

Abstract
Background:
Our previous studies have shown that regulatory factor X5 (RFX5), a classical transcription regulator of MHCII genes, was obviously overexpressed in hepatocellular carcinoma (HCC) tumors. However, the role of RFX5 in the carcinogenesis and progress of HCC remains unknown. This study aimed to reveal its biological significance and the underlying mechanism in HCC.
Methods:
RFX5 mRNA expression level and copy number variation in HCC tumors and cell lines were determined by analyzing deposited data sets in the Cancer Genome Atlas and Gene Expression Omnibus database. The biological significance of RFX5 in HCC was investigated by monitoring the colony formation and subcutaneous tumor growth capacity when RFX5 was silenced with lentiviral short hairpin RNA and CRISPR/Cas9 system in HCC cell lines. The downstream gene transcriptionally activated by RFX5 in HCC cells was determined by chromatin immunoprecipitation and luciferase reporter assay. The involvement of tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein theta (YWHAQ) in HCC development was further determined by performing colony formation rescue assay and subcutaneous tumor growth rescue experiment. The association of YWHAQ with recurrence-free survival of patients with HCC was assessed by Kaplan-Meier analysis. Moreover, apoptosis level and the protein level of p53 pathway were determined to reveal the mechanism of RFX5 in driving HCC development.
Results:
RFX5 was amplified and highly overexpressed in HCC tumor tissues compared with the corresponding non-tumor tissues. The mRNA expression level of RFX5 was significantly correlated with its DNA copy number (r = 0.4, P < 0.001). Functional study demonstrated that RFX5 was required for both clonogenic forming in vitro and subcutaneous tumor growth in vivo of HCC cells. Further study identified YWHAQ, namely 14-3-3 tau, as a key downstream transcriptional target gene of RFX5, which was tightly regulated by RFX5 in HCC. Moreover, overexpression of YWHAQ largely rescued the clonogenic growth of HCC cells that was suppressed by RFX5 knockdown. In addition, overexpression of YWHAQ in primary tumor was linked to poor prognosis of patients with HCC. These results demonstrated that YWHAQ was a downstream effector of RFX5 in HCC. Notably, RFX5-YWHAQ pathway could protect cells from apoptosis by suppressing the p53 and Bax in HCC.
Conclusion:
RFX5 is a putative HCC driver gene that plays an important role in the development and progression of HCC by transactivating YWHAQ and suppressing apoptosis.
Keywords: Hepatocellular carcinoma, Transcription factor, Apoptosis, P53
Introduction
Hepatocellular carcinoma (HCC) accounts for a large portion of cancer-related deaths, and is a major public health challenge worldwide.[1] However, the 5-year survival rate of HCC is unsatisfied due to the lack of cutting-edge treatment options. In order to improve the treatment response of HCC, it is imperative to elucidate the underlying mechanisms in the process of carcinogenesis and progression of HCC.
Regulatory factor X5 (RFX5) is a winged-helix transcription factor that comprises the transcription factor complex RFX together with other two subunits, RFXANK/B and RFXAP. RFX5 has been reported as a dominant regulator of MHCII gene transcription.[2–4] And we have previously shown that RFX5 overexpression in HCC cells did not affect MHCII expression, but transactivated non-MHCII target genes, such as tripeptidyl peptidase 1 (TPP1).[5] However, the biological significance of overexpressed RFX5 has not yet been determined in HCC.
Hence, we proposed to investigate the biological significance of RFX5 in HCC in this study. Our major aim was to ascertain the downstream target genes of RFX5 and revealed the molecular mechanism of RFX5 in the development and progression of HCC.
Methods
Ethical approval
The study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of the Affiliated Hospital of Guilin Medical University and Peking University People's Hospital. Informed written consent was obtained from all patients before their enrollment in this study.
Gene expression data and copy number variation
Gene expression data and DNA copy number variation (CNV) data of HCC tumor tissues and cell lines were obtained from the liver hepatocellular carcinoma (LIHC) database of the Cancer Genome Atlas (TCGA) project and Gene Expression Omnibus database (GSE38207). Data analysis and visualization were performed as previously reported.[6–8]
HCC tissue samples and cell lines
All HCC tumor tissues were collected after written informed consent signed by each patient at the affiliated hospital of Guilin Medical University (Guilin cohort). MHCC-97H and MHCC-97L cell lines were kindly provided by the Academy of Military Medical Science (Beijing, China). Other cell lines (HepG2, Hep3B, SK-HEP-1, PLC/PRF/5, Huh7, Li7, HEK293T, and GP2-293) were either purchased from the American Type Culture Collection (USA) or from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China).
Plasmid constructs, lentiviral productions, and stable cell lines establishments
The pGL4-Tyrosine 3-monooxygenase/tryptophan5-monooxygenase activation protein theta (pGL4-YWHAQ) luciferase reporter vector was constructed by cloning the promoter region of YWHAQ gene into the pGL4.10 vector (Progmega, Madison, Wisconsin, USA). Human YWHAQ open reading frame (ORF) sequence and RFX5 ORF sequence were respectively cloned into the retroviral vector pMSCVneo, named pMSCV-YWHAQ and pMSCV-RFX5. SgRNA specifically targeting human RFX5 (RFsg1: GGTTTAGATGACCGTTCCCG; RFsg3: GCCTCCACCAGTTCATCTCG) were cloned into LentiCRISPRv2 (Addgene plasmid # 52961, a gift from Feng Zhang) following provider's guide. Lentiviral vector expressing FLAG-tagged RFX5 ORF, short hairpin RNA (shRNA) specifically targeting RFX5 (RFK1 and RFK2) and control shRNA (NS) were cloned as previously reported.[5] The recombinant retroviral and lentiviral production, purification, and stable cell lines establishment were performed according to our previous report.[6]
Chromatin immunoprecipitation and luciferase reporter assay
The chromatin immunoprecipitation (ChIP) assay was conducted according to previous report.[5] Free DNA fragments eluted from RFX5 antibody or IgG (control) were subjected to ChIP-PCR analysis with primers covering YWHAQ promoter (YWHAQ-PF: TTAGGCTTTCCAGGTTATGCA; YWHAQ-PR: CGCTTCAGCAGGGTGTTCC). Cells co-transfected with firefly luciferase plasmid and RFX5 expression plasmid were subjected to luciferase reporter assay following procedure described in our previous publication.[5]
Tissue microarray and immunohistochemistry
The tissue microarray (TMA) consisted of 128 pairs of primary HCC tumor tissues and corresponding non-tumor tissues from Guilin cohort. Antigen retrieval was performed with EDTA buffer (pH 8.0) at 100°C for 15 min. Rabbit anti-human RFX5 antibody (Novus Biological, San Diego, California, USA) at a working dilution of 1:100 or rabbit anti-human YWHAQ antibody (Proteintech, Rosemont, Pennsylvania, USA) at a working dilution of 1:1000 was incubated at 4°C overnight, followed by incubating horseradish peroxidase (HRP)-conjugated secondary antibody (Dako, Copenhagen, Denmark) for 1 h at 37°C. Then DAB reaction was used to reveal antibody binding with EnVision detection system (Dako). RFX5 protein expression was scored by two pathologists independently according to the percentage of tumor cells with nuclear staining (0, nuclear staining in less than 5% of tumor cells [N−]; 1, nuclear staining in 5% to 30% of tumor cells [N+]; 2, nuclear staining in 31% to 50% of tumor cells [N++]; 3, nuclear staining in >51% of tumor cells [N+++]).
Quantitative real-time polymerase chain reaction
In order to validate the RFX5 overexpression in HCC, the RFX5 mRNA expression level in Guilin cohort and eight HCC cell lines (MHCC-97H, MHCC-97L, HepG2, Hep3B, SK-HEP-1, PLC/PRF/5, Huh7, and Li7) was measured by quantitative real-time polymerase chain reaction (QRT-PCR) analysis. The QRT-PCR amplification was performed with a mixture containing 50 ng cDNA template and SYBR Green I Master on a LightCycler 480 (Roche Applied Science, Rotkreuz, Germany). The primer sequences used for QRT-PCR were as follows: RFX5, 5′-GATGAGCCTGATGCTAAGAGC-3′ and 5′-CCCTCTACTTTGTTCTGCACG-3′; and GAPDH, 5′-CCACATCGCTCAGACACCAT-3′ and 5′-GGCAACAATATCCACTTTACCAGA-3′. Relative gene expression was calculated and normalized against GAPDH.
Clonogenicity assay
HCC cell lines stably transduced with lentivirus or retrovirus were subjected to clonogenicity assay. Each cell lines were plated in six-well plates for 1000 cells in each well in triplicates. These cells were cultured after 14 days and then fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 30 min. The colonies were stained with 1% crystal violet solution for 1 h. After washing with PBS, the plates were air-dried. Cell colonies were counted and subjected to statistical analysis.
Xenograft tumor growth assay
Xenograft tumor growth assay was established by subcutaneous injection of 2 × 106 HCC cells (Consg1-MHCC-97H, RFsg1-MHCC-97H, Consg1-SK-HEP-1, RFsg1-SK-HEP-1, YWHAQ-SK-HEP-1, and YWHAQ-SK-HEP-1 with RFX5 knockdown by RFsg1) into the dorsal flank of BALB/c Nude mice (male, 4 weeks). Tumors growth were measured with vernier calipers after 3 weeks and tumor volumes were calculated with the following formula: (W2 × L)/2, where W is width and L is length. All experiments were performed under protocols approved by the Experimental Animal Center of Peking University People's Hospital.
Western blotting analysis
Total protein was extracted from HCC cells using RIPA lysis buffer and quantified using Pierce BCA Protein Assay kit according to the manufacturer's protocol (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Protein extracts (10 μg) were separated by SDS/PAGE gel (Thermo Fisher Scientific) and then transferred to a nitrocellulose membrane (Thermo Fisher Scientific). After membrane was blocked with 5% non-fat dry milk dissolved in Tris-buffered saline with Tween-20 buffer for 1 h at room temperature, it was incubated at 4°C overnight with primary antibodies against RFX5 (1:1000; Proteintech), YWHAQ (1:10,000; Proteintech), p53 antibody (1:2000; Proteintech), and Bax antibody (1:1500; Proteintech) followed by incubation with HRP-conjugated secondary antibodies (1:5000; Proteintech) for 1 h at room temperature. The band were detected using a Western Lighting Plus-ECL (PerkinElmer, Waltham, Massachusetts, USA) and visualized on X-ray film.
Flow cytometry analysis
Annexin V and 7-AAD staining were performed to determine the cell apoptosis rate by using the Annexin V-FITC apoptosis Detection Kit (BD Biosciences, San Jose, California, USA). Flow cytometry analysis was performed on FACSAria II (BD Biosciences, USA). Following data analysis was performed with FlowJo analysis software (Tree Star, USA).
Statistical analysis
Data clean and statistical analysis were performed with GraphPad Prism 7.0 (GraphPad Software, USA) and R Statistical Software (Foundation for Statistical Computing, Austria).[9] All data that subjected to statistical analysis was from at least three independent experiments. The differences among multiple groups were compared by one-way analysis of variance. The mRNA level of RFX5 in HCC tumor tissues and adjacent non-tumor tissues were compared using a paired Student's t test. The unpaired t test was used to calculate a two-tailed P value. Survival analyses in TCGA LIHC cohort were assessed by Kaplan-Meier plots and log-rank tests. The Pearson correlation analysis was used to determine the association between the mRNA expression level and DNA copy number of RFX5. A P < 0.05 was considered statistically significant.
Results
RFX5 is frequently amplified and overexpressed in HCC
RFX5, a gene located at 1q21, was amplified in 24.8% (92/371) of patients with HCC with 1.5-fold increase in tumor vs. normal tissue by analyzing the LIHC CNV date from TCGA [Figure 1A]. The mRNA level of RFX5 was dramatically higher in HCC tumor tissues compared with normal tissues according to the RNA-seq date from TCGA LIHC dataset (0.140 ± 0.052 vs. −1.062 ± 0.084, t = 8.240, P < 0.001) [Figure 1B]. Remarkably, the expression of RFX5 mRNA significantly correlated with its DNA copy number (r = 0.4, P < 0.001) [Figure 1C], which indicated that increased copy number of RFX5 may be one of the reasons leading to its overexpression in HCC.
Figure 1.
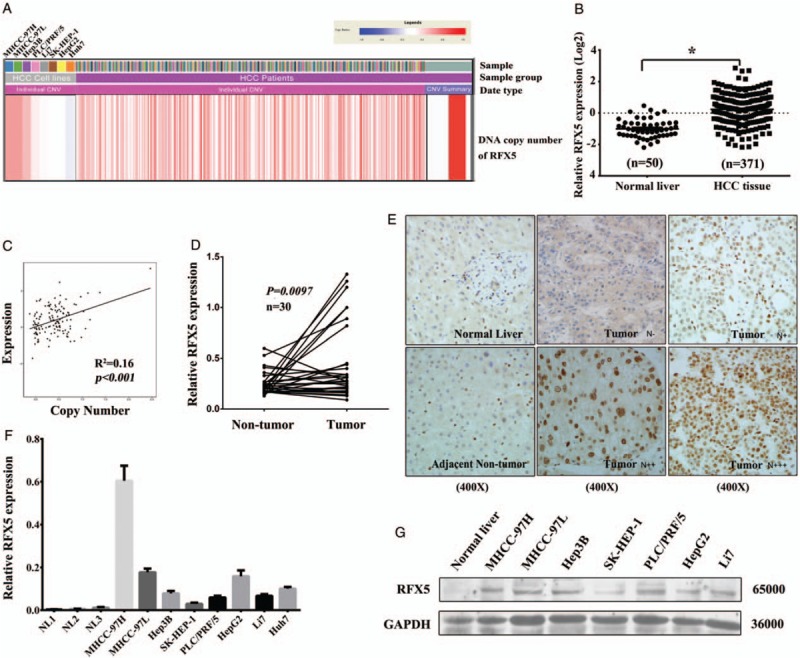
RFX5 is amplified and overexpressed in HCC. (A) The CNV of RFX5 locus in HCC tissues and cell lines. (B) The mRNA expression of RFX5 in HCC tissues determined by RNA-seq data from TCGA LIHC dataset. (C) The correlation between the mRNA expression and DNA copy number of RFX5 in patients with HCC from TCGA LIHC dataset. (D) The mRNA level of RFX5 in HCC tumor tissues and adjacent non-tumor tissues was determined by QRT-PCR analysis. (E) The expression of RFX5 protein in HCC tumor and adjacent non-tumor tissues was determined by IHC analysis (original magnification: ×400. N−, nuclear staining in less than 5% of tumor cells; N+, nuclear staining in 5% to 30% of tumor cells; N++, nuclear staining in 31% to 50% of tumor cells; N+++, nuclear staining in >51% of tumor cells). (F) The mRNA level of RFX5 in HCC cell lines was determined by QRT-PCR analysis. Data were shown as mean ± SD (n = 3). (G) The expression of RFX5 protein in HCC cell lines was determined by Western blotting analysis. ∗P < 0.001. CVN: Copy number variation; HCC: Hepatocellular carcinoma; IHC: Immunohistochemistry; LIHC: Liver hepatocellular carcinoma; N: Nucleus; QRT-PCR: Quantitative real-time polymerase chain reaction; RFX5: Regulatory factor X-5; RNA-seq: RNA sequencing; SD: Standard deviation; TCGA: The Cancer Genome Atlas.
In consistent with TCGA LIHC cohort, overexpression of RFX5 mRNA was detected in HCC tumor tissues as compared with the paired non-tumor tissues of Guilin cohort (P = 0.0097) [Figure 1D]. The expression of RFX5 protein in HCC was further determined by immunohistochemistry (IHC) staining a TMA containing 128 primary HCC specimens with anti-RFX5 antibody. In comparison with the corresponding non-tumor tissue, RFX5 overexpression was found in 91/128 (71.1%) informative HCC cases [Figure 1E]. RFX5 protein was mostly located in the nucleus of tumor cells and displayed a heterogeneous expression pattern in HCC (N−, 28.90% [37/128]; N+, 31.25% [40/128]; N++, 30.47% [39/128]; N+++, 9.38% [12/128]) [Figure 1E]. However, there was no correlation between the expression level of RFX5 and the prognosis of HCC. Maybe, the small number of cases enrolled in this analysis limited the statistic power.
Moreover, the amplification and overexpression of RFX5 in HCC cell lines were prevalent. The expression level of RFX5 mRNA in most HCC cell lines (MHCC-97H, MHCC-97L, HepG2, Hep3B, SK-HEP-1, PLC/PRF/5, Huh7, and Li7) was determined by QRT-PCR. These HCC cell lines (8/8) showed amplification in RFX5 locus [Figure 1F], while MHCC-97H had the highest RFX5 mRNA level. Furthermore, we found that RFX5 protein was also overexpressed in most HCC cell lines (7/7) by Western blotting analysis [Figure 1G].
RFX5 oncogene dependence in human HCC cell lines
In order to determine the biological significance of RFX5 in HCC, cell lines transduced with shRNAs targeting RFX5 (RFK1 and RFK2) were subjected to clonogenic assay. Knocking down RFX5 greatly reduced the clone-forming ability in MHCC-97H (P < 0.001), HepG2 (P < 0.001), and SK-HEP-1 (P < 0.001), but not in MHCC97L [Figure 2A and 2B]. Moreover, HepG2 cells transduced with lentiviral vector expressing FLAG-RFX5 were able to form more colonies in comparison with controls (P < 0.05) [Figure 2C and 2D].
Figure 2.
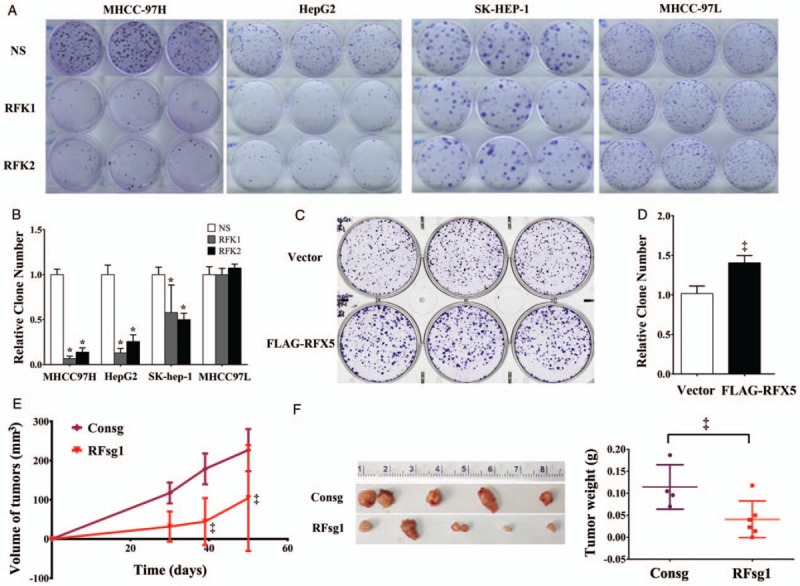
Oncogene dependence of RFX5 in HCC. (A and B) Clonogenicity assay of MHCC-97H, HepG2, SK-HEP-1 and MHCC-97L infected with lentiviral control shRNA (NS) and RFX5 specific shRNAs (RFK1 and RFK2) (A) and the quantification of colonies of four HCC cell lines from three independent repeats. Data were shown as mean ± SD (B). (C and D) Clonogenicity assay of HepG2 cells infected lentivirus Vector and FLAG-RFX5 (C), and the quantification of clone numbers from three independent repeats. Data were shown as mean ± SD (D). (E and F) MHCC-97H cells treated with lentivirus expressing espCas9 and sgRNAs against non-targeting control (Consg) and RFX5 (RFsg1) were subcutaneously implanted in BALB/c Nude. Tumor volumes were measured with calipers and calculated (E). Tumors were harvested and weighted after MHCC-97H implantation for 50 days (F). ∗P < 0.001, ‡P < 0.05. HCC: Hepatocellular carcinoma; RFX5: Regulatory factor X-5; SD: Standard deviation; shRNA: Short hairpin RNA.
To disclose the oncogene dependency of RFX5 in HCC, we determined the growth of subcutaneous tumors from MHCC-97H cells infected with RFX5 specific lentiviral sgRNAs (RFsg1) or sgRNA control (Consg) based on CRISPR/Cas9 system. The averaged tumor volumes were decreased over time in the RFsg1 group compared with the Consg group (P < 0.05) [Figure 2E]. After implantation for 50 days, the mice were sacrificed and the size and weight of tumors were measured. The results showed that the tumors developed from RFsg1-transfected MHCC-97H cells displayed dramatically smaller volume and lighter weight than that developed from control cells (the averaged tumor volume: 94.00 ± 45.81 mm3vs. 226.8 ± 26.86 mm3, t = 2.500, P = 0.0387; the averaged tumor weight: 0.041 ± 0.017 g vs. 0.115 ± 0.053 g, t = 2.520, P = 0.0358) [Figure 2E and 2F]. It showed that knocking down RFX5 with sgRNA1 dramatically suppressed the tumor growth.
RFX5 positively regulates YWHAQ expression in HCC
In our previous study, RFX5 could regulate TPP1.[5] However, subsequent function study proved that TPP1 had no effect on regulating the apoptosis, proliferation, and subcutaneous tumor growth in HCC (data not shown).
Further, the transcriptional target of RFX5 in HCC cells was determined by ChIP-seq analysis in HepG2. We found that there was a strong RFX5 binding peak existing in the promoter region (−1020 to +2036 bp) of YWHAQ [Figure 3A] and then validated it by ChIP-PCR. The data showed that it stably yielded specific amplification of the immunoprecipitated DNA fragments in HepG2, MHCC-97H, and SK-HEP-1 by the primer recognizing YWHAQ promoter region [Figure 3B]. These results suggested that RFX5 could directly bind the YWHAQ promoter region in HCC cells. We further determined whether the binding of RFX5 to the YWHAQ promoter could promote the transcription of YWHAQ. Co-transfection of FLAG-RFX5 plasmid with reporter plasmid pGL4-YWHAQ in HEK293T greatly enhanced the transcription levels of Luciferase, controlled by the promoter region (−1020 to +2036 bp) of YWHAQ [Figure 3C]. The results suggest that RFX5 increases the transcriptional activity of the YWHAQ promoter.
Figure 3.
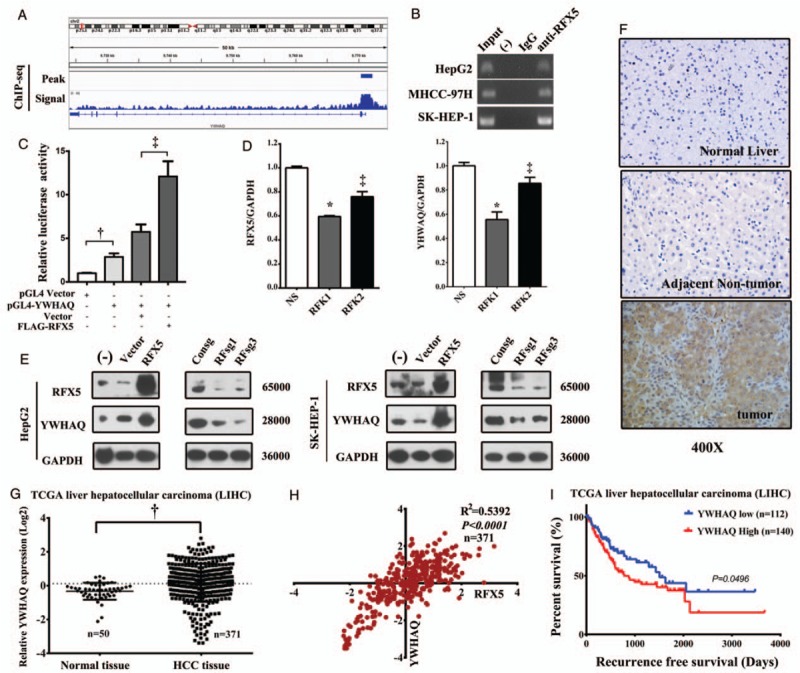
RFX5 positively regulates YWHAQ expression in HCC. (A) The ChIP-seq binding peak, signal of RFX5 in the promoter region of YWHAQ. (B) Immunoprecipitated DNA fragments with anti-RFX5 antibody were subjected to PCR detection of YWHAQ promoter region in HCC cells. (C) The transcriptional activity of YWHAQ promoter reporter constructs (pGL4-YWHAQ) was measured in HEK293T by RLA, the ratio of firefly to Renilla luciferase activities. The effect of RFX5 expression on the transcriptional activity of YWHAQ was determined by co-transfection of FLAG-RFX5 with pGL4-YWHAQ in HEK293T. Data were shown as mean ± SD (n = 3). (D) HepG2 cells transduced with shRNA targeting RFX5 were subjected to QRT-PCR analysis to determine the expression level of YWHAQ mRNA. Data were shown as mean ± SD (n = 3). (E) HepG2 and SK-HEP-1 cells transduced with lentiviral RFX5 or sgRNA targeting RFX5 were subjected to Western blotting analysis to determine the expression level of YWHAQ protein. (F) The expression of YWHAQ protein in HCC tumor and adjacent non-tumor tissues was determined by IHC. (G) The mRNA expression of YWHAQ in HCC tissues determined by RNA-seq data from TCGA LIHC dataset. (H) The correlation of the mRNA expression of RFX5 and YWHAQ in patients with HCC determined by RNA-seq data from TCGA LIHC dataset. (I) The correlation of YWHAQ mRNA expression level with recurrence-free survival time determined by RNA-seq in TCGA LIHC dataset. ‡P < 0.05, †P < 0.01, ∗P < 0.001. ChIP: Chromatin immunoprecipitation assay; HCC: Hepatocellular carcinoma; LIHC: Liver hepatocellular carcinoma; PCR: Polymerase chain reaction; QRT-PCR: Quantitative real-time polymerase chain reaction; RFX5: Regulatory factor X-5; RLA: Relative luciferase activity; RNA-seq: RNA sequencing; SD: Standard deviation; shRNA: Short hairpin RNA; TCGA: The Cancer Genome Atlas.
We knocked down RFX5 by lentiviral shRNAs (RFK1 and RFK2) or sgRNA (RFsg1 and RFsg3), and overexpressed it by retroviral pMSCV-RFX5 in HCC cells. We then determined the expression level of YWHAQ by QRT-PCR and Western blotting, to further identify whether modulating RFX5 could regulate the expression of YWHAQ. In HepG2 cells, RFX5 knockdown by shRNAs significantly inhibited the transcription levels of YWHAQ (P < 0.05) [Figure 3D]. In HepG2 and SK-HEP-1 cells, RFX5 overexpression significantly upregulated the protein level of YWHAQ; while knocking down RFX5 with RFsg1 or RFsg3 greatly decreased the protein level of YWHAQ [Figure 3E]. These results indicated that RFX5 closely regulated the expression level of YWHAQ.
YWHAQ is overexpressed in HCC and linked to poor prognosis
Similarly to RFX5, the level of YWHAQ mRNA dramatically elevated in HCC tumor tissues compared with adjacent non-tumor tissues by analyzing the TCGA LIHC RNA-seq data (0.058 ± 0.058 vs. −0.450 ± 0.071, t = 3.153, P = 0.0017) [Figure 3G]. Strikingly, the mRNA expression level of YWHAQ was tightly correlated with that of RFX5 (r = 0.734, R2 = 0.540, P < 0.001) in LIHC RNA-seq data [Figure 3H].
Furthermore, to identify the expression pattern of YWHAQ protein in HCC, we performed IHC analysis of YWHAQ with the TMA of Guilin cohort containing 51 primary HCC specimens. In comparison with the corresponding adjacent non-tumor tissue, YWHAQ overexpression was detected in tumor tissue of 70.59% (36/51) informative HCC cases [Figure 3F]. Intriguingly, the mRNA expression level of YWHAQ was significantly associated with the prognosis of patients with HCC in TCGA LIHC dataset. Patients with higher YWHAQ mRNA expression level tend to have a shorter recurrence-free survival time (P = 0.0496) [Figure 3I].
RFX5-YWHAQ pathway promotes the clonogenic growth and tumorigenicity of HCC by suppressing apoptosis
To determine whether the downstream event led to the suppression of clonogenic growth of HCC cell lines after RFX5 silencing, retroviral YWHAQ (pMSCV-YWHAQ) was transduced to recover the YWHAQ level in RFsg-treated HepG2 and SK-HEP-1 cell lines. The colony formation rescue assay showed that overexpression of YWHAQ fully recovered the colony numbers of RFsg-treated HepG2 [Figure 4A and 4B] and SK-HEP-1 cells [Figure 4C and 4D].
Figure 4.

YWHAQ overexpression largely rescues the growth inhibitory effects of RFX5 knockdown in HCC. (A and B) Clonogenicity assay of HepG2 cells infected with RFX5 sgRNAs (RFsg1 and RFsg3) and rescued with lentiviral control vector or lentiviral YWHAQ expressing vector (A), and the quantification of clone numbers from three independent repeats (B). Data were shown as mean ± SD. (C and D) Clonogenicity assay of SK-HEP-1 cells infected with RFX5 sgRNAs (RFsg1 and RFsg3) and rescued with lentiviral control vector or lentiviral YWHAQ expressing vector (C), and the quantification of clone numbers from three independent repeats (D). Data were shown as mean ± SD. (E and F) SK-HEP-1 cells treated with lentivirus expressing espCas9 and sgRNAs against non-targeting control (Consg) and RFX5 (RFsg1) alone, or transduced Consg and RFsg1 together with lentiviral YWHAQ, were implanted subcutaneously in BALB/c nude for three different mice in each group. Tumor volume was measured with calipers and calculated (E). ‡: Vector + RFsg1 vs. Vector + Consg; §: YWHAQ + RFsg1 vs. Vector + RFsg1. Tumors were harvest and weighted after MHCC-97H implantation for 60 days (F). §P < 0.05, ‡P < 0.05, †P < 0.01, ∗P < 0.001. HCC: Hepatocellular carcinoma; RFX5: Regulatory factor X-5; SD: Standard deviation.
Moreover, the rescue experiment was also performed with SK-HEP-1 cells in tumor xenograft model. We found that the tumor growth was inhibited by RFX5 knockdown (RFsg1 group) and this inhibition effect could be rescued by YWHAQ overexpression (YWHAQ-RFsg1 group), as shown by tumor volume over time after implementation [Figure 4E]. After implantation for 60 days, the mice were sacrificed and the size and weight of tumors were measured. The tumors of RFsg1 group were dramatically smaller and lighter than that of Consg group (the averaged tumor volume: 365.3 ± 29.42 mm3vs. 589.0 ± 22.87 mm3, P < 0.05; the averaged tumor weight: 0.059 ± 0.001 g vs. 0.100 ± 0.008 g, P < 0.05), while the tumors developed from YWHAQ-RFsg1 group were significantly larger and heavier than that developed from RFsg1 group (the averaged tumor volume: 807.0 ± 156.90 mm3vs. 365.3 ± 29.42 mm3, P < 0.05; the averaged tumor weight: 0.199 ± 0.051 g vs. 0.059 ± 0.001 g, P < 0.05) [Figure 4E and 4F]. The data demonstrated that the tumor size and weight of subcutaneous tumor were significantly rescued by transduced with lentiviral YWHAQ after RFX5 silencing. All these results showed that YWHAQ overexpression largely attenuated the growth inhibitory effects of RFX5 depletion in HCC cells, indicating that YWHAQ is a downstream effector of RFX5.
Next, we examined the effect of RFX5-YWHAQ pathway on apoptosis in HCC. Following the treatment of cells with Dactinomycin D (ActD) for 6 h, overexpression of RFX5 or YWHAQ dramatically inhibited ActD-induced apoptosis in both HepG2 [Figure 5A] and SK-HEP-1 [Figure 5B], while knockdown of RFX5 with RFsg (RFsg1 or RFsg3) increased the apoptotic level in both HepG2 [Figure 5C] and SK-HEP-1 [Figure 5D].
Figure 5.
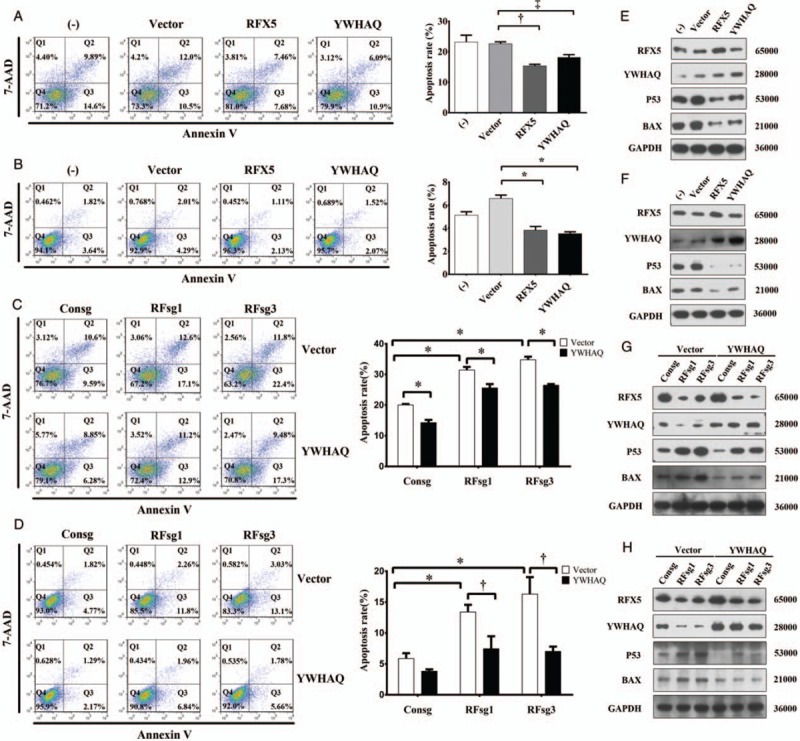
RFX5-YWHAQ inhibits apoptosis by regulating p53 and downstream pathway in HCC. (A and B) HepG2 (A) and SK-HEP-1 cells (B) transduced lentiviral vector control, RFX5 or YWHAQ were treated with 5 nmol/L ActD and subjected to apoptosis analysis by one-way ANOVA. Data were shown as mean ± SD (n = 3). (C and D) HepG2 (C) and SK-HEP-1 cells (D) transduced lentiviral sgRNA or YWHAQ were treated with 5 nmol/L ActD and subjected to apoptosis analysis by one-way analysis of variance. Data were shown as mean ± SD (n = 3). (E and F) Total proteins from HepG2 (E) and SK-HEP-1 cells (F) transduced lentiviral vector control, RFX5 or YWHAQ were subjected to Western blotting analysis of indicated proteins. (G and H) Total proteins from HepG2 (G) and SK-HEP-1 cells (H) transduced lentiviral sgRNA or YWHAQ were subjected to Western blotting analysis of indicated proteins. ‡P < 0.05, †P < 0.01, ∗P < 0.001. ActD: Dactinomycin D; HCC: Hepatocellular carcinoma; RFX5: Regulatory factor X-5; SD: Standard deviation.
To further study if RFX5 protected HCC cells from apoptosis via YWHAQ, we overexpressed YWHAQ in RFsg-treated HCC cells and monitored the alteration in apoptotic level. As expected, overexpression of YWHAQ significantly suppressed the apoptosis induced by RFX5 knockdown in both HepG2 [Figure 5C] and SK-HEP-1 [Figure 5D]. These data supported the finding that RFX5 could suppress apoptosis in HCC via RFX5-YWHAQ pathway.
RFX5-YWHAQ pathway negatively regulates p53 and its downstream signaling pathway
To reveal the mechanism of RFX5-YWHAQ pathway in protecting HCC cells from apoptosis, the expression of p53 and Bax was further detected by Western blotting. When overexpression of RFX5 could upregulate the protein level of YWHAQ, the protein level of p53 and Bax was greatly reduced in both HepG2 [Figure 5E] and SK-HEP-1 [Figure 5F] cells. Besides, directly overexpression of YWHAQ induced similar regulation effect on the protein level of p53 and Bax [Figure 5E and 5F], which was consistent with the results mentioned above.
Moreover, when RFX5 silencing dramatically downregulated the protein level of YWHAQ, the protein level of p53 and Bax was upregulated in both HepG2 [Figure 5G] and SK-HEP-1 [Figure 5H] cells. While overexpressing YWHAQ in RFX5 knockdown cells could reverse the protein level of p53 and Bax to similar level in parent HepG2 [Figure 5G] and SK-HEP-1 [Figure 5H] cells.
Discussion
In this study, we revealed that RFX5 plays an important role in HCC as a putative tumor driver gene. It was frequently amplified and overexpressed in HCC and was essential for HCC progression. We also identified that increased copy number of RFX5 may be one of the reasons leading to its overexpression in HCC. However, the expression of RFX5 protein by IHC had no correlation with clinicopathological parameters (gender, age, alcohol, tumor size, AFP, HBsAg, cirrhosis, TNM stage, multi-nodularity, lympho-invasion, venous infiltration, and metastasis and invasion) of 128 patients with HCC of Guilin cohort. Furthermore, there was no correlation between the expression level of RFX5 and the prognosis of HCC. It suggested that RFX5 might function together with its transactivated target genes as transcription factor in HCC, but not only by itself. Though RFX5 has been known as a crucial regulator of MHCII gene transcription,[2–4] our study proved that RFX5 also plays a key role in tumor, which gave us a clue about a possible crosstalk between immune response and tumor development that should be further explored in the further studies.
YWHAQ belongs to 14-3-3 family proteins, which form homo- or hetero-dimeric structures and mainly bind to their target proteins through phosphorylation-dependent interactions which function as chaperones, adapters, and regulators, enabling them to play an essential role in multiple cellular processes.[10–12] Several previous studies have shown that 14-3-3 proteins contributed to initiation, oncogenesis, and progression of several human tumors.[13] YWHAQ was frequently overexpressed in primary human breast cancers and correlated with shorter overall survival.[14] Hodgkinson et al proposed a potential role for YWHAQ as predictive biomarkers of neoadjuvant chemotherapy resistance in breast cancer.[15] In our study, we explore the role of YWHAQ in HCC and found YWHAQ overexpression in patients with HCC correlated with the recurrence-free survival.
Next, we proved that the transcription of YWHAQ was largely regulated by RFX5 in HCC. We then showed that YWHAQ was a dominant downstream effector of RFX5 in HCC, and RFX5-YWHAQ pathway could protect HCC cells from apoptosis. We further identified that overexpression of both RFX5 and YWHAQ significantly down-regulated the protein level of p53 and Bax and subsequently inhibited DNA damage-induced apoptosis. Moreover, overexpression of YWHAQ in RFX5 knockdown cells could rescue the phenotype induced by RFX5 depletion. These results demonstrated that RFX5 transcriptionally activated YWHAQ and closely regulated the p53-Bax apoptosis pathway, which is consistent with previous studies in many aspects. Such as, the overexpression of 14-3-3 proteins could promote survival of cancer cells likely through the negative regulation of p53.[16] In addition, under normal cellular condition, 14-3-3 proteins could interact with Bax and sequester it in the cytoplasm in order to protect the cell from apoptosis.[17]
However, there were several limitations in our study. First, the small number of cases enrolled in this analysis limited the statistic power. Second, we need to collect more cases and then explore the role of RFX5 in HBV or HCV-related HCC tissues. Third, due to lack of the TMA, we did not have enough data to analyze the relationships between YWHAQ expression by IHC staining and clinicopathological parameter of patients with HCC. These issues should be conducted in further studies.
In conclusion, our results revealed that RFX5 plays an important role in development of HCC. It transcriptionally activates YWHAQ expression, which promotes HCC progression via regulating p53-Bax pathway and inhibiting apoptosis.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81201569 and No. 81541151), the Beijing Natural Science Foundation (No. 7132186), and the National Key Sci-Tech Special Project of China (No. 2018ZX10302207, No. 2017ZX10203202).
Conflicts of interest
None.
Footnotes
How to cite this article: Chen DB, Zhao YJ, Wang XY, Liao WJ, Chen P, Deng KJ, Cong X, Fei R, Wu X, Shao QX, Wei L, Xie XW, Chen HS. Regulatory factor X5 promotes hepatocellular carcinoma progression by transactivating tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein theta and suppressing apoptosis. Chin Med J 2019;00:00–00. doi: 10.1097/CM9.0000000000000296
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018; 68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Steimle V, Durand B, Barras E, Zufferey M, Hadam MR, Mach B, et al. A novel DNA-binding regulatory factor is mutated in primary MHC class II deficiency (bare lymphocyte syndrome). Genes Dev 1995; 9:1021–1032. doi: 10.1101/gad.9.9.1021. [DOI] [PubMed] [Google Scholar]
- 3.Scholl T, Mahanta SK, Strominger JL. Specific complex formation between the type II bare lymphocyte syndrome-associated transactivators CIITA and RFX5. Proc Natl Acad Sci USA 1997; 94:6330–6334. doi: 10.1073/pnas.94.12.6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clausen BE, Waldburger JM, Schwenk F, Barras E, Mach B, Rajewsky K, et al. Residual MHC class II expression on mature dendritic cells and activated B cells in RFX5-deficient mice. Immunity 1998; 8:143–155. doi: 10.1016/S1074-7613(00)80467-7. [DOI] [PubMed] [Google Scholar]
- 5.Zhao Y, Xie X, Liao W, Zhang H, Cao H, Fei R, et al. The transcription factor RFX5 is a transcriptional activator of the TPP1 gene in hepatocellular carcinoma. Oncol Rep 2017; 37:289–296. doi: 10.3892/or.2016.5240. [DOI] [PubMed] [Google Scholar]
- 6.Xie X, Wang X, Liao W, Fei R, Wu N, Cong X, et al. PPPDE1 promotes hepatocellular carcinoma development by negatively regulate p53 and apoptosis. Apoptosis 2019; 24:135–144. doi: 10.1007/s10495-018-1491-6. [DOI] [PubMed] [Google Scholar]
- 7.Wang K, Lim HY, Shi S, Lee J, Deng S, Xie T, et al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology 2013; 58:706–717. doi: 10.1002/hep.26402. [DOI] [PubMed] [Google Scholar]
- 8.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol 2011; 29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.R Core Team. R: A Language and Environment for Statistical Computing. Vienna: Austria: R Foundation for Statistical Computing; 2013. [Google Scholar]
- 10.Martinez JD. 14-3-3 proteins: do they have a role in human cancer? Future Oncol 2005; 1:631–633. doi: 10.2217/14796694.1.5.631. [DOI] [PubMed] [Google Scholar]
- 11.Morrison DK. The 14-3-3 proteins: integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol 2009; 19:16–23. doi: 10.1016/j.tcb.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tzivion G, Gupta VS, Kaplun L, Balan V. 14-3-3 proteins as potential oncogenes. Semin Cancer Biol 2006; 16:203–213. doi: 10.1016/j.semcancer.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 13.Khorrami A, Sharif Bagheri M, Tavallaei M, Gharechahi J. The functional significance of 14-3-3 proteins in cancer: focus on lung cancer. Horm Mol Biol Clin Investig 2017; 32:1–17. doi: 10.1515/hmbci-2017-0032. [DOI] [PubMed] [Google Scholar]
- 14.Wang B, Liu K, Lin HY, Bellam N, Ling S, Lin WC. 14-3-3Tau regulates ubiquitin-independent proteasomal degradation of p21, a novel mechanism of p21 downregulation in breast cancer. Mol Cell Biol 2010; 30:1508–1527. doi: 10.1128/MCB.01335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hodgkinson VC, ELFadl D, Agarwal V, Garimella V, Russell C, Long ED, Fox JN, et al. Proteomic identification of predictive biomarkers of resistance to neoadjuvant chemotherapy in luminal breast cancer: a possible role for 14-3-3 theta/tau and tBID? J Proteomics 2012; 75:1276–1283. doi: 10.1016/j.jprot.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 16.Vazquez A, Grochola LF, Bond EE, Levine AJ, Taubert H, Muller TH, et al. Chemosensitivity profiles identify polymorphisms in the p53 network genes 14-3-3tau and CD44 that affect sarcoma incidence and survival. Cancer Res 2010; 70:172–180. doi: 10.1158/0008-5472.CAN-09-2218. [DOI] [PubMed] [Google Scholar]
- 17.Samuel T, Weber HO, Rauch P, Verdoodt B, Eppel JT, McShea A, et al. The G2/M regulator 14-3-3sigma prevents apoptosis through sequestration of Bax. J Biol Chem 2001; 276:45201–45206. [DOI] [PubMed] [Google Scholar]