-mCD8-GFP; FRT40A,TubP-GAL80/CyO; +;OK107 crossed with FRT40A/CyO; UAS-VENUS-Ank2-L4/TM6B males. Clones were generated as previously described [26]; briefly, first-instar larvae from the above crosses were heat shocked at 37°C for 5 minutes in a water bath.
Mushroom body neuron counting utilized the UAS-nls-mCherry fly stock, which was obtained from the BDSC. 201Y-GAL4 was used to express UAS-nls-mCherry in control flies (w+; 201Y- GAL4/+; UAS-nls-mCherry/+ and w+; 201Y-GAL4/+; UAS-nls-mCherry/UAS-GFP) and flies overexpressing Ank2-L4 (w+; 201Y-GAL4/+; UAS-nls-mCherry/UAS-VENUS-Ank2-L4).
Immunohistochemistry
Late third-instar larval brains were dissected in ice-cold PBS (pH 7.4) (Invitrogen) and fixed in 4% paraformaldehyde (PFA) for 25 minutes, and post-fixed for 25 minutes in 4% PFA plus 0.5% TritonX-100 for 25 minutes [20]. Primary antibodies included mouse anti-Fasciclin 2 (Fas2; mAb 1D4; 1:200; Developmental Studies Hybridoma Bank, Iowa City, IA), rat anti-HA (mAb 3F10; 1:1000; Sigma-Aldrich, St. Louis, MO) and rabbit anti-HA (mAb C29F4, 1:200; Cell Signaling, Danvers MA). Due to the presence of VENUS on all Ank2 constructs, Alexa-fluor secondary antibodies (A568 and A633) were used for immunofluorescent detection of markers in experiments expressing these transgenes (1:500, Molecular Probes/Life Technologies, Grand Island, NY). In order to prevent samples from being squished, coverslips were set on number 1 glass chips. Prepared samples were imaged under 40X-oil on a Zeiss NLO510 confocal microscope and analyzed using the Imaris Surpass View application.
AIS Quantification
The cell bodies of gamma neurons in the mushroom body are located at variable distances from the axonal compartment; thus, we characterized a uniform zero reference point for measurements of the AIS boundary position [20]. First, the 3D visualization function of Imaris (Bitplane, version 7.5.2) was used to orient all MB image stacks identically, with the MB peduncle in the plane of the computer screen. A nerve that crosses the proximal axons of MB neurons at a stereotyped location, and that is labeled by anti-Fas2 staining, was identified as a fiducial landmark, and positions of molecular markers were measured relative to this feature. Clipping planes were used to mask staining in regions adjacent to the peduncle, and the ‘Add spots’ function was used to measure distances between points. As the boundaries of compartment-specific markers is graded rather than abrupt [31,32], the average of three measurements were taken (spanning the width of the peduncle) from the caudal edge of the fiducial nerve to the proximal boundary of Fas2 staining. The zero point was defined as the average location of the proximal boundary of Fas2 staining in control samples, relative to this ‘crossover’ nerve. Boundary positions of experimental samples were then reported relative to the zero point.
Mushroom Body Neuron Counting
Male flies expressing 201Y-GAL4 and UAS-nls-mCherry alone or with UAS-GFP or UAS-VENUS-Ank2-L4 were aged to 3-, 10-, 30-, or 45-days old. Whole brains were microdissected and fixed in 4% paraformaldehyde, then mounted on slides with VectaShield (Vector Laboratories, Burlingame, CA). Microscopy was performed on a Zeiss NLO510 confocal microscope, with images acquired using a 40-X oil objective. Z-stacks were collected from individual brain hemispheres and were analyzed using IMARIS and its ‘Add spots’ function to automatically count the labeled neurons.
qRT-PCR
RNA was isolated from 25 Drosophila heads using TRI reagent, and 1000ng of RNA was used for synthesis of cDNA using High Capacity cDNA Reverse Transcription Kit (both reagents from ThermoFisher Scientific, used following manufacturer’s protocol). qRT-CR reactions were prepared using the Affymetrix VeriQuest Probe qPCR Master Mix (Santa Clara, CA) with specific TaqMan gene primers: Ank (Probe# Dm01835806_g1 Cat# 4351372), and control: Rpl32 (Probe# Dm02151827_g1 Cat# 4331182). QuantStudio 6 Flex Real-Time PCR System was used for quantifying expression level.
Statistical Analysis
Statistical analyses were completed using Prism (GraphPad, version 7.04). Differences in the size of the AIS were assessed via one-way ANOVA with Tukey multiple comparisons testing. An initial, non-blinded AIS quantification was validated by doing a blinded re-analysis; blinded quantifications were not statistically different from the original values for all genotypes discussed in the paper (data not shown). For the lethality screen, significant differences in ratios of offspring were assessed by chi-square analysis. For quantification of AIS defects (swelling and ‘beads-on-a-string’) samples were counted blind to genotype. Significant changes in the prevalence of different degenerative phenotypes in MARCM clones were determined by chi-square analysis with Yates’ correction. Significant differences in neuron loss were tested by two-way ANOVA with Tukey multiple comparisons testing.
RESULTS
Overexpression of the C-terminal tail of Ank2-L results in a severely shortened AIS
To identify regulators of the axon initial segment, we conducted a small screen of candidate genes to search for modifiers of AIS size. The screen revealed that expression of constructs encoding some, or all, of the C-terminal tail of the Ank2-L isoform can alter the size of the AIS. In control flies, Fasciclin 2 (Fas2) labels the axonal lobes and peduncle of mushroom body (MB) neurons but is excluded from the AIS (Fig. 1a; [20]). Measuring the shift of the border of Fas2 staining towards or away from the somatodendritic region of MB neurons (relative to a fixed landmark, the stereotyped position of an unrelated Fas2+ nerve in the larval brain; see Methods, also [20]), we find that expression of Ank2-L fragments modulates the size of the AIS. Specifically, expression of a portion of the L4 exons of Ankyrin2 (UAS-VENUS-Ank2-L4, corresponding to a fragment of the C-terminal domain of Ank2 isoform Ank2-L; amino acids 1530–3005) in MB neurons results in a shortening of the AIS (UAS-VENUS-Ank2-L4 mean±SEM: −17.34±0.66μm, p<0.0001, relative to the mean position in controls, which was defined as 0; control SEM = 0.44μm; Fig. 1a-b, e). Expression of Ank2-L8, corresponding to the complete C-terminal domain of Ank2-L (UAS-VENUS-Ank2-L8; amino acids 1530–4083), resulted in a rather different phenotype suggestive of gross axonal reorganization (−18.91±0.46μm, p<0.0001; Fig. 1c, e). Fas2 accumulation, which stops at the boundary between the peduncle and calyx in Ank2-L4 samples, was observed in the calyx of Ank2-L8-expressing samples (ie., within the somatodendritic compartment), perhaps consistent with a role of Ank2- L8 in dendritic targeting. Indeed, Weiner et al found that Ank2-L8 localizes to dendrite branch points in ddaE neurons [33]. Given the severe nature of the Ank2-L8 induced phenotype, Ank2- L8 was excluded from all further experiments. Ank2-mediated modulation of the AIS was specific to the variant C-terminal portions of the Ank2 locus, as expressing Ank2-S (UAS-VENUS-Ank2-S; amino acids 1–1159), the N-terminal domain of Ank2, had no effect on the AIS (−0.92±0.27μm, p = 0.999; Fig. 1d, e). Thus, expression of the C-terminal tail of Ank2-L is sufficient to induce shortening of the AIS. All Ank2 derivatives assayed (Ank2-S, Ank2-L4 and Ank2-L8) showed uniform localization throughout the cell bodies, dendrites, peduncle and axonal lobes of 201Y+ MB neurons (Suppl Fig 1).
Fig. 1. Overexpression of the partial or complete C-terminal tail of Ank2-L severely shortens the AIS.

Z-projected confocal images of late third-instar larval brains stained with anti-Fas2. (a-d) Pink arrow indicates a stereotypical nerve that crosses over in front of the MB peduncle, and is used as a fiducial landmark to measure position of AIS boundary. The proximal border of the axonal Fas2 accumulation in the MB peduncle is indicated by the light blue arrow; the mean position of this proximal border in 201Y control samples is defined as zero. All scale bars are equal to 10μm. MB-specific GAL4 driver 201Y is present in all samples. (a) control, (b) UAS-VENUS- Ank2-L4 (U-A2L4), (c) UAS-VENUS-Ank2-L8 (U-A2L8), and (d) UAS-VENUS-Ank2-S (U-A2S). (e) Quantification of the distance of the proximal edge of the Fas2-positive boundary from the average position of the control boundary. Bars are presented as mean+SEM; statistical significance is relative to the 201Y-GAL4 control (****=p<0.0001). For each genotype, the number of MBs analyzed is presented at the bottom of the bar
In addition to Fas2, we also assayed the AIS with other markers shown previously to be diagnostic for different subcellular domains within MB neurons. In control neurons, actin tagged with RFP (Act-RFP) is present throughout MB neurons, but at significantly lower levels in the AIS than in the somatodendritic and axonal compartments (Fig. 2a; [20]). Similarly, tagged forms of synaptotagmin localize to the axonal lobes and calyx of control neurons while being selectively excluded from the AIS (Fig. 2g). In flies expressing Ank2-L4, the boundary of each of these markers at the proximal border of the axonal compartment was consistently and coordinately shifted towards the calyx, indicating that it is the AIS border per se that has been shifted and not simply the response of any single class of markers (Fig. 2d-f, j-l). Analysis of a marker that is concentrated in the AIS, an HA-tagged version of the voltage-gated potassium channel, Shal [20], also gave consistent results (data not shown). Antibody staining of the somatodendritic-specific protein Futsch/MAP1B did not reveal any alterations in localization following overexpression of Ank2-L4 (data not shown), indicating that neuronal polarity is maintained, as is the distal boundary of the somatodendritic compartment, even when the AIS is severely shortened.
Fig. 2. Ank2-L4 overexpression relocalizes multiple AIS markers.
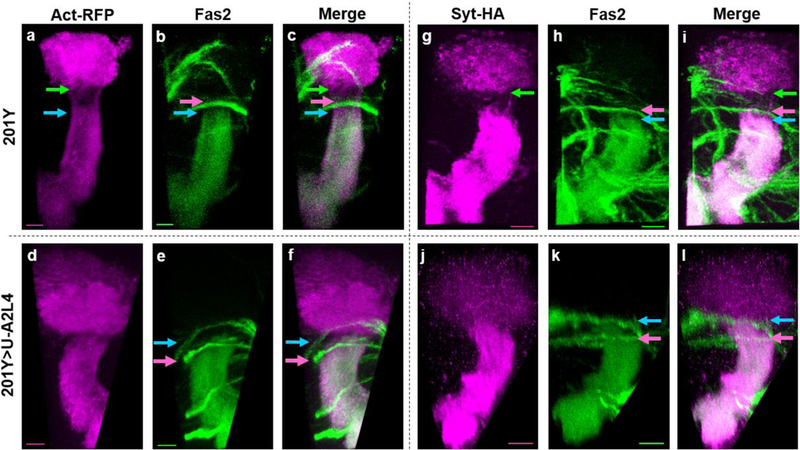
Projected confocal images of late third-instar larval brains expressing markers that delineate the AIS. (a-c, g-i) 201Y-GAL4 controls; (d-f, j-l) 201Y-driven expression of Ank2-L4 (U-A2L4); (a) and (d) show Actin-RFP expression, and (g) and (j) outline the AIS using Synaptotagmin-HA; the same samples were co-stained with anti-Fas2 (b, e, h, and k), with panels (c, f, i, and l) showing the resulting merged images. In control samples, the somatodendritic boundary is marked by the green arrow; in Ank2-L4 samples, this boundary is indistinguishable from the proximal border of the axonal signal (indicated by the blue arrow). The pink arrow indicates a stereotypical nerve that crosses over in front of the MB peduncle, and is used as a landmark to quantify position of AIS boundary. All scale bars are equal to 10μm
Reduction of Ank2L expression level results in a shortened AIS
We next investigated the mechanism underlying Ank2-L4-mediated shortening of the AIS. As expression of Ank2-L4 shortened the AIS, we hypothesized that decreasing Ank2 levels would elongate the AIS. We tested this two ways, by measuring the AIS in an Ank2 mutant background, and by expressing Ank2-RNAi in the MB. Unexpectedly, reducing Ank2-L levels shortened the AIS rather than extending it. Ank22001 is selectively null for the Ank2-L isoform [24], and larvae homozygous for Ank22001 exhibited a shortened AIS phenotype in MB gamma neurons (−6.56 ± 0.72μm, p<0.01; Fig. 3b,g). To confirm the mutant phenotype, we used RNAi to reduce Ank2 levels. Expression of multiple Ank2-RNAi constructs resulted in a partially shortened AIS (BDSC #29438, −5.60 ± 1.44μm, p<0.001; Fig. 3e,g; BDSC #33414, data not shown). As antibodies against Ank2 were unavailable, RNAi efficacy was tested genetically. Complete loss of Ank2 results in late larval/early pupal lethality (Pielage, Cheng et al. 2008). Thus, we screened offspring resulting from pan-neuronal expression of Ank2-RNAi (BDSC #29438), which revealed lethality similar to Ank2-null mutants (Table 1; p<0.0001). Furthermore, flies co-expressing Ank2-L4 and Ank2-RNAi exhibited an AIS that was not significantly different from Ank2-RNAi alone, as expected if the RNAi leads to efficient degradation of the Ank2-L transgene RNA (−3.72 ± 0.85μm, p = 0.90; Fig. 3f,g). It was striking that neither the mutant nor the RNAi reduced the length of the AIS as severely as did overexpression of the Ank2-L tail domain. Neither the presence of Dicer2 nor enhancing RNAi levels by raising flies at elevated temperatures to increase GAL4 activity was sufficient to generate an aberrant AIS phenotype as severe as that observed with overexpression of Ank2-L4 (data not shown). This suggests that Ank2-L4 acts as a dominant negative agent in regulation of AIS length. We note, however, that Ank2-L4 is apparently not dominant-negative for all functions of Ank2; for example, pan-neural expression of Ank2-L4 did not cause lethality (Table 1; p=0.722). We will consider this pattern of phenotypes further in the Discussion. Together, these findings demonstrate that decreased Ank2 levels shorten the AIS, and that expression of Ank2-L4 shortens it even more.
Fig. 3. Reduction of Ank2-L expression levels results in a shortened AIS.

Z-projected confocal images of late third-instar larval brains stained with anti-Fas2. (a-f) Pink arrow indicates a stereotypical nerve that crosses over in front of the MB peduncle, and is used as a fiducial landmark to measure position of AIS boundary. The proximal border of the axonal Fas2 accumulation in the MB peduncle is indicated by the light blue arrow. All scale bars are equal to 10μm. (a) w1118 control samples were compared to (b) samples homozygous for the Ank22001 mutation and thus null for Ank2-L. (c-f) MB-specific GAL4 driver 201Y is present in all samples. (c) control, (d) UAS-VENUS-Ank2-L4 (U-A2L4), (e) UAS-Ank2-RNAi (U-A2RNAi), or (f) both UAS-VENUS-Ank2-L4 and UAS-Ank2-RNAi. (g) Quantification of the shift in Fas2 accumulation was performed as in Fig. 1e. Bars are presented as mean±SEM; statistical significance is relative to the w1118 or 201Y-GAL4 control (**=p<0.01, ***=p<0.001, ****=p<0.0001). For each genotype, the number of MBs analyzed is presented at the bottom of the bar
Table 1. Ank2-L4 overexpression does not cause lethality.
Results from a genetic lethality screen to assess viability upon pan-neuronal expression of UAS- Ank2-RNAi (U-A2RNAi) or UAS-VENUS-Ank2-L4 (U-A2L4). Virgins heterozygous for the pan- neuronal driver ELAV-GAL4 and a balancer chromosome (CyO – curly wing) were crossed with males carrying the respective UAS construct. The ratio of balancer to non-balancer offspring was quantified; significant differences in ratios were assessed by chi-square analysis
| CyO offspring | Non-CyO offspring | p-value | |
|---|---|---|---|
| ELAV-Gal4/CyO X U-A2RNAi | 140 | 62 | p<0.0001 |
| ELAV-Gal4/CyO X U-A2L4 | 122 | 130 | p=0.722 |
Ank2-L4 and Cdk5α regulate the AIS through parallel genetic pathways
Data above show that overexpression of Ank2-L4 in MB neurons drastically shortens the AIS. Similarly, Cdk5/Cdk5α has also been shown to regulate AIS length, with loss of function shortening the AIS while gain of function extends the AIS [20]. As both Ank2-L4 and Cdk5/Cdk5α alter the size of the AIS, we used genetic epistasis to investigate whether they do so through common or parallel pathways. Overexpression of UAS-Cdk5α in MB neurons shifts the boundary of Fas2 staining distally, away from the calyx (3.47±0.68μm, p<0.05; Fig. 4b,e; [20]). In contrast, MB-specific expression of Ank2-L4 shifts the Fas2 staining proximally (−17.34 ± 0.66μm, p<0.0001; Fig. 1b; 2e,k; 4c,e). Co-expression of both constructs yields an intermediate phenotype, with the Fas2 boundary restored to approximately its wild-type position (0.26 ± 0.89μm, p = 0.999; Fig. 4d,e), corresponding to an AIS of approximately wild-type length, indicating that Ank2-L4 and Cdk5α do not have an epistatic relationship, but suggesting rather that Ank2-L4 and Cdk5α act through parallel pathways to modulate the AIS. qRT-PCR verified that UAS-driven co-expression of Cdk5α did not affect the level of expression of Ank2-L4 in experimental samples (Ank2-L4 alone, 1.65 ± 0.09 relative to 201Y control (mean ± SEM) vs 1.55 ± 0.03 when co-expressing Cdk5α; difference not significant (p = 0.38)).
Fig. 4. Ank2-L4 and Cdk5α regulate the AIS through parallel genetic pathways.

Projected confocal images of late third-instar larval brains stained with anti-Fas2. (A-D) Pink arrow indicates a stereotypical nerve that crosses over in front of the MB peduncle, and is used as a fiducial landmark to measure position of AIS boundary. The proximal border of the axonal Fas2 accumulation in the MB peduncle is indicated by the light blue arrow; the mean position of this proximal border in 201Y control samples is defined as zero. All scale bars are equal to 10μm. MB-specific GAL4 driver 201Y is present in all samples. (a) control, (b) UAS-Cdk5α (U- Cdk5α), (c) UAS-VENUS-Ank2-L4 (U-A2L4), and (d) both UAS-VENUS-Ank2-L4 and UAS- Cdk5α. (e) Quantification of the observed shift in Fas2 accumulation was completed as in Fig. 1e. Bars are presented as mean±SEM; statistical significance is relative to the 201Y-GAL4 control (*=p<0.05, ****=p<0.0001). For each genotype, the number of MBs analyzed is presented at the bottom of the bar
Modulation of the AIS is associated with axonal degeneration
Disruption of the AIS from Cdk5α deletion was associated with development of swellings of the portion of the proximal axon corresponding to the location of the AIS in ~17% of single MB gamma-neurons by ~45 days of age, and in some cases was accompanied by fragmentation of the axon [26]. We therefore tested if overexpression of Ank2-L4 also induced degenerative phenotypes in the axon. Individual MB neurons were labeled by MARCM and assayed at 30 days for the presence of morphological phenotypes. Flies with MB-specific expression of Ank2-L4 showed a significant increase in the prevalence of degenerative phenotypes, such as swelling of the axon at the region corresponding to the wild-type location of the AIS, and/or a ‘beads-on-a-string’ phenotype with a row of large swellings separated by narrow constrictions (control: 9.1%, Ank2-L4: 72.7%; p=0.002; Fig. 5b-c, g). Specifically, 40.9% of Ank2-L4-expressing neurons showed evidence of swelling, 9.1% exhibited a beads-on-a-string phenotype, and 22.7% showed both; in controls, the only aberrant morphology observed was swelling at the site of the AIS, and these were noticeably less severe than the swellings observed with Ank2-L4 overexpression (Fig. 5a, g).
Fig. 5. AIS perturbation is associated with morphological degeneration and neuronal loss.

(a-f) Confocal images of single MARCM clones from 30-day old brains. MB-specific GAL4 driver 201Y is present in all samples. All scale bars are equal to 10μm. (a,d) control, (b-c) UAS-VENUS-Ank2-L4, and (e-f) UAS-Cdk5α. Representative images show (b, e) swelling or (c, f) a “beads-on-a-string” phenotype in the proximal axon. (g) Quantification of the prevalence of degenerative morphologies; significance assessed by χ2 test, with Yates’ correction (**p<0.01). For each genotype, the total number of clones analyzed is presented at the bottom of the bar. (h) Quantification of the number of MB neurons per brain hemisphere in aged flies with MB-specific expression of UAS-nls-mCherry alone, UAS-nls-mCherry plus UAS-GFP, or UAS-nls-mCherry plus UAS-VENUS-Ank2-L4. The number of 201Y>nls-mCherry positive MB neurons per hemisphere is presented as mean±SEM. Significant differences were assessed by two-way ANOVA; differences labeled with asterisks (*) are relative to the Day 3 UAS-nls-mCherry alone control; differences marked with “#” are relative to the Day 3 sample co-expressing UAS-GFP and UAS-nls-mCherry. *p<0.05; ***p<0.001; #p<0.05
We also tested if overexpression of Cdk5α and the consequent extension of the AIS led to any morphological phenotypes. Similar to expression of Ank2-L4, MB-specific expression of UAS-Cdk5α caused a significant increase in degenerative phenotypes, as 73.3% of Cdk5α-expressing neurons were affected, compared to 9.1% of control neurons (p=0.004; Fig. 5e-g). 26.7% of Cdk5α-expressing neurons had swelling, while 33.3% had a beads-on-a-string phenotype, and 13.3% had both. These data show that multiple manipulations that perturb the axon initial segment can induce axonal degeneration.
Finally, we asked whether the axonal degeneration associated with AIS structural changes was accompanied by cell loss, as assayed by counting the nuclei of MB gamma-neurons. We expressed a nuclear-localized mCherry (nls-mCherry) under the control of a MB-specific GAL4 driver (201Y-GAL4) in neurons that were otherwise wild-type, or that also expressed Ank2-L4, or as a control, expressed a cytosolic GFP (UAS-GFP). Flies expressing UAS-nls-mCherry alone (mCh alone) showed relatively consistent levels of 201Y-positive MB neurons from Day (D) 3 to D30 before declining at D45 (mean±SEM: D3 mCh alone=827.4 ± 112.8, D30 mCh alone=727.3 ± 80.9, D45 mCh alone=582.1 ± 58.5 neurons/MB; Fig. 5h). Flies co-expressing UAS-nls-mCherry with UAS-GFP (mCh+GFP) exhibited lower numbers of MB neurons at all time points, although these decreases were not significantly different from the three-day old controls until Day 45 (D45 mCh+GFP=484.1 ± 44.7 neurons/MB, p<0.05). Expression of Ank2-L4 (mCh+A2L4) resulted in significant loss of MB neurons by D30, and an even further reduction at D45 (D30 mCh+A2L4=505.2 ± 37.2, p<0.05; D45 mCh+A2L4=400.4 ± 33.7 neurons/MB, p<0.001). Thus, a treatment that shortens the axon initial segment results in accelerated cell loss among affected neurons.
DISCUSSION
Previous experiments by us and others have shown that defects in the axon initial segment are sometimes observed in neurons that have been subjected to neurodegenerative insults, but whether the AIS defects are themselves causal for degeneration has remained unknown. We have here identified a novel reagent to manipulate the AIS in Drosophila central brain neurons. We show that overexpression of a C-terminal portion of the Ank2-L isoform drastically shortens the AIS in MB neurons, as measured with multiple markers, and that this modulation of the AIS occurs through a genetic pathway that is parallel to Cdk5α-mediated regulation of the AIS. Further, we demonstrate that dysregulation of either mechanism triggers axonal degeneration, and in some cases, age-dependent loss of MB neurons.
Based on structural similarities and the presence of a giant exon, Drosophila Ank2-L has been proposed to be the ortholog of the mammalian AnkG and serve as a master organizer of the AIS [23]. Our results confirm an important role of Ank2-L for AIS formation and maintenance, but suggest that other components can compensate for its function in regulating the size of the AIS, at least in part. In mammals, deletion of AnkG results in the complete loss of the AIS. Consistent with this, we observe a partial shortening of the AIS when we reduce Ank2-L levels, either genetically, with a null mutant, or with RNAi. However, we fail to see complete absence of the AIS from reduced Ank2-L levels, even when those levels are decreased enough to cause lethality. This discrepancy could stem from the differing sets of ankyrin genes between flies and mammals. Drosophila only has two identified ankyrins [22,34] while mammals carry three ankyrin genes [35–37]. It may be that as mammals have an expanded ankyrin repertoire with more specialized functions for each gene, the loss of one completely ablates the associated function. In Drosophila, the neuron-specific Ank2 is accompanied by Ank1, which is also enriched at the AIS [20]. It is possible that deletion of Ank2 disrupts the AIS to a certain degree, but there is compensation by Ank1, or by another Ank2 isoform, such as Ank2-XL, preventing the complete dissolution of the AIS. Interplay between multiple ankyrins at the AIS has been shown in mammals, as overexpression of AnkB in cultured hippocampal neurons resulted in a more restricted distribution of AnkG at the AIS [38]. Another possibility is that Ank1 plays a significant role in the organization of the Drosophila AIS. However, Ank1 lacks the giant exons common to mammalian AnkG, so if it were to be capable of acting as the sole organizer of the AIS, it would likely do so through somewhat different mechanisms. Experiments reducing Ank1 levels selectively in the MB will be informative to reveal the specific contributions of Ank1 in establishment and maintenance of the AIS. Furthermore, it will be interesting to see if Ank1 is altered in MB neurons when Ank2-L levels are perturbed or Ank2-L4 is overexpressed.
It was shown recently that Ank2-L localizes to the AIS in multipolar ddaE neurons, and Ank2-null mutants exhibit loss of a diffusion barrier and loss of potassium channel localization in these neurons [23]. One potential explanation for the apparent difference in AIS phenotype in MB vs ddaE neurons upon Ank2-deletion could be cell type differences. At the neuromuscular junction (NMJ) of Drosophila motoneurons, Ank2-L was localized to the axon and pre-synaptic terminal [24]. In ddaE neurons, in contrast, Ank2-L4 accumulated in the proximal axon and regulated the diffusion barrier, while Ank2-S appeared ubiquitously throughout the entire neuron [23]. In our hands, neither Ank2-S, Ank2-L4, nor Ank2-L8 showed any specific localization within MB gamma neurons, as accumulation of all three was observed in the cell bodies, dendrites, and axonal lobes (Supplemental Figure 1). Thus, it seems that different neuronal subtypes may use different targeting paradigms. A second possible explanation for the differences between these results could be the sensitivity of the assays used to measure AIS defects. Measuring changes in the boundary of antibody staining is largely qualitative, whereas intermediate effects might be easier to recognize when assaying a quantitative measure, such as diffusion rates. Regardless, both our data here and the study by Jegla et al support the conclusion that Ank2 contributes to the proper maintenance of the axon initial segment.
Overexpression of the C-terminal portion of Ank2-L drastically shortened the AIS, as did genetic reduction or removal of Ank2-L. This was initially unexpected, as overexpression of AnkG in cultured hippocampal neurons nearly tripled the length of the AIS [38], and suggests that the isolated C-terminal domain of Ank2-L acts as a dominant negative and not as a gain of function. Indeed, expression of the Ank2-L C-terminus had a significantly stronger effect on the AIS than even the Ank2-L null mutant, a property known genetically as an “antimorph” [39]. It may be, for example, that Ank2-L4 acts by mislocalizing the normal binding partners of Ank2-L, either directly or by changing the localization of the endogenous Ank2-L, or that it mislocalizes or inactivates components that would normally be able to compensate for the absence of Ank2. Formally, we also cannot rule out the possibility that Ank2-L4 acts as a neomorph with regards to AIS regulation, as the total absence of Ank2-L in homozygous mutants failed to cause the severely shortened phenotype seen with Ank2-L4 overexpression. It has been demonstrated previously that the complete C-terminal domain of Ank2 exhibits neomorphic activity during neuromuscular junction development, as expression of Ank2-L8 in presynaptic neurons at the NMJ results in the formation of small, highly ramified satellite boutons at the synapse [24]. It is also striking that the antimorphic effect of Ank2-L is assay dependent; the AIS phenotype is stronger than that of the null mutant, but unlike the genetic mutant, the Ank2-L C-terminus does not induce lethality even when expressed pan-neuronally at high level. Such assay-specific differences in phenotypic strength are not uncommon, and may arise, for example, from differences in the spectrum of proteins that bind a particular domain in different tissues [40].
The cell loss observed from expression of Ank2-L was far less extensive than the axonal morphological phenotypes. This finding raises questions about the relationship of axonal degeneration to loss of the cell soma. Deletion of Cdk5α results in a shortened AIS [20], as well as causing degenerative phenotypes and neuronal loss that stem, in part, from an acceleration of physiological aging [27] and hyperactivation of the immune system [41]. However, the level of neuron loss in Cdk5α-null flies was greater than what was observed here. This suggests that some aging-associated degenerative pathways are more strongly productive of soma loss, while axonal degeneration, which one would expect to be equally effective at disrupting neuronal function, may not always lead to death of the soma, at least in Drosophila.
Disruption of the AIS has been observed in association with multiple neurodegenerative stimuli, including altered expression of Cdk5α in Drosophila [20] and tau mutation or acetylation in mice [16]. The data here show that a manipulation that directly targets a structural component of the AIS leads to axonal degeneration, as Ank2-L4 overexpression resulted in a significant increase in the prevalence of axonal swelling within the proximal axon, and blebbing of the axon in aged flies, in concert with shortening the AIS. Additionally, Ank2-L4 overexpression resulted in a significant increase in cell loss relative to control. The observed degenerative phenotypes and cell loss do not appear to be due to non-specific detrimental effects on neuronal morphology or physiology during development. Expression of neither Ank2-L4 nor Ank2-L8, which had a more severe effect on the AIS, significantly altered the gross morphological structure of the MB neurons of 3rd instar larva (data not shown), indicating that any phenotypes are largely restricted to subcellular organization. Moreover, it is worth noting that even though the AIS was shortened to the point that we could not detect it with our molecular markers when Ank2-L4 was overexpressed, Futsch remained restricted to the somatodendritic region suggesting maintenance of a diffusion barrier, just as was observed in Cdk5α-null mutant flies [20]. Thus, it is unlikely that the observed neurodegeneration stems from gross disruption of neuronal polarity or morphology.
The data reported here show that the structure of the AIS is regulated by at least two parallel pathways, one defined by Cdk5α and one by Ank2-L, and that perturbation of either pathway leads to axonal degeneration and neuron loss. As such, the most parsimonious interpretation is that a shared feature, such as the altered AIS, is responsible for the observed degenerative phenotypes. However, we cannot exclude the possibility that overexpression of the dominant- negative Ank2-L4 disrupts some other aspect(s) of cell structure or physiology that it also shares with Cdk5α. Modest structural plasticity of the AIS does not always lead to degeneration; indeed, modulation of AIS length is a vital part of fine-tuning neuronal excitability [42]. Thus, shortening of the AIS by overexpression of Ank2-L4 may be only one aspect of disturbed AIS function and neuronal physiology from this manipulation. For example, our data also demonstrate alteration of actin organization in the AIS, and the localized swelling of the axon is likely associated with defects in axonal transport [43]. Moreover, the drastic reduction we observe in the extent of the AIS is expected to have severe consequences for neuronal excitability. Finally, Ank2-L4 expression may cause unrecognized changes in other parts of the cell. The key point, however, is that our data seems to exclude the hypothesis that AIS loss is simply a secondary, late-stage marker of neurodegeneration, but rather suggests that it is associated with the earliest stages of the process, as targeting either of two independent regulators of the AIS, one a core structural component, is sufficient to induce degeneration.
Using a novel reagent, we have unlocked a new method for modulating the AIS independent of Cdk5/Cdk5α, the only known AIS regulator in Drosophila MB neurons. The regulation of the AIS presented here is likely to be direct as Ank2-L4 acts as an antimorph of the essential ankyrin isoform that helps construct the AIS. Remarkably, we show that a genetic treatment that disrupts the AIS by manipulation of one of its core components is also sufficient to cause axon degeneration and even neuron loss in flies, supporting the hypothesis that the AIS disruption observed in association with several neurodegenerative mechanisms contributes causally to the process of neurodegeneration.
Supplementary Material
ACKNOWLEDGEMENTS
We thank each member of our lab, and also Chi-Hon Lee and Ela Serpe, for helpful discussions during the course of this work. We thank Jan Pielage, Chi-Hon Lee, Andreas Prokop, Melissa Rolls, and the Bloomington Drosophila Stock Center for fly stocks used in these experiments. We are grateful to Stephen Wincovitch of the NHGRI Cytogenetics and Microscopy Core Facility for his assistance with microscopy. These experiments were supported by the Basic Neuroscience Program of the Intramural Research Program of the National Institute of Neurological Disorders and Stroke, National Institutes of Health, Z01 NS003106.
References
- 1.Rasband MN (2010) The axon initial segment and the maintenance of neuronal polarity. Nature Reviews Neuroscience 11 (8):552–562. doi: 10.1038/nrn2852 [DOI] [PubMed] [Google Scholar]
- 2.Palmer LM, Stuart GJ (2006) Site of action potential initiation in layer 5 pyramidal neurons. J Neurosci 26 (6):1854–1863. doi: 10.1523/JNEUROSCI.4812-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans MD, Dumitrescu AS, Kruijssen DLH, Taylor SE, Grubb MS (2015) Rapid Modulation of Axon Initial Segment Length Influences Repetitive Spike Firing. Cell Rep 13 (6):1233–1245. doi: 10.1016/j.celrep.2015.09.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kole MH, Ilschner SU, Kampa BM, Williams SR, Ruben PC, Stuart GJ (2008) Action potential generation requires a high sodium channel density in the axon initial segment. Nat Neurosci 11 (2):178–186. doi: 10.1038/nn2040 [DOI] [PubMed] [Google Scholar]
- 5.Khaliq ZM, Raman IM (2006) Relative contributions of axonal and somatic Na channels to action potential initiation in cerebellar Purkinje neurons. J Neurosci 26 (7):1935–1944. doi: 10.1523/JNEUROSCI.4664-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shah MM, Migliore M, Valencia I, Cooper EC, Brown DA (2008) Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A 105 (22):7869–7874. doi: 10.1073/pnas.0802805105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galiano MR, Jha S, Ho TS, Zhang C, Ogawa Y, Chang KJ, Stankewich MC, Mohler PJ, Rasband MN (2012) A distal axonal cytoskeleton forms an intra-axonal boundary that controls axon initial segment assembly. Cell 149 (5):1125–1139. doi: 10.1016/j.cell.2012.03.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou D, Lambert S, Malen PL, Carpenter S, Boland LM, Bennett V (1998) AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol 143 (5):1295–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garrido JJ, Giraud P, Carlier E, Fernandes F, Moussif A, Fache MP, Debanne D, Dargent B (2003) A targeting motif involved in sodium channel clustering at the axonal initial segment. Science 300 (5628):2091–2094. doi: 10.1126/science.1085167 [DOI] [PubMed] [Google Scholar]
- 10.Pan Z, Kao T, Horvath Z, Lemos J, Sul JY, Cranstoun SD, Bennett V, Scherer SS, Cooper EC (2006) A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci 26 (10):2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wallace RH, Wang DW, Singh R, Scheffer IE, George AL Jr., Phillips HA, Saar K, Reis A, Johnson EW, Sutherland GR, Berkovic SF, Mulley JC (1998) Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet 19 (4):366–370. doi: 10.1038/1252 [DOI] [PubMed] [Google Scholar]
- 12.Leussis MP, Madison JM, Petryshen TL (2012) Ankyrin 3: genetic association with bipolar disorder and relevance to disease pathophysiology. Biol Mood Anxiety Disord 2:18. doi: 10.1186/2045-5380-2-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cruz DA, Eggan SM, Azmitia EC, Lewis DA (2004) Serotonin1A receptors at the axon initial segment of prefrontal pyramidal neurons in schizophrenia. Am J Psychiatry 161 (4):739–742. doi: 10.1176/appi.ajp.161.4.739 [DOI] [PubMed] [Google Scholar]
- 14.Codina-Sola M, Rodriguez-Santiago B, Homs A, Santoyo J, Rigau M, Aznar-Lain G, Del Campo M, Gener B, Gabau E, Botella MP, Gutierrez-Arumi A, Antinolo G, Perez-Jurado LA, Cusco I (2015) Integrated analysis of whole-exome sequencing and transcriptome profiling in males with autism spectrum disorders. Mol Autism 6:21. doi: 10.1186/s13229-015-0017-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Kumar Y, Zempel H, Mandelkow E-M, Biernat J, Mandelkow E (2011) Novel diffusion barrier for axonal retention of Tau in neurons and its failure in neurodegeneration. The EMBO journal 30 (23):4825–4837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sohn PD, Tracy TE, Son H-I, Zhou Y, Leite REP, Miller BL, Seeley WW, Grinberg LT, Gan L (2016) Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Molecular Neurodegeneration 11 (1). doi: 10.1186/s13024-016-0109-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanchez-Mut JV, Aso E, Panayotis N, Lott I, Dierssen M, Rabano A, Urdinguio RG, Fernandez AF, Astudillo A, Martin-Subero JI, Balint B, Fraga MF, Gomez A, Gurnot C, Roux JC, Avila J, Hensch TK, Ferrer I, Esteller M (2013) DNA methylation map of mouse and human brain identifies target genes in Alzheimer’s disease. Brain 136 (Pt 10):3018–3027. doi: 10.1093/brain/awt237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marin MA, Ziburkus J, Jankowsky J, Rasband MN (2016) Amyloid-beta plaques disrupt axon initial segments. Exp Neurol 281:93–98. doi: 10.1016/j.expneurol.2016.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benusa SD, George NM, Sword BA, DeVries GH, Dupree JL (2017) Acute neuroinflammation induces AIS structural plasticity in a NOX2-dependent manner. J Neuroinflammation 14 (1):116. doi: 10.1186/s12974-017-0889-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trunova S, Baek B, Giniger E (2011) Cdk5 Regulates the Size of an Axon Initial Segment- Like Compartment in Mushroom Body Neurons of the Drosophila Central Brain. Journal of Neuroscience 31 (29):10451–10462. doi: 10.1523/JNEUROSCI.0117-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hortsch M (2002) The Axonal Localization of Large Drosophila Ankyrin2 Protein Isoforms Is Essential for Neuronal Functionality. Molecular and Cellular Neuroscience 20 (1):43–55. doi: 10.1006/mcne.2002.1113 [DOI] [PubMed] [Google Scholar]
- 22.Bouley M, Tian MZ, Paisley K, Shen YC, Malhotra JD, Hortsch M (2000) The L1-type cell adhesion molecule neuroglian influences the stability of neural ankyrin in the Drosophila embryo but not its axonal localization. J Neurosci 20 (12):4515–4523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jegla T, Nguyen MM, Feng C, Goetschius DJ, Luna E, van Rossum DB, Kamel B, Pisupati A, Milner ES, Rolls MM (2016) Bilaterian Giant Ankyrins Have a Common Evolutionary Origin and Play a Conserved Role in Patterning the Axon Initial Segment. PLOS Genetics 12 (12):e1006457. doi: 10.1371/journal.pgen.1006457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pielage J, Cheng L, Fetter RD, Carlton PM, Sedat JW, Davis GW (2008) A Presynaptic Giant Ankyrin Stabilizes the NMJ through Regulation of Presynaptic Microtubules and Transsynaptic Cell Adhesion. Neuron 58 (2):195–209. doi: 10.1016/j.neuron.2008.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stephan R, Goellner B, Moreno E, Frank CA, Hugenschmidt T, Genoud C, Aberle H, Pielage J (2015) Hierarchical Microtubule Organization Controls Axon Caliber and Transport and Determines Synaptic Structure and Stability. Developmental Cell 33 (1):5–21. doi: 10.1016/j.devcel.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trunova S, Giniger E (2012) Absence of the Cdk5 activator p35 causes adult-onset neurodegeneration in the central brain of Drosophila. Disease Models & Mechanisms 5 (2):210–219. doi: 10.1242/dmm.008847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spurrier J, Shukla AK, McLinden K, Johnson K, Giniger E (2018) Altered expression of the Cdk5 activator-like protein, Cdk5alpha, causes neurodegeneration, in part by accelerating the rate of aging. Dis Model Mech 11 (3). doi: 10.1242/dmm.031161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Connell-Crowley L, Le Gall M, Vo DJ, Giniger E (2000) The cyclin-dependent kinase Cdk5 controls multiple aspects of axon patterning in vivo. Current Biology 10 (10):599–603 [DOI] [PubMed] [Google Scholar]
- 29.Connell-Crowley L, Vo D, Luke L, Giniger E (2007) Drosophila lacking the Cdk5 activator, p35, display defective axon guidance, age-dependent behavioral deficits and reduced lifespan. Mechanisms of Development 124 (5):341–349. doi: 10.1016/j.mod.2007.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith-Trunova S, Prithviraj R, Spurrier J, Kuzina I, Gu Q, Giniger E (2015) Cdk5 regulates developmental remodeling of mushroom body neurons in Drosophila: CDK5 Regulates Neuronal Remodeling. Developmental Dynamics 244 (12):1550–1563. doi: 10.1002/dvdy.24350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winckler B, Forscher P, Mellman I (1999) A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature 397 (6721):698–701. doi: 10.1038/17806 [DOI] [PubMed] [Google Scholar]
- 32.Nakada C, Ritchie K, Oba Y, Nakamura M, Hotta Y, Iino R, Kasai RS, Yamaguchi K, Fujiwara T, Kusumi A (2003) Accumulation of anchored proteins forms membrane diffusion barriers during neuronal polarization. Nat Cell Biol 5 (7):626–632. doi: 10.1038/ncb1009 [DOI] [PubMed] [Google Scholar]
- 33.Weiner AT, Seebold DY, Michael NL, Guignet M, Feng C, Follick B, Yusko BA, Wasilko NP, Torres-Gutierrez P, Rolls MM (2018) Identification of Proteins Required for Precise Positioning of Apc2 in Dendrites. G3 (Bethesda) 8 (5):1841–1853. doi: 10.1534/g3.118.200205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dubreuil RR, Yu J (1994) Ankyrin and beta-spectrin accumulate independently of alpha- spectrin in Drosophila. Proc Natl Acad Sci U S A 91 (22):10285–10289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lambert S, Bennett V (1993) Postmitotic expression of ankyrinR and beta R-spectrin in discrete neuronal populations of the rat brain. J Neurosci 13 (9):3725–3735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kordeli E, Lambert S, Bennett V (1995) AnkyrinG. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem 270 (5):2352–2359 [DOI] [PubMed] [Google Scholar]
- 37.Otto E, Kunimoto M, McLaughlin T, Bennett V (1991) Isolation and characterization of cDNAs encoding human brain ankyrins reveal a family of alternatively spliced genes. J Cell Biol 114 (2):241–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galiano Mauricio R, Jha S, Ho Tammy S-Y, Zhang C, Ogawa Y, Chang K-J, Stankewich Michael C, Mohler Peter J, Rasband Matthew N (2012) A Distal Axonal Cytoskeleton Forms an Intra-Axonal Boundary that Controls Axon Initial Segment Assembly. Cell 149 (5):1125–1139. doi: 10.1016/j.cell.2012.03.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muller HJ (1932) Further studies of the nature and causes of gene mutations. Proceedings of the 6th International Congress of Genetics:213–255 [Google Scholar]
- 40.Herskowitz I (1987) Functional inactivation of genes by dominant negative mutations. Nature 329 (6136):219–222. doi: 10.1038/329219a0 [DOI] [PubMed] [Google Scholar]
- 41.Shukla AK, Spurrier J, Kuzina I, Giniger E (2018–19) Hyperactive innate immunity causes degeneration of dopamine neurons upon altering activity of Cdk5. Cell Reports (In Press) [DOI] [PMC free article] [PubMed]
- 42.Yamada R, Kuba H (2016) Structural and Functional Plasticity at the Axon Initial Segment. Front Cell Neurosci 10:250. doi: 10.3389/fncel.2016.00250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH Jr., Brown H, Tiwari A, Hayward L, Edgar J, Nave KA, Garberrn J, Atagi Y, Song Y, Pigino G, Brady ST (2009) Axonal transport defects in neurodegenerative diseases. J Neurosci 29 (41):12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.