

Abstract
Supplemental Digital Content is available in the text
To the Editor: Late-onset multiple acyl-CoA dehydrogenase deficiency (MADD) is clinically characterized by a fluctuating or progressive proximal myopathy, exercise intolerance but good responsive to riboflavin. ETFDH mutations are a major cause of late-onset MADD. We analyzed the clinical course, biochemical studies, and muscle magnetic resonance imaging (MRI) and pathologies of two late-onset MADD adult male patients who were misdiagnosed as polymyositis and presented with serious clinical symptoms of rhabdomyolysis and respiratory insufficient after using large dosage of intravenous glucocorticoids. Our current report broadens the clinical phenotypes spectrum of MADD and reminds clinicians to be cautious about using large dosage glucocorticoids in metabolic compromised patients.
MADD (OMIM 231680) is an autosomal recessive inborn error of fatty acids, amino acids, and choline metabolic disturbance.[1] The clinical phenotypes of MADD can be divided into the following categories: neonatal onset with congenital anomalies (type I), neonatal onset without congenital anomalies (type II), and late onset (type III).[2] Late-onset MADD presents as fluctuating proximal muscle weakness with heterogeneous extramuscular symptoms, such as intermittent episodes of nauseating and vomiting, diarrhea, hypoglycemia, hepatomegaly, or metabolic acidosis.[2] Late-onset MADD can be triggered by fasting, cold, infection, or gestation.[3] Intriguingly, most patients with late-onset MADD are respond well to a pharmacologic dosage of riboflavin and described as riboflavin responsive MADD (RR-MADD).[4] ETFDH mutation is a major genetic cause of RR-MADD and hot-spot mutations of this gene are c.250G>A and c.770A>G in China.[4–6] The clinical spectrums of patients with late-onset MADD is often mild. Here, we describe two patients presented with lethal rhabdomyolysis and respiratory insufficiency, but carried common homozygous ETFDH-c.250G>A mutation and compound heterozygous mutation of ETFDH-c.959C>T and ETFDH-c.250G>A, respectively.
Patient 1 was a 46-year-old male referred to local hospital with complaints of progressive limbs muscle weakness and general fatigue after a cold for 3 months. He presented with difficulties in standing up from a squatting state, climbing stairs and walking long distances, accompanied by exercise intolerance, intermittent abdominal distension, nausea, and vomiting. Gradually, he was unable to raise his head, open mouth and chew foods. Muscle strength measurements are shown in Table 1. Polymyositis was first suspected, thus intravenous glucocorticoids (methylprednisolone, initial dosage was 60 mg/d) was applied. However, neck and masticatory muscle weakness was much more serious and he developed into dysphagia. The patient refused nasogastric intubation and was in a state of stravation, which resulted in completely bedridden and dyspnea.
Table 1.
Manual muscle testing of two patients on muscle strength examination.

To assess the patient's condition objectively, a series of auxiliary examination and biochemical test were performed. His plasma creatine kinase (CK) fluctuated from 528 to 1595 IU/L (reference interval [RI]: 38–174 IU/L). With muscle weakness got worse, his CK level suddenly increased to 23,500 IU/L, serum myoglobin (Mb) exceeded 1000 IU/L, urinary myohemoglobin increased to 282.46 μg/L. He complained of generalized myalgia and pigmenturia, which indicated rhabdomyolysis was occurred. With the condition progressed, patient suffered type II respiratory insufficiency (PCO2, 62.5 mmHg, RI: 35–45 mmHg; PO2, 59.0 mmHg, RI: 80–100 mmHg, pH was 7.516). Plasma lactic acid was 5.0 mmol/L (RI: 0.7–2.1 mmol/L). Alanine aminotransferase (ALT), 1343 IU/L (RI: 9–50 IU/L), aspartate aminotransferase (AST), 7241 IU/L (RI: 15–40 IU/L), lactate dehydrogenase (LDH), 13,508 IU/L (RI: 109–245 IU/L). All these biochemical tests indicated patient suffered rhabdomyolysis and respiratory insufficiency. Assistant ventilation was used to relieve his hypercapnia. Combined therapy including intravenous immunoglobulin (20 g/day) and methylprednisolone (120 mg/day) was used to suppress the immune response. Hepatomegaly and fatty infiltration in live was detected by abdominal Doppler ultrasound. Electromyography showed myogenic abnormality.
Patient 2 was a 60-year-old male complained of difficulty in walking long distances, climbing stairs, and standing up for over 8 months. The patient initially exhibited recurrent shortness of breath with chest distress, especially after physical labor. He was referred to the local hospital and diagnosed with “hypertension and cardiac insufficiency.” His symptoms were progressively deteriorated and presented with symmetrical limbs muscle weakness, fatigability, myalgia, and head drop syndrome. Other concomitant symptoms of dysphagia, loss of appetite, intermittent nausea, and vomiting extremely reduced his food intake. Muscle strength measurements are shown in Table 1. Polymyositis was first considered, and he was treated with intravenous methylprednisolone (initial dose at 60 mg/day). However, within 1 week, plasma CK increased to 16,444.6 IU/L, CK-MB was 519.3 IU/L, plasma lactic acid was 9.38 mmol/L, AST 1501 IU/L, and ALT 250 IU/L. Urinary myohemoglobin increased to 635.62 μg/L. In such serious condition, the patient was sent to our department. We performed an emergency hemodialysis to rescue hyperkalemia (K+: 7.25 mmol/L) and hypocalcemia (Ca2+: 2.86 mmol/L). Blood gas analysis revealed that PO2 was 56.0 mmHg, PaCO2 was 19.3 mmHg, and pH was 7.263. Therefore, 3 L/min oxygen inhalation with a facemask was used to improve the level of oxygen saturation. A computed tomography scan showed mild pulmonary inflammation and bilateral pleural thickening. Abdominal Doppler ultrasound showed hepatomegaly and fatty infiltration of the liver. EMG revealed a myogenic abnormality with partial nerve conduction impairment.
Metabolic spectroscopy analysis
In patient 1, the blood acyl-carnitine analysis revealed typical MADD (C8: 0.95 μmol/L, RI: 0.01–0.30 μmol/L; C10: 1.49 μmol/L, RI: 0.02–0.50 μmol/L; C12: 0.42 μmol/L, RI: 0.02–0.20 μmol/L; C14: 0.43 μmol/L, RI: 0.01–0.30 μmol/L) with normal free carnitine in serum. A urinary organic acid assay showed that the concentrations of 3-hydroxybutyrate acid and 4-hydroxyphenyl-lactic acid were elevated. Patient 2 shared similar results in urinary organic acid analysis and plasma acyl-carnitine profiling (C4, C6, C8, and C10 were obviously elevated).
Muscle MRI
Muscle MRI of Patient 1 showed a filamentous high signal in gluteus maximus, a punctate high signal and muscle atrophy in sartorius, gracilis, and biceps femoris were observed in the T1 phase [Supplementary Figure 1A and 1B]. The short-time inversion recovery phase showed obvious edema in affected muscles [Supplementary Figure 1C and 1D]. The degree of fat infiltration in the thigh muscle was Grade 2 and mildly abnormal (fatty streaks detected below 30% muscle volume) according to a previous report.[7] Muscle MRI of Patient 2 showed a filamentous high signal in gluteus maximus, gracilis, and biceps femoris, as well as some punctate high signals in adductor magnus and semi-tendinosus in the T1 phase [Supplementary Figure 1E and 1F]. A broad high signal was observed in the STIR phase, indicating a muscle edema [Supplementary Figure 1G and 1H]. Muscle atrophy was observed in semi-membranosus, semi-tendinosus and biceps femoris, while rectus femoris, vastus lateralis, vastus medialis and vastus intermedius were considered normal or slightly hypertrophic [Supplementary Figure 1E–1H].
Muscle histopathology
In Patient 1, the hematoxylin-eosin (HE) staining revealed a mild variation in fiber size and numerous vacuoles in muscle fibers. Vacuoles were predominantly in type I fibers, as demonstrated by acid and alkaline adenosine triphosphatase (ATPase) staining. Oil red O (ORO) staining confirmed that these vacuoles were lipid droplets. Ragged red fibers were not detected in modified gomori trichrome (MGT). Some muscle fibers presented architecture distortion in nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR). Cyclo-oxygenase and succinate dehydrogenase (SDH) enzyme activities were diffuse and decreased in type I fibers [Figure 1A]. The histopathology of Patient 2 was similar to that of Patient 1, with the exception of multiple necrotic muscle fibers, which was due to rhabdomyolysis [Figure 1B].
Figure 1.
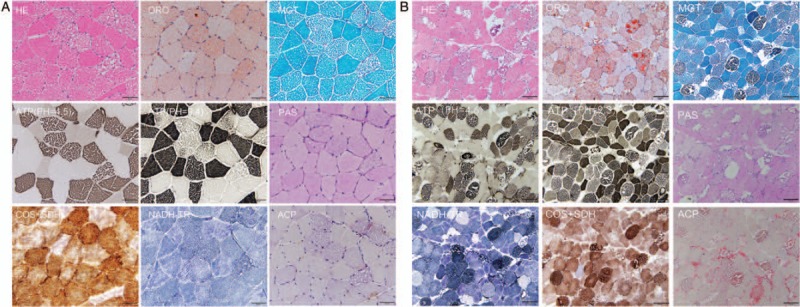
Muscle pathology of the Patient 1 (A) and Patient 2 (B). (A) HE staining showed a numerous of large vacuoles deposited in muscle fibers. ORO staining revealed that vacuoles were stained by ORO dye, indicating the accumulation of lipid droplets. NADH-TR staining revealed a slightly abnormal structure in type II fibers. Cyclo-oxygenase and SDH recombination staining showed enzymatic deficiency in some fibers. Scale bar = 50 μm. (B) The characteristic features of Patient 2 were highly similar to those of Patient 1. However, there were many scattered necrotic fibers in the type I myofibers. MGT staining displayed many dark blue necrotic fibers. Scale bar = 100 μm. HE: Hematoxylin-eosin; MGT: Modified gomori trichrome; NADH-TR: Nicotinamide adenine dinucleotide-tetrazolium reductase; ORO: Oil red O; SDH: Succinate dehydrogenase.
Genetic analysis and Western blot analysis
The Patient 1 was harbored a homozygous mutation of c.250G>A (p.A84T) in exon 3 of ETFDH. Co-segregation analysis revealed that his father and three siblings were all carried a heterozygous mutation of c.250G>A, while his mother's DNA sample was unavailable [Figure 2 A-a].
Figure 2.
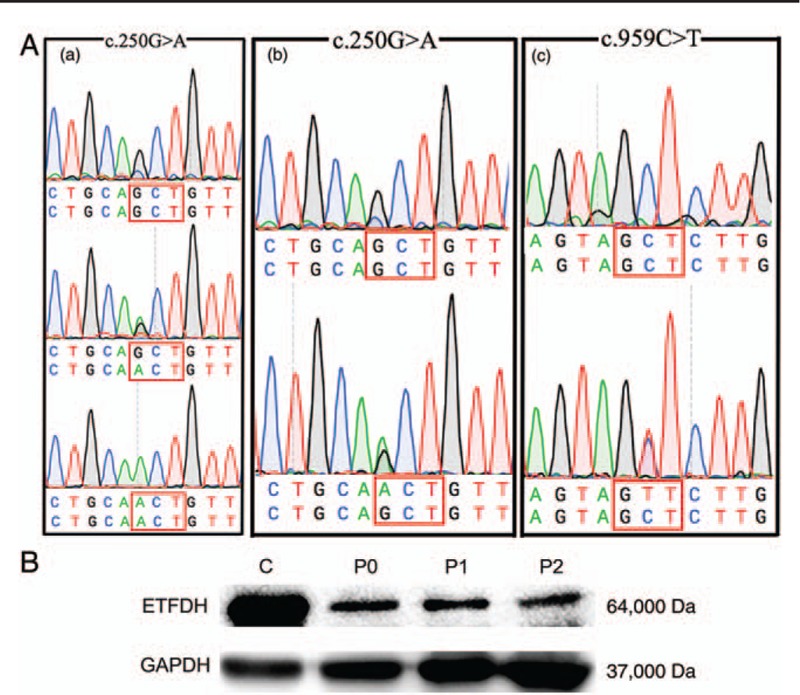
(A-a) Identification of ETFDH mutation of c.250G>A in Patient 1. The upper panel depicts the wild-type sequence, the lower panel represents the homozygous mutated sequence. The middle panel of the box shows a heterozygous sequence. (A-b, A-c) Identification of ETFDH-c.250G>A and c.959C>T in Patient 2. (B) Western blot analysis of muscular ETFDH proteins. The upper panel shows a 64-kDa ETFDH protein. The bottom panel shows a reference GAPDH protein. C indicates a normal control, P0 is a patient with positive MADD, P1 is Patient 1 and P2 is Patient 2. GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; MADD: Late-onsetmultiple acyl-CoAdehydrogenase deficiency.
The Patient 2 harbored a compound heterozygous mis-sense mutation of c.250G>A [Figure 2A-b] and a novel variant locus c.959C>T in ETFDH [Figure 2A-c]. The novel variant of c.959C>T was absented in 1000 genomes, only one case of a heterozygous mutation was found in ExAC that was not present in 100 control subjects. The functional disruption of proteins due to amino acid change was predicted by the SIFT (score: 0.25) and PolyPhen-2 (score: 0.998) software programs.
Western blot analysis showed that ETFDH expression in the muscle specimens was significantly decreased in both patients [Figure 2B].
Treatment and follow-up study
Since these two patients were finally diagnosed as late-onset MADD by biochemically and genetically, intravenous glucocorticoid was replaced by riboflavin (150 mg/day), l-carnitine (90 mg/day), and coenzyme Q (60 mg/day). Patient 1 was gradually withdrawn from mechanically assisted ventilation and nasogastric intubation in 1 week and he achieved good recovery of myodynamia and daily living ability in 3 weeks. Patient 2 recovered to asymptomatic and could walk independently within 1 month. In the follow-up study, after more than 6 months, Patient 1 presented with normal strength and was able to raise up from a sitting squatting state without any help. The CK level and blood acyl-carnitine analysis decreased to normal levels. After more than 3 months, the CK, LDH, liver enzymes, and Mb levels of Patient 2 was returned to normal range. Both patients took riboflavin (30 mg/day) and coenzyme Q as a maintenance treatment to prevent disease recurrence.
Typical clinical phenotype of late-onset MADD was presented with recurrent or progressive proximal myopathy, muscle weakness and myalgia, and recurrent episodes of hypoglycemia with hypoketosis, as well as gastrointestinal disorders and hepatic dysfunction. Many factors, including cold, infection and starvation, increase metabolic stress, and exacerbate muscle weakness. When patients described their symptoms were muscle weakness or myalgia, clinicians were first impressed by polymyositis, glycogen storage diseases, or progressive muscular dystrophy. Our patients were initially presented with polymyositis-like myopathy but rapidly developed into an acute metabolic crisis. Notably, the patients exhibited some clinical clues indicating metabolic disorders rather than polymyositis, such as exercise intolerance, dropped head syndrome, hyperlactatemia, high levels of LDH, and myogenic damage in EMG.
Rhabdomyolysis accompanied by respiratory deficiency has been reported in very few patients with MADD.[8] To date, no literature in English reported rhabdomyolysis and respiratory failure resulted from the common mutations of c.250G>A in ETFDH. Patient suffered rhabdomyolysis was characterized by a marked increase in the plasma CK level with or without myoglobinuria.[9] The muscle MRI of our patients showed that several muscles were infiltrated by fatty tissue. In addition, extensive swelling and edema were observed in the gluteus femoris muscle, which were resulted from rhabdomyolysis. Both patients were presented with hypertrophy in anterior parts of the thigh muscle, which might be a compensation for the atrophy and weakness in the posterior thigh muscle. A variable degree of regenerating fibers was seen, if muscle biopsy was performed after a metabolic crisis episode, which suggested that mild rhabdomyolysis events occurred during metabolic decompensation in MADD.[10] In patient 1, necrotic and regeneration muscle fibers were not observed, because muscle biopsy was performed before rhabdomyolysis occured. Another serious complication in our patients was acute respiratory insufficiency, we hypothesized that respiratory insufficiency was the outcome of the interaction between myopathic and metabolic factors, including the acid-base disturbance and internal environment disturbance caused by rhabdomyolysis.
Glucocorticoids can promote the treatment effects to a certain extent in some patients with RR-MADD, but cannot achieve the same efficacy as the riboflavin. Glucocorticoids played a good role in supplying the increased metabolic demand of body through promote catabolic and liberate energy substrates and alleviate oxidative stress, as well.[11] However, the condition of our patients deteriorated sharply after intravenous glucocorticoids therapy, we assumed that glucocorticoids aggravated the oxidative stress injury when patients in a state of extremely fatigue, starvation, and infection. Thus, patients could not get through the metabolic crisis and developed into rhabdomyolysis and respiratory insufficiency.
These special cases expanded the spectrums of the variable phenotypes and nature history of late-onset RR-MADD.
Declaration of patient consent
The authors certify that all of the participations have written informed consent and this study was approved by the local Ethical Committees.
Funding
This work was supported by grants from National Natural Science Foundation of China (U1505222, 81271254, Beijing).
Conflicts of interest
None.
Supplementary Material
Footnotes
How to cite this article: Chen HZ, Jin M, Cai NQ, Lin XD, Liu XY, Xu LQ, Lin MT, Lin F, Wang N, Wang ZQ, Xu GR. Rhabdomyolysis and respiratory insufficiency due to the common ETFDH mutation of c.250G>A in two patients with late-onset multiple acyl-CoA dehydrogenase deficiency. Chin Med J 2019;00:00–00. doi: 10.1097/CM9.0000000000000288
References
- 1.Bruno C, Dimauro S. Lipid storage myopathies. Curr Opin Neurol 2008; 21:601–606. doi: 10.1097/WCO.0b013e32830dd5a6. [DOI] [PubMed] [Google Scholar]
- 2.Olsen RKJ, Andresen BS, Christensen E, Bross P, Skovby F, Gregersen N. Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple Acyl-CoA dehydrogenation deficiency. Hum Mutat 2003; 22:12–23. doi: 10.1002/humu.10226. [DOI] [PubMed] [Google Scholar]
- 3.Zhuo ZH, Jin PN, Li FY, Li HY, Chen XX, Wang HL. A case of late-onset riboflavin responsive multiple acyl-CoA dehydrogenase deficiency (MADD) with a novel mutation in ETFDH gene. J Neurol Sci 2015; 353:84–86. doi: 10.1016/j.jns.2015.04.011. [DOI] [PubMed] [Google Scholar]
- 4.Lan MY, Fu MH, Liu YF, Huang CC, Chang YY, Liu JS, et al. High frequency of ETFDH c.250G > A mutation in Taiwanese patients with late-onset lipid storage myopathy. Clin Genet 2010; 78:565–569. doi: 10.1111/j.1399-0004.2010.01421.x. [DOI] [PubMed] [Google Scholar]
- 5.Wang ZQ, Chen XJ, Murong SX, Wang N, Wu ZY. Molecular analysis of 51 unrelated pedigrees with late-onset multiple acyl-CoA dehydrogenation deficiency (MADD) in southern China confirmed the most common ETFDH mutation and high carrier frequency of c.250G > A. J Mol Med 2011; 89:569–576. doi: 10.1007/s00109-011-0725-7. [DOI] [PubMed] [Google Scholar]
- 6.Xi JY, Wen B, Lin J, Zhu WH, Luo SS, Zhao CB, et al. Clinical features and ETFDH mutation spectrum in a cohort of 90 Chinese patients with late-onset multiple acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 2014; 37:399–404. doi: 10.1007/s10545-013-9671-6. [DOI] [PubMed] [Google Scholar]
- 7.Liu XY, Jin M, Wang ZQ, Wang DN, He JJ, Lin MT, et al. Skeletal muscle magnetic resonance imaging of the lower limbs in late-onset lipid storage myopathy with electron transfer flavoprotein dehydrogenase gene mutations. Chin Med J 2016; 129:1425–1431. doi: 10.4103/0366-6999.183423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olsen RKJ, Pourfarzam M, Morris AAM, Dias RC, Knudsen I, Andresen BS, et al. Lipid-storage myopathy and respiratory insufficiency due to ETFQO mutations in a patient with late-onset multiple acyl-CoA dehydrogenation deficiency. J Inherit Metab Dis 2004; 27:671–678. doi: 10.1023/B:BOLI.0000042986.10291.e9. [DOI] [PubMed] [Google Scholar]
- 9.Zutt R, van der Kooi AJ, Linthorst GE, Wanders RJA, de Visser M. Rhabdomyolysis: review of the literature. Neuromuscular Disord 2014; 24:651–659. doi: 10.1016/j.nmd.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 10.Song Y, Selak MA, Watson CT, Coutts C, Scherer PC, Panzer JA, et al. Mechanisms underlying metabolic and neural defects in zebrafish and human multiple acyl-CoA dehydrogenase deficiency (MADD). PLoS One 2009; 4:e8329.doi: 10.1371/journal.pone.0008329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peckett AJ, Wright DC, Riddell MC. The effects of glucocorticoids on adipose tissue lipid metabolism. Metabolism 2011; 60:1500–1510. doi: 10.1016/j.metabol.2011.06.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.