

Abstract
Rhodopsin mislocalization is frequently observed in retinitis pigmentosa (RP) patients. For example, class I mutant rhodopsin is deficient in the VxPx trafficking signal, mislocalizes to the plasma membrane (PM) of rod photoreceptor inner segments (ISs), and causes autosomal dominant RP. Mislocalized rhodopsin causes photoreceptor degeneration in a manner independent of light-activation. In this manuscript, we took advantage of Xenopus laevis models of both sexes expressing wild-type human rhodopsin or its class I Q344ter mutant fused to Dendra2 fluorescent protein to characterize a novel light-independent mechanism of photoreceptor degeneration caused by mislocalized rhodopsin. We found that rhodopsin mislocalized to the PM is actively internalized and transported to lysosomes where it is degraded. This degradation process results in the downregulation of a crucial component of the photoreceptor IS PM: the sodium-potassium ATPase α-subunit (NKAα). The downregulation of NKAα is not because of decreased NKAα mRNA, but due to cotransport of mislocalized rhodopsin and NKAα to lysosomes or autophagolysosomes. In a separate set of experiments, we found that class I mutant rhodopsin, which causes NKAα downregulation, also causes shortening and loss of rod outer segments (OSs); the symptoms frequently observed in the early stages of human RP. Likewise, pharmacological inhibition of NKAα led to shortening and loss of rod OSs. These combined studies suggest that mislocalized rhodopsin leads to photoreceptor dysfunction through disruption of the PM protein homeostasis and compromised NKAα function. This study unveiled a novel role of lysosome-mediated degradation in causing inherited disorders manifested by mislocalization of ciliary receptors.
SIGNIFICANCE STATEMENT Retinal ciliopathy is the most common form of inherited blinding disorder frequently manifesting rhodopsin mislocalization. Our understanding of the relationships between rhodopsin mislocalization and photoreceptor dysfunction/degeneration has been far from complete. This study uncovers a hitherto uncharacterized consequence of rhodopsin mislocalization: the activation of the lysosomal pathway, which negatively regulates the amount of the sodium-potassium ATPase (NKAα) on the inner segment plasma membrane. On the plasma membrane, mislocalized rhodopsin extracts NKAα and sends it to lysosomes where they are co-degraded. Compromised NKAα function leads to shortening and loss of the photoreceptor outer segments as observed for various inherited blinding disorders. In summary, this study revealed a novel pathogenic mechanism applicable to various forms of blinding disorders caused by rhodopsin mislocalization.
Keywords: primary cilia, protein trafficking, retina, retinitis pigmentosa, rhodopsin, rod photoreceptor
Introduction
Rhodopsin mislocalization is commonly observed among various inherited blinding disorders and other forms of visual impairments (Edward et al., 1993; Li et al., 1995; Fariss et al., 1997; Milam et al., 1998; Adams et al., 2007; Fliegauf et al., 2007). Genetic evidence indicates that mislocalized rhodopsin is the causative agent for rod photoreceptor degeneration; class I rhodopsin mutations disrupt the putative ciliary targeting signal VxPx, induce rhodopsin mislocalization, and result in some of the most severe forms of autosomal dominant retinitis pigmentosa (RP; Sung et al., 1993, 1994; Tam et al., 2000; Deretic et al., 2005; Nemet et al., 2015b). Before the onset of rod degeneration in these disorders, rod outer segments (OSs) first become shorter and then lost entirely, thus compromising photoreceptor function and vision (Milam et al., 1998). Mislocalized rhodopsin causes photoreceptor degeneration in both light-dependent and -independent manners (Alfinito and Townes-Anderson, 2002; Tam et al., 2006; Concepcion and Chen, 2010). Light activation of rhodopsin leads to ectopic activation of G-protein-mediated cascade and subsequent photoreceptor degeneration (Alfinito and Townes-Anderson, 2002). Although light exposure may hasten the onset of photoreceptor degeneration, the underlying mechanism of light-independent photoreceptor cell death remains unsolved. Light-independent effects include morphological changes in the plasma membrane (PM), which leads to neurite sprouting, suggesting the direct effect of rhodopsin mislocalization is on the PM (Milam et al., 1996; Tam et al., 2006). Therefore, we sought to understand how rhodopsin mislocalization induces PM toxicity and results in dysfunction of rod photoreceptors.
Photoreceptor inner segments (ISs), where rhodopsin mislocalizes, play critical roles in maintaining the dark current. In the OSs, photon absorption by rhodopsin triggers a series of biochemical events, which result in hydrolysis of cGMP, closure of cGMP-gated channels, and reduced inward flow of Na+ and Ca2+. In the IS, Na+/K+-ATPase pumps out Na+ in exchange for K+, which is then conducted through K+ channels. Phototransduction-mediated changes in the net flow of Na+ and K+ constitute the basis for signal transmission from OS to IS, eventually leading to reduced glutamate release from the synaptic terminus (Yau and Baylor, 1989). Rhodopsin is the major component of the OS, which comprises a modified ciliary structure (Palczewski, 2006). In individual vertebrate rods, 106–108 new rhodopsin molecules are synthesized daily to maintain the OS (Nathans, 1992). In ciliopathies represented by patients and animal models with class I mutations, an equivalently massive amount of rhodopsin becomes targeted to the IS PM. Such a massive amount of rhodopsin incorrectly delivered to the IS PM would likely affect the local concentration of other transmembrane proteins, and may also disturb the cell's ion homeostasis (Demontis et al., 1995).
Rod photoreceptors have an intrinsic mechanism to remove IS-mislocalized rhodopsin. For example, in photo-damaged rat retinas, rod photoreceptors initially exhibit IS-mislocalized rhodopsin, which disappears following the recovery (Edward et al., 1993). More recently, we found that class I mutant rhodopsin mistrafficked to the IS PM is continuously eliminated (Lodowski et al., 2013). Rhodopsin is normally eliminated through phagocytosis of rod OSs by the retinal pigment epithelium (RPE; Young, 1967). However, rhodopsin mislocalized on the IS PM has no direct access to the RPE, and therefore the mechanisms of mislocalized rhodopsin elimination have been unclear. In this manuscript, we explored the intracellular mechanisms rod photoreceptors use to remove mislocalized rhodopsin. Intracellular class I mutant rhodopsin partially colocalized with markers of early endosome (EEA1) and lysosome (LAMP1; Lodowski et al., 2013). These observations led us to question whether mislocalized rhodopsin is continuously transported to IS lysosomes for degradation. By combining genetic and pharmacological approaches, we tested the hypothesis that perturbation of IS PM protein homeostasis forms the basis for photoreceptor dysfunction manifested by shortening and loss of OS structures. Our studies revealed a novel disease-causative mechanism that will be applicable to a wide spectrum of ciliopathies caused by defects in machinery or signals associated with ciliary receptor transport (Nemet et al., 2015b).
Materials and Methods
Animals.
All animal procedures were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University. Adult female and male frogs were purchased from Nasco and housed at 16°C under a 12 h light/dark cycle. All tadpoles used for experiments were housed at 16°C in 24 h darkness. Tadpoles were fed spirulina (http://www.Nuts.com) until metamorphosis was complete, and then were fed pelleted frog brittle (Nasco). Both male and female tadpoles were used for all experiments.
Reagents.
Unless otherwise specified, all reagents were purchased from either Fischer Scientific or Sigma-Aldrich.
Molecular cloning.
DNA expression vectors were generated by standard methods combining PCR, DNA recombination, and site-directed mutagenesis. The expression vector containing the Xenopus opsin promotor (XOP) followed by GFP–sodium-potassium ATPase α-subunit (NKAα; Laird et al., 2015) was a kind gift from Dr. Sheila Baker (University of Iowa). The vector contained an AgeI site upstream of GFP-NKAα. Therefore, site-directed mutagenesis was used to create an additional AgeI site after the region encoding GFP; GFP region was then removed using AgeI enzyme. To introduce PSmOrange (Subach et al., 2011) upstream of the Xenopus laevis NKAα3 coding region and generate PSmOrange-NKAα, cDNA encoding PSmOrange (Plasmid 31898, Addgene; RRID:Addgene_31898) was amplified using a pair of primers: NKA-mOrange-IF-F (5′-GCT TCT AGG GAT CCA CCG GTG CCA CCA TGG TGA GCA AGG GCG AGG-3′) and NKA-mOrange-IF-R (5′-CGT CCG TAC CCC ATA CCG GTC TTG TAC AGC TCG TCC ATG C-3′). The resulting DNA fragment was introduced downstream of XOP and upstream of NKAα using In-Fusion cloning kit (Takara). The plasmid vectors containing XOP-Rho-Dend2-1D4 and XOP-RhoQ344ter-Dend2 were previously generated (Lodowski et al., 2013). XOP-Rho-Dend2-1D4 is designed to express human rhodopsin followed by Dendra2 fluorescent protein and the last 8 aa of rhodopsin, which serve as the epitope for the monoclonal antibody 1D4 (Xie et al., 2011). The addition of the 1D4 epitope does not inhibit the function or localization of rhodopsin, and results in proper targeting of the fusion protein to the OSs (Xie et al., 2011; Lodowski et al., 2013). XOP-RhoQ344ter-Dend2 is designed to express class I mutant (Q344ter) of human rhodopsin missing the last five amino acids (QVAPA) followed by Dendra2 fluorescent protein. All the vectors contained polyadenylation signals following the coding and noncoding regions.
Transgenesis of Xenopus laevis.
Transgenic Xenopus laevis were produced using the intracytoplasmic sperm injection (ICSI) method following the previously published procedure (Sparrow et al., 2000; Smith et al., 2006; Lodowski et al., 2013). Expression vectors for PSmOrange-NKAα, Rho-Dend2-1D4, and RhoQ344ter-Dend2 were purified from bacteria cultures using EndoFree Plasmid Midi Prep Kit (Qiagen), and then used to isolate DNA fragments containing the XOP promoter, coding/noncoding regions, and polyadenylation signal for transgenesis. Fully developed tadpoles were screened for the presence of Dendra2 fluorescence in their eyes at 7 d postfertilization (7 DPF). Tadpoles were immersed in 6% methylcellulose to prevent movement while probed for green fluorescence in their eyes using a Leica MZ16F stereoscope (Leica Microsystems). Tadpoles were classified into one of the three following categories based on the expression level of the transgene: low, medium, or high as assessed by intensity of green fluorescence in their eyes. To prevent photobleaching, light-dependent photoreceptor degeneration, and unintended photoconversion of fluorescent protein Dendra2, tadpoles were reared in 24 h darkness. A male animal expressing high-level of RhoQ344ter-Dend2 was raised to maturity (>1 year old) and killed to collect sperm, which were then used to create second generation animals (Q1). This sperm was also used for generating tadpoles that coexpressed PSmOrange-NKAα and RhoQ344ter-Dend2. For this purpose, the sperms were subjected to ICSI method described here with linearized DNA containing XOP-PSmOrange-NKAα.
Photoconversion of Dendra2 in tadpole retina.
Dendra2 fluorescent protein in Xenopus tadpole eye was photoconverted as described previously (Lodowski et al., 2013; Lodowski and Imanishi, 2015). Xenopus tadpoles were anesthetized in 0.026% tricaine methanesulfonate and then immobilized in 6% methylcellulose covered with a protective light filter to prevent tissue damage. Light from a 405 nm laser placed 10 cm above the tadpole was directed at both eyes for a total of 15–20 min with 30 s breaks between each minute. The successful and nearly complete photoconversion was confirmed via inspection of green and red fluorescence by Leica MZ16F stereoscope. After photoconversion, tadpoles were subjected to small molecule treatment or killed and imaged immediately.
Immunohistochemistry.
Xenopus laevis eyes were fixed as previously described (Lodowski et al., 2013). Tadpoles were first killed in a 0.26% tricaine and 0.52% sodium bicarbonate solution (in dH2O) and decapitated. Their heads were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) in 0.1 m phosphate buffer (PB, NaHPO4, and Na2HPO4 dissolved in MilliQ H2O, pH 7.4) for 6 h at RT. Fixed heads were incubated in 5% sucrose in phosphate buffer for 30 min, and then subsequently washed in 10, 15, and 20% sucrose solutions in 0.1 m PB. The heads were then incubated overnight at 4°C in a solution containing 20% sucrose and optimum cutting temperature compound (OCT compound, Sakura Finetek) in a 2:1 ratio. The following day, heads were aligned in Tissue-Tek cryomolds (Sakura Finetek) and frozen in isopentane cooled by liquid N2. Frozen tissue sections (12 μm) were cut using a Leica CM1850 cryostat (Leica Microsystems) and collected on Superfrost Plus microscopy slides. Sections were dried and stored at −80°C until use. Slides were removed from −80°C storage conditions, warmed using a 37°C incubator (CCC 0.5d, Boekel Scientific), and then sections were encircled using an ImmEdge pen (Vector Laboratories). Sections were rehydrated and blocked in a 1.5% normal goat serum diluted in PBS solution (in mm: 137 NaCl, 2.7 KCl, 8 Na2HPO4, 2 KH2PO4) for 30 min at RT. Blocking solution was then removed and primary antibody (1:100) diluted in PBS containing 0.1% Triton X-100 (PBST) was added; sections were incubated at RT for ∼14 h and then washed three times with PBST for 10 min each. After the washes, sections were incubated in PBST containing anti-mouse Cy3 (1:100) and Hoechst dye (1:1000) for 1 h at RT followed by an additional three washes with PBST for 10 min each. After these washes, one drop of VECTASHIELD mounting medium (Vector Laboratories) was added to a square coverslip and placed over the section. Nail polish (Electron Microscopy Sciences) was used to seal the coverslip, and the sections were dried at RT for >30 min before imaging. Antibodies and fluorescent dyes used for immunofluorescence imaging were as follows: mouse anti-Na+/K+ ATPase (Lebovitz et al., 1989; Hybridoma Bank, RRID:AB_2166869), mouse anti-peripherin/rds (anti-xrds 2D4, a kind gift from Dr. Robert Molday, University of British Columbia), mouse XAP-2 (clone 5B9, Hybridoma Bank), Hoechst (ThermoFisher Scientific, 33342), and anti-mouse-Cy3 conjugate (Jackson ImmunoResearch Laboratories, Inc., 115-165-166).
SDS-PAGE and immunoblotting.
Tadpoles were killed in water containing 0.26% tricaine and 0.52% sodium bicarbonate before decapitation and eyeball removal. After removal of the lens and RPE cell layer, both isolated retinas from a single tadpole were homogenized via sonication in 100 μl of SDS buffer (0.1% SDS v/w in PBS) containing cOmplete Mini Protease Inhibitor Cocktail (Roche) and 0.1% β-mercaptoethanol. Proteins in these samples were resolved by SDS-PAGE using a standard method using a Mini-PROTEAN Tetra Cell (Bio-Rad) powered by a Powerpac HC (Bio-Rad). Transfers were performed by a semidry transfer cell (Trans-Blot SD Semi-Dry Transfer Cell, Bio-Rad) following the manufacturer's recommended protocol. Membranes were blocked in a 5% (w/v) dried milk (Nestle, Carnation) in PBS for 30 min before antibody incubation. Primary antibodies were added to PBS containing 0.1% Tween and incubated with membranes for 16 h at 4°C. Membranes were washed in PBS containing 0.1% Tween three times for 10 min each, and then incubated in secondary antibody solution for 1 h at RT. Antibodies and dilutions used were as follows; anti-β tubulin (Chu and Klymkowsky, 1989; Hybridoma Bank, RRID:AB_2315513; 1:500), anti-Na+/K+ ATPase α (Santa Cruz Biotechnology, M7-PB-E9; 1:500), and anti-rhodopsin (B6-30, 2 μg/ml; a kind gift from Dr. David Salom, Case Western Reserve University; RRID:AB_2572379; 1:3000). Secondary antibody used for immunoblotting was anti-mouse IgG-HRP (SC-2005, Santa Cruz Biotechnology; 1:1000). After secondary antibody incubation, membranes were washed three times for 10 min each before being exposed to HyGLO Chemiluminescent HRP Antibody Detection Reagent Quick Spray (Denville Scientific). After HRP exposure to substrate, membranes were immediately imaged by a Tanon 5200 developer (Tanon Science and Technology).
Reverse transcription and quantitative PCR.
Total RNA was isolated from Xenopus retinas using TRIzol (Invitrogen) according to the manufacturer's instructions. Approximately 5 μg of RNA was subjected to reverse transcription using iScript Reverse Transcription Supermix for RT-qPCR (Biorad). The resulting cDNA was used as a template for real-time PCR analyses. Following oligonucleotide primers were designed for amplifying NKAα and β-tubulin transcripts: NKAα-F (5′-GTC AGA CAG TTA CCG CGT AGC CAC C-3′); NKAα-R (5′-CTT TAT CCA CTC AGG GGT TGT GGG AGG-3′); β-tubulin-F (5′-ACA CGG CAT TGA TCC TAC AG-3′); β-tubulin-R (5′-AGC TCC TTC GGT GTA ATG AC-3′). NKAα and β-tubulin transcripts were amplified and quantified using iQ SYBR Green Supermix (Biorad) and a StepOnePlus Real-Time PCR System (Applied Biosystems). StepOne software (v2.3) was used for analysis.
Treatment of tadpoles by lysosome or NKA inhibitor.
Animals were treated in 0.1× Mark's Modified Ringer (MMR) solution (10× MMR composition: 1 m NaCl, 20 mm KCl, 20 mm CaCl2, 10 mm MgCl2, 50 mm HEPES, pH 7.5; Lodowski et al., 2013) containing 0.1% DMSO and either bafilomycin A1 (lysosome inhibitor, 100 nm), digoxin (NKA inhibitor, 3.5 μm), ouabain (NKA inhibitor, 1 μm), wedelolactone (NKA inhibitor, 3.5 μm), istaroxime (NKA inhibitor, 3 μm), or 3,4,5,6-tetrahydroxyxanthone hydrate (NKA inhibitor, 10 μm). All inhibitors were initially diluted in DMSO, and then applied to 5 ml of 0.1× MMR buffer containing a single tadpole. Bafilomycin A1 treatments lasted for 24 h while NKA inhibitor treatments lasted for 3–7 d. As controls, animals were placed in 0.1× MMR containing 0.1% DMSO. For all treatments that lasted >24 h, the 0.1× MMR solutions containing the drug in 0.1% DMSO were replaced daily in experimental groups, whereas 0.1× MMR solutions containing 0.1% DMSO were replaced daily in control groups.
Preparation of retina explant for confocal imaging.
Whole eyes were extracted from tadpole heads using surgical tools after euthanasia and decapitation. Retinas were immediately placed into a glass bottom dish (P35G-1.5-14-C, MatTek Corporation) containing equilibrated modified Wolf medium (d-glucose, 700 mg/L, 30 mm NaHCO3, 55% MEM, 31% sodium-free BBS, 10% FBS; Lodowski et al., 2013). Equilibration of Wolf medium was achieved by incubating the dish containing the medium in a gas chamber supplied with 5% CO2 and 95% O2 (Airgas; Nemet et al., 2014). The glass bottom dishes were coated with Cell-Tak Cell and Tissue Adhesive (Corning). RPE were carefully removed from the neural retinas, which were then placed onto the dish, and then a circular glass coverslip (12 mm diameter and 0.13–0.17 mm thickness) was used to seal and flatten the retinas before imaging. Retinas were kept alive in Wolf medium with constant flow of 5% CO2 and 95% O2 in a humid imaging chamber (Tokai Hit). For labeling of lysosomes, retinas were incubated in equilibrated modified Wolf medium containing 1:100 dilution of LysoTracker Red DND-99 (ThermoFisher Scientific) for 30 min prior mounting on a glass bottom dish and imaging.
Confocal microscopy.
Live and fixed retinas were imaged using a Leica TCS SP2 laser scanning confocal/multiphoton microscope system equipped with four lasers for excitation: a 488 nm argon ion, 543 nm HeNe, 633 nm HeNe, and tunable Chameleon XR Ti:sapphire laser (Leica Microsystems). An HCX PL APO CS 40X/1.25 oil UV objective lens was used for imaging. Images were acquired using Leica Confocal Software v2.61 (Leica Microsystems). For images that required fluorescence quantification and comparison, the exact same settings and parameters, including laser power, zoom factor, image averaging, resolution, and photomultiplier tube (PMT) voltage were used for both control and experimental groups. These parameters were adjusted to maximize signal brightness while minimizing fluorescence saturation.
Image analysis and quantification.
Relative amounts of rhodopsin fused to Dendra2, PSmOrange-NKAα, or immunofluorescently-labeled NKAα were quantified using ImageJ (Schneider et al., 2012). Images for quantification were taken under the exact same acquisition conditions including laser power, voltage, and PMT settings. Therefore, mean pixel intensity values were directly compared among the specimens. Individual optical section images (z slices) were used for quantification. Images were loaded in ImageJ and the freehand selection tool was used to encompass the region(s)-of-interest to be measured, such as vesicles or entire IS cytoplasmic regions. For the selection of the cytoplasmic regions, IS PM regions were excluded to focus on the intracellular fluorescence intensities. For all fluorescence quantification, background measurements were taken and subtracted from all values. All measurements were copied into Microsoft Excel 2016 (v16.0, 32-bit, Microsoft) or SigmaPlot (v12.0, Systat Software) for analysis. For the experiment where Dendra2/Cy3 levels were measured on the PM and compared, the IS PM region near the base of the OS was highlighted using the straight-line tool and measurements were taken in both green (Dendra2) and red (Cy3) channels. To determine whether the magnitude of NKAα loss was dependent on the amounts of RhoQ344ter-Dend2, individual rod photoreceptor cells were categorized based on their expression levels of RhoQ344ter-Dend2. For this purpose, Dendra2 signal on the PM was measured and only rods that exhibited fluorescence in the top 10% of all cells were categorized into the high-expression group (High Q), whereas all other rods analyzed were categorized into the low-expression group (Low Q). To accurately quantify the number and length of OSs as well as number of nuclei, the same region of each retina was imaged by centering the retina 100 μm from the optic nerve head (to either nasal or temporal side) and using zoom factor 2 to analyze the rods in these regions. The number of OSs and nuclei (Table 1) were counted across 180 μm of retina length on both the sides (±90 μm) of this center. Images taken in the red channel were converted to magenta by loading the original image (TIFF format) into ImageJ, using the LUT Menu function, and then saving as a TIFF file.
Table 1.
Summary of effects caused by mislocalized rhodopsin or drug treatment
| Condition | OS length, μm | OS/180 μm retina length | Nuclei/180 μm retina length |
|---|---|---|---|
| Rho-Dend2-1D4 | 20.0 ± 4.3 | 23.6 ± 1.1 | 115.7 ± 8.0 |
| RhoQ344Ter-Dend2 | 11.2 ± 2.6 | 17.5 ± 2.5 | 104.2 ± 5.7 |
| Untreated | 15.6 ± 4.4 | 24.9 ± 1.4 | 108.8 ± 9.1 |
| Digoxin | 11.1 ± 4.7 | 16.5 ± 2.6 | 106.9 ± 7.8 |
| Ouabain | 12.0 ± 4.6 | 19.3 ± 5.4 | 105.5 ± 10.0 |
| Wedelolactone | 14.4 ± 7.1 | 18.5 ± 1.5 | 103.0 ± 5.6 |
| Istaroxime | 13.0 ± 4.5 | 22.0 ± 2.2 | 114.5 ± 4.5 |
| TTX | 13.5 ± 4.5 | 19.1 ± 2.9 | 103.3 ± 12.6 |
Experimental design and statistical analysis.
For comparison between a single experimental group and a control group, Student's t-test was used to determine statistical significance. To compare significance among multiple experimental groups, ANOVA was used. For ANOVA, a Tukey post hoc test was used for multiple comparison and identification of pairs with significant differences. The p values for these analyses were calculated using the Analysis function of the SigmaPlot software. Statistical significance is denoted as *p < 0.05, **p < 0.01, ***p < 0.001, and NS (not significant, p > 0.05) in the Results, figures, and figure legends. The p values are indicated along with statistical parameters such as number of cells/vesicles counted and number of animals used in each experiment in the Results and figure legends. For calculations in which the average was generated, values are represented by mean ± SD. For the purposes of qPCR and immunoblot in which unfixed retinas were homogenized and analyzed, tadpoles with high amounts of Dendra2 fluorescence were selected to highlight differences from nontransgenic retinas.
Results
Class I mutant rhodopsin is actively degraded by lysosomes in rod photoreceptors
We previously demonstrated that class I mutant rhodopsin is actively eliminated from rod photoreceptor IS PM (Lodowski et al., 2013). To characterize the removal process of class I mutant rhodopsin and compare it to that of wild-type rhodopsin in living cells, Q344ter, a class I mutant human rhodopsin with the 5 terminal amino acids truncated, and wild-type human rhodopsin were fused to Dendra2 fluorescent protein and individually expressed in Xenopus laevis rod photoreceptors. As reported previously for endogenous rhodopsin, wild-type rhodopsin fused to Dendra2 specifically localized to rod OSs (Fig. 1A, Rho-Dend2-1D4, Control; Lodowski et al., 2013). As reported for the localization of untagged class I mutant rhodopsin in transgenic animal models (Sung et al., 1993, 1994; Tam et al., 2006; Concepcion and Chen, 2010), the majority of class I mutant rhodopsin fused to Dendra2 mislocalized to the IS PM (Fig. 1A, RhoQ344ter-Dend2, Control; Lodowski et al., 2013). Wild-type rhodopsin correctly localizes to the rod OS and is removed via phagocytosis by the neighboring RPE cells (Young, 1967; Besharse et al., 1977). However, because IS PM mislocalized rhodopsin has no direct access to the RPE, we asked whether RhoQ344ter-Dend2 is degraded within lysosomes of rod photoreceptor cells.
Figure 1.
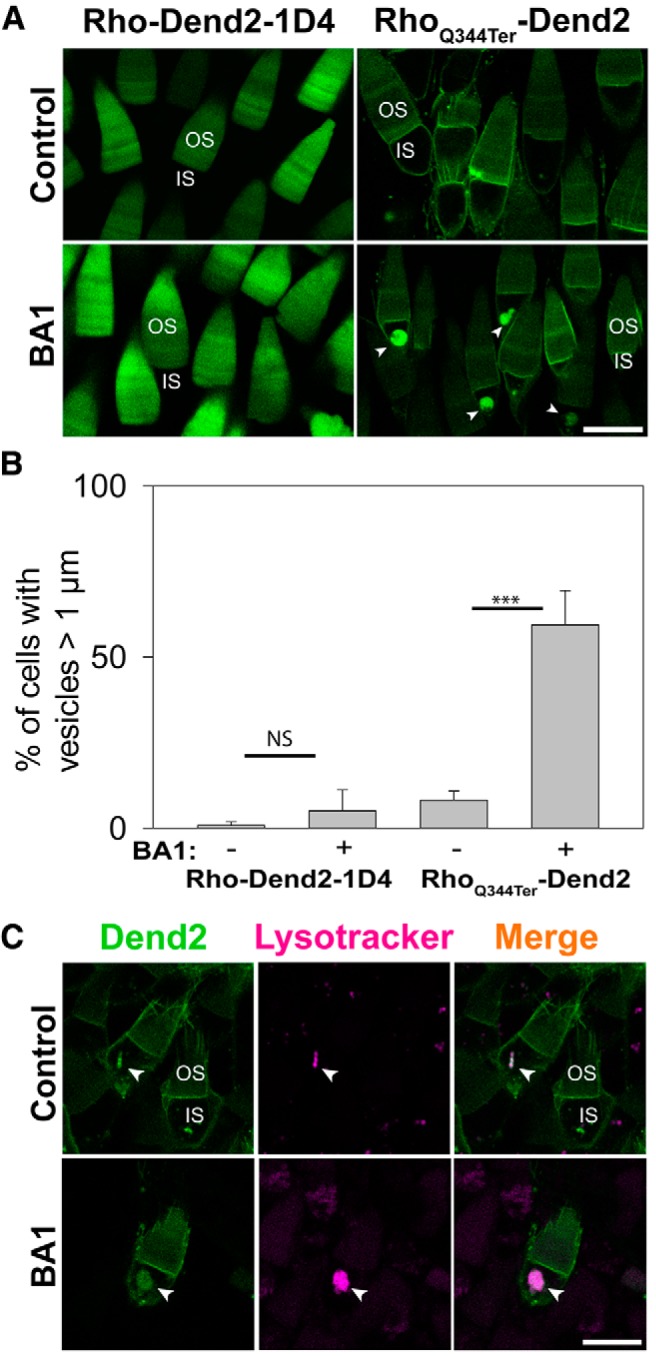
Class I mutant rhodopsin accumulates in lysosomes after inhibition of lysosome-mediated degradation. A, Confocal imaging of live Xenopus laevis rod photoreceptors expressing either Rho-Dend2-1D4 (left) or RhoQ344ter-Dend2 (right). OS and IS structures are labeled in a representative rod cell of each image. Before the imaging, the animals were treated with DMSO (Control) or 100 nm BA1 (BA1) for 24 h. In control retinas, neither Rho-Dend2-1D4 nor RhoQ344ter-Dend2 accumulated significantly in vesicular structures of the ISs. After BA1 treatment, RhoQ344ter-Dend2 accumulated in vesicular structures (arrowheads) in the ISs, but Rho-Dend2-1D4 did not. B, In retinas expressing either Rho-Dend2-1D4 or RhoQ344ter-Dend2, we assessed the percentage of rod photoreceptor cells that exhibited vesicles >1 μm in diameter. BA1 treatment of photoreceptors expressing Rho-Dend2-1D4 increased this percentage from 0.9 ± 1.0% (untreated) to 5.1 ± 6.2% (NS, p = 0.23). BA1 treatment of photoreceptors expressing RhoQ344ter-Dend2 increased this percentage from 8.2 ± 2.7% to 59.4 ± 10.0% (p < 0.001), indicating that RhoQ344ter-Dend2, but not Rho-Dend2-1D4, accumulates upon BA1 treatment. C, Confocal imaging of live Xenopus laevis rod photoreceptors expressing RhoQ344ter-Dend2 and labeled with LysoTracker Red dye. LysoTracker Red signal colocalized with RhoQ344ter-Dend2 in untreated cells (Control, arrowheads), and the amount of RhoQ344ter-Dend2 colocalized with LysoTracker Red increased after pretreatment with BA1 (arrowheads). Scale bars, 10 μm. ***p < 0.001, NS, not significant.
Lysosome-mediated degradation was inhibited by bafilomycin A1 (BA1; 100 nm) in Xenopus laevis rods either expressing Rho-Dend2-1D4 or RhoQ344ter-Dend2 (Fig. 1A). After 24 h of treatment, RhoQ344ter-Dend2 frequently accumulated in vesicular structures within the rod ISs (Fig. 1A, RhoQ344ter-Dend2, BA1, arrowheads) with a size distribution (diameter) of 2.07 ± 0.63 μm (n = 126 vesicles from 7 animals). In the absence of BA1 treatment, RhoQ344ter-Dend2 rarely accumulated in vesicular structures with diameters >1 μm (Fig. 1A, RhoQ344ter-Dend2, Control). In animals expressing RhoQ344ter-Dend2 and treated with BA1, 59.4 ± 10.0% of rod photoreceptors exhibited vesicular structures with diameter >1 μm (Fig. 1B; based on 64 cells each from n = 4 animals), whereas untreated animals exhibited these structures in only 8.2 ± 2.7% of rod photoreceptors (p < 0.001; Fig. 1B; based on 61 cells each from n = 4 animals). These observations suggest that RhoQ344ter-Dend2 is prone to lysosome-mediated degradation, and that inhibition of this degradation via BA1 treatment resulted in the intravesicular accumulation of RhoQ344ter-Dend2.
As a control, animals expressing Rho-Dend2-1D4 did not significantly accumulate in intracellular structures (>1 μm) after BA1 treatment (Fig. 1A, Rho-Dend2-1D4, BA1). Untreated rods expressing Rho-Dend2-1D4 exhibited intracellular structures (>1 μm) at a low-frequency of 0.9 ± 1.0% (Fig. 1B; based on 57 cells each from n = 4 animals). After BA1 treatment, the number of rods exhibiting these structures increased to 5.1 ± 6.2% (Fig. 1B; based on 64 cells each from n = 4 animals), but not in a statistically significant manner (NS, p = 0.23). These observations indicate that Rho-Dend2-1D4 is marginally degraded by lysosomes within rod photoreceptors, and confirm previous findings that the majority of wild-type rhodopsin is degraded by RPE cells (Young, 1967; Besharse et al., 1977).
To identify these intracellular organelles containing RhoQ344ter-Dend2, LysoTracker Red (Pierzyńska-Mach et al., 2014) was used to specifically label lysosomes in photoreceptor cells. In rod ISs of untreated animals incubated with LysoTracker Red dye, LysoTracker-positive lysosomes frequently contained RhoQ344ter-Dend2 (Fig. 1C, Control, arrowheads). The majority of LysoTracker-positive lysosomes contained RhoQ344ter-Dend2 (68.0 ± 16.2% based on 32 lysosome-positive cells each from n = 4 animals). BA1 prevents acidification of lysosomes/autophagolysosomes through inhibition of the V-ATPase of the lysosome, which is responsible for pumping H+ ions into the compartment (Yamamoto et al., 1998). Because LysoTracker Red only labels low-pH organelles, it did not reproducibly label lysosomes of BA1-treated cells. Because BA1 is a reversible inhibitor of the V-ATPase (Yamamoto et al., 1998), we improved labeling of lysosomes by lifting BA1 treatment for 1 h before labeling with LysoTracker Red (Fig. 1C, BA1, arrowheads). Under these conditions, accumulated RhoQ344ter-Dend2 colocalized with LysoTracker Red in 84.3 ± 5.3% of all lysosome-labeled cells observed (based on 31 lysosome-positive cells each from n = 4 animals). Therefore, the majority of these intracellular organelles containing RhoQ344ter-Dend2 are lysosomes or autophagolysosomes.
Mislocalized rhodopsin is trafficked from the plasma membrane to intracellular lysosomes
Two alternative explanations are plausible regarding the source of RhoQ344ter-Dend2 in lysosomes: one possibility is that RhoQ344ter-Dend2 is continuously removed from the PM and trafficked to lysosomes, and another possibility is that RhoQ344ter-Dend2 is sent to lysosomes directly after its synthesis in the Golgi apparatus. To distinguish between these two possibilities, we took advantage of the photoconversion property of Dendra2. Dendra2 is a photoconvertible fluorescent protein engineered to irreversibly shift its emission peak from shorter (507 nm, green) to longer (573 nm, red) wavelength (Nemet et al., 2015a). This photoconversion technique allowed us to study the destiny of photoconverted red RhoQ344ter-Dend2 by discriminating it from newly synthesized green RhoQ344ter-Dend2 coming from the Golgi apparatus. If RhoQ344ter-Dend2 is actively transferred from the PM to lysosomes, then we would expect a statistically significant increase of red proteins in the IS after 24 h of BA1 treatment (Fig. 2A, Model i). If, however, PM-localized RhoQ344ter-Dend2 is not being transferred to the IS lysosomes (Fig. 2A, Model ii), then we would not expect a significant increase of intracellular red proteins after BA1 treatment.
Figure 2.

Mislocalized rhodopsin is trafficked from the plasma membrane to intracellular lysosomes. A, Experimental design to determine the origin of proteins accumulating in the ISs of RhoQ344ter-Dend2 cells. Before photoconversion, the majority of RhoQ344ter-Dend2 is localized to the IS PM. After photoconversion and BA1 treatment (24 h), red protein should accumulate if RhoQ344ter-Dend2 is being trafficked from the PM to the IS lysosomes (Model i). If RhoQ344ter-Dend2 is not internalized, then red RhoQ344ter-Dend2 will not accumulate significantly in the IS lysosomes (Model ii). B, Immediately following photoconversion, rods expressing RhoQ344ter-Dend2 were imaged (0 h). Photoconversion was efficient as confirmed by the loss of green fluorescence (0 h, green). The majority of red RhoQ344ter-Dend2 was observed on the IS PM (0 h, red, PM, arrowhead). After 24 h of BA1 treatment, RhoQ344ter-Dend2 accumulated intracellularly in the ISs of rods (BA1, arrowheads). A nominal amount of green RhoQ344ter-Dend2 was observed after 24 h (BA1 and Control, Green). This observation indicates that new RhoQ344ter-Dend2 was synthesized in these retinas. C, Average red fluorescence intensities (a.u./area) were measured for intracellular regions of ISs in individual rods. The histograms indicate frequency of cells (y-axis) for each fluorescence intensity range (x-axis). Gray arrowheads indicate the average fluorescence intensity of each condition (0 h, BA1, or Control). Red fluorescence intensity values in the ISs were 36.7 ± 13.9 a.u. for time = 0 h condition, 29.4 ± 16.8 a.u. for 24 h DMSO-treated Control group, and 60.2 ± 16.8 a.u. for the BA1-treated group. Animals were aged 9 DPF upon photoconversion (0 h) and were killed and imaged at 10 DPF (BA1 and Control), with a total of 250 cells for each condition measured from n = 5 animals. Scale bar, 10 μm.
Photoconversion of rod photoreceptors expressing RhoQ344ter-Dend2 allowed us to label existing fusion proteins in red (Fig. 2B, 0 h, Red). Most red RhoQ344ter-Dend2 in these rods was observed on the IS PM (Fig. 2B, 0 h, Red, PM, arrowhead). Photoconversion of RhoQ344ter-Dend2 was highly efficient; significant green fluorescence was not observed after photoconversion (Fig. 2B, 0 h, green). After 24 h of BA1 treatment following the photoconversion, both green and red RhoQ344ter-Dend2 accumulated in intracellular vesicular structures (Fig. 2B, BA1, arrowheads). The majority of Dendra2 present in these structures exhibited red fluorescence (Fig. 2B, BA1, red, arrowheads), thus supporting our model in which PM-localized RhoQ344ter-Dend2 was internalized (Fig. 2A, Model i).
As controls, a set of animals expressing RhoQ344ter-Dend2 were photoconverted and then placed in 0.1× MMR buffer containing 0.1% DMSO (the vehicle we used for BA1) for 24 h. As expected, RhoQ344ter-Dend2 did not significantly accumulate in the ISs of untreated rods (Fig. 2B, Control). Because RhoQ344ter-Dend2 did not accumulate under these conditions, the accumulation of RhoQ344ter-Dend2 observed in the BA1-treated group must have been due to lysosome inhibition and not due to other experimental conditions. After 24 h, newly synthesized green RhoQ344ter-Dend2 was observed on the PM of both treated and untreated rods (Fig. 2B, BA1 and Control, green). Green protein was also observed in the intracellular structures of BA1-treated rods (Fig. 2B, BA1, green, arrowheads). Therefore, under our experimental conditions, rod photoreceptors were healthy and capable of synthesizing and trafficking new RhoQ344ter-Dend2.
We confirmed through quantitative analysis that red RhoQ344ter-Dend2 accumulated after inhibition of lysosomes. Red fluorescence within the IS cytoplasm of individual rods from each experimental group was measured, and rods were categorized based on intracellular fluorescence intensities within their ISs (Fig. 2C). Overall, there were variable amounts of red fluorescence in the ISs for all the conditions tested; this was likely because our transgenic system results in variable expression of the transgene in individual rods (Lodowski et al., 2013). Despite the variance, BA1-treated rod cells (Fig. 2C, BA1) more frequently exhibited high amounts of red fluorescence in their ISs (Fig. 2C, compare BA1 to 0 h and Control). On average, BA1-treated rods exhibited significantly (p < 0.001) more red fluorescence in their ISs than in control rods; BA1-treated rods had 2.03 ± 0.89 times more red fluorescence than control rods and 1.62 ± 0.71 times more red fluorescence than 0 h rods (based on n = 250 cells from 5 animals for each condition). We believe that the increased amount of red Dendra2 observed in BA1-treated rods was largely contributed by vesicular accumulated proteins (Fig. 2B, BA1, red, arrowheads), whereas red protein observed in untreated rods were contributed by proteins existing diffusively in the IS (e.g., endoplasmic reticulum). Red fluorescence in the IS decreased after 24 h in untreated rods, suggesting that red protein existing at 0 h had exited structures such as ER after 24 h. In summary, these results statistically support Model i (Fig. 2A) in which PM-localized RhoQ344ter-Dend2 is actively removed from the PM and translocated to IS lysosomes for degradation.
The Na+/K+ ATPase is downregulated in cells expressing class I mutant rhodopsin
In healthy rod photoreceptors, rhodopsin does not accumulate on the IS PM. In rods afflicted by ciliopathy, the daily accumulation, removal, and degradation of PM-mislocalized rhodopsin would likely have negative effects on the PM protein homeostasis. An essential component of the photoreceptor IS PM is the Na+/K+-ATPase, which plays important roles in maintaining the Na+ and K+ gradients required for the photoresponses of cone and rod cells (Wetzel et al., 1999). In our experiments, RhoQ344ter-Dend2 and Rho-Dend2-1D4 are expressed under the regulation of the Xenopus rhodopsin promoter, which drives expression specifically in rods (Mani et al., 2001). By immunofluorescence microscopy, robust and consistent expression of NKAα was observed in cone cells (Fig. 3A, white asterisks). In contrast, the expression levels of NKAα were attenuated in the rod cells expressing RhoQ344ter-Dend2 (Fig. 3A, RhoQ344ter-Dend2, arrowheads). The amount of NKAα on the IS PM of rods expressing Rho-Dend2-1D4 appeared to be higher than that of rods expressing RhoQ344ter-Dend2 (Fig. 3A, Rho-Dend2-1D4 and RhoQ344ter-Dend2, arrowheads), indicating the Q344ter class I mutation is the cause of NKAα downregulation.
Figure 3.

NKAα is downregulated in rods expressing RhoQ344ter-Dend2. A, Immunofluorescence labeling of NKAα (magenta) in retinas expressing either Rho-Dend2-1D4 (left column) or RhoQ344ter-Dend2 (right column). NKAα signal on the PM of cells expressing wild-type rhodopsin was relatively uniform (Rho-Dend2-1D4, NKAα, arrowheads), whereas the NKAα signal was noticeably reduced or absent from cells expressing class I mutant rhodopsin (RhoQ344ter-Dend2, NKAα, arrowheads). NKAα expression was robust in cone cells (white asterisks). B, Representative immunoblot comparing the expression levels of NKAα and β-tubulin in NT or Q1 retinas. Molecular weight (MW) is indicated in kilodaltons. C, Left side of graph (IB, delimited by dotted line): in NT or Q1 retinas, the protein amounts of NKAα were quantified and normalized to the amounts of β-tubulin protein. NT value was standardized to 1.00. (Exact values were 1.00 ± 0.0.19 for NT and 0.50 ± 0.12 for RhoQ344ter-Dend2, based on n = 6 animals; p < 0.01). Right side of graph (qPCR): the amount of NKAα transcript relative to β-tubulin transcript was assessed by quantitative PCR. ΔCt values of nontransgenic and high-expression RhoQ344ter-Dend2 retinas were compared (4.60 ± 0.27 for RhoQ344ter-Dend2 retinas and 4.42 ± 0.78 for nontransgenic retinas based on n = 5 animals; p = 0.63). Scale bar, 10 μm. **p < 0.01, NS, not significant.
NKAα downregulation was independently confirmed via immunoblot analysis, which revealed an ∼50% decrease in NKAα in retinas expressing RhoQ344ter-Dend2 (Q1) compared with nontransgenic (NT) retinas (p < 0.01; Fig. 3B,C, IB). This modest decrease, compared with the decrease observed in individual rods, was consistent with the presence of NKAα in secondary retinal neurons which were not affected by RhoQ344ter-Dend2. Based on quantitative PCR analysis, we found that downregulation of NKAα protein is not because of reduced mRNA levels. The ΔCt value compares relative expression of a pair of genes (Mahanty et al., 2017). When we quantified NKAα transcript amounts relative to β-tubulin's, the ΔCt values were not significantly different between NT and Q1 retinas (Fig. 3C, qPCR). Because of the lack of significant change in gene expression and the significant difference in the protein level, we surmised that NKAα is downregulated by one or more posttranslational processes, and likely by lysosome-mediated degradation.
NKAα downregulation is correlated to the expression level of RhoQ344ter-Dend2 in individual rods
In F0 transgenic retinas expressing either RhoQ344ter-Dend2 or Rho-Dend2-1D4, individual rods express varying amounts of the protein product likely because of position-effect variegation (Moritz et al., 2001). Individual rods were categorized based on the expression levels (low and high) of RhoQ344ter-Dend2. We found that the higher the expression level of RhoQ344ter-Dend2, the less NKAα was present on the IS PM (Fig. 4A, Low Q and High Q, arrowheads). As controls, nontransgenic rods (NT) and rods expressing Rho-Dend2-1D4 (WT) did not exhibit downregulations of NKAα (Fig. 4A, NT and WT, arrowheads). Quantification of NKAα in the apical region of the IS PM (Fig. 4A, NKAα, arrowheads) revealed that whereas rods with low expression (Low Q) of RhoQ344ter-Dend2 had reduced NKAα presence on the IS PM compared with nontransgenic and Rho-Dend2-1D4 transgenic rods, rods with high expression (High Q) of RhoQ344ter-Dend2 had the lowest amounts of NKAα compared with all groups tested (p < 0.001; Fig. 3B, compare High Q to Low Q, NT, and WT). The inverse relationship between the expression levels of RhoQ344ter-Dend2 and NKAα is consistent with a model in which these two proteins are co-degraded: higher expression of RhoQ344ter-Dend2 would result in more active lysosomal degradation, which would coincidentally lower the NKAα levels.
Figure 4.
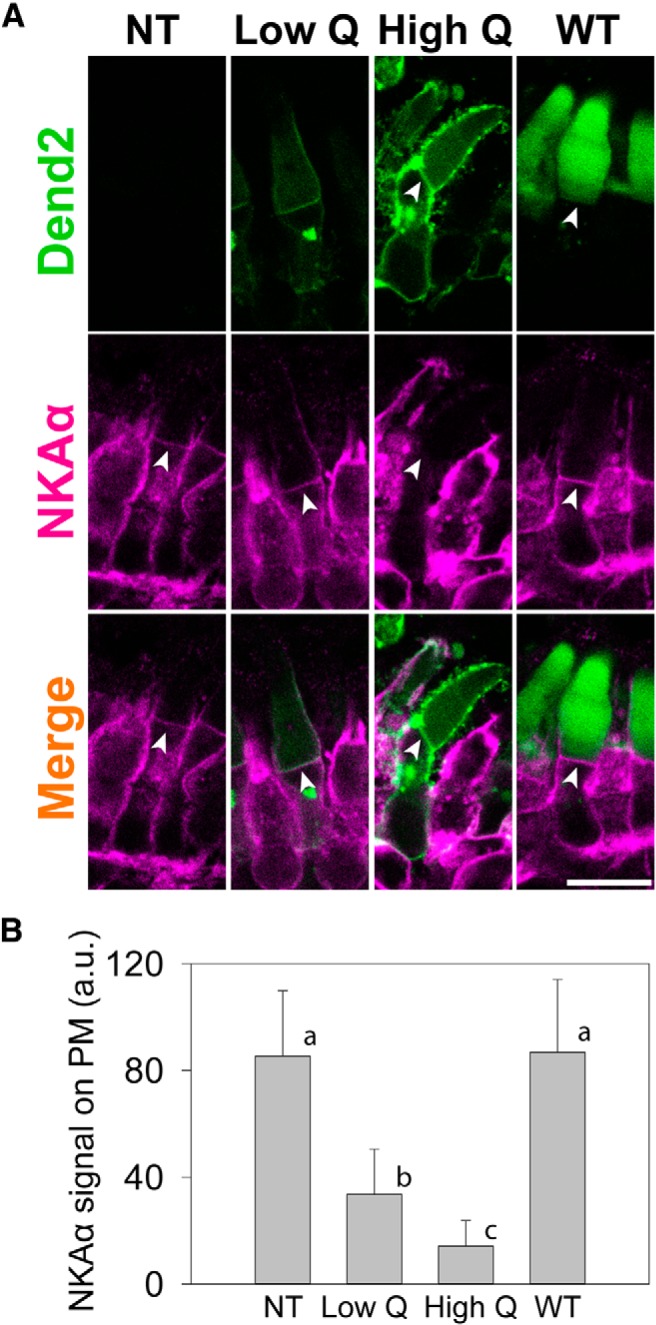
Degree of NKAα downregulation is dependent on the amount of class I mutant rhodopsin. A, Direct comparison of NKAα immunofluorescence in a rod cell not expressing RhoQ344ter-Dend2 (NT), a rod cell expressing low amount of RhoQ344ter-Dend2 (Low Q), a rod cell expressing high amount of RhoQ344ter-Dend2 (High Q), and a rod cell expressing Rho-Dend2-1D4 (WT). Arrowheads indicate the apical region of the IS PM. Scale bar, 10 μm. B, Quantification of NKAα immunofluorescence observed in A. Compared with NT and WT rods, NKAα was downregulated in rods expressing class I mutant rhodopsin in a manner depending on the expression level of RhoQ344ter-Dend2. Rods with low expression of RhoQ344ter-Dend2 (Low Q) had higher quantities of NKAα on the PM than rods with high expression of RhoQ344ter-Dend2 (High Q). By ANOVA Tukey post hoc test, all the pairs (a vs b, a vs c, and b vs c) were significantly different (p < 0.001; based on 52 cells from n = 4 animals for each condition) except for between WT and NT rods (a, p = 0.58), which indicated that the presence of the wild-type rhodopsin transgene did not affect the expression of NKAα.
NKAα is cotrafficked with RhoQ344ter-Dend2 to lysosomes in vivo
Co-degradation of RhoQ344ter-Dend2 and NKAα would require these two proteins to be in the same lysosomal compartments. To visualize the possible colocalization in vivo, we fused NKAα to fluorescent protein PSmOrange (Subach et al., 2011), which emits orange fluorescence distinguishable from green Dendra2 fluorescence. PSmOrange-NKAα and RhoQ344ter-Dend2 were coexpressed in Xenopus tadpoles, which were then treated with BA1 to inhibit lysosome-mediated degradation (Fig. 5A). After 24 h of BA1 treatment, PSmOrange-NKAα and RhoQ344ter-Dend2 accumulated together in intracellular vesicles of rod ISs (Fig. 5A, BA1, arrowheads). Based on the fluorescence intensities, the amount of PSmOrange-NKAα in the ISs was ∼2.15 times higher (p < 0.001) in BA1-treated rods than in untreated rods [Fig. 5B, compare NKAα+Q344ter with (+) and without (−) BA1]. To test whether lysosomal accumulation of PSmOrange-NKAα was dependent on RhoQ344ter-Dend2, we repeated the experiment using animals expressing PSmOrange-NKAα, but not RhoQ344ter-Dend2 (Fig. 5C). After BA1 treatment, PSmOrange-NKAα did not significantly accumulate (NS, p = 0.35) in the ISs over untreated rods (Fig. 5B, compare NKAα with (+) and without (−) BA1). These results indicate that lysosomal localization of PSmOrange-NKAα was induced by RhoQ344ter-Dend2.
Figure 5.
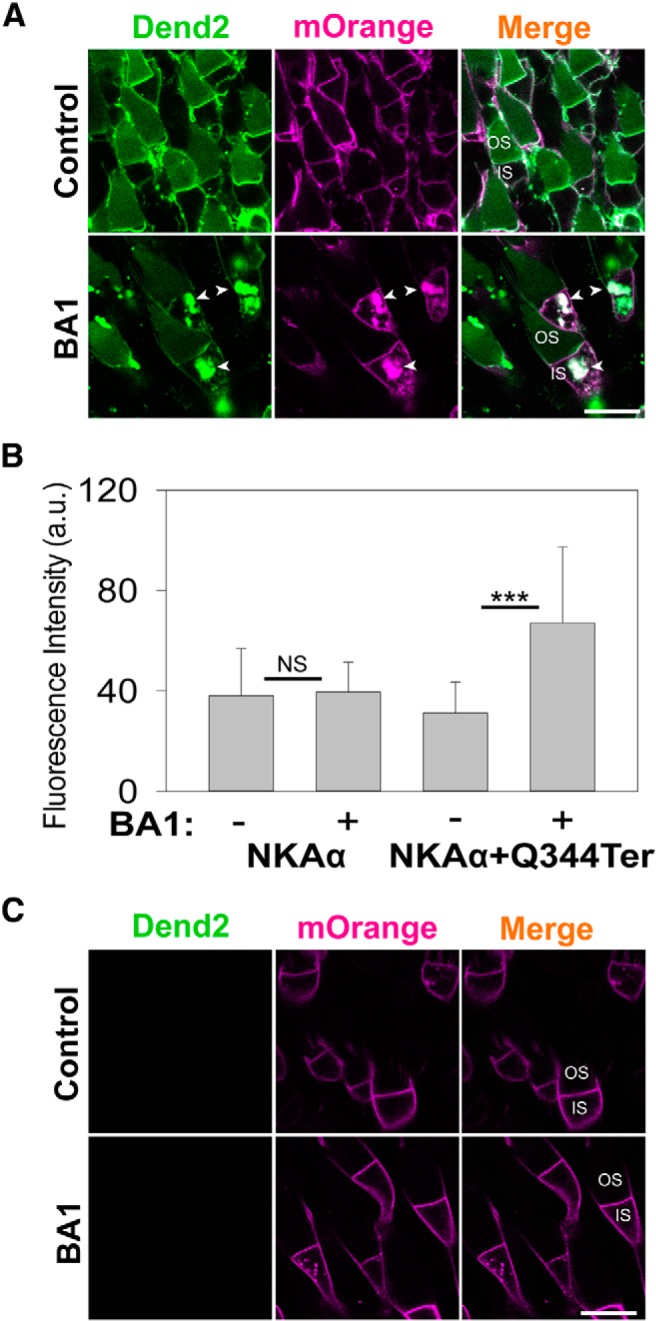
RhoQ344ter-Dend2 induces internalization of NKAα. A, RhoQ344ter-Dend2 (green) was expressed together with NKAα fused to PSmOrange fluorescent protein (magenta) in rod photoreceptors. These transgenic tadpoles aged 9 DPF were treated for 24 h with either DMSO (Control) or 100 nm BA1 (BA1). BA1-treated rods exhibited vesicles containing both RhoQ344ter-Dend2 and PSmOrange-NKAα in the ISs. B, Comparative quantification of intracellular fluorescence from PSmOrange-NKAα in either the presence (+) or absence (−) of 100 nm BA1 and either with (+Q344ter) or without coexpression of RhoQ344ter-Dend2. Nearly equal amounts of PSmOrange-NKAα were observed among untreated and BA1-treated cells expressing only PSmOrange-NKAα (NS, p = 0.35; based on 80 untreated cells and 140 BA1-treated cells from n = 4 animals each). Conversely, BA1-treated cells expressing both PSmOrange-NKAα and RhoQ344ter-Dend2 had significantly increased amounts of PSmOrange-NKAα in the ISs compared with untreated cells expressing both PSmOrange-NKAα and RhoQ344ter-Dend2 (p < 0.001; based on 160 untreated cells and 173 BA1-treated cells from n = 4 animals each). This difference suggests that the presence of RhoQ344ter-Dend2 was required to bring PSmOrange-NKAα to the IS lysosomes for degradation. C, Cells expressing only the PSmOrange-NKAα fusion protein without RhoQ344ter-Dend2 did not significantly accumulate PSmOrange-NKAα upon treatment with BA1. Scale bars, 10 μm. ***p < 0.001, NS, not significant.
Rod photoreceptors expressing RhoQ344ter-Dend2 have shorter, disorganized OSs
In RP, the first observable histopathologic change is the shortening and loss of the OSs (Milam et al., 1998) followed by loss of the photoreceptors themselves (Milam et al., 1998; Koch et al., 2015). We then asked whether rhodopsin mislocalization, which is commonly observed in various forms of RP (Li et al., 1995; Milam et al., 1998; Gao et al., 2002), is the cause of these OS defects. We labeled RhoQ344ter-Dend2 transgenic retinas (21 DPF) with anti-peripherin/rds (P/rds), a marker for rod and cone OS disk membranes (Arikawa et al., 1992; Tian et al., 2014). Rod cells expressing RhoQ344ter-Dend2 had noticeably disorganized OSs compared with those expressing Rho-Dend2-1D4 (Fig. 6A). RhoQ344ter-Dend2 retinas exhibited fewer and shorter P/rds-positive OSs compared with retinas expressing Rho-Dend2-1D4 (Fig. 6A, P/rds). In addition to anti-P/rds, which labels both cone and rod OSs, we also used a XAP-2 antibody that specifically labels Xenopus rod OSs (Fig. 6B; Viczian et al., 2009; Choi et al., 2011). Consistent with the results obtained for P/rds-positive OSs, RhoQ344ter-Dend2 transgenic retinas contained ∼30% fewer (p < 0.001; based on retinas from n = 6 animals) XAP-2-positive OSs (per 180 μm retina length; Table 1) than Rho-Dend2-1D4 transgenic retinas (Fig. 6B, XAP-2). Additionally, RhoQ344ter-Dend2 rods were ∼44% shorter than Rho-Dend2-1D4 rods (p < 0.001; based on 65 OSs from n = 6 animals for each condition; Table 1). To clarify the relationship between NKAα downregulation and OS defects, we measured the NKAα levels and OS lengths in the same individual rods expressing RhoQ344ter-Dend2. Rods expressing RhoQ344ter-Dend2 had variable degrees of NKAα downregulation (Fig. 6C). For this analysis, we categorized individual rods (192 measurements from n = 6 individual animals) into three groups depending on the severity of the downregulation. Rods with NKAα levels similar (80–100%) to the nontransgenic values had OSs that were 16.0 ± 4.0 μm long, and thus comparable to OS lengths of nontransgenic rod photoreceptors (Table 1, untreated). Rods with NKAα downregulation demonstrated significantly shorter rod OSs (p < 0.001); rods that contained 40–60% of the normal NKAα level had OSs that were 11.3 ± 4.2 μm long, whereas rods that expressed < 20% of the average nontransgenic NKAα level had OSs that were 6.1 ± 3.5 μm long (Fig. 6C). This analysis indicated that NKAα downregulation coincides with the shortening of rod OSs.
Figure 6.

Rods expressing RhoQ344ter-Dend2 exhibit disorganized rod OSs. Immunofluorescence labeling of OSs using (A) anti-P/rds (magenta) and (B) XAP-2 (magenta) antibodies. Retina sections (21 DPF) expressing either Rho-Dend2-1D4 (green) or RhoQ344ter-Dend2 (green) were labeled. C, Rods with lower NKAα expression demonstrate shorter OSs. NKAα expression levels in RhoQ344ter-Dend2-positive rods (21 DPF) were normalized to average NKAα level (100%) in nontransgenic wild-type (wt) rods (21 DPF). Rods with three different ranges of NKA expression levels (80–100, 40–60, and 0–20%) were analyzed. D, Representative immunoblot comparing 21 DPF retina samples of either NT or Q1 animals. Primary antibodies against β-tubulin and P/rds were used in combination with anti-mouse HRP for chemiluminescence detection of the signals. E, Representative blot comparing 21 DPF retina samples of either NT or Q1 retinas. Primary antibody against rhodopsin (B6–30) was used in combination with anti-mouse HRP for chemiluminescence detection of the signals originating from endogenous Xenopus rhodopsin. Molecular weight (MW) marker is in kilodaltons for both D and E. F, Quantification of immunopositive signals in D and E. NT average value was standardized to 1.00 for both P/rds (normalized to β-tubulin) and total endogenous rhodopsin, for readily comparison with groups expressing RhoQ344ter-Dend2 (Q1). For immunoblot against P/rds, the relative values were 1.00 ± 0.23 for NT and 0.52 ± 0.42 for Q1 (based on n = 6 animals; p = 0.025). For immunoblot against total endogenous rhodopsin, the relative values were 1.00 ± 0.21 for NT and 0.14 ± 0.37 for Q1 (based on n = 6 animals; p < 0.001). Scale bars, 10 μm. *p < 0.05, ***p < 0.001.
As additional tests to gauge OS loss, retinas from 21 DPF transgenic tadpoles were subjected to immunoblotting analyses using antibodies against P/rds and rhodopsin (Fig. 6D,E). The amount of P/rds, normalized to the expression levels of β-tubulin, was nearly 50% lower (p = 0.025) in Q1 retinas compared with NT retinas (Fig. 6D,F; representative blot based on n = 6 animals). The amount of endogenous Xenopus rhodopsin was > 85% lower (p < 0.001) in Q1 retinas than in NT retinas (Fig. 6E,F; n = 6 animals). While there were significant differences in OS structure and number between RhoQ344ter-Dend2 and Rho-Dend2-1D4 retinas, rod nuclei were only marginally lost in RhoQ344ter-Dend2 retinas at this stage: the average number of nuclei was only ∼10% lower in retinas expressing RhoQ344ter-Dend2 than in retinas expressing Rho-Dend2-1D4 (Table 1; p = 0.017 based on n = 6 animals). This minor decrease suggests that OS shortening and loss both precede rod cell death. These results indicate that rhodopsin mislocalization, as well as NKAα downregulation, contribute to OS shortening and loss, which are the beginning signs of retinal disease.
Pharmacological inhibition of NKAα with digoxin mimics the degenerative phenotype observed in RhoQ344ter-Dend2 retinas
To examine whether compromised NKAα activity can be causative to the shortening or loss of rod OSs, we treated nontransgenic animals (starting age 14 DPF) with digoxin, which specifically inhibits NKAα. The integrity of the OS structures was assessed by P/rds (Fig. 7A) and XAP-2 (Fig. 7B) immunofluorescence. After 3 d, retinas from digoxin-treated animals exhibited shorter and fewer OS structures than untreated animals. On average, the rod OSs of 3 d treated retinas were ∼20.0% shorter (p < 0.001; based on analyzing 96 OSs from n = 4 untreated animals and 120 OSs from n = 4 treated animals) and 30% fewer (p < 0.03; based on retinas from n = 4 animals each for treated and untreated) than those of untreated retinas. After 7 d of treatment, this effect was exacerbated. On average, the rod OSs of 7 d digoxin-treated retinas were ∼29% shorter (p < 0.001; based on analyzing 250 OSs from n = 5 untreated animals and 170 OSs from n = 5 treated animals; Table 1) and 34% fewer (p < 0.001; based on retinas from n = 5 animals each for treated and untreated; Table 1) than those of untreated retinas. The magnitude of OS shortening and loss observed in 7 d digoxin-treated animals, which were 21 DPF at the time of analysis, was similar to that seen in RhoQ344ter-Dend2 animals, which were also analyzed at 21 DPF (Table 1). Although OS structures were affected by the digoxin treatment, the number of nuclei per retina area (180 μm) remained nearly identical between treated and untreated animals for both 3 (p = 0.96; based on n = 4 animals) and 7 (p = 0.56; based on n = 5 animals) day treatments (Table 1). This result is similar to the pathogenic process of RP beginning with OS shortening and loss before the onset of rod cell death (Milam et al., 1998). Immunoblotting revealed a 30% decrease in the total endogenous rhodopsin levels in digoxin-treated (7 d) retinas (Fig. 7C), which is consistent with the occurrence of OS loss and shortening. These results suggest that compromised NKAα function, whether via pharmacological inhibition or co-degradation with RhoQ344ter-Dend2, leads to compromised rod OSs and reduced amounts of rod OS membrane proteins as represented by P/rds and rhodopsin.
Figure 7.
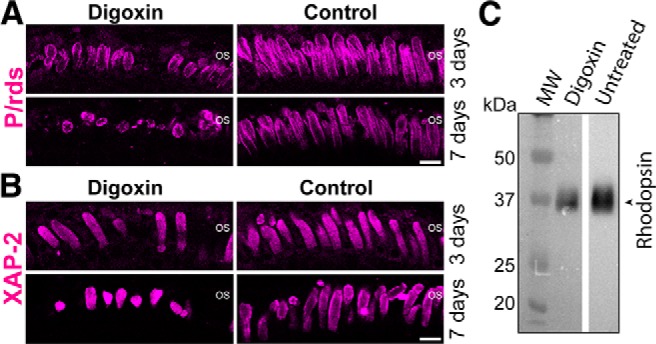
Inhibition of NKA results in disorganized rod OSs of Xenopus laevis. Xenopus tadpoles (NT) were treated with digoxin, which is a specific inhibitor of NKA. Control animals were treated with 0.1% DMSO and were housed in the same conditions. A, Immunofluorescence labeling of photoreceptor OSs using anti-P/rds after 3 or 7 d of either 3.5 μm digoxin treatment or no treatment (Control). Digoxin-treated retinas had fewer and shorter OS structures compared with control retinas; this effect was exacerbated with longer treatment time (compare 3 and 7 d). B, Immunofluorescence labeling of rod OSs using XAP-2 antibody after 3 or 7 d of digoxin treatment. Treated retinas (digoxin) had shorter and fewer OS structures than untreated retinas (Control). C, Representative immunoblot comparing endogenous rhodopsin levels between digoxin-treated and untreated (Untreated) retinas. Mean rhodopsin signal in untreated retinas was standardized to 1.00 (a.u.). The relative values were 1.00 ± 0.31 (n = 6 animals) for untreated retinas and 0.69 ± 0.14 (n = 6 animals) for digoxin-treated retinas. Molecular weight (MW) marker is in kilodaltons. Scale bars, 10 μm.
To rule out possible nonspecific effects of digoxin, we tested four additional NKA inhibitors that are structurally distinct from digoxin (Fig. 8; Table 1) and hence unlikely to share off-targets. Treatments with these four NKA inhibitors, for up to 7 d, resulted in time-dependent shortening and loss of rod OSs as observed for digoxin-treated retina (Fig. 8A–D, compare 3 and 7 d). As with the studies of digoxin, photoreceptor outer segments were visualized by P/rds and XAP-2 immunofluorescence. Ouabain is a steroidal glycoside more potent and selective than digoxin (Gable et al., 2017), whereas istaroxime is a steroidal but non-glycoside molecule (De Munari et al., 2003). After 7 d of treatment with ouabain, rod OSs were 17–23.5% (p < 0.001) shorter and 12–22.5% fewer (p < 0.05) on average compared with age-matched untreated retinas (Table 1). We also tested the effects of NKA inhibitors that are unrelated to steroidal or glycoside NKA inhibitors, and thus do not share the mechanism of action with digoxin. Wedelolactone and 3,4,5,6-tetrahydroxyxanthone hydrate (TTX) are coumestan and xanthone-based structures, respectively (Wang and O'Doherty, 2012), and do not share a structural backbone with digoxin. In inhibiting NKA, these classes of molecules demonstrate different mechanisms of action and binding to NKA (Pôças et al., 2003; Zhang et al., 2010; Wang and O'Doherty, 2012). After wedelolactone or TTX treatment, the rod OSs were 8–13.7% shorter (p = 0.043 for wedelolactone and p < 0.001 for TTX) and 23.3–25.7% fewer in number (p < 0.001) than untreated retinas. Various degrees of OS shortening and loss were observed among these inhibitors, potentially because of their different pharmacokinetics and other pharmacological properties. Nevertheless, these treatments invariably did not result in the statistically significant changes in the number of nuclei (Table 1; p > 0.05 for all untreated vs treated combinations). These experiments generally support the notion that NKA inhibition emulates the symptoms of early stage RP: OS shortening and loss without the significant loss of rod photoreceptor cells.
Figure 8.
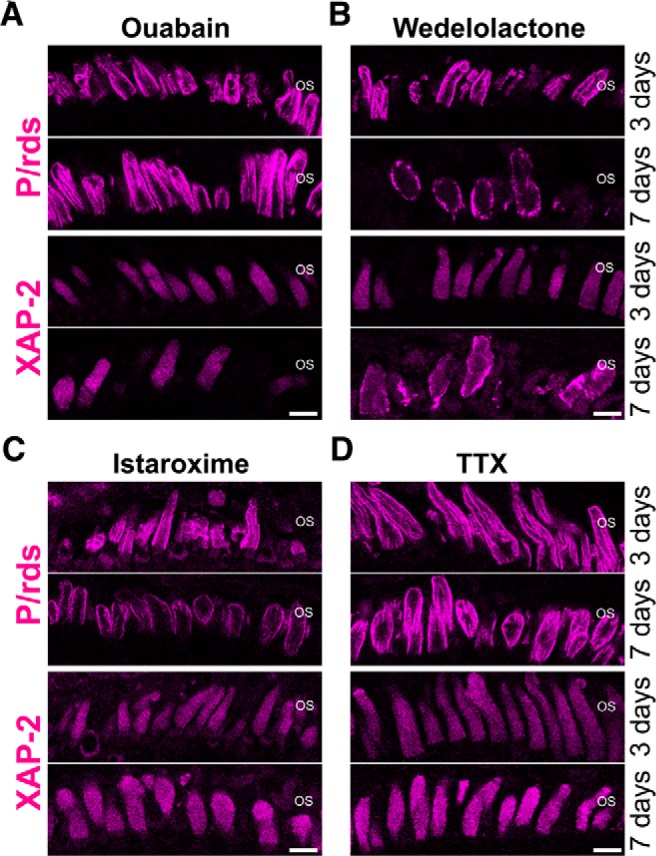
NKA inhibitors structurally distinct from digoxin induce disorganization of rod OSs. Photoreceptor OSs were labeled by anti-P/rds antibody (magenta), which binds to both cone and rod OSs, or by XAP-2 antibody, which specifically binds to rod OSs (magenta). A, Animals were treated with 1.0 μm ouabain for 3 or 7 d. B, Animals were treated with 3.5 μm wedelolactone for 3 or 7 d. C, Animals were treated with 3.0 μm istaroxime for 3 or 7 d. D, Animals were treated with 10.0 μm TTX for 3 or 7 d. Rod OSs were shorter and fewer after treatment with all the NKA inhibitors. Scale bars, 10 μm.
Discussion
By taking advantage of class I mutant rhodopsin, which is deficient in ciliary targeting, we proved a novel mechanism of photoreceptor toxicity caused by mislocalized rhodopsin in retinal ciliopathies. This paper provided evidence that mislocalized rhodopsin is continuously internalized, transported to lysosomes, and degraded. This lysosome-mediated degradation results in disruption of the protein homeostasis of the rod IS PM where class I mutant rhodopsin is locally added and removed at rapid rates. Class I mutant rhodopsin causes photoreceptor degeneration in a manner independent of rhodopsin activation by light (Tam et al., 2006; Concepcion and Chen, 2010). In this study, all the animals were reared in 24 h darkness until subjected to live microscopy experiments, and therefore served as a model for light-independent disruption of photoreceptor structure and function. Mislocalized rhodopsin induces co-degradation of NKAα, and this degradation led to severe downregulation of NKAα on the IS PM. In our experiments, the transgene is not overexpressed, and is only observed at levels between 1 and 5% of endogenous rhodopsin (Lodowski et al., 2013). Transgenic expression of wild-type Rho-Dend2-1D4 did not alter the amount of NKAα. Therefore, downregulation of NKAα was not due to overexpression of transgene in our system, but due to specific effect of class I mutant RhoQ344ter-Dend2 mislocalized to the IS PM. The effect we observed in our class I model may also be occurring in other inherited blinding disorders that exhibit rhodopsin mislocalization. Rhodopsin mislocalization is commonly observed in a subset of Joubert syndrome (Lessieur et al., 2017), Bardet–Biedl syndrome (Nishimura et al., 2004; Simons et al., 2011), as well as many other forms of retinitis pigmentosa in which rhodopsin is not the defective gene (Hagstrom et al., 2001; Gao et al., 2002; Adamian et al., 2006). Therefore, further studies are warranted to determine whether NKAα downregulation is occurring in these diseases.
Separate lines of evidence indicate that compromised NKAα function, either because of pharmacological inhibition or downregulation by mislocalized rhodopsin, triggers shortening and loss of rod OSs. This finding is significant because rhodopsin mislocalization, OS shortening, and loss of OS structures are the most commonly observed events in retinitis pigmentosa patients (Li et al., 1995; Milam et al., 1998), but their cause–effect relationships have been unclear until this study. The finding that NKAα downregulation causes photoreceptor dysfunction underscores the important role of NKAα in photoreceptor cell health and function. NKAα is expressed in high density on the IS PM of rod photoreceptors (Schneider et al., 1991; Wetzel et al., 1999). NKAα is an essential component of rod photoreceptors, and is responsible for generating the dark current and maintaining ion homeostasis within these cells (Demontis et al., 1995; Roy et al., 2013). Shortening of OSs is possibly a homeostatic mechanism rods use to maintain the current per OS plasma membrane area after the corresponding current in the IS is reduced due to NKA downregulation. In the retina of Drosophila, RNAi-mediated knockdown of NKAα resulted in loss of photoresponse and rapid degeneration of photoreceptors (Luan et al., 2014). Similarly, in the retina of mice, inhibition of NKAα by digoxin caused photoreceptor-specific cell death (Landfried et al., 2017). In our model, NKAα became downregulated because of constant co-degradation with mislocalized rhodopsin. This downregulation of NKAα resulted in shortening and loss of rod OSs; these defects were recapitulated by pharmacological inhibition of NKAα pumping function. The degree of NKAα downregulation was 83.4 ± 11.2% in rods expressing a high-level of RhoQ344ter-Dend2. This degree of downregulation was similar to the extent of NKA inhibition (∼ 80%; Gable et al., 2017) by the inhibitor concentrations (3.5 μm for digoxin and 1 μm for ouabain) introduced in this study. Thus, those studies strongly suggest that NKA downregulation induced by rhodopsin mislocalization is sufficient to cause rod OS shortening and loss. NKAα inhibitors with distinct physicochemical and pharmacological properties invariably caused rod OS loss and shortening without appreciable loss of rod photoreceptors, emulating the conditions of early stage RP patients.
In addition to maintaining ion homeostasis, an additional function of NKA proteins has been proposed: as a binding partner for retinoschisin (Molday et al., 2007), which is a photoreceptor adhesion molecule. Such adhesion is likely critical for photoreceptor–Muller glia and photoreceptor–bipolar cell interactions (Molday et al., 2007; Friedrich et al., 2011). Neurite sprouting accompanies rhodopsin mislocalization in retinitis pigmentosa (Li et al., 1995; Tam et al., 2006), which is suggestive of defective cell–cell interaction and adhesion. Therefore, unlike pharmacological inhibition of NKAα, physical removal of NKAα induced by rhodopsin mislocalization may result in additional adverse effects to the PM because of the structural roles of Na+/K+-ATPase proteins.
In retinitis pigmentosa patients, shortening and loss of OSs are observed before photoreceptor cell death and compromise their vision (Milam et al., 1996; Oh et al., 2014). Class I mutant rhodopsin, mislocalized to the IS PM, causes shortening and loss of rod OSs at the early stages of pathogenesis. Our observation that total endogenous Xenopus rhodopsin is reduced by ∼85% in RhoQ344ter-Dend2 retinas is consistent with previous finding that retinas expressing class I mutant rhodopsin had low amount of endogenous rhodopsin (Tam et al., 2006; Lodowski et al., 2013). Similar to RP patients, our Xenopus model did not demonstrate significant death of rod photoreceptors. Sustained downregulation or inhibition of NKA, however, would lead to rod photoreceptor cell death as observed for NKAβ2 knock-out mouse (Molthagen et al., 1996), digoxin-treated mouse (Landfried et al., 2017), and NKA-deficient Drosophila (Luan et al., 2014) models. In the early stages of pathogenesis, as tested in our study, the occurrence of OS loss/shortening is well-separated from the onset of cell death events. The number of nuclei per same retinal area stayed fairly consistent between retinas expressing Rho-Dend2–1D or RhoQ344ter-Dend2, and also among untreated retinas and retinas treated with NKA inhibitors, indicating that compromised NKAα function initially results in OS loss/shortening. This result is supported by our previous study in which only a nominal number of TUNEL-positive nuclei were observed in retinas expressing class I mutant rhodopsin (Lodowski et al., 2013). Therefore, our Xenopus model will be useful to understand the early pathogenesis process of human retinitis pigmentosa.
In summary, we demonstrated that mislocalized rhodopsin disrupts the PM homeostasis of rod photoreceptors through concomitant lysosome-mediated degradation of IS PM membrane protein, NKA. Lysosome-mediated degradation is associated with various photoreceptor degenerative disorders (Chen et al., 2013; Bogéa et al., 2015; Yao et al., 2018), but the context and consequences of the degradation differ among these diseases. For example, in a light-dependent photoreceptor degeneration model, activation of lysosomes and autophagy is neuroprotective (Chen et al., 2013), but in a mouse model of a class II rhodopsin gene mutation (Sakami et al., 2011, 2014), activation exacerbates the photoreceptor degeneration (Yao et al., 2018). In the case of retinal diseases caused by mislocalized rhodopsin, our study suggests lysosome-mediated degradation is both beneficial and detrimental. On one hand, lysosomes are used to remove and eliminate toxic rhodopsin species mislocalized on the PM, but on the other hand, lysosomes cause collateral damage by co-eliminating NKAα. Other than NKAα, additional IS membrane proteins, which are physiologically relevant and essential, might also be degraded together with mislocalized rhodopsin and therefore downregulated on the IS PM. Thus, in relation to rhodopsin mislocalization, additional global consequences of rhodopsin degradation to the photoreceptor proteome should be characterized to fully comprehend the mechanisms of retinal ciliopathies.
Footnotes
This work was supported by the U.S. National Institutes of Health (NIH) Grants R01EY028884, R21EY027292, and T32EY007157. We thank Dr. Sheila Baker (University of Iowa) for the plasmid vector containing GFP-NKAα, Dr. Robert Molday (University of British Columbia) for the anti-peripherin/rds antibody (anti-xrds 2D4), and Dr. David Salom (Case Western Reserve University) for the anti-rhodopsin B6–30 monoclonal antibody. The monoclonal antibodies against Na+/K+ ATPase α subunit (deposited by Douglas Fambrough, Johns Hopkins University), the monoclonal antibody against β tubulin (deposited by Michael Klymkowsky, University of Colorado), and the XAP-2 monoclonal antibody (deposited by D.S. Sakaguchi and W.A. Harris, University of California) were obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at the Department of Biology of the University of Iowa, Iowa City. The DNA vector containing PSmOrange, deposited by Vladislav Verkhusha, was obtained from Addgene. Confocal microscopy using a Leica TCS SP2 MP was conducted at the light microscopy imaging core facility of Case Western Reserve University.
The authors declare no competing financial interests.
References
- Adamian M, Pawlyk BS, Hong DH, Berson EL (2006) Rod and cone opsin mislocalization in an autopsy eye from a carrier of X-linked retinitis pigmentosa with a Gly436Asp mutation in the RPGR gene. Am J Ophthalmol 142:515–518. 10.1016/j.ajo.2006.03.061 [DOI] [PubMed] [Google Scholar]
- Adams NA, Awadein A, Toma HS (2007) The retinal ciliopathies. Ophthalmic Genet 28:113–125. 10.1080/13816810701537424 [DOI] [PubMed] [Google Scholar]
- Alfinito PD, Townes-Anderson E (2002) Activation of mislocalized opsin kills rod cells: a novel mechanism for rod cell death in retinal disease. Proc Natl Acad Sci U S A 99:5655–5660. 10.1073/pnas.072557799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arikawa K, Molday LL, Molday RS, Williams DS (1992) Localization of peripherin/rds in the disk membranes of cone and rod photoreceptors: relationship to disk membrane morphogenesis and retinal degeneration. J Cell Biol 116:659–667. 10.1083/jcb.116.3.659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besharse JC, Hollyfield JG, Rayborn ME (1977) Turnover of rod photoreceptor outer segments: II. Membrane addition and loss in relationship to light. J Cell Biol 75:507–527. 10.1083/jcb.75.2.507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogéa TH, Wen RH, Moritz OL (2015) Light induces ultrastructural changes in rod outer and inner segments, including autophagy, in a transgenic Xenopus laevis P23H rhodopsin model of retinitis pigmentosa. Invest Ophthalmol Vis Sci 56:7947–7955. 10.1167/iovs.15-16799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Sawada O, Kohno H, Le YZ, Subauste C, Maeda T, Maeda A (2013) Autophagy protects the retina from light-induced degeneration. J Biol Chem 288:7506–7518. 10.1074/jbc.M112.439935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi RY, Engbretson GA, Solessio EC, Jones GA, Coughlin A, Aleksic I, Zuber ME (2011) Cone degeneration following rod ablation in a reversible model of retinal degeneration. Invest Ophthalmol Vis Sci 52:364–373. 10.1167/iovs.10-5347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu DT, Klymkowsky MW (1989) The appearance of acetylated alpha-tubulin during early development and cellular differentiation in Xenopus. Dev Biol 136:104–117. 10.1016/0012-1606(89)90134-6 [DOI] [PubMed] [Google Scholar]
- Concepcion F, Chen J (2010) Q344ter mutation causes mislocalization of rhodopsin molecules that are catalytically active: a mouse model of Q344ter-induced retinal degeneration. PLoS One 5:e10904. 10.1371/journal.pone.0010904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis GC, Ratto GM, Bisti S, Cervetto L (1995) Effect of blocking the Na+/K+ ATPase on Ca2+ extrusion and light adaptation in mammalian retinal rods. Biophys J 69:439–450. 10.1016/S0006-3495(95)79917-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Munari S, Cerri A, Gobbini M, Almirante N, Banfi L, Carzana G, Ferrari P, Marazzi G, Micheletti R, Schiavone A, Sputore S, Torri M, Zappavigna MP, Melloni P (2003) Structure-based design and synthesis of novel potent Na+, K+-ATPase inhibitors derived from a 5α,14α-androstane scaffold as positive inotropic compounds. J Med Chem 46:3644–3654. 10.1021/jm030830y [DOI] [PubMed] [Google Scholar]
- Deretic D, Williams AH, Ransom N, Morel V, Hargrave PA, Arendt A (2005) Rhodopsin C terminus, the site of mutations causing retinal disease, regulates trafficking by binding to ADP-ribosylation factor 4 (ARF4). Proc Natl Acad Sci U S A 102:3301–3306. 10.1073/pnas.0500095102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edward DP, Lim K, Sawaguchi S, Tso MO (1993) An immunohistochemical study of opsin in photoreceptor cells following light-induced retinal degeneration in the rat. Graefes Arch Clin Exp Ophthalmol 231:289–294. 10.1007/BF00919107 [DOI] [PubMed] [Google Scholar]
- Fariss RN, Molday RS, Fisher SK, Matsumoto B (1997) Evidence from normal and degenerating photoreceptors that two outer segment integral membrane proteins have separate transport pathways. J Comp Neurol 387:148–156. [DOI] [PubMed] [Google Scholar]
- Fliegauf M, Benzing T, Omran H (2007) When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol 8:880–893. 10.1038/nrm2278 [DOI] [PubMed] [Google Scholar]
- Friedrich U, Stöhr H, Hilfinger D, Loenhardt T, Schachner M, Langmann T, Weber BH (2011) The Na/K-ATPase is obligatory for membrane anchorage of retinoschisin, the protein involved in the pathogenesis of X-linked juvenile retinoschisis. Hum Mol Genet 20:1132–1142. 10.1093/hmg/ddq557 [DOI] [PubMed] [Google Scholar]
- Gable ME, Ellis L, Fedorova OV, Bagrov AY, Askari A (2017) Comparison of digitalis sensitivities of Na(+)/K(+)-ATPases from human and pig kidneys. ACS Omega 2:3610–3615. 10.1021/acsomega.7b00591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Cheon K, Nusinowitz S, Liu Q, Bei D, Atkins K, Azimi A, Daiger SP, Farber DB, Heckenlively JR, Pierce EA, Sullivan LS, Zuo J (2002) Progressive photoreceptor degeneration, outer segment dysplasia, and rhodopsin mislocalization in mice with targeted disruption of the retinitis pigmentosa-1 (Rp1) gene. Proc Natl Acad Sci U S A 99:5698–5703. 10.1073/pnas.042122399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagstrom SA, Adamian M, Scimeca M, Pawlyk BS, Yue G, Li T (2001) A role for the tubby-like protein 1 in rhodopsin transport. Invest Ophthalmol Vis Sci 42:1955–1962. [PubMed] [Google Scholar]
- Koch SF, Tsai YT, Duong JK, Wu WH, Hsu CW, Wu WP, Bonet-Ponce L, Lin CS, Tsang SH (2015) Halting progressive neurodegeneration in advanced retinitis pigmentosa. J Clin Invest 125:3704–3713. 10.1172/JCI82462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird JG, Pan Y, Modestou M, Yamaguchi DM, Song H, Sokolov M, Baker SA (2015) Identification of a VxP targeting signal in the flagellar Na+ /K+-ATPase. Traffic 16:1239–1253. 10.1111/tra.12332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landfried B, Samardzija M, Barben M, Schori C, Klee K, Storti F, Grimm C (2017) Digoxin-induced retinal degeneration depends on rhodopsin. Cell Death Dis 8:e2670. 10.1038/cddis.2017.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebovitz RM, Takeyasu K, Fambrough DM (1989) Molecular characterization and expression of the (Na+ + K+)-ATPase alpha-subunit in Drosophila melanogaster. EMBO J 8:193–202. 10.1002/j.1460-2075.1989.tb03364.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessieur EM, Fogerty J, Gaivin RJ, Song P, Perkins BD (2017) The ciliopathy gene ahi1 is required for zebrafish cone photoreceptor outer segment morphogenesis and survival. Invest Ophthalmol Vis Sci 58:448–460. 10.1167/iovs.16-20326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZY, Kljavin IJ, Milam AH (1995) Rod photoreceptor neurite sprouting in retinitis pigmentosa. J Neurosci 15:5429–5438. 10.1523/JNEUROSCI.15-08-05429.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodowski KH, Imanishi Y (2015) Monitoring of rhodopsin trafficking and mistrafficking in live photoreceptors. Methods Mol Biol 1271:293–307. 10.1007/978-1-4939-2330-4_19 [DOI] [PubMed] [Google Scholar]
- Lodowski KH, Lee R, Ropelewski P, Nemet I, Tian G, Imanishi Y (2013) Signals governing the trafficking and mistrafficking of a ciliary GPCR, rhodopsin. J Neurosci 33:13621–13638. 10.1523/JNEUROSCI.1520-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan Z, Reddig K, Li HS (2014) Loss of Na+/K+-ATPase in Drosophila photoreceptors leads to blindness and age-dependent neurodegeneration. Exp Neurol 261:791–801. 10.1016/j.expneurol.2014.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanty A, Purohit GK, Mohanty S, Nayak NR, Mohanty BP (2017) Suitable reference gene for quantitative real-time PCR analysis of gene expression in gonadal tissues of minnow Puntius sophore under high-temperature stress. BMC Genomics 18:617. 10.1186/s12864-017-3974-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SS, Batni S, Whitaker L, Chen S, Engbretson G, Knox BE (2001) Xenopus rhodopsin promoter: identification of immediate upstream sequences necessary for high level, rod-specific transcription. J Biol Chem 276:36557–36565. 10.1074/jbc.M101685200 [DOI] [PubMed] [Google Scholar]
- Milam AH, Li ZY, Cideciyan AV, Jacobson SG (1996) Clinicopathologic effects of the Q64ter rhodopsin mutation in retinitis pigmentosa. Invest Ophthalmol Vis Sci 37:753–765. [PubMed] [Google Scholar]
- Milam AH, Li ZY, Fariss RN (1998) Histopathology of the human retina in retinitis pigmentosa. Prog Retin Eye Res 17:175–205. 10.1016/S1350-9462(97)00012-8 [DOI] [PubMed] [Google Scholar]
- Molday LL, Wu WW, Molday RS (2007) Retinoschisin (RS1), the protein encoded by the X-linked retinoschisis gene, is anchored to the surface of retinal photoreceptor and bipolar cells through its interactions with a Na/K ATPase-SARM1 complex. J Biol Chem 282:32792–32801. 10.1074/jbc.M706321200 [DOI] [PubMed] [Google Scholar]
- Molthagen M, Schachner M, Bartsch U (1996) Apoptotic cell death of photoreceptor cells in mice deficient for the adhesion molecule on glia (AMOG, the β2-subunit of the Na,K-ATPase). J Neurocytol 25:243–255. 10.1007/BF02284800 [DOI] [PubMed] [Google Scholar]
- Moritz OL, Tam BM, Papermaster DS, Nakayama T (2001) A functional rhodopsin-green fluorescent protein fusion protein localizes correctly in transgenic Xenopus laevis retinal rods and is expressed in a time-dependent pattern. J Biol Chem 276:28242–28251. 10.1074/jbc.M101476200 [DOI] [PubMed] [Google Scholar]
- Nathans J. (1992) Rhodopsin: structure, function, and genetics. Biochemistry 31:4923–4931. 10.1021/bi00136a001 [DOI] [PubMed] [Google Scholar]
- Nemet I, Tian G, Imanishi Y (2014) Submembrane assembly and renewal of rod photoreceptor cGMP-gated channel: insight into the actin-dependent process of outer segment morphogenesis. J Neurosci 34:8164–8174. 10.1523/JNEUROSCI.1282-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemet I, Ropelewski P, Imanishi Y (2015a) Applications of phototransformable fluorescent proteins for tracking the dynamics of cellular components. Photochem Photobiol Sci 14:1787–1806. 10.1039/C5PP00174A [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemet I, Ropelewski P, Imanishi Y (2015b) Rhodopsin trafficking and mistrafficking: signals, molecular components, and mechanisms. Prog Mol Biol Transl Sci 132:39–71. 10.1016/bs.pmbts.2015.02.007 [DOI] [PubMed] [Google Scholar]
- Nishimura DY, Fath M, Mullins RF, Searby C, Andrews M, Davis R, Andorf JL, Mykytyn K, Swiderski RE, Yang B, Carmi R, Stone EM, Sheffield VC (2004) Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci U S A 101:16588–16593. 10.1073/pnas.0405496101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Kim SH, Kim YJ, Lee H, Cho JH, Cho YH, Kim CK, Lee TJ, Lee S, Park KH, Yu HG, Lee HJ, Jun SC, Kim JH (2014) Detection of retinitis pigmentosa by differential interference contrast microscopy. PLoS One 9:e97170. 10.1371/journal.pone.0097170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K. (2006) G protein-coupled receptor rhodopsin. Annu Rev Biochem 75:743–767. 10.1146/annurev.biochem.75.103004.142743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierzyńska-Mach A, Janowski PA, Dobrucki JW (2014) Evaluation of acridine orange, LysoTracker red, and quinacrine as fluorescent probes for long-term tracking of acidic vesicles. Cytometry A 85:729–737. 10.1002/cyto.a.22495 [DOI] [PubMed] [Google Scholar]
- Pôças ES, Costa PR, da Silva AJ, Noël F (2003) 2-methoxy-3,8,9-trihydroxy coumestan: a new synthetic inhibitor of Na+,K+-ATPase with an original mechanism of action. Biochem Pharmacol 66:2169–2176. 10.1016/j.bcp.2003.08.005 [DOI] [PubMed] [Google Scholar]
- Roy M, Sivan-Loukianova E, Eberl DF (2013) Cell-type-specific roles of Na+/K+ ATPase subunits in Drosophila auditory mechanosensation. Proc Natl Acad Sci U S A 110:181–186. 10.1073/pnas.1208866110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakami S, Maeda T, Bereta G, Okano K, Golczak M, Sumaroka A, Roman AJ, Cideciyan AV, Jacobson SG, Palczewski K (2011) Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J Biol Chem 286:10551–10567. 10.1074/jbc.M110.209759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakami S, Kolesnikov AV, Kefalov VJ, Palczewski K (2014) P23H opsin knock-in mice reveal a novel step in retinal rod disc morphogenesis. Hum Mol Genet 23:1723–1741. 10.1093/hmg/ddt561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider BG, Shyjan AW, Levenson R (1991) Co-localization and polarized distribution of Na,K-ATPase α3 and β2 subunits in photoreceptor cells. J Histochem Cytochem 39:507–517. 10.1177/39.4.1848572 [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons DL, Boye SL, Hauswirth WW, Wu SM (2011) Gene therapy prevents photoreceptor death and preserves retinal function in a Bardet–Biedl syndrome mouse model. Proc Natl Acad Sci U S A 108:6276–6281. 10.1073/pnas.1019222108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SJ, Fairclough L, Latinkic BV, Sparrow DB, Mohun TJ (2006) Xenopus laevis transgenesis by sperm nuclear injection. Nat Protoc 1:2195–2203. 10.1038/nprot.2006.325 [DOI] [PubMed] [Google Scholar]
- Sparrow DB, Latinkic B, Mohun TJ (2000) A simplified method of generating transgenic Xenopus. Nucleic Acids Res 28:E12. 10.1093/nar/28.4.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subach OM, Patterson GH, Ting LM, Wang Y, Condeelis JS, Verkhusha VV (2011) A photoswitchable orange-to-far-red fluorescent protein, PSmOrange. Nat Methods 8:771–777. 10.1038/nmeth.1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CH, Davenport CM, Nathans J (1993) Rhodopsin mutations responsible for autosomal dominant retinitis pigmentosa: clustering of functional classes along the polypeptide chain. J Biol Chem 268:26645–26649. [PubMed] [Google Scholar]
- Sung CH, Makino C, Baylor D, Nathans J (1994) A rhodopsin gene mutation responsible for autosomal dominant retinitis pigmentosa results in a protein that is defective in localization to the photoreceptor outer segment. J Neurosci 14:5818–5833. 10.1523/JNEUROSCI.14-10-05818.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam BM, Moritz OL, Hurd LB, Papermaster DS (2000) Identification of an outer segment targeting signal in the COOH terminus of rhodopsin using transgenic Xenopus laevis. J Cell Biol 151:1369–1380. 10.1083/jcb.151.7.1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam BM, Xie G, Oprian DD, Moritz OL (2006) Mislocalized rhodopsin does not require activation to cause retinal degeneration and neurite outgrowth in Xenopus laevis. J Neurosci 26:203–209. 10.1523/JNEUROSCI.3849-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G, Ropelewski P, Nemet I, Lee R, Lodowski KH, Imanishi Y (2014) An unconventional secretory pathway mediates the cilia targeting of peripherin/rds. J Neurosci 34:992–1006. 10.1523/JNEUROSCI.3437-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viczian AS, Solessio EC, Lyou Y, Zuber ME (2009) Generation of functional eyes from pluripotent cells. PLoS Biol 7:e1000174. 10.1371/journal.pbio.1000174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, O'Doherty GA (2012) Modulators of Na/K-ATPase: a patent review. Expert Opin Ther Pat 22:587–605. 10.1517/13543776.2012.690033 [DOI] [PubMed] [Google Scholar]
- Wetzel RK, Arystarkhova E, Sweadner KJ (1999) Cellular and subcellular specification of Na,K-ATPase α and β isoforms in the postnatal development of mouse retina. J Neurosci 19:9878–9889. 10.1523/JNEUROSCI.19-22-09878.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, D'Antona AM, Edwards PC, Fransen M, Standfuss J, Schertler GF, Oprian DD (2011) Preparation of an activated rhodopsin/transducin complex using a constitutively active mutant of rhodopsin. Biochemistry 50:10399–10407. 10.1021/bi201126r [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y (1998) Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 23:33–42. 10.1247/csf.23.33 [DOI] [PubMed] [Google Scholar]
- Yao J, Qiu Y, Frontera E, Jia L, Khan NW, Klionsky DJ, Ferguson TA, Thompson DA, Zacks DN (2018) Inhibiting autophagy reduces retinal degeneration caused by protein misfolding. Autophagy 14:1226–1238. 10.1080/15548627.2018.1463121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau KW, Baylor DA (1989) Cyclic GMP-activated conductance of retinal photoreceptor cells. Annu Rev Neurosci 12:289–327. 10.1146/annurev.ne.12.030189.001445 [DOI] [PubMed] [Google Scholar]
- Young RW. (1967) The renewal of photoreceptor cell outer segments. J Cell Biol 33:61–72. 10.1083/jcb.33.1.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Li Z, Tian J, Jiang W, Wang Y, Zhang X, Li Z, You Q, Shapiro JI, Si S, Xie Z (2010) Identification of hydroxyxanthones as Na/K-ATPase ligands. Mol Pharmacol 77:961–967. 10.1124/mol.110.063974 [DOI] [PMC free article] [PubMed] [Google Scholar]