

Abstract
The Na+/K+-ATPase (NKA) is a ubiquitous membrane-bound enzyme responsible for generating and maintaining the Na+ and K+ electrochemical gradients across the plasmalemma of living cells. Numerous studies in non-neuronal tissues have shown that this transport mechanism is reversibly regulated by phosphorylation/dephosphorylation of the catalytic α subunit and/or associated proteins. In neurons, Na+/K+ transport by NKA is essential for almost all neuronal operations, consuming up to two-thirds of the neuron's energy expenditure. However, little is known about its cellular regulatory mechanisms. Here we have used an electrophysiological approach to monitor NKA transport activity in male rat hippocampal neurons in situ. We report that this activity is regulated by a balance between serine/threonine phosphorylation and dephosphorylation. Phosphorylation by the protein kinases PKG and PKC inhibits NKA activity, whereas dephosphorylation by the protein phosphatases PP-1 and PP-2B (calcineurin) reverses this effect. Given that these kinases and phosphatases serve as downstream effectors in key neuronal signaling pathways, they may mediate the coupling of primary messengers, such as neurotransmitters, hormones, and growth factors, to the NKAs, through which multiple brain functions can be regulated or dysregulated.
SIGNIFICANCE STATEMENT The Na+/K+-ATPase (NKA), known as the “Na+ pump,” is a ubiquitous membrane-bound enzyme responsible for generating and maintaining the Na+ and K+ electrochemical gradients across the plasma membrane of living cells. In neurons, as in most types of cells, the NKA generates the negative resting membrane potential, which is the basis for almost all aspects of cellular function. Here we used an electrophysiological approach to monitor physiological NKA transport activity in single hippocampal pyramidal cells in situ. We have found that neuronal NKA activity is oppositely regulated by phosphorylation and dephosphorylation, and we have identified the main protein kinases and phosphatases mediating this regulation. This fundamental form of NKA regulation likely plays a role in multiple brain functions.
Keywords: CA1 pyramidal cell, Na+/K+-ATPase, protein kinases, protein phosphatases, slow afterhyperpolarization, sodium pump
Introduction
The Na+/K+-ATPase (NKA; commonly referred to as the “Na+ pump”) is a ubiquitous membrane-bound enzyme responsible for generating and maintaining the Na+ and K+ electrochemical gradients across the plasma membrane of living cells (Matchkov and Krivoi, 2016). In neurons, these gradients underlie the negative resting plasma membrane potential (Vm), and mediate numerous cellular processes, such as the generation of actions potentials, synaptic transmission, transport of essential compounds, and more. For each ATP hydrolyzed, NKAs export three Na+ ions in exchange of two imported K+ ions, thereby generating an outward current, which increases resting Vm beyond the value dictated by the electrochemical equilibrium. As the rate of electrogenic Na+/K+ transport by NKAs increases with the rise of intracellular Na+ concentration (Therien and Blostein, 2000), intense spike activity augments NKA current, further hyperpolarizing the neurons until excess intracellular Na+ is pumped out of the cell. This activity-dependent hyperpolarization leads to spike frequency adaptation and even to cessation of firing. Engagement of NKAs in this type of feedback inhibition was previously described in multiple types of vertebrate neurons (Koike et al., 1972; Gordon et al., 1990; Morita et al., 1993; Parker et al., 1996; Pulver and Griffith, 2010; Kim and von Gersdorff, 2012; Zhang et al., 2015; Kueh et al., 2016).
The ion transport activity of NKAs is subjected to acute and chronic modulation by multiple endogenous steroids and primary messengers. Whereas the former agents act generally by binding directly to specific sites at the catalytic α subunit of the NKA molecule, the latter agents, namely, neurotransmitters, hormones, and growth factors, modulate the pump by engaging plasma membrane G-protein-coupled receptors linked to intracellular signaling cascades that affect the phosphorylation state of the α NKA subunit (or intermediate proteins functionally linked to NKA) (Therien and Blostein, 2000; Poulsen et al., 2010). Phosphorylation of these target proteins by protein kinases (PKs) can inhibit or facilitate NKA transport activity in a species- and tissue-specific manner, and its dephosphorylation by protein phosphatases (PPs) exerts the reverse action.
Numerous studies have addressed the phosphorylation/dephosphorylation regulation of NKAs in non-neuronal cell types (Therien and Blostein, 2000; Poulsen et al., 2010). Enigmatically, not much is known about this type of regulation in neurons, even though NKA activity consumes up to two-thirds of the neuron's energy expenditure (Erecinska and Silver, 1994; Howarth et al., 2012) and is required directly or indirectly for almost all neuronal functions.
In CA1 pyramidal cells maintained at near physiological conditions (35°C; extracellular free Ca2+ concentration 1.2 mm), a distinct slow (up to 20–30 s long) afterhyperpolarization (sAHP) is readily evoked by trains of ≥5 closely spaced spikes (Gulledge et al., 2013; Tiwari et al., 2018). An early small component of this sAHP is generated by Ca2+-gated K+ channels (the KCa-sAHP component) and can be eliminated by blockers of voltage-gated Ca2+ channels (Alger and Nicoll, 1980; Hotson and Prince, 1980; Gustafsson and Wigström, 1981; Madison and Nicoll, 1984; Lancaster and Adams, 1987). Several lines of evidence have indicated that the predominant KCa-independent sAHP component is generated by NKAs (Gulledge et al., 2013; Tiwari et al., 2018). First, this component (the NKA-sAHP component) was not associated with a membrane conductance change. Second, although decreasing with hyperpolarization, it did not reverse even at very negative Vm (−115 mV), as expected for NKA current (Rakowski et al., 1997). Third, it was suppressed by ouabain and by a K+-deficient extracellular solution, known to inhibit NKA activity (Glynn and Karlish, 1975). A similar dual-component sAHP can be evoked in CA1 pyramidal cells also by focal application of glutamate (Thompson and Prince, 1986).
The stability of the NKA-sAHP during prolonged (>60 min) intracellular recordings (Tiwari et al., 2018) enables its use as a reliable readout of ongoing NKA activity in single identified neurons. Here we have used such recordings in CA1 pyramidal cells to investigate the regulation of neuronal NKAs by phosphorylation/dephosphorylation and to identify key players in this fundamental process.
Materials and Methods
Ethical approval.
All animal experiments were conducted in accordance with the guidelines of the Animal Care Committee of the Hebrew University.
Hippocampal slice preparation.
Adult (125–175 g) male Wistar rats were decapitated under isoflurane anesthesia, and transverse dorsal hippocampal slices (400 μm) were prepared with a vibratome and transferred to a storage chamber perfused with oxygenated (95% O2/5% CO2) aCSF at room temperature. For recording, slices were placed one at a time in an interface chamber and superfused (flow rate 1 ml/min) with warmed (35°C) oxygenated aCSF containing blockers of synaptic transmission. The temperature was measured with a thermal probe juxtaposed to the slice and maintained at 35°C with a feedback controller (NPI).
Electrophysiology.
Intracellular current-clamp recordings were performed within the entire width of the pyramidal layer of hippocampal subarea CA1b using sharp glass microelectrodes (depth of recording 100–300 μm from slice surface). Using such microelectrodes avoided inadvertent rundown of NKA activity associated with whole-cell patch pipette recordings (due to washout of soluble ingredients critical for NKA function) (Gadsby and Nakao, 1989; Dobretsov et al., 1999; Wang and Huang, 2006). This recording method also enabled the intracellular pulsatile injections of Na+ to evoke NKA-sAHPs in conditions of blocked excitability. For standard intracellular recordings, the microelectrodes were filled with 4 m K+-acetate and 100 mm KCl. For intracellular recordings combined with Na+ injections, the microelectrodes were filled with 3 m Na+-acetate and 100 mm KCl. The filled microelectrodes had resistances of 90–110 mΩ. A bridge amplifier (Axoclamp 2A or Axoclamp 900A, Molecular Devices) was used, allowing simultaneous injection of current and measurement of membrane potential. The bridge balance was carefully monitored and adjusted before each measurement. The pyramidal cells included in this study had stable resting potentials of ≥−60 mV, and overshooting action potentials. The intracellular signals were filtered online at 10 kHz, digitized at a sampling rate of ≥10 kHz, and stored by a personal computer using a data acquisition system (Digidata 1440A) and pCLAMP10.4 software (Molecular Devices).
Apparent input resistance (RN) was measured using a series of 500 ms hyperpolarizing square current pulses of 50 pA increments. Rin was provided by the slope of the relationship of voltage deviation versus current intensity in the linear part of the hyperpolarizing range. Spikes were evoked by injecting brief (2 ms), suprathreshold depolarizing current pulses via the intracellular microelectrode. Spike trains of various durations were elicited by repetitive stimulation at 50 Hz. The size of an sAHP was assessed by measuring its amplitudes at distinct time points after cessation of stimulation and its area (“area under the curve”). Steady positive or negative currents were injected to depolarize or hyperpolarize membrane potential (Vm), as required by the experimental protocol.
Intracellular Na+ injection.
Slices were perfused with aCSF containing TTX (0.5 μm) and 4-ethylphenylamino-1,2-dimethyl-6-methylaminopyrimidinium chloride (ZD7288; 50 μm). The Na+-acetate-filled microelectrodes were used to deliver trains of 150 short (2 ms) pulses of 4–6 nA at 100 Hz to neurons. In some experiments, apamin (100 nm) and 1-[(2-chlorophenyl)diphenylmethyl]-1H-pyrazole (TRAM-34; 5 μm) were also added to prevent potential activation of Ca2+-activated K+ channels secondary to Na+-induced rise in intracellular Ca2+ concentration ([Ca2+]i) (Song et al., 2013), but the evoked sAHPs appeared the same.
Solutions and chemicals.
The normal aCSF comprised the following (in mm): 124 NaCl, 3.5 KCl, 1 MgCl2, 1.6 CaCl2, 26 NaHCO3, and 10 glucose, pH 7.35 (osmolarity 305 mOsm) to which CNQX (15 μm), d-APV (50 μm), picrotoxin (100 μm), and 3-aminopropyl(diethoxymethyl) phosphinic acid hydrate (CGP-55845; 1 μm) were added to block, respectively, AMPA, NMDA, GABAA, and GABAB receptors mediating synaptic transmission. We used 1.6 mm CaCl2 in the aCSF because, in the bicarbonate buffer system, this concentration yields 1.2 mm free Ca2+, corresponding to the baseline interstitial brain Ca2+ concentration measured in vivo (Jones and Keep, 1988). In most experiments, as indicated, the aCSFs contained also the HCN channel blocker ZD7288 (50 μm). The aCSFs designed to block voltage-gated Ca2+ channels contained also NiCl2 (200 μm) and CdCl2 (200 μm). In K+-free aCSF, KCl was omitted. The aCSF designed to enable Ca2+ spikes contained TTX (0.5 μm), a voltage-gated Na+ channel blocker, and 4-AP (3 mm), a blocker of fast voltage-gated K+ channels, as well as 10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone dihydrochloride (XE991; 5 μm) and apamin (100 nA) to block, respectively, hyperpolarizing KV7/M and SK channels that are also activated by the Ca2+ spike (Yue and Yaari, 2004; Chen et al., 2014).
Picrotoxin, CGP-55845, NiCl2, CdCl2, l-arginine, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), PDBu, TTX, TRAM-34, Na-acetate, 4-AP, and ouabain were obtained from Sigma-Aldrich. Noradrenaline (NA) was obtained from Hospira. Chelerythrine chloride, CNQX, apamin, and APV were obtained from Alomone Labs. ZD7288, 8-bromoadenosine 3′,5′-cyclic adenosine monophosphate (8Br-cAMP), tautomycetin, fostriecin, and XE991 were obtained from Tocris Bioscience. (9S,10R,12R)-2,3,9,10,11,12-hexahydro-10-methoxy-2,9-dimethyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzo-diazocine-10-carboxylic acid, methyl ester (KT5823), okadaic acid (OA), and N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide dihydrochloride (H89) were obtained from Abcam-Zotal.
In some experiments, as indicated, slices were preincubated with drugs in the storage chamber for 1–2 h. The same drugs were also included in the perfusing aCSF in the recording chamber. In other experiments, as indicated, drugs were added only to the aCSF perfusing the recording chamber. Reversal of drug effects was not attempted due to their persistent actions.
Data analysis.
The data were obtained from experiments performed on 199 neurons in 196 slices (∼1 neuron per slice) obtained from 134 rats. Each rat supplied 1 or 2 slices (average 1.46) for the experiments. Results are presented as mean ± SEM. Assessment of statistical significance of differences between means was performed with paired two-tailed Student's t test or, for multiple comparisons, with one-way ANOVA using two-sided Dunnett t test. In all tests, the significance level was set to p < 0.05.
Results
Isolation of the NKA-sAHP for the study of NKA regulation
To study the regulation of NKA activity by PKs and PPs in single identified neurons, we performed intracellular sharp microelectrode recordings in CA1 pyramidal cells in adult rat hippocampal slices. The slices were superfused with aCSF containing Cd2+ and Ni2+ (200 μm each) to block the KCa-sAHP, which under our near-physiological conditions (35°C; free extracellular Ca2+ concentration 1.2 mm) contributes only partially to sAHPs evoked by spike trains (Tiwari et al., 2018). We used NKA-sAHPs evoked by long spike trains (150 spikes, triggered by 2-ms-long depolarizing intracellular current pulses, delivered at 50 Hz) (Tiwari et al., 2018) as readouts of NKA transport activity. Evidence that these sAHPs are generated entirely by NKAs has been provided recently (Gulledge et al., 2013; Tiwari et al., 2018). The aCSF contained, in addition, 50 μm ZD7288 to block sAHP shunting by the opening of h-conductance (i.e., HCN channels) (Kaczorowski, 2011; Tiwari et al., 2018), as well as blockers of fast glutamatergic and GABAergic synaptic transmission (see Materials and Methods). Experimental drugs were added to the aCSF before and/or during recordings, as specified. The NKA-sAHPs were monitored for at least 30 min.
In the control aCSF, the mean Vm, apparent RN, and spike amplitude of the sampled neurons were −67.3 ± 3.7 mV, 68.5 ± 15.7 mΩ, and 94.9 ± 6.7 mV, respectively (n = 102). While resting Vm varied between −62.0 and −76.5 mV, all recordings of NKA-sAHPs were made from Vm of −70 mV, maintained by constant current injection. This procedure reduced the variability in size of the NKA-sAHPs conferred by their voltage dependence (Tiwari et al., 2018). We measured the size of an NKA-sAHP by two parameters (Fig. 1a): (1) amplitude at 1 s after the spike train, at which time the medium AHP has ended (Storm, 1989) and (2) area (i.e., integral, or “area under the curve”). Mean values in control recordings were −6.2 ± 0.1 mV and −76.4 ± 1.5 mV * s (n = 102), respectively.
Figure 1.
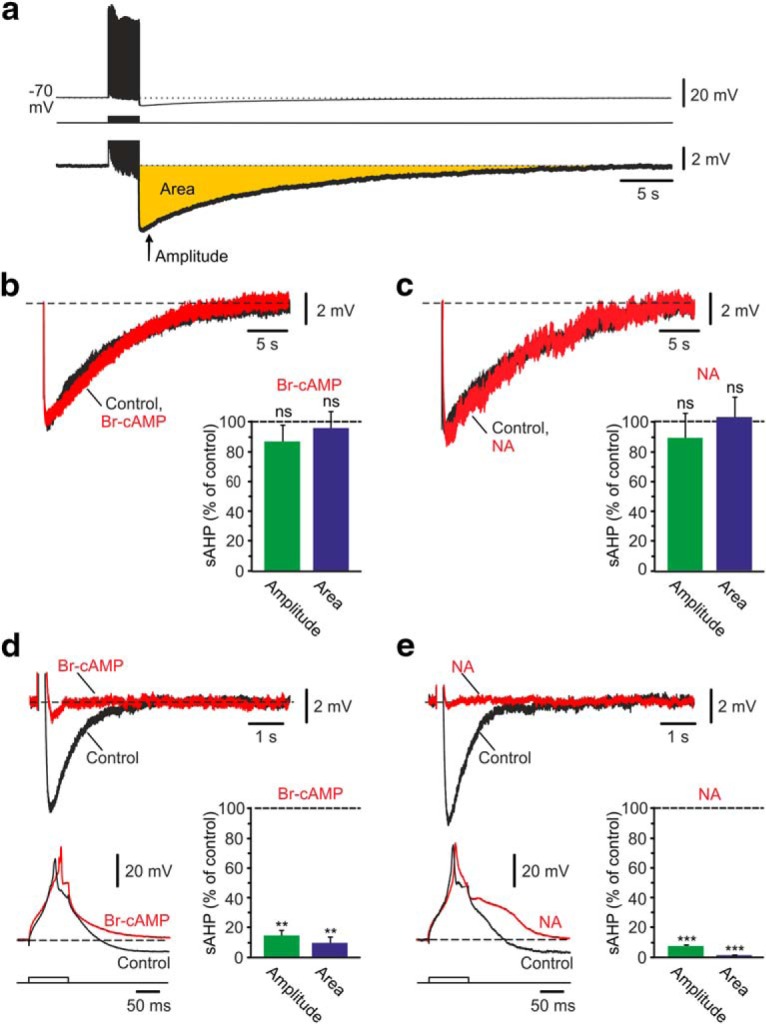
Effects of PKA activation on the NKA-sAHPs. a, A representative sAHP (top) evoked by a 50 Hz train of 150 spikes. Here and below, each sAHP trace is the average of three consecutive recordings obtained at 3 min intervals. Bottom, The enlarged NKA-sAHP trace represents the two measured parameters for NKA-sAHP size: peak amplitude and “area” delineated by the voltage trace and baseline Vm (yellow). Dashed line indicates baseline Vm (−70 mV). b, Activation of PKA by Br-cAMP. The two overlaid traces represent the NKA-sAHPs in the same neuron before (black) and after perfusing the slice with the PKA activator Br-cAMP (2 mm; red). Bar represents the normalized results of six experiments (mean ± SEM) for NKA-sAHP amplitudes (green) and areas (blue). c, Activation of PKA by NA. Same as in b, but applying NA (10 μm) that activates the cAMP/PKA signaling pathway (n = 5). Neither drug affected the size of the NKA-sAHPs, implying that phosphorylation by PKA does not regulate the NKAs. d, Activation of PKA by Br-cAMP. The two overlaid top traces represent the K(Ca)-sAHP (black) before and after perfusing the slice with 2 mm Br-cAMP for 20 min (red). The two overlaid bottom traces represent the respective Ca2+ spikes that evoked these sAHPs. Bar represents the normalized results of seven experiments (mean ± SEM) for K(Ca)-sAHP amplitudes (green) and areas (blue). e, Activation of PKA by NA. Same as in d, but applying 10 μm NA (n = 5). Expectedly, both drugs markedly reduced the K(Ca)-sAHPs without diminishing the Ca2+ spikes. **p < 0.01, ***p < 0.001, ns, not significant.
Effects of PKA modulators
The PKs most frequently implicated in NKA regulation in non-neuronal tissues are the cAMP-dependent PKA, the cyclic-GMP (cGMP)-dependent PKG, and PKC (Therien and Blostein, 2000). To test whether PKA activation affects NKA activity in CA1 pyramidal cells, we monitored the effects of Br-cAMP, a stable, cell-permeable cAMP analog (Robison et al., 1968) on the NKA-sAHPs. Application of Br-cAMP at concentrations up to 2 mm for 30 min had no effect on the amplitude (n = 6; p = 0.4089) or area (n = 6; p = 0.703) of the NKA-sAHPs (Fig. 1b). Likewise, applying 10 μm NA, an activator of the cAMP/PKA pathway in CA1 pyramidal cells (Madison and Nicoll, 1986; Pedarzani and Storm, 1993), affected neither the amplitude (n = 5; p = 0.663) nor the area (n = 5; p = 0.8247) of the NKA-sAHPs (Fig. 1c).
To assure that Br-cAMP and NA activate the cAMP/PKA pathway under our experimental conditions, we examined whether they suppress the IK(Ca)-sAHP, as previously described (Madison and Nicoll, 1986; Pedarzani and Storm, 1993). To that end, Ca2+ spikes followed by “pure” IK(Ca)-sAHP were triggered by 90-ms-long depolarizing current pulses in CA1 pyramidal cells superfused with standard aCSF containing 0.5 μm TTX and 3 mm 4-AP. Br-cAMP, even at a low concentration (2 μm), almost completely abolished the IK(Ca)-sAHP (Fig. 1d; amplitude and area reduced to 14.8% and 9.8% of control, respectively; p = 0.0022 and p = 0.0092, respectively; n = 7) without reducing the Ca2+ spike. A similar effect was exerted by 10 μm NA (Fig. 1e; amplitude and area reduced to 7.3% and 1.0% of control, respectively; p = 0.0001 and p = 0.001, respectively; n = 5).
To test whether the NKA-sAHPs in control conditions are already reduced by the cAMP/PKA pathway, so that they cannot be further affected, we incubated slices for ≥1 h with 10 μm H89, a PKA inhibitor (Chijiwa et al., 1990). The NKA-sAHPs recorded in these slices (n = 10) were similar in amplitude and in area to those in control slices (p = 0.659 and p = 0.988, respectively; see Fig. 7).
Figure 7.
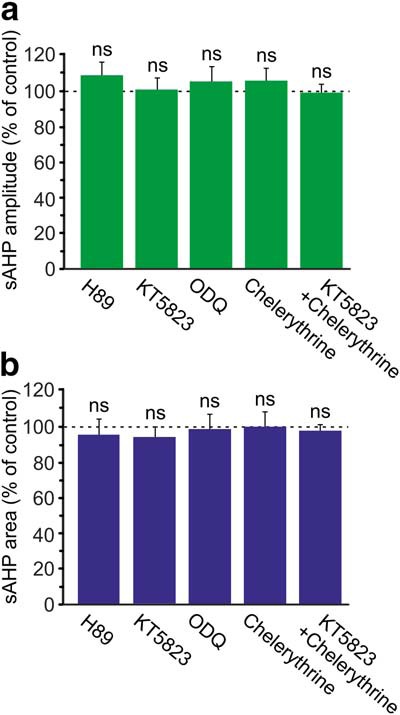
Effects of inhibitors of PK signaling pathways on the NKA-sAHPs. a, Bar diagram represents the effects of various blockers of signaling pathways, applied for ≥1 h, on the amplitudes of the NKA-sAHPs. Amplitude values are normalized with respect to mean control value (n = 102) and depicted as mean ± SEM. The blockers applied are H89 (n = 10), KT5823 (n = 17), ODQ (n = 11), chelerythrine (n = 18), and KT5823 together with chelerythrine (n = 16). Statistical significance was tested with one-way ANOVA (α = 0.05) with two-sided Dunnett t test. b, Same as in a, but for NKA-sAHPs areas. Neither one of the blockers applied affected the amplitude or area of the NKA-sAHPs, ns, not significant.
Together, these data suggest that the cAMP/PKA pathway does not regulate NKA activity in CA1 pyramidal cells. They also indicate that, of the two sAHP components, namely, IK(Ca)-sAHP and NKA-sAHP, only the former is regulated by cAMP/PKA signaling.
Effects of PKG modulators
A second key regulator of NKA activity in many non-neural tissues is the cGMP-dependent PKG (Therien and Blostein, 2000). To test whether PKG activation affects NKA activity in CA1 pyramidal cells, we monitored the effects of Br-cGMP, a cell-permeable cGMP analog resistant to degradation by cyclic GMP phosphodiesterase (Muneyama et al., 1971), on the size of the NKA-sAHPs. Application of 10 μm Br-cGMP markedly suppressed the NKA-sAHPs (Fig. 2a; amplitudes and areas reduced to 35.1% and 38.1% of control, respectively; p = 0.001 and p = 0.0046, respectively; n = 7). In another series of experiments, applying a higher dose of Br-cGMP (100 μm) caused an even greater reduction in NKA-sAHP size (Fig. 2b; amplitudes and areas reduced to 25.3% and 14.1% of control, respectively; p = 0.0001 and p = 0.002, respectively; n = 6).
Figure 2.

Effects of PKG activation on the NKA-sAHPs. a, Activation of PKG by Br-cGMP. The two overlaid traces represent the NKA-sAHPs in the same neuron before (black) and after perfusing the slice with the PKG activator Br-cGMP (10 μm; red). Bar represents the normalized results of seven experiments (mean ± SEM) for NKA-sAHP amplitudes (green) and areas (blue). b, Same as in a, but applying a higher concentration of Br-cGMP (100 μm; n = 6). c, Same as in a, but in slices pretreated with the PKG inhibitor KT5823 (2 μm; n = 6). The strong inhibition of the NKA-sAHPs by Br-cGMP indicates that phosphorylation by PKG downregulates NKA activity. d, Activation of the GC/cGMP/PKG pathway. The two overlaid traces represent the NKA-sAHPs in the same neuron before (black) and after perfusing the slice with the NO precursor l-arginine (100 μm; red). Bar represents the normalized results of nine experiments (mean ± SEM) for NKA-sAHP amplitudes (green) and areas (blue). e, Same as in d, but in slices pretreated with the GC inhibitor ODQ (10 μm; n = 5). f, Same as in d, but in slices pretreated with the PKG inhibitor KT5823 (2 μm; n = 5). l-Arginine suppressed the NKA-sAHPs provided that GC and PKG were operative, supporting the notion that phosphorylation by PKG downregulates NKA activity. **p < 0.01, ***p < 0.001, ns, not significant.
Br-cGMP (100 μm) depolarized the neurons by 3.0 ± 0.9 mV (p = 0.023; n = 6) but had no significant effect on RN (p = 0.2689) or spike amplitude (p = 0.1249). Therefore, its effect on the NKA-sAHPs cannot be due to shunting by cGMP-activated channels (Kuzmiski and MacVicar, 2001). Pretreating slices for 30–60 min with 2 μm KT5823, a PKG inhibitor (Kase et al., 1987), entirely prevented the Br-cGMP-induced suppression of the NKA-sAHPs (Fig. 2c; p = 0.255 and p = 0.793 for amplitudes and areas, respectively; n = 6), indicating that the Br-cGMP-induced NKA-sAHPs suppression is due to NKA inhibition via the cGMP/PKG pathway.
The synthesis of cGMP by guanylate cyclase (GC) and, consequently, downstream PKG activation, is strongly enhanced by nitric oxide (NO) (Arnold et al., 1977; Miki et al., 1977). Application of 100 μm l-arginine, the biological substrate of NO synthase (NOS) (Knowles et al., 1989), markedly suppressed the NKA-sAHPs (Fig. 2d; amplitude and area reduced to 28.4% and 32.8%, of control; p = 0.001 and p = 0.0008, respectively; n = 9). This effect was absent in slices pretreated for 1 h with 10 μm ODQ, a blocker of soluble GC (Garthwaite et al., 1995) (Fig. 2e; p = 0.5716 and p = 0.9214 for amplitudes and areas, respectively; n = 5). It was also absent in slices pretreated with the PKG inhibitor KT5823 (Fig. 2f; p = 0.3468 and p = 0.9410 for amplitudes and areas, respectively; n = 5). These results indicate that NKA activity is inhibited by NO via GC stimulation and cGMP production, rather than by direct nitrosylation of the NKA molecule (Sato et al., 1997).
The NKA-sAHPs recorded in KT5823-pretreated slices (n = 17) were similar in size to those in control slices (p = 1.000 and p = 0.884 for amplitudes and areas, respectively; see Fig. 7). The NKA-sAHPs recorded in ODQ-pretreated slices (n = 11) were also similar in size to those in control slices (p = 0.941 and p = 1.000 for amplitudes and areas, respectively; see Fig. 7).
Effects of PKC modulators
A third key regulator of NKA activity in many non-neural tissues is the Ca2+- and phospholipid-dependent PKC (Therien and Blostein, 2000). In neural tissue, PKC activation by the phorbol-ester PDBu was shown to modestly inhibit NKA activity in cultured striatal neurons (Nishi et al., 1999). To test whether PKC activation affects NKAs in CA1 pyramidal cells, we monitored the effects of PDBu on NKA-sAHPs in single CA1 pyramidal cells. Application of 5 μm PDBu strongly reduced these potentials (Fig. 3a; amplitudes and areas reduced to 20.0% and 18.4% of control, respectively; p = 0.0088 and p = 0.0078, respectively; n = 5). The suppression of the NKA-sAHPs by PDBu was prevented by preincubation of the slices in aCSF containing 5 μm chelerythrine, a PKC inhibitor (Herbert et al., 1990) (Fig. 3b; p = 0.0993 and p = 0.6035 for amplitudes and areas, respectively; n = 5), confirming that PDBu suppresses the NKA-sAHPs via PKC.
Figure 3.

Effects of PKC activation on the NKA-sAHPs. a, Activation of PKC by PDBu. The two overlaid traces represent the NKA-sAHPs in the same neuron before (black) and after perfusing the slice with the PKC activator PDBu (5 μm; red). Bar represents the normalized results of five experiments (mean ± SEM) for NKA-sAHP amplitudes (green) and areas (blue). b, Same as in a, but in slices pretreated with the PKC inhibitor chelerythrine (5 μm; n = 5). c, Same as in a, but in slices pretreated with the PKG inhibitor KT5823 (2 μm; n = 6). The strong inhibition of the NKA-sAHPs by PDBu, independent of PKG activity, indicates that phosphorylation by PKC downregulates NKA activity. **p < 0.01, ***p < 0.001, ns, not significant.
PDBu depolarized the neurons by 9.4 ± 2.3 mV (p = 0.009; n = 5) and decreased spike amplitude by 8.9 ± 2.4% (p = 0.0273) without significantly affecting RN. Intriguingly, these effects were noted also in slices pretreated with chelerythrine, and therefore may be due to a PKC-independent action (Doerner et al., 1990). Despite these changes, chelerythrine completely prevented the PDBu-induced suppression of the NKA-sAHPs. However, it did not, by itself, affect the size of the NKA-sAHPs, compared with those in control slices (p = 0.780 and p = 0.996 for amplitudes and areas, respectively).
These results suggest that PKC activation leads to NKA inhibition in CA1 pyramidal cells. It has been shown that NKAs contain several possible phosphorylation sites for PKC (Therien and Blostein, 2000). However, it has also been shown that PKC activation by PDBu enhances NOS catalytic activity by direct phosphorylation or via intermediary proteins, causing enhanced NO production (Okada, 1995; Wang and Huang, 2006). Such an action would lead to NKA inhibition via the GC/cGMP/PKG pathway. We tested this possible mechanism by applying PDBu to slices pretreated with the PKG inhibitor KT5823 (2 μm). Under this condition, adding 5 μm PDBu also remarkably suppressed the NKA-sAHPs (Fig. 3c; amplitudes and areas reduced to 10.0% and 6.1% of control, respectively; p = 0.0003 and p = 0.0001, respectively; n = 6), mimicking its effect on these potentials in slices with untampered PKG activity (Fig. 3a). These data indicate that PKC activation inhibits the NKAs directly rather than via PKG activation.
Effects of PP inhibitors
The results so far have identified two PKs (i.e., PKG and PKC) that potentially regulate neuronal NKA activity. To test whether NKA activity in “resting” condition is affected by these PKs, we inhibited the activity of PPs that reverse the action of these PKs. We reasoned that, if PKG and/or PKC are tonically active, then inhibiting the PPs will suppress the NKA-sAHPs. The main PPs that dephosphorylate NKAs in non-neural tissues are the serine/threonine PPs PP-1, PP-2A, and PP-2B (calcineurin) (Therien and Blostein, 2000). We applied PP inhibitors at concentrations known to effectively and selectively inhibit these PPs.
Applying 25 nm tautomycetin, a PP-1 inhibitor (Mitsuhashi et al., 2001), had no effect on the NKA-sAHPs (Fig. 4a; p = 0.759 and p = 0.5298 for amplitudes and areas, respectively; n = 7). Applying 50 nm fostriecin, a PP-2A inhibitor (Walsh et al., 1997), also had no effect on the NKA-sAHPs (Fig. 4b; p = 0.9544 and p = 0.7504 for amplitudes and areas, respectively; n = 6). Applying 10 μm FK506, a PP-2B inhibitor (Liu et al., 1991), also had no effect as well on the NKA-sAHPs (Fig. 4c; p = 0.8673 and p = 0.6503 for amplitudes and areas, respectively; n = 5). However, applying 1 μm OA, an inhibitor of both PP-1 and PP-2A (Takai et al., 1992), together with FK506 (OA+FK506), to simultaneously block all three PPs, almost completely suppressed the NKA-sAHPs (Fig. 4d; amplitudes and areas reduced to 5.9% and 6.8% of control; p = 0.0002 and p = 0.0003, respectively; n = 6). None of these drug treatments significantly affected resting Vm, apparent RN and spike amplitude. These results indicate that NKAs are tonically inhibited and disinhibited by phosphorylation and dephosphorylation, respectively. Furthermore, the results suggest that dephosphorylation by more than one of the three tested PPs can independently disinhibit the NKAs, as inhibition of one only had no effect on the NKA-sAHPs Figure 4a–c).
Figure 4.
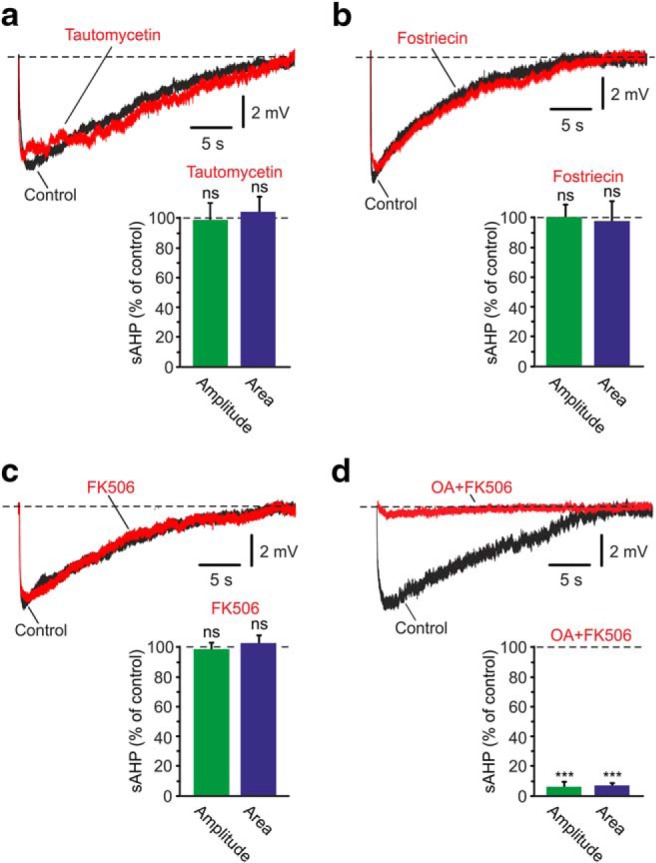
Effects of PP inhibitors on the NKA-sAHPs. a, Inhibition of PP-1. The two overlaid traces represent the control NKA-sAHPs in the same neuron before (Control; black) and after perfusing the slice with the PP-1 inhibitor tautomycetin (25 nm; red). Bar diagrams represent the normalized results of seven experiments (mean ± SEM) for NKA-sAHP amplitudes (green) and areas (blue). b, Inhibition of PP-2A. Same as in a, but applying the PP-2A inhibitor fostriecin (50 nm; n = 6). c, Inhibition of PP-2B. Same as in a, but applying the PP-2B inhibitor FK506 (10 μm; n = 5). d, Simultaneous PP-1, PP-2A, and PP-2B inhibition. Same as in a, but applying together the PP-1 and PP-2A inhibitor OA (1 μm; red) and FK506 (n = 6). Inhibiting either one of the three PPs alone had no effect on the NKA-sAHPs. Inhibiting all three together strongly suppressed the NKA-sAHPs, indicating that at least two of the PPs can each independently dephosphorylate the NKAs. ***p < 0.001, ns, not significant.
We next tested whether phosphorylation by PKG and/or PKC exerts the tonic inhibitory effect on the NKAs. To that end, we applied OA+FK506 (to inhibit all three PPs) in slices pretreated with KT5823 and chelerythrine (to inhibit both PKG and PKC). In this condition, PP inhibition had no effect on the NKA-sAHPs (Fig. 5a; p = 0.0762 and p = 0.5487 for amplitudes and areas, respectively; n = 5), indicating that either PKG or PKC, or both together, mediate NKA tonic inhibition.
Figure 5.

Effects of PP inhibitors combined with PK inhibitors on the NKA-sAHPs. a, Simultaneous PP-1, PP-2A, and PP-2B inhibition in condition of inhibited PKs. The slices were pretreated with 2 μm KT5823 and 5 μm chelerythrine to inhibit both PKG and PKC. The two overlaid traces represent the control NKA-sAHPs in the same neuron before (Control; black) and after applying together 1 μm OA and 10 μm FK506 (red). Bar represents the normalized results of five experiments (mean ± SEM) for NKA-sAHP amplitudes (green) and areas (blue). b, Same as in a, but in slices pretreated only with the PKC inhibitor chelerythrine (5 μm; n = 6). c, Same as in a, but in slices pretreated only with the PKG inhibitor KT5823 (2 μm; n = 6). Inhibiting all three PPs, which normally suppressed the NKA-sAHPs, had no effect on condition of inhibited PKG and PKC. When only one of these two PKs was inhibited, then subsequent inhibition of all three PPs suppressed the NKA-sAHPs. Thus, both PKG and PKC mediate tonic phosphorylation of NKAs. **p < 0.01, ***p < 0.001, ns, not significant.
To differentiate between these options, we monitored the effects of the PP inhibitors after blocking only one of the two PKs. In slices pretreated with chelerythrine to inhibit PKC only, subsequent application of OA+FK506 suppressed the NKA-sAHPs (Fig. 5b; amplitudes and areas reduced to 7.9% and 3.0% of control, respectively; p = 0.0001 and p = 0.0029, respectively; n = 6). In slices pretreated with KT5823 to inhibit PKG only, subsequent application of OA+FK506 together also suppressed the NKA-sAHPs (Fig. 5c; amplitudes and areas reduced to 8.7% and 7.9% of control, respectively; p = 0.0007 and p = 0.0002, respectively; n = 6). These results indicate that phosphorylation by both PKG and PKC exerts a tonic inhibitory effect on the NKAs.
We finally sought to identify which of the three PPs plays a role in tonically disinhibiting the NKAs. To that end, we monitored the effects of inhibiting different pairs of the PPs on the NKA-sAHPs. Applying OA (to inhibit PP-1 and PP-2A) had no effect (Fig. 6a; p = 0.3406 and p = 0.3924 for amplitudes and areas, respectively; n = 9), indicating that PP-2B alone disinhibits the NKAs. Applying fostriecin and FK-506 in combination to inhibit both PP-2A and PP-2B also had no effect (Fig. 6b; p = 0.3726 and p = 0.104 for amplitudes and areas, respectively; n = 6), indicating that also PP-1 alone disinhibits the NKAs. In contrast, applying tautomycetin and FK-506 together to inhibit both PP-1 and PP-2B, but not PP-2A, almost completely suppressed the NKA-sAHPs (Fig. 6c; amplitudes and areas reduced to 2.9% and 3.5%, respectively; p = 0.0001 and p = 0.0006, respectively; n = 5), indicating that PP-2A alone cannot disinhibit the NKAs. These data thus show that, in CA1 pyramidal cells, dephosphorylation by PP-1 or by PP-2B, but not by PP-2A, disinhibits the NKAs. Furthermore, our finding that either PP-1 or PP-2B, acting alone, can reverse tonic NKA inhibition induced by PKG and PKC, suggests that either one of the two PPs can reverse the phosphorylation mediated by either PKG or PKC.
Figure 6.

Effects of different combinations of PP inhibitors on the NKA-sAHPs. a, Inhibition of PP-1 and PP-2A. The two overlaid traces represent NKA-sAHPs in the same neuron before (Control; black) and after perfusing the slice with the PP-1 and PP-2A inhibitor OA (1 μm; red). Bar diagrams represent the normalized results of nine experiments (mean ± SEM) for NKA-sAHP amplitudes (green) and areas (blue). b, Inhibition of PP-2A and PP-2B. Same as in a, but applying the PP-2A inhibitor fostriecin (50 nm) together with the PP-2B inhibitor FK506 (10 μm; n = 6). c, Inhibition of PP-1 and PP-2B. Same as in a, but applying the PP-1 inhibitor tautomycetin (25 nm) together with the PP-2B inhibitor FK506 (10 μm; n = 5). Inhibiting PP-1and PP-2A, or PP-2A and PP-2B, had no effect on the NKA-sAHPs, implying that both PP-1 and PP-2B can independently dephosphorylate the NKAs. Inhibiting PP-1 and PP-2B completely suppressed the NKA-sAHPs, implying that PP-2A is not effective in dephosphorylating the NKAs. ***p < 0.001, ns, not significant.
The size of the NKA-sAHPs, compared with the amplitudes and areas of these potentials in untreated neurons (n = 102), was not modified by merely blocking PKC with chelerythrine (p = 0.780 and p = 1.000, n = 18) or by blocking PKC and PKG together with chelerythrine and KT5823 (p = 1.000 and p = 0.998, n = 16; Fig. 7). The latter observation suggests that the tonic inhibitory action of these PKs is low in “resting” slices, and is readily reversed by the PPs. Stronger activation of these signaling pathways likely is required to cause functional inhibition of NKA activity.
Effects of modulators of PKs and PPs on sAHPs evoked by Na+ injections
The experiments described thus far were conducted using NKA-sAHPs evoked by spike trains as readouts of NKA activity. To verify our main results using an alternative approach, which does not depend on spikes, we monitored the effects of modulators of PKs and PPs on NKA-sAHPs evoked by intracellular Na+ injections. To that end, slices were perfused with aCSF containing TTX (0.5 μm) and ZD7288 (50 μm). The microelectrodes were filled with a solution comprised of 3 m Na-acetate and 100 mm KCl. Injections of Na+ were performed by delivering trains of 150 short (2 ms) pulses of 4–6 nA at 100 Hz to the neurons. The evoked sAHPs attained a peak amplitude of 7.9 ± 1.9 mV and an area of 43.7 ± 7.8 mV/s, and lasted ∼10 ± 2 s (Fig. 8a). Continuously applying 50-ms-long hyperpolarizing current pulses (−50 pA at 2 Hz) to monitor changes in membrane conductance (Tiwari et al., 2018) did not disclose any changes during these sAHPs (Fig. 8a; hyperpolarizing pulse amplitudes 2 s before and 1 s after Na+ injection; p = 0.552; n = 5). Changing to K+-free aCSF, a procedure that inhibits NKA activity (Glynn and Karlish, 1975; Tiwari et al., 2018), led within 20 min to a strong suppression of the Na+ injection-induced sAHPs (Fig. 8b; amplitudes and areas reduced to 19.3% and 15.5% of control, respectively; p = 0.0407 and p = 0.0005, respectively; n = 5). These findings confirm that the Na+ injection-evoked sAHPs are also pure NKA-sAHPs.
Figure 8.
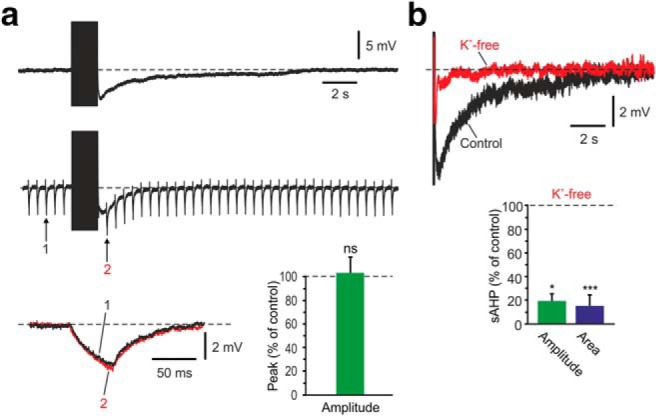
NKA-sAHPs can be evoked by intracellular Na+ injections. a, Top, Representative sAHP evoked by intracellular Na+ injection (150 pulses of 4–6 nA, each 2 ms long, delivered at 100 Hz) in TTX-containing aCSF. Bottom, Another sAHP evoked by Na+ injection, but negative current pulses (50 ms, −50 pA) were delivered at 2 Hz continuously. Two hyperpolarizing voltage responses elicited ∼2 s before Na+ injection onset (1) and 1 s after end of injection (2) are overlaid below. The responses are similar, indicating lack of change in membrane conductance during the sAHP. Bar diagram represents the peak amplitudes of the hyperpolarizing voltage responses at Time point 2 normalized to that obtained at Time point 1 (mean ± SEM; n = 5). b, Overlaid traces of sAHPs evoked by intracellular Na+ injection before (Control) and 20 min after slice perfusion with K+-free aCSF (K+-free) to block NKA activity. Summary bar diagram represents the peak amplitudes and areas of the sAHPs in K+-free aCSF (percentage of control; mean ± SEM; n = 5). *p < 0.05, ***p < 0.001, ns, not significant.
We tested the effects of the PK activators Br-cAMP, Br-cGMP, and PDBu on the Na+ injection-evoked NKA-sAHPs. Whereas Br-cAMP (2 mm) had no significant effect on these potentials (Fig. 9a; p = 0.3432 and p = 0.7452 for amplitudes and areas, respectively; n = 4), they were markedly suppressed by 100 μm Br-cGMP (Fig. 9b; amplitude and areas reduced to 38.4% and 19.1% of control, respectively; p = 0.0077 and p = 0.0041, respectively; n = 4) and by 5 μm PDBu (Fig. 9c; amplitude and areas reduced to 18.0% and 6.0% of control, respectively; p = 0.0039 and p = 0.002, respectively; n = 5). Likewise, applying OA+FK506 strongly suppressed the sAHPs (Fig. 9d; amplitudes and areas reduced to 13.3% and 9.3% of control, respectively; p = 0.0044 and p = 0.0002, respectively; n = 4). These data confirm our conclusion that phosphorylation by PKC or PKG, but not by PKA, inhibits NKA activity, whereas PP-1 and PP-2B reverse this effect.
Figure 9.
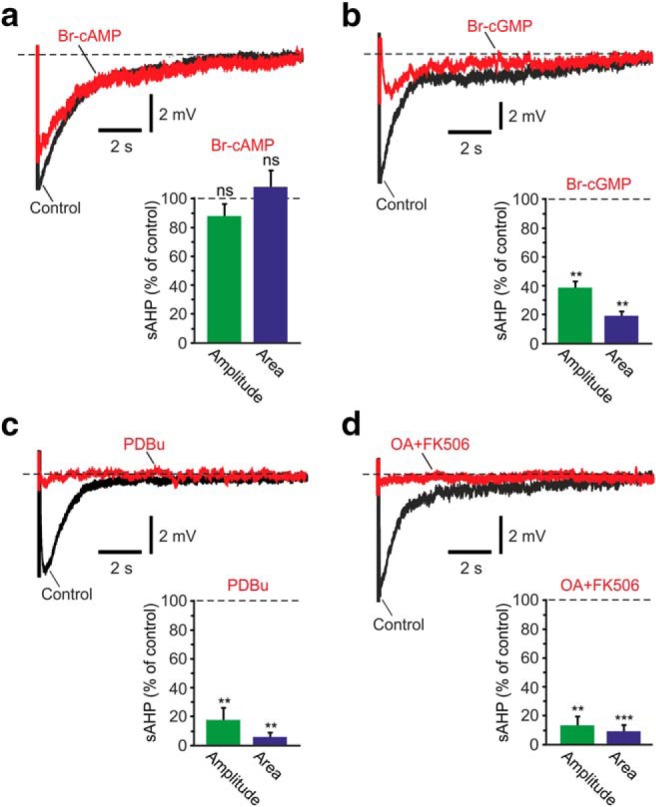
NKA-sAHPs evoked by Na+ injections are regulated by PKs and PPs. a, NKA-sAHPs were evoked by intracellular Na+ injections in TTX-containing aCSF. Activation of PKA: The two overlaid traces represent the NKA-sAHPs in the same neuron before (black) and after perfusing the slice with the PKA activator Br-cAMP (2 mm; red). Bar represents the normalized results of four experiments (mean ± SEM) for NKA-sAHP amplitudes (green) and areas (blue). b, Activation of PKG: Same as in a, but applying the PKG activator Br-cGMP (100 μm; n = 4). c, Activation of PKC: Same as in a, but applying the PKC activator PDBu (5 μm; n = 5). d, Inhibition of PPs. Same as in a, but applying the PP inhibitors OA (1 μm) and FK506 (10 μm; n = 4). Activation of PKA had no significant effect on the NKA-sAHPs, whereas all other treatments strongly suppressed these potentials, implicating PKG, PKC, and the PPs in NKA activity regulation by phosphorylation/dephosphorylation. **p < 0.01, ***p < 0.001, ns, not significant.
Discussion
Here we have used an electrophysiological approach to investigate systematically the regulation of NKA activity in single identified neurons in situ. Our data demonstrate the regulation of NKA transport activity by phosphorylation/dephosphorylation in single neurons, and identify two PKs and two PPs participating in this fundamental process.
Neuronal NKA activity is not regulated by PKA
Modulation of NKA transport activity by cAMP/PKA signaling has been noted in many non-neuronal tissues, where it can be either facilitatory or inhibitory, depending on tissue type and animal species. In some tissues, NKA inhibition by cAMP/PKA signaling is mediated by direct phosphorylation of the α subunit, reducing its affinity to intracellular Na+, whereas in others it involves phosphorylation of an intermediate protein (Therien and Blostein, 2000). In brain tissue, PKA phosphorylation of an intermediary protein modestly inhibited NKA activity in synaptosomes prepared from whole rat brains (Lingham and Sen, 1982). Likewise, the cAMP-PKA pathway mediated a partial reduction in NKA activity induced by dopaminergic agonists in rat striatal slices (Pinto Ferreira et al., 1998) and by prostaglandin E2 in rat hippocampal slices (Oliveira et al., 2009). However, because brain slices are comprised of multiple cell types, the latter results do not necessarily implicate NKAs in neurons.
Surprisingly, we found that activation of the cAMP-PKA pathway does not modify the NKA-sAHPs in CA1 pyramidal cells. This contrasts markedly with abolishment of the KCa-sAHP component by PKA activation (Madison and Nicoll, 1986; Pedarzani and Storm, 1993), as shown also here. Because both α1 and α3 NKA isoforms contribute to the generation of the NKA-sAHPs (Tiwari et al., 2018), our results suggest that neither of the two neuronal NKA isoforms is subjected to regulation by PKA.
Neuronal NKA activity is inhibited by PKG
Both facilitatory and inhibitory effects of cGMP/PKG signaling on NKAs transport activity have been previously observed in various mammalian tissues, including brain slices, attesting to the multiplicity of mechanisms involved in NKA regulation by PKG (Therien and Blostein, 2000). Here we found that direct PKG activation strongly suppresses the NKA-sAHPs, thus indicating that cGMP/PKG signaling inhibits NKAs in CA1 pyramidal cells. Furthermore, we show that NKA activity is inhibited by PKG activation via NOS/GC/cGMP/PKG signaling. This conclusion is congruent with a previous biochemical study showing that spermidine inhibits NKAs in hippocampal slices via the latter pathway (Carvalho et al., 2012). However, a previous biochemical study reported that cGMP/PKG signaling enhances α3-NKA activity in cerebellar Purkinje neurons (Nathanson et al., 1995). Thus, the regulation of neuronal NKAs by PKG-induced phosphorylation may also be tissue- and subunit-specific, as observed in non-neuronal tissues (Therien and Blostein, 2000).
The molecular mechanism by which cGMP-activated PKG causes NKA inhibition in CA1 pyramidal cells remains unclear. Direct phosphorylation of NKA α-subunit by PKG is a likely possibility, although so far it was demonstrated in mammalian kidneys and in Xenopus oocytes, where it led to enhanced NKA activity (Fotis et al., 1999). Alternatively, PKG may exert its action via intermediary proteins. For example, PKG-induced DARPP-32 phosphorylation, likely by inhibiting PP-1 (Hemmings et al., 1984), reduced renal NKA activity (Eklöf et al., 2001). However, this mechanism alone is not applicable to CA1 pyramidal cells, as pharmacological PP-1 inhibition did not modify the NKA-sAHPs. A more plausible mechanism has been described in ocular ciliary muscle, where PKG inhibited NKA activity by stimulating the Src family of protein tyrosine kinases (Shahidullah et al., 2014). Inhibition of NKA activity by direct tyrosine phosphorylation of the α1-NKA subunit was demonstrated in several other non-neural tissues (e.g., El-Beialy et al., 2010; Mandal et al., 2011). On the other hand, direct tyrosine phosphorylation of the α3 NKA subunit was shown to enhance NKA activity in cortical neurons (Wang and Yu, 2005).
Neuronal NKA activity is inhibited by PKC
As is the case with PKA and PKG, the effects of PKC activation on NKA transport activity are varied, causing either enhancement or inhibition in a tissue-specific manner (Therien and Blostein, 2000). Part of this variation may depend on the isoform of the PKC involved (Efendiev et al., 1999), or on the concentration of intracellular Ca2+ (Cheng et al., 1999), or on ambient temperature (Féraille et al., 2000). Here we show that PKC activation strongly suppresses NKA activity in CA1 pyramidal cells.
In non-neuronal tissues, PKC was shown to inhibit NKA activity by direct phosphorylation of serine/threonine residues at the catalytic α subunit (Therien and Blostein, 2000). The general consensus is that PKC phosphorylation occurs at the N terminus (Feschenko and Sweadner, 1995; Béguin et al., 1996), although PKC phosphorylation sites at the C terminus can be exposed in some conditions (Mahmmoud and Cornelius, 2002). In rats, both α1 and α3 NKA isoforms were inhibited through such a mechanism (Béguin et al., 1996). A similar mechanism may account for PKC-mediated NKA inhibition in acutely dissociated striatal neurons (Nishi et al., 1999) and in serotonin-induced NKA inhibition in choroid plexus (Fisone et al., 1995).
We considered also that activated PKC may cause NKA inhibition indirectly by enhancing NOS catalytic activity, causing enhanced NO production and activation of the NOS/GC/cGMP/PKG pathway (Marin et al., 1992; Okada, 1995). However, this option was refuted by showing that PDBu suppresses the NKA-sAHPs also in slices pretreated with the PKG inhibitor KT5823.
Neuronal NKA activity is regulated by phosphorylation/dephosphorylation
Numerous cellular processes are regulated by a balance between phosphorylation and dephosphorylation of critical proteins. Congruently, the effects of PKs on NKAs were shown to be reversed by PPs in some non-neuronal tissues. The major PPs that have been implicated in this regulatory process are PP-1 and PP-2B (for review, see Therien and Blostein, 2000). Neurons, CA1 pyramidal cells included, express these two PPs, as well as PP-2A, and all three participate in multiple regulatory functions (e.g., Yao et al., 2013; Lucas and Armstrong, 2015; Williams et al., 2015; Hell, 2016). Indeed, they are the most abundant neuronal serine/threonine PPs and dephosphorylate >90% of neuronal phosphoproteins (Mansuy and Shenolikar, 2006). However, only little is known about their role in neuronal NKA activity regulation. In cultured cerebellar neurons, activation of PP-2B was shown to dephosphorylate NKAs that were previously phosphorylated by PKC, leading to reversal of NKA inhibition (Marcaida et al., 1996).
Although blocking separately each one of the three PPs did not affect the NKA-sAHPs, blocking all three together almost entirely suppressed these potentials. The latter effect was seen also when either PKG or PKC was inhibited, but not when the two PKs were simultaneously inhibited. These results indicated that NKA activity in “resting” conditions requires tonic dephosphorylation of NKAs (or intermediate proteins) to reverse the inhibitory action of tonic phosphorylation by PKG and PKC. Further analysis indicated that tonic dephosphorylation by PP-1 or by PP-2B, but not by PP-2A, secure normal NKA activity in the face of tonic phosphorylation by PKG and PKC. Our finding that either one of the two PPs can reverse the effect of either one of the two PKs suggests that they all might converge on the same phosphorylation sites of the α-NKA protein.
CA1 pyramidal cells express both α1 and α3 NKA isoforms (McGrail et al., 1991; Juhaszova and Blaustein, 1997), and both isoforms contribute synergistically to the generation of the NKA-sAHPs (Tiwari et al., 2018). Our results, showing almost full suppression of the NKA-sAHPs by PKC and PKG activators and PP-1 and PP2B inhibitors, suggest that both NKA isoforms are similarly regulated by these PKs and PPs.
Functional implications
Here we show, in CA1 pyramidal cells, that phosphorylation by PKG or PKC inhibits NKA activity. In either case, this inhibition is only partial because even prolonged exposures (>1 h) to PKG or PKC activators did not induce spreading depression or other signs of neuronal deterioration that accompany substantial NKA inhibition (Balestrino et al., 1999). This conclusion is also compatible with biochemical studies showing 40%–50% inhibition of purified NKAs by PKC phosphorylation (Bertorello et al., 1991; Cheng et al., 1999). Such inhibition likely would enhance CA1 pyramidal cell excitability, as happens when the KCa-sAHP component is suppressed (Madison and Nicoll, 1984). Congruently, partial inhibition of NKAs in CA1 pyramidal cells using ouabain-like NKA antagonists caused enhanced intrinsic excitability (McCarren and Alger, 1987) and a decrease in GABAA receptor-mediated currents recorded in these neurons (Vaillend et al., 2002).
We have previously shown that the NKA-sAHP in CA1 pyramidal cells is predominantly generated by α1-NKAs (Tiwari et al., 2018). It is not yet known whether NKA regulation by phosphorylation/dephosphorylation occurs also in other types of neurons. In particular, it would be interesting to explore this issue in interneurons, which, unlike pyramidal cells, preferentially express α3-NKAs (Richards et al., 2007). Intriguingly, excitation of dentate interneurons by glutamate elicited a sustained depolarization due to partial NKA inhibition (Ross and Soltesz, 2000). This effect was dependent on a rise in [Ca2+]i and therefore possibly mediated by PKG and/or PKC signaling. It is likely that NKA inhibition in interneurons would enhance synaptic inhibition of principal neurons, thereby reducing network excitability. The consequences of simultaneous NKA inhibition in pyramidal cells and inhibitory interneurons are difficult to predict. In two studies, applying ouabain-like NKA antagonists to hippocampal slices, thereby partially inhibiting NKAs in all cell types, induced or augmented epileptiform bursting (Vaillend et al., 2002; Krishnan et al., 2015).
Partial NKA inhibition or NKA deficiency in brain has also been implicated in spatial learning and memory deficits (Sato et al., 2004; Moseley et al., 2007), as well as in the pathophysiology of Alzheimer's disease (e.g., Ohnishi et al., 2015; Petrushanko et al., 2016). Thus, the ability of activated PKG and PKC to reversibly inhibit neuronal NKAs suggests that these kinases may be downstream effectors of signaling pathways that couple primary messengers to NKAs, through which multiple brain functions can be regulated or dysregulated.
Footnotes
This work was supported by the Deutsch-Israelische Projectkooperation program of the Deutsche Forschungsgemeinschaft, the Israeli Science Foundation, and the Erna and Henri Leir Foundation.
The authors declare no competing financial interests.
References
- Alger BE, Nicoll RA (1980) Epileptiform burst afterhyperpolarization: calcium-dependent potassium potential in hippocampal CA1 pyramidal cells. Science 210:1122–1124. 10.1126/science.7444438 [DOI] [PubMed] [Google Scholar]
- Arnold WP, Mittal CK, Katsuki S, Murad F (1977) Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci U S A 74:3203–3207. 10.1073/pnas.74.8.3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestrino M, Young J, Aitken P (1999) Block of (Na+, K+)ATPase with ouabain induces spreading depression-like depolarization in hippocampal slices. Brain Res 838:37–44. 10.1016/S0006-8993(99)01674-1 [DOI] [PubMed] [Google Scholar]
- Béguin P, Peitsch MC, Geering K (1996) alpha 1 but not alpha 2 or alpha 3 isoforms of Na, K-ATPase are efficiently phosphorylated in a novel protein kinase C motif. Biochemistry 35:14098–14108. 10.1021/bi960516o [DOI] [PubMed] [Google Scholar]
- Bertorello AM, Aperia A, Walaas SI, Nairn AC, Greengard P (1991) Phosphorylation of the catalytic subunit of Na+, K+-ATPase inhibits the activity of the enzyme. Proc Natl Acad Sci U S A 88:11359–11362. 10.1073/pnas.88.24.11359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho FB, Mello CF, Marisco PC, Tonello R, Girardi BA, Ferreira J, Oliveira MS, Rubin MA (2012) Spermidine decreases Na+,K+-ATPase activity through NMDA receptor and protein kinase G activation in the hippocampus of rats. Eur J Pharmacol 684:79–86. 10.1016/j.ejphar.2012.03.046 [DOI] [PubMed] [Google Scholar]
- Chen S, Benninger F, Yaari Y (2014) Role of small conductance Ca2+-activated K+ channels in controlling CA1 pyramidal cell excitability. J Neurosci 34:8219–8230. 10.1523/JNEUROSCI.0936-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SX, Aizman O, Nairn AC, Greengard P, Aperia A (1999) [Ca2+]i determines the effects of protein kinases A and C on activity of rat renal Na+, K+-ATPase. J Physiol 518:37–46. 10.1111/j.1469-7793.1999.0037r.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H (1990) Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma. J Biol Chem 265:5267–5272. [PubMed] [Google Scholar]
- Dobretsov M, Hastings SL, Stimers JR (1999) Functional Na+/K+ pump in rat dorsal root ganglia neurons. Neuroscience 93:723–729. 10.1016/S0306-4522(99)00122-0 [DOI] [PubMed] [Google Scholar]
- Doerner D, Abdel-Latif M, Rogers TB, Alger BE (1990) Protein kinase C-dependent and -independent effects of phorbol esters on hippocampal calcium channel current. J Neurosci 10:1699–1706. 10.1523/JNEUROSCI.10-05-01699.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efendiev R, Bertorello AM, Pedemonte CH (1999) PKC-beta and PKC-zeta mediate opposing effects on proximal tubule Na+, K+-ATPase activity. FEBS Lett 456:45–48. 10.1016/S0014-5793(99)00925-4 [DOI] [PubMed] [Google Scholar]
- Eklöf AC, Holtbäck U, Svennilson J, Fienberg A, Greengard P, Aperia A (2001) Increased blood pressure and loss of anp-induced natriuresis in mice lacking DARPP-32 gene. Clin Exp Hypertens 23:449–460. 10.1081/CEH-100104236 [DOI] [PubMed] [Google Scholar]
- El-Beialy W, Galal N, Deyama Y, Yoshimura Y, Suzuki K, Tei K, Totsuka Y (2010) Regulation of human and pig renal Na+, K+-ATPase activity by tyrosine phosphorylation of their alpha(1)-subunits. J Membr Biol 233:119–126. 10.1007/s00232-010-9231-z [DOI] [PubMed] [Google Scholar]
- Erecinska M, Silver IA (1994) Ions and energy in mammalian brain. Prog Neurobiol 43:37–71. 10.1016/0301-0082(94)90015-9 [DOI] [PubMed] [Google Scholar]
- Féraille E, Béguin P, Carranza ML, Gonin S, Rousselot M, Martin PY, Favre H, Geering K (2000) Is phosphorylation of the alpha1 subunit at ser-16 involved in the control of Na, K-ATPase activity by phorbol ester-activated protein kinase C? Mol Biol Cell 11:39–50. 10.1091/mbc.11.1.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschenko MS, Sweadner KJ (1995) Structural basis for species-specific differences in the phosphorylation of Na, K-ATPase by protein kinase C. J Biol Chem 270:14072–14077. 10.1074/jbc.270.23.14072 [DOI] [PubMed] [Google Scholar]
- Fisone G, Snyder GL, Fryckstedt J, Caplan MJ, Aperia A, Greengard P (1995) Na+, K+-ATPase in the choroid plexus: regulation by serotonin/protein kinase C pathway. J Biol Chem 270:2427–2430. 10.1074/jbc.270.6.2427 [DOI] [PubMed] [Google Scholar]
- Fotis H, Tatjanenko LV, Vasilets LA (1999) Phosphorylation of the alpha-subunits of the Na+/K+-ATPase from mammalian kidneys and Xenopus oocytes by cGMP-dependent protein kinase results in stimulation of ATPase activity. Eur J Biochem 260:904–910. 10.1046/j.1432-1327.1999.00237.x [DOI] [PubMed] [Google Scholar]
- Gadsby DC, Nakao M (1989) Steady-state current-voltage relationship of the Na/K pump in guinea pig ventricular myocytes. J Gen Physiol 94:511–537. 10.1085/jgp.94.3.511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B (1995) Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol Pharmacol 48:184–188. [PubMed] [Google Scholar]
- Glynn IM, Karlish SJ (1975) The sodium pump. Annu Rev Physiol 37:13–55. 10.1146/annurev.ph.37.030175.000305 [DOI] [PubMed] [Google Scholar]
- Gordon TR, Kocsis JD, Waxman SG (1990) Electrogenic pump (Na+/K+-ATPase) activity in rat optic nerve. Neuroscience 37:829–837. 10.1016/0306-4522(90)90112-H [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Dasari S, Onoue K, Stephens EK, Hasse JM, Avesar D (2013) A sodium-pump-mediated afterhyperpolarization in pyramidal neurons. J Neurosci 33:13025–13041. 10.1523/JNEUROSCI.0220-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson B, Wigström H (1981) Evidence for two types of afterhyperpolarization in CA1 pyramidal cells in the hippocampus. Brain Res 206:462–468. 10.1016/0006-8993(81)90548-5 [DOI] [PubMed] [Google Scholar]
- Hell JW. (2016) How Ca2+-permeable AMPA receptors, the kinase PKA, and the phosphatase PP2B are intertwined in synaptic LTP and LTD. Sci Signal 9:e2. 10.1126/scisignal.aaf7067 [DOI] [PubMed] [Google Scholar]
- Hemmings HC Jr, Greengard P, Tung HY, Cohen P (1984) DARPP-32, a dopamine-regulated neuronal phosphoprotein, is a potent inhibitor of protein phosphatase-1. Nature 310:503–505. 10.1038/310503a0 [DOI] [PubMed] [Google Scholar]
- Herbert JM, Augereau JM, Gleye J, Maffrand JP (1990) Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun 172:993–999. 10.1016/0006-291X(90)91544-3 [DOI] [PubMed] [Google Scholar]
- Hotson JR, Prince DA (1980) A calcium-activated hyperpolarization follows repetitive firing in hippocampal neurons. J Neurophysiol 43:409–419. 10.1152/jn.1980.43.2.409 [DOI] [PubMed] [Google Scholar]
- Howarth C, Gleeson P, Attwell D (2012) Updated energy budgets for neural computation in the neocortex and cerebellum. J Cereb Blood Flow Metab 32:1222–1232. 10.1038/jcbfm.2012.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones HC, Keep RF (1988) Brain fluid calcium concentration and response to acute hypercalcaemia during development in the rat. J Physiol 402:579–593. 10.1113/jphysiol.1988.sp017223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhaszova M, Blaustein MP (1997) Na+ pump low and high ouabain affinity alpha subunit isoforms are differently distributed in cells. Proc Natl Acad Sci U S A 94:1800–1805. 10.1073/pnas.94.5.1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowski CC. (2011) Bidirectional pattern-specific plasticity of the slow afterhyperpolarization in rats: role for high-voltage activated Ca2+ channels and I h. Eur J Neurosci 34:1756–1765. 10.1111/j.1460-9568.2011.07899.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M (1987) K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem Biophys Res Commun 142:436–440. 10.1016/0006-291X(87)90293-2 [DOI] [PubMed] [Google Scholar]
- Kim JH, von Gersdorff H (2012) Suppression of spikes during posttetanic hyperpolarization in auditory neurons: the role of temperature, Ih currents, and the Na+-K+-ATPase pump. J Neurophysiol 108:1924–1932. 10.1152/jn.00103.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles RG, Palacios M, Palmer RM, Moncada S (1989) Formation of nitric oxide from l-arginine in the central nervous system: a transduction mechanism for stimulation of the soluble guanylate cyclase. Proc Natl Acad Sci U S A 86:5159–5162. 10.1073/pnas.86.13.5159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike H, Mano N, Okada Y, Oshima T (1972) Activities of the sodium pump in cat pyramidal tract cell studied with intracellular injection of sodium ions. Exp Brain Res 14:449–462. [DOI] [PubMed] [Google Scholar]
- Krishnan GP, Filatov G, Shilnikov A, Bazhenov M (2015) Electrogenic properties of the Na+/K+ ATPase control transitions between normal and pathological brain states. J Neurophysiol 113:3356–3374. 10.1152/jn.00460.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kueh D, Barnett WH, Cymbalyuk GS, Calabrese RL (2016) Na+/K+ pump interacts with the h-current to control bursting activity in central pattern generator neurons of leeches. Elife 5:e19322. 10.7554/eLife.19322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmiski JB, MacVicar BA (2001) Cyclic nucleotide-gated channels contribute to the cholinergic plateau potential in hippocampal CA1 pyramidal neurons. J Neurosci 21:8707–8714. 10.1523/JNEUROSCI.21-22-08707.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Adams NR (1987) Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J Physiol 389:187–203. 10.1113/jphysiol.1987.sp016653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingham RB, Sen AK (1982) Regulation of rat brain (Na+ + K+)-ATPase activity by cyclic AMP. Biochim Biophys Acta 688:475–485. 10.1016/0005-2736(82)90359-5 [DOI] [PubMed] [Google Scholar]
- Liu J, Farmer JD Jr, Lane WS, Friedman J, Weissman I, Schreiber SL (1991) Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66:807–815. 10.1016/0092-8674(91)90124-H [DOI] [PubMed] [Google Scholar]
- Lucas SJ, Armstrong DL (2015) Protein phosphatase modulation of somatostatin receptor signaling in the mouse hippocampus. Neuropharmacology 99:232–241. 10.1016/j.neuropharm.2015.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA (1984) Control of the repetitive discharge of rat CA1 pyramidal neurones in vitro. J Physiol 354:319–331. 10.1113/jphysiol.1984.sp015378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA (1986) Actions of noradrenaline recorded intracellularly in rat hippocampal CA1 pyramidal neurones, in vitro. J Physiol 372:221–244. 10.1113/jphysiol.1986.sp016006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmmoud YA, Cornelius F (2002) Protein kinase C phosphorylation of purified Na,K-ATPase: C-terminal phosphorylation sites at the alpha- and gamma-subunits close to the inner face of the plasma membrane. Biophys 82:1907–1919. 10.1016/S0006-3495(02)75540-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal A, Shahidullah M, Beimgraben C, Delamere NA (2011) The effect of endothelin-1 on src-family tyrosine kinases and Na, K-ATPase activity in porcine lens epithelium. J Cell Physiol 226:2555–2561. 10.1002/jcp.22602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansuy IM, Shenolikar S (2006) Protein serine/threonine phosphatases in neuronal plasticity and disorders of learning and memory. Trends Neurosci 29:679–686. 10.1016/j.tins.2006.10.004 [DOI] [PubMed] [Google Scholar]
- Marcaida G, Kosenko E, Miñana MD, Grisolía S, Felipo V (1996) Glutamate induces a calcineurin-mediated dephosphorylation of Na+, K+-ATPase that results in its activation in cerebellar neurons in culture. J Neurochem 66:99–104. 10.1046/j.1471-4159.1996.66010099.x [DOI] [PubMed] [Google Scholar]
- Marin P, Lafon-Cazal M, Bockaert J (1992) A nitric oxide synthase activity selectively stimulated by NMDA receptors depends on protein kinase C activation in mouse striatal neurons. Eur J Neurosci 4:425–432. 10.1111/j.1460-9568.1992.tb00892.x [DOI] [PubMed] [Google Scholar]
- Matchkov VV, Krivoi II (2016) Specialized functional diversity and interactions of the Na, K-ATPase. Front Physiol 7:179. 10.3389/fphys.2016.00179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarren M, Alger BE (1987) Sodium-potassium pump inhibitors increase neuronal excitability in the rat hippocampal slice: role of a Ca2+-dependent conductance. J Neurophysiol 57:496–509. 10.1152/jn.1987.57.2.496 [DOI] [PubMed] [Google Scholar]
- McGrail KM, Phillips JM, Sweadner KJ (1991) Immunofluorescent localization of three Na, K-ATPase isozymes in the rat central nervous system: both neurons and glia can express more than one Na, K-ATPase. J Neurosci 11:381–391. 10.1523/JNEUROSCI.11-02-00381.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki N, Kawabe Y, Kuriyama K (1977) Activation of cerebral guanylate cyclase by nitric oxide. Biochem Biophys Res Commun 75:851–856. 10.1016/0006-291X(77)91460-7 [DOI] [PubMed] [Google Scholar]
- Mitsuhashi S, Matsuura N, Ubukata M, Oikawa H, Shima H, Kikuchi K (2001) Tautomycetin is a novel and specific inhibitor of serine/threonine protein phosphatase type 1, PP1. Biochem Biophys Res Commun 287:328–331. 10.1006/bbrc.2001.5596 [DOI] [PubMed] [Google Scholar]
- Morita K, David G, Barrett JN, Barrett EF (1993) Posttetanic hyperpolarization produced by electrogenic Na+-K+ pump in lizard axons impaled near their motor terminals. J Neurophysiol 70:1874–1884. 10.1152/jn.1993.70.5.1874 [DOI] [PubMed] [Google Scholar]
- Moseley AE, Williams MT, Schaefer TL, Bohanan CS, Neumann JC, Behbehani MM, Vorhees CV, Lingrel JB (2007) Deficiency in Na, K-ATPase alpha isoform genes alters spatial learning, motor activity, and anxiety in mice. J Neurosci 27:616–626. 10.1523/JNEUROSCI.4464-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muneyama K, Bauer RJ, Shuman DA, Robins RK, Simon LN (1971) Chemical synthesis and biological activity of 8-substituted adenosine 3′,5′-cyclic monophosphate derivatives. Biochemistry 10:2390–2395. 10.1021/bi00788a033 [DOI] [PubMed] [Google Scholar]
- Nathanson JA, Scavone C, Scanlon C, McKee M (1995) The cellular Na+ pump as a site of action for carbon monoxide and glutamate: a mechanism for long-term modulation of cellular activity. Neuron 14:781–794. 10.1016/0896-6273(95)90222-8 [DOI] [PubMed] [Google Scholar]
- Nishi A, Fisone G, Snyder GL, Dulubova I, Aperia A, Nairn AC, Greengard P (1999) Regulation of Na+, K+-ATPase isoforms in rat neostriatum by dopamine and protein kinase C. J Neurochem 73:1492–1501. 10.1046/j.1471-4159.1999.0731492.x [DOI] [PubMed] [Google Scholar]
- Ohnishi T, Yanazawa M, Sasahara T, Kitamura Y, Hiroaki H, Fukazawa Y, Kii I, Nishiyama T, Kakita A, Takeda H, Takeuchi A, Arai Y, Ito A, Komura H, Hirao H, Satomura K, Inoue M, Muramatsu S, Matsui K, Tada M, et al. (2015) Na, K-ATPase α3 is a death target of Alzheimer patient amyloid-β assembly. Proc Natl Acad Sci U S A 112:E4465–E4474. 10.1073/pnas.1421182112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada D. (1995) Protein kinase C modulates calcium sensitivity of nitric oxide synthase in cerebellar slices. J Neurochem 64:1298–1304. 10.1046/j.1471-4159.1995.64031298.x [DOI] [PubMed] [Google Scholar]
- Oliveira MS, Furian AF, Rambo LM, Ribeiro LR, Royes LF, Ferreira J, Calixto JB, Otalora LF, Garrido-Sanabria ER, Mello CF (2009) Prostaglandin E2 modulates Na+, K+-ATPase activity in rat hippocampus: implications for neurological diseases. J Neurochem 109:416–426. 10.1111/j.1471-4159.2009.05961.x [DOI] [PubMed] [Google Scholar]
- Parker D, Hill R, Grillner S (1996) Electrogenic pump and a Ca2+- dependent K+ conductance contribute to a posttetanic hyperpolarization in lamprey sensory neurons. J Neurophysiol 76:540–553. 10.1152/jn.1996.76.1.540 [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF (1993) PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron 11:1023–1035. 10.1016/0896-6273(93)90216-E [DOI] [PubMed] [Google Scholar]
- Petrushanko IY, Mitkevich VA, Anashkina AA, Adzhubei AA, Burnysheva KM, Lakunina VA, Kamanina YV, Dergousova EA, Lopina OD, Ogunshola OO, Bogdanova AY, Makarov AA (2016) Direct interaction of beta-amyloid with Na, K-ATPase as a putative regulator of the enzyme function. Sci Rep 6:27738. 10.1038/srep27738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto Ferreira M, DeLucia R, Luiz Aizenstein M, Glezer I, Scavone C (1998) Fencamfamine modulates sodium, potassium-ATPase through cyclic AMP and cyclic AMP-dependent protein kinase in rat striatum. J Neural Transm 105:549–560. 10.1007/s007020050078 [DOI] [PubMed] [Google Scholar]
- Poulsen H, Morth P, Egebjerg J, Nissen P (2010) Phosphorylation of the Na+, K+-ATPase and the H+, K+-ATPase. FEBS Lett 584:2589–2595. 10.1016/j.febslet.2010.04.035 [DOI] [PubMed] [Google Scholar]
- Pulver SR, Griffith LC (2010) Spike integration and cellular memory in a rhythmic network from Na+/K+ pump current dynamics. Nat Neurosci 13:53–59. 10.1038/nn.2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakowski RF, Gadsby DC, De Weer P (1997) Voltage dependence of the Na/K pump. J Membr Biol 155:105–112. 10.1007/s002329900162 [DOI] [PubMed] [Google Scholar]
- Richards KS, Bommert K, Szabo G, Miles R (2007) Differential expression of Na+/K+-ATPase alpha-subunits in mouse hippocampal interneurones and pyramidal cells. J Physiol 585:491–505. 10.1113/jphysiol.2007.144733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison GA, Butcher RW, Sutherland EW (1968) Cyclic AMP. Annu Rev Biochem 37:149–174. 10.1146/annurev.bi.37.070168.001053 [DOI] [PubMed] [Google Scholar]
- Ross ST, Soltesz I (2000) Selective depolarization of interneurons in the early posttraumatic dentate gyrus: involvement of the Na+/K+-ATPase. J Neurophysiol 83:2916–2930. 10.1152/jn.2000.83.5.2916 [DOI] [PubMed] [Google Scholar]
- Sato T, Kamata Y, Irifune M, Nishikawa T (1997) Inhibitory effect of several nitric oxide-generating compounds on purified Na+, K+-ATPase activity from porcine cerebral cortex. J Neurochem 68:1312–1318. 10.1046/j.1471-4159.1997.68031312.x [DOI] [PubMed] [Google Scholar]
- Sato T, Tanaka K, Ohnishi Y, Teramoto T, Irifune M, Nishikawa T (2004) Effects of steroid hormones on (Na+, K+)-ATPase activity inhibition-induced amnesia on the step-through passive avoidance task in gonadectomized mice. Pharmacol Res 49:151–159. 10.1016/j.phrs.2003.09.006 [DOI] [PubMed] [Google Scholar]
- Shahidullah M, Mandal A, Wei G, Delamere NA (2014) Nitric oxide regulation of Na, K-ATPase activity in ocular ciliary epithelium involves Src family kinase. J Cell Physiol 229:343–352. 10.1002/jcp.24454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Thompson SM, Blaustein MP (2013) Nanomolar ouabain augments Ca2+ signalling in rat hippocampal neurones and glia. J Physiol 591:1671–1689. 10.1113/jphysiol.2012.248336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. (1989) An after-hyperpolarization of medium duration in rat hippocampal pyramidal cells. J Physiol 409:171–190. 10.1113/jphysiol.1989.sp017491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai A, Murata M, Torigoe K, Isobe M, Mieskes G, Yasumoto T (1992) Inhibitory effect of okadaic acid derivatives on protein phosphatases: a study on structure-affinity relationship. Biochem J 284:539–544. 10.1042/bj2840539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therien AG, Blostein R (2000) Mechanisms of sodium pump regulation. Am J Physiol Cell Physiol 279:C541–C566. 10.1152/ajpcell.2000.279.3.C541 [DOI] [PubMed] [Google Scholar]
- Thompson SM, Prince DA (1986) Activation of electrogenic sodium pump in hippocampal CA1 neurons following glutamate-induced depolarization. J Neurophysiol 56:507–522. 10.1152/jn.1986.56.2.507 [DOI] [PubMed] [Google Scholar]
- Tiwari MN, Mohan S, Biala Y, Yaari Y (2018) Differential contributions of Ca2+-activated K+ channels and Na+/K+-ATPases to the generation of the slow afterhyperpolarization in CA1 pyramidal cells. Hippocampus 28:338–357. 10.1002/hipo.22836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillend C, Mason SE, Cuttle MF, Alger BE (2002) Mechanisms of neuronal hyperexcitability caused by partial inhibition of Na+-K+-ATPases in the rat CA1 hippocampal region. J Neurophysiol 88:2963–2978. 10.1152/jn.00244.2002 [DOI] [PubMed] [Google Scholar]
- Walsh AH, Cheng A, Honkanen RE (1997) Fostriecin, an antitumor antibiotic with inhibitory activity against serine/threonine protein phosphatases types 1 (PP1) and 2A (PP2A), is highly selective for PP2A. FEBS Lett 416:230–234. 10.1016/S0014-5793(97)01210-6 [DOI] [PubMed] [Google Scholar]
- Wang XQ, Yu SP (2005) Novel regulation of Na, K-ATPase by Src tyrosine kinases in cortical neurons. J Neurochem 93:1515–1523. 10.1111/j.1471-4159.2005.03147.x [DOI] [PubMed] [Google Scholar]
- Wang YC, Huang RC (2006) Effects of sodium pump activity on spontaneous firing in neurons of the rat suprachiasmatic nucleus. J Neurophysiol 96:109–118. 10.1152/jn.01369.2005 [DOI] [PubMed] [Google Scholar]
- Williams AD, Jung S, Poolos NP (2015) Protein kinase C bidirectionally modulates Ih and hyperpolarization-activated cyclic nucleotide-gated (HCN) channel surface expression in hippocampal pyramidal neurons. J Physiol 593:2779–2792. 10.1113/JP270453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao W, Zou HJ, Sun D, Ren SQ (2013) Aβ induces acute depression of excitatory glutamatergic synaptic transmission through distinct phosphatase-dependent mechanisms in rat CA1 pyramidal neurons. Brain Res 1515:88–97. 10.1016/j.brainres.2013.03.049 [DOI] [PubMed] [Google Scholar]
- Yue C, Yaari Y (2004) KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. J Neurosci 24:4614–4624. 10.1523/JNEUROSCI.0765-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HY, Picton L, Li WC, Sillar KT (2015) Mechanisms underlying the activity-dependent regulation of locomotor network performance by the Na+ pump. Sci Rep 5:16188. 10.1038/srep16188 [DOI] [PMC free article] [PubMed] [Google Scholar]