

Abstract
Sleep deprivation (SD) interferes with hippocampal structural and functional plasticity, formation of long-term memory and cognitive function. The molecular mechanisms underlying these effects are incompletely understood. Here, we show that SD impaired synaptic tagging and capture and behavioral tagging, two major mechanisms of associative learning and memory. Strikingly, mutant male mice lacking the p75 neurotrophin receptor (p75NTR) were resistant to the detrimental effects of SD on hippocampal plasticity at both cellular and behavioral levels. Mechanistically, SD increased p75NTR expression and its interaction with phosphodiesterase. p75NTR deletion preserved hippocampal structural and functional plasticity by preventing SD-mediated effects on hippocampal cAMP–CREB-BDNF, cAMP-PKA-LIMK1-cofilin, and RhoA-ROCK2 pathways. Our study identifies p75NTR as an important mediator of hippocampal structural and functional changes associated with SD, and suggests that targeting p75NTR could be a promising strategy to limit the memory and cognitive deficits that accompany sleep loss.
SIGNIFICANCE STATEMENT The lack of sufficient sleep is a major health concern in today's world. Sleep deprivation (SD) affects cognitive functions such as memory. We have investigated how associative memory mechanisms, synaptic tagging and capture (STC), was impaired in SD mice at cellular and behavioral level. Interestingly, mutant male mice that lacked the p75 neurotrophin receptor (p75NTR) were seen to be resistant to the SD-induced impairments in hippocampal synaptic plasticity and STC. Additionally, we elucidated the molecular pathways responsible for this rescue of plasticity in the mutant mice. Our study has thus identified p75NTR as a promising target to limit the cognitive deficits associated with SD.
Keywords: late-LTP, metaplasticity, p75NTR, sleep deprivation, synaptic capture, synaptic tagging
Introduction
Sleep deprivation (SD) is particularly prevalent in modern societies where many sacrifice sleep to meet work demands and social commitments. Insufficient sleep affects cognitive and emotional functions (Banks and Dinges, 2007; Rasch and Born, 2013). SD is known to impact several psychiatric disorders such as schizophrenia, anxiety, addiction disorders, and also neurodegenerative diseases such as Alzheimer's disease (Wulff et al., 2010; Krause et al., 2017; Shokri-Kojori et al., 2018). One of the most profound effects of SD is the marked impairment of learning and memory, reflected in the alterations of synaptic plasticity and function (Havekes et al., 2012; Abel et al., 2013). Among the various synaptic plasticity mechanisms, long-term potentiation (LTP) is considered to be the cellular correlate of learning and memory (Bliss and Lømo, 1973). The late-associative properties of LTP, such as synaptic tagging and capture (STC), provide a cellular basis for associative plasticity (Frey and Morris, 1997; Martin et al., 2000). In STC, interaction between a synaptic tag and plasticity related products (PRPs) within a specific time interval leads to long-lasting plasticity in weakly tetanized synaptic inputs. The synaptic tag is created by a specific type of weak stimulation and PRPs are generated because of strong stimulus in two separate and independent synaptic inputs of the same neuronal population (Frey and Morris, 1997; Ballarini et al., 2009; Redondo and Morris, 2011). Several molecules have been implicated in this process, including Ca2+/calmodulin-dependent protein kinase II and IV (CaMKII, CaMKIV), protein kinase M ζ (PKMζ), and brain-derived neurotrophic factor (BDNF; Redondo and Morris, 2011). BDNF acts as an important PRP, whereas its receptor Tropomyosin receptor kinase B (TrkB) plays a critical role in setting the synaptic tag (Lu et al., 2011). Long-term plasticity and memory are known to be affected in the SD brain, with accompanying functional and structural changes in neuronal circuitry (Campbell et al., 2002; McDermott et al., 2003; Guan et al., 2004; Vecsey et al., 2009; Havekes et al., 2016b). Hippocampal-dependent synaptic plasticity is particularly vulnerable to the lack of sleep (Vecsey et al., 2009). Understanding how sleep affects synaptic plasticity and efficacy is critical to unravel the contribution of sleep to cognition.
The p75 neurotrophin receptor (p75NTR) is a low-affinity receptor that can bind to different neurotrophic factors, including nerve growth factor, neurotrophin-3, neurotrophin-4, and BDNF (Dechant and Barde, 2002; Roux and Barker, 2002; Chao, 2003; Gentry et al., 2004; Underwood and Coulson, 2008). Some of the intracellular signaling pathways regulated by p75NTR in response to neurotrophins binding include NF-kB, c-Jun kinase, and various caspases pathways (Carter et al., 1996; Yoon et al., 1998; Friedman, 2000; Troy et al., 2002). p75NTR plays an important role in controlling the survival mechanisms of neurons during early neuronal development (Dechant and Barde, 2002; Lu et al., 2005; Ibáñez and Simi, 2012). In the adult nervous system, p75NTR is re-expressed under different pathological conditions, including epilepsy, axotomy, and neurodegeneration (Dechant and Barde, 2002), where it can contribute to neuronal death and axonal degeneration and dysfunction (Ibáñez and Simi, 2012). In addition, p75NTR has been shown to be involved in the modulation of synaptic transmission and plasticity, specifically in the facilitation of long-term depression (LTD; Rösch et al., 2005; Woo et al., 2005), a mechanism that opposes LTP, leading to reduced synaptic efficacy (Bliss and Cooke, 2011). The loss of p75NTR in mutant mice has no effect on LTP, but impairs hippocampal LTD (Rösch et al., 2005). p75NTR is important in maintaining homeostasis for hippocampal-dependent synaptic plasticity and efficacy (Rösch et al., 2005; Woo et al., 2005). Interestingly, p75NTR has been shown to act as a regulator of circadian clock oscillation (Baeza-Raja et al., 2013). However, its potential role in SD and its effects on synaptic plasticity and cognition have not been investigated. Here, we used genetically modified p75NTR knock-out (KO) mice to study the potential role of this receptor in the synaptic changes resulting from SD. Our data demonstrate that long-term plasticity, associative plasticity, and long-term memory are resistant to the detrimental effects of SD in p75NTR KO mice. We also provide mechanistic insights into the molecular processes that contribute to these effects.
Materials and Methods
Animals and SD.
A total of 84 male C57BL/6J wild-type (WT) and 72 male p75NTR KO 5–7 weeks mice were used for experiments in this study. p75NTR KO mice (exon III; Lee et al., 1992) were obtained from The Jackson Laboratory and back crossed to the C57BL/6J background for at least 10 generations. The animals were housed in standard cages and maintained on a 12 h light/dark cycle (light from 7:00 A.M. to 7:00 P.M.) with food and water ad libitum. All experiments were approved by the Institutional Animal Care and Use Committees at the National University of Singapore and can be produced upon request. For SD procedure, the mice were subjected to 5 h SD (from 7:30 A.M. to 12:30 P.M.) by gentle handling technique before the experiments (Ledoux et al., 1996; van der Borght et al., 2006; Vecsey et al., 2009). In this method, mice were kept awake by tapping or gentle shaking of the cage, or by direct contact with the animal either through a soft brush or by hand if they were not responding to earlier disturbances (Ledoux et al., 1996; van der Borght et al., 2006; Vecsey et al., 2009). Control mice, however, were left totally undisturbed until experiments.
Electrophysiology.
The mice were decapitated after anesthetization using CO2 and the brains were quickly removed and placed into cold (4°C) artificial CSF (ACSF). The ACSF contained the following (in mm): 124 NaCl, 3.7 KCl, 1.0 MgSO4.7H2O, 2.5 CaCl2.2H2O, 1.2 KH2PO4, 24.6 NaHCO3, and 10 d-glucose, equilibrated with 95% O2–5% CO2 (carbogen; total consumption 16 L/h; Shetty et al., 2015). From each mouse, 6–8 transverse hippocampal slices (400 μm thick) were prepared from the right hippocampus by using a manual tissue chopper. The slices were incubated at 32°C in an interface chamber (Scientific System Design) at an ACSF flow rate of 1 ml/min. In all the electrophysiological recordings, two-pathway experiments were performed. Two monopolar, lacquer-coated stainless steel electrodes (5 MΩ; AM Systems) were positioned at an adequate distance within the stratum radiatum of the CA1 region for stimulating two independent synaptic inputs, S1 and S2, of a neuronal population, thus evoking field EPSP (fEPSP) from Schaffer collateral–commissural–CA1 synapses. Pathway specificity was tested using the paired pulse facilitation protocol described by Sajikumar and Korte (2011) and Li et al. (2014). For recording, one electrode was placed in the CA1 apical dendritic layer. The signals were amplified by a differential amplifier (Model 1700, AM Systems) and were digitized using a CED 1401 analog-to-digital converter (Cambridge Electronic Design). After the pre-incubation period of 3 h, an input/output curve (afferent stimulation vs fEPSP slope) was plotted before experiments. To set the test stimulus intensity, a fEPSP of 40% of its maximal amplitude was determined. Biphasic constant current pulses were used for stimulation. Late-LTP was induced using three stimulus trains of 100 pulses strong tetanus (STET), 100 Hz; duration, 0.2 ms/polarity; intertrain interval, 10 min. A weak tetanization (WTET) protocol consisting of one 100 Hz train (21 biphasic constant-current pulses; pulse duration per half-wave, 0.2 ms) was used to induce early-LTP. The slopes of fEPSPs were monitored online and a baseline was recorded for 30 min. For baseline recording and testing at each time point, four 0.2 Hz biphasic constant-current pulses (0.1 ms/polarity) were used. More details about the preparation, incubation and electrophysiological procedures were described by Shetty et al. (2015).
Western blotting.
Whole hippocampi were isolated, snap frozen in liquid nitrogen and stored at −80°C. The tissues were then lysed in Tissue Protein Extraction reagent (T-PER; ThermoFisher Scientific) with protease and phosphatase inhibitor (ThermoFisher Scientific), followed by centrifugation for 5 min at 10,000 rpm at 4°C. The protein concentration was determined using Bradford protein assay (Bio-Rad). The appropriate protein concentration was determined and the protein was added to the sample buffer, heated at 95°C for 10 min before loading onto SDS-polyacrylamide gels. The gels were then transferred to PVDF membranes (Bio-Rad) in a semidry transfer cell (Bio-Rad) for 1 h at 15 V. Membranes were incubated with 5% w/v dry milk or bovine serum albumin in 1× TBST and immunoblotted with primary antibodies against phosphodiesterase (PDE4A5; 1:500; Abcam), p75NTR (1:1000, Neuromics), total or phospho-ERK (1:1000; Cell Signaling Technology), total or phospho-CREB (1:1000; Cell Signaling Technology), RhoA (1:1000, Abcam), ROCK2 (1:1000; Cell Signaling Technology), total LIMK1 (1:1000; Cell Signaling Technology), phospho-LIMK1 (1:1000; Abcam), total or phospho-cofilin (1:1000; Cell Signaling Technology), and tubulin (1:20,000; Sigma-Aldrich) followed by secondary peroxidase-conjugated antibodies directed against mouse (tubulin), rabbit (ERK, CREB, RhoA, ROCK2, LIMK1, and cofilin), and goat (p75NTR). Signals were generated by using the SuperSignal West Pico Chemiluminescent Substrate (ThermoFisher Scientific). The amount of protein was quantified by densitometric measurement of Western blots using ImageJ (NIH software). The densitometric values of each blot were normalized to the amounts of tubulin, which served as loading control, and were calculated in relation to the control group.
Immunoprecipitation.
Hippocampi were microdissected post-euthanasia and snap-frozen. Snap-frozen tissue was lysed using a bead homogenizer (Bertin Instruments) in lysis buffer containing the following (in mm): 60 octyl-β-glucoside, 50 Tris-HCl, pH 7.6, 1 EDTA, 150 NaCl, 270 sucrose, and 0.5% Triton X-100 in the presence of protease and phosphatase inhibitors (Nacalai Tesque). The tissue extracts were then centrifuged at 10,000 × g and the pellet discarded. Protein concentrations were determined by Bradford protein assay (Bio-Rad). One milligram lysates were incubated with 0.8 μg anti-p75NTR (extracellular domain) antibody (Alomone, ANT-007) or anti-PDE4A5 antibody (Abcam) overnight, followed by Protein G Fast Flow Sepharose beads (GE Healthcare) for 2 h. The beads were collected and extensively washed in lysis buffer excluding octyl-β-glucoside, before the addition of Laemmli sample buffer and analysis by SDS-PAGE and Western blotting.
ELISA.
The cAMP levels in the mouse hippocampal samples were determined using a commercially available ELISA kit (Enzo Life Sciences) in accordance with the manufacturer's instructions. In brief, standards and samples were added to wells coated with an IgG antibody. A cAMP conjugated to alkaline phosphatase was then added, followed by a rabbit polyclonal antibody against cAMP. The antibody binds to cAMP in the sample or to the conjugate in a competitive manner. The plate was washed, leaving only bound cAMP. pNpp substrate solution was added and produced a yellow color when catalyzed by the alkaline phosphatase on the cAMP conjugate. The stop solution was then added and the yellow color was read at 405 nm. The amount of signal present is indirectly proportional to the amount of cAMP in the sample. The BDNF levels in the mouse hippocampal samples were determined using a commercially available ELISA kit (Biosensis) in accordance with the manufacturer's instructions. In brief, standards and samples were added to wells precoated with a rabbit anti-BDNF polyclonal antibody and then followed by a biotinylated anti-BDNF detection antibody and horseradish peroxidase-conjugated streptavidin. The plate was sequentially added with the TMB substrate, which yields a colored reaction product and read at 450 nm. The amount of signal present is directly proportional to the concentration of BDNF present in the samples and standards.
Immunohistochemistry.
Mice were deeply anesthetized and intracardially perfused with cold PBS followed by 4% formaldehyde (Sigma-Aldrich). Brains were dissected and postfixed for 16 h at 4°C. After cryoprotection in 40% sucrose for 16 h, 40 μm coronal sections were collected on a cryostat. The sections were then blocked in 5% donkey serum in TBS before incubation with primary antibodies against RhoA, ROCK2, LIMK1, and cofilin (Abcam), followed by fluorophore-conjugated secondary antibodies (Invitrogen). Coverslips were mounted onto microscopy slides using ProLong Diamond anti-fade mounting media (Invitrogen). Confocal laser scanning microscopy was performed on a SP8 confocal microscope (Leica).
Golgi staining.
To characterize neuronal processes and spines, Golgi staining was performed using the superGolgi Kit (Bioenno) as per the manufacturer's specifications. Briefly, after deep anesthesia, mice were intracardially perfused with saline. Brains were removed from the mice and immersed in Golgi-Cox solution for 10 d at 4°C and then transferred into the post-impregnation buffer, and sectioned with a vibratome at a thickness of 200 μm. Slices were mounted, stained, and developed as per the manufacturer's instructions. Microscopy images of the CA1 pyramidal layers of the hippocampus were captured using a 63× oil-immersion objective on a SP8 confocal microscope (Leica) using confocal reflection microscopy for 3D visualization. The number of dendritic spines were quantified by manual counting along consecutive 100 μm sections within each image and tabulated.
Home cage and sleep monitoring.
Home cage activity including sleep pattern and amount of time spent for sleeping from WT and p75NTR KO mice was assessed by HomeCageScan (CleverSys). This system enables studying unconstrained mice sleeping behaviors in a home cage for extended periods of time (24 h in this study). Sleep time (seconds) across 24 h during both light-on and light-off periods was compared between WT and p75NTR KO mice.
Behavioral tagging study.
The mice were placed in an open field (OF) for 10 min within 1 h before weak inhibitory avoidance (IA) training. The OF is a plastic box with the dimension of 35 (width) × 35 (length) × 35 cm (height). A weak IA training, which induces short-term memory can be consolidated into long-term memory by an exploration to a novel environment occurring close in time to the training session (Moncada and Viola, 2006, 2007; Moncada et al., 2015). Because the hippocampus is important in detecting and responding to new stimuli (Knight, 1996; Grunwald et al., 1998; Moncada and Viola, 2006), the effects of the interaction between the spatial novel exploration and the IA training in the mice hippocampus is a suitable behavioral tagging study. The IA apparatus is a 50 (width) × 25 (height) × 25 cm (length) Plexiglas box with a 5 (height) × 8 (width) × 25 cm (length) platform on the left end of a series of bars, which constitutes the floor of the box. During the training session, mice were placed on the platform facing the left rear corner of the box. When they stepped down, putting their four paws on the bars, they received a weak foot shock (0.3 mA, 2 s). After which, they were removed from the box and returned to their home cage. As a control experiment, the mice were also tested with weak IA without OF. Memory was measured by comparing the step-down latency in the training session to that in the test session. The cutoff time for step-down latency was 4 min. A high percentage of step-down latency indicates that the animal has better associative memory. Memory was tested at three different time points: 1 h, 24 h, and 7 d after training sessions.
Pharmacology.
TrkB/Fc chimera human recombinant (TrkB/Fc; 688-TK, R&D Systems) was dissolved in sterile PBS and stored at −20°C. The stocks were stored for not more than a week. The cell-permeable p75NTR signaling inhibitor TAT-Pep5 (Calbiochem) was dissolved in DMSO and stored at −20°C. The stocks were stored for not more than a month. Just before application, the stocks were diluted to the final concentration in ACSF and bubbled with carbogen to be bath applied for specified durations. The final concentration used for TrkB/Fc was 1 μg/ml and for TAT-Pep5 was 0.1 μm.
Statistical analysis.
All data are represented as mean ± SEM. The fEPSP slope value expressed as percentages of average baseline values per time point were subjected to statistical analysis using GraphPad Prism 6.0. Nonparametric tests were used as the normality variations at small sample sizes. Wilcoxon signed rank test (Wilcox test) was used to compare within a group and the Mann–Whitney U test (U test) was used to compare data between groups. Statistical comparisons for the behavioral tagging, Western blot, ELISA, and spine count data were performed using unpaired Student's t test or one-way ANOVA. p < 0.05 was considered as the cutoff for statistically significant difference.
Results
Deletion of p75NTR prevents late-LTP deficits caused by SD
First, we evaluated whether there is a change in the sleep pattern in p75NTR KO mice compared with WT mice. The amount of time spent resting and sleeping was comparable between WT and p75NTR KO mice when left undisturbed during the light cycle (Fig. 1). However, it was reported earlier that neurotrophin factors modulate the electrical activity of neurons in the rostral pontine tegmentum, which is responsible for the generation of rapid eye movement sleep, by acting on neurotrophin receptors including p75NTR (Yamuy et al., 1995, 2000). It would be worthwhile to examine sleep staging in future studies given that p75NTR is highly expressed in brainstem regions implicated in sleep staging and arousal (Yamuy et al., 1995). p75NTR is widely expressed in the peripheral nervous system, with a wide range of functions in metabolism (Baeza-Raja et al., 2012), bone formation (Tomlinson et al., 2017), and thermosensitivity (Niklison-Chirou et al., 2013). However, we could not observe differences in body weight between the WT and p75NTR KO mice (C57BL/6J WT 21.83 ± 0.67g and p75NTR KO, 21.26 ± 0.24g), similar to a previous finding (Yeo et al., 1997). Previous studies have shown that SD is associated with deficits in hippocampal LTP (Campbell et al., 2002; McDermott et al., 2006; Vecsey et al., 2009). We hence performed experiments to discern the effects of SD on early-LTP (which is protein synthesis-independent) and late-LTP (which depends on protein synthesis) in Schaffer collateral–CA1 synapses of hippocampal slices derived from either non-SD (NSD) or SD WT mice. A two-pathway experimental design (Fig. 2A) was used to study two independent synaptic inputs to the same CA1 pyramidal neuron population. A single WTET (100 Hz, 21 pulses) resulted in early-LTP, which gradually decayed to baseline after 1–2 h (Fig. 2B, closed and open red circles for NSD and SD mice, respectively). The first mean fEPSP slope value after WTET was 157.93 ± 11.88% (Wilcox test, p = 0.01) and 159.45 ± 7.11% (Wilcox test, p = 0.008) for NSD and SD mice, respectively. The potentiation in S1 (closed red circles) was statistically significant up to 55 min (Wilcox test, p = 0.03) or up to 110 min (U test, p = 0.04) for NSD mice. Whereas in SD mice, significant potentiation in S1 (open red circles) was observed until 55 min (Wilcox test, p = 0.008; U test, p = 0.04). Next, we evaluated late-LTP by providing STET (Fig. 2C). In NSD WT mice, three spaced trains of STET resulted in long-lasting late-LTP (closed red circles). The mean fEPSP slope value immediately after first tetanization was 157.08 ± 9.70% (Wilcox test, p = 0.008) and the potentiation was maintained for up to 4 h (159.95 ± 15.44%, Wilcox test, p = 0.02; U test, p = 0.009). The same stimulation paradigm also resulted in the induction of LTP in SD WT mice (Fig. 2C, open red circles) with mean fEPSP slope value at 149.32 ± 9.83% (Wilcox test, p = 0.02). However, the LTP decayed to baseline within 35 min (Wilcox test, p = 0.04) or 50 min (U test, p = 0.04) and stayed stable until the end of the recording (94.39 ± 8.39%, Wilcox test, p = 0.56). The control synaptic input S2 (closed and open blue circles for NSD and SD mice, respectively) in both early- and late-LTP experiments in NSD and SD groups (Fig. 2B,C) remained stable at baseline throughout the entire recording period. In summary, these findings show that SD impairs late plasticity without affecting early plasticity, in agreement with earlier reports (Guan et al., 2004; Kopp et al., 2006; Vecsey et al., 2009; Havekes et al., 2016b).
Figure 1.
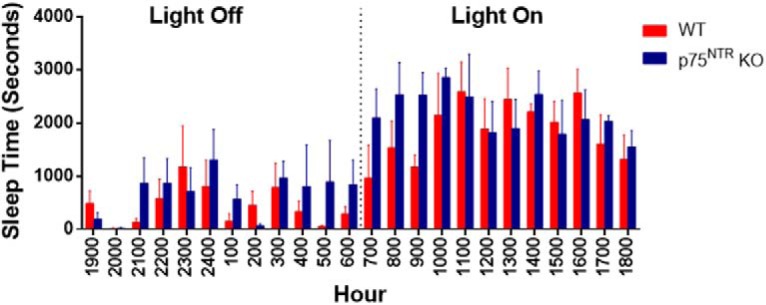
Monitoring of sleep pattern in WT and p75NTR KO mice. Sleep pattern and amount of time spent for sleeping from WT and p75NTR KO mice were assessed by HomeCageScan (CleverSys). Histograms showing sleep time (seconds) across 24 h during both light-on and light-off periods were compared between WT and p75NTR KO mice; N = 4 for each group. We could not observe any difference in the amount of sleeping time between WT and p75NTR KO mice during light-on and light-off periods. Error bars indicate ±SEM.
Figure 2.

p75NTR KO displays normal late-LTP regardless of SD and hippocampal p75NTR protein level increases after sleep loss. A, Schematic representation of a hippocampal slice showing the location of electrodes in the CA1 region. Recording electrode (rec) was positioned onto CA1 apical dendrites flanked by two stimulating electrodes S1 and S2 in the stratum radiatum (sr) to stimulate two independent Schaffer collateral (sc) synaptic inputs to the same neuronal populations. The WTET in synaptic input S1 in B resulted in early-LTP that gradually decayed to baseline in NSD (closed red circles; N = 7) and SD (open red circles; N = 7) WT mice. C, The STET in S1 resulted in a significant potentiation that maintained for 4 h in NSD WT mice (closed red circles; N = 6). The STET in S1 (open red circles) resulted in late-LTP that was gradually decayed to baseline potentials in SD WT mice (N = 7). The WTET in S1 in D resulted in early-LTP that gradually decayed to baseline in NSD (closed red circles; N = 7) and SD (open red circles; N = 6) p75NTR KO mice. E, The STET in S1 resulted in a significant potentiation that maintained for 4 h in NSD p75NTR KO mice (closed red circles; N = 7). STET in S1 (open red circles) resulted in a significant potentiation that also maintained for 4 h in SD p75NTR KO mice (N = 7). Control potentials from S2 (closed and open blue circles) remained stable during the recording period in all experiments. Analog traces represent typical fEPSPs of inputs S1 and S2 15 min before (solid line), 60 min after (dashed line) tetanization, and at the end of the recording (dotted line). Single arrow represents the time of induction of early-LTP by WTET. Three solid arrows represent STET for the induction of late-LTP. Scale bars: vertical, 2 mV; horizontal, 3 ms. Error bars indicate ±SEM. F, Western blot analysis of hippocampal p75NTR level between NSD and SD WT mice. G, Ratio of fold-change from Western blot. The p75NTR protein level was significantly increased (p = 0.028) in SD WT mice compared with NSD WT mice; N = 4 for each group. The values of the individual groups were calculated in relation to the control group, whereas tubulin serves as a loading control. Asterisk indicates significant differences between groups (unpaired t test, *p < 0.05). Error bars indicate ±SEM.
As p75NTR has been shown to contribute to different disease states in the nervous system, we investigated whether this receptor plays a role in the detrimental effects of SD on hippocampal circuitry and function. We found that SD significantly increased (p = 0.0286) the expression of p75NTR in the WT mouse hippocampus compared with control NSD mice (Fig. 2F,G). To investigate whether p75NTR plays a role in synaptic plasticity changes associated with SD, we compared early-LTP between NSD and SD p75NTR KO mice. Similar to WT mice, early-LTP induction in the hippocampal slices derived from both NSD and SD p75NTR KO mice led to a decaying form of LTP. The first fEPSP slope value after WTET in NSD (Fig. 2D, closed red circles) was 160.60 ± 10.79% (Wilcox test, p = 0.02) and for SD (Fig. 2D, open red circles) was 152.58 ± 6.99% (Wilcox test, p = 0.008). The increase in fEPSPs in S1 (closed red circles) stayed statistically significant up to 80 min (Wilcox test, p = 0.03) or up to 100 min (U test, p = 0.007) after WTET for NSD mice. S1 (open red circles) showed a statistically significant potentiation that lasted until 75 min (Wilcox test, p = 0.02; U test, p = 0.02) for SD mice. NSD p75NTR KO mice displayed unaltered late-LTP (Fig. 2E). The mean fEPSP value immediately after the first tetanization (closed red circles), 153.72 ± 8.44%, was statistically different from the baseline (Wilcox test, p = 0.03) and the potentiation maintained for up to 4 h (Wilcox test, p = 0.02; U test, p = 0.005). These data further support normal LTP in p75NTR KO mice (Rösch et al., 2005; Woo et al., 2005). To test whether SD impairs late-LTP in p75NTR KO, we applied STET to induce late-LTP at synaptic input S1 in SD p75NTR KO mice (Fig. 2E, open red circles). The fEPSP slope value after the first tetanization was 170.73 ± 8.47% (Wilcox test, p = 0.02). In contrast to SD WT mice, late-LTP from SD p75NTR KO mice remained statistically significant throughout the entire recording period of 4 h (160.20 ± 6.46%, Wilcox test, p = 0.02; U test, p = 0.005). The baseline in synaptic input S2 (closed and open blue circles for NSD and SD mice, respectively) were stable throughout the recording period in all experiments (Fig. 2D,E). These results show that the deletion of p75NTR prevents the deficits in late-LTP caused by SD, suggesting an essential role of p75NTR in mediating the adverse impact of SD on hippocampal neuronal plasticity.
Deletion of p75NTR prevents STC deficits caused by SD
Although the effects of SD on LTP have been known for some time, it is unclear whether SD would also affect associative plasticity as elicited by the STC protocol. To investigate STC, we used the “strong before weak” (SBW) experimental paradigm (Frey and Morris, 1998), in which late-LTP is induced in S1 by STET 30 min before the induction of early-LTP by WTET in S2 (Fig. 3A–D). First, we investigated STC in hippocampal slices from NSD WT mice. After recording a stable baseline of 30 min in S1 and S2, late-LTP was induced in S1 by STET. After 30 min, WTET was applied to induce early-LTP in S2 (Fig. 3A, red and blue circles). The post-tetanization potentiation in S1 was statistically significant immediately after tetanization (156.90 ± 4.88%, Wilcox test, p = 0.03) and maintained at significant levels until the end of recording (144.62 ± 6.46%, Wilcox test, p = 0.03). WTET at S2 also led to a statistically significant potentiation (164.91 ± 6.18%, Wilcox test, p = 0.02) immediately after tetanization and stayed up to 4 h (140.40 ± 9.37%, Wilcox test, p = 0.02). Thus, STC was expressed in NSD WT mice. However, the same experimental protocol did not result in STC in the hippocampal slices of SD WT mice (Fig. 3B, red and blue circles). The post-tetanization potentiation in S1 was statistically significant (147.22 ± 10.90%, Wilcox test, p = 0.03) immediately after STET but stayed only up to 125 min (Wilcox test, p = 0.02). The fEPSP gradually decayed to the baseline values toward the end of recording (97.43 ± 3.30%, Wilcox test, p = 0.57). The post-tetanization potentials in S2 showed statistically significant potentiation until 95 min (Wilcox test, p = 0.03) and the potentiation gradually decayed to baseline at the end of recording (98.66 ± 12.27%, Wilcox test, p = 0.4688). These results indicate that SD disrupts the expression of STC in hippocampal neurons.
Figure 3.

p75NTR KO displays normal STC regardless of SD. SBW paradigm was used to study STC. A, Both STET in S1 (closed red circles) and WTET in S2 (closed blue circles) resulted in a significant potentiation that maintained until the end of recording in NSD WT mice (N = 6). B, Both STET in S1 (open red circles) and WTET in S2 (open blue circles) resulted in early-LTP that was not reinforced into late-LTP in SD WT mice (N = 7). C, Both STET in S1 (closed red circles) and WTET in S2 (closed blue circles) resulted in a significant potentiation that maintained until the end of experiment in NSD p75NTR KO mice (N = 7). D, Both STET in S1 (open red circles) and WTET in S2 (open blue circles) resulted in a significant potentiation that maintained until the end of recording in SD p75NTR KO mice (N = 7). E, The STET in S1 resulted in a significant potentiation that maintained for 4 h in WT mice with gentle handling (GH; open red circles; N = 6) and those without (closed red circles; N = 6). F, STET in S1 resulted in a significant potentiation that maintained for 4 h in p75NTR KO mice with GH (open red circles; N = 5) and those without (closed red circles; N = 5) p75NTR KO mice. Control potentials from S2 (closed and open blue circles) remained stable during the recording period in all experiments. E, F, Experiments were conducted from the hippocampal slices obtained from animals maintained at dark cycle. Scale bars: vertical, 2 mV; horizontal, 3 ms. Error bars indicate ±SEM. Symbols and analog traces as in Figure 1.
Given the resistance of p75NTR KO mice to the effects of SD on late-LTP, we investigated the expression of STC in these mice. In NSD p75NTR KO mice, induction of late-LTP in S1 by STET led to statistically significant potentiation (Fig. 3C, red circles; 138.73 ± 11.04%, Wilcox test, p = 0.02) immediately after tetanization and the potentiation remained stable until the end of recording (Wilcox test, p = 0.009). Similarly, induction of early-LTP by WTET also resulted in a statistically significant potentiation in S2 after the first tetanization (Fig. 3C, blue circles; 170.87 ± 1.73%, Wilcox test, p = 0.02), which remained stable up to 4 h (141.22 ± 6.84%, Wilcox test, p = 0.02). Interestingly, SD p75NTR KO mice displayed normal STC similar to both the NSD p75NTR KO mice and NSD WT mice (Fig. 3D, red and blue circles). Statistically significant potentiation was observed in S1 (140.88 ± 8.10%, Wilcox test, p = 0.03) after STET and the potentiation remained stable up to the end of recording (137.19 ± 9.41%, Wilcox test, p = 0.03). In S2, WTET yielded statistically significant potentiation immediately after induction (157.87 ± 9.15%, Wilcox test, p = 0.03) and the potentiation maintained until the end of the recording (130.93 ± 5.39%, Wilcox test, p = 0.03). These results show that deletion of p75NTR prevents the deficits in associative plasticity that are caused by SD.
To confirm that the observed changes in synaptic plasticity are because of SD and not from gentle handling itself, both WT and p75NTR KO mice were either left undisturbed or subjected to 5 h of gentle handling during the dark cycle before late-LTP recording. In undisturbed WT mice, three spaced trains of STET resulted in long-lasting late-LTP (closed red circles) that maintained for up to 4 h (154.30 ± 4.52%, Wilcox test, p = 0.03; U test, p = 0.002; Fig. 3E). The same stimulation paradigm also resulted in late-LTP (open red circles) that maintained toward the end of recording in WT mice subjected to gentle handling (162.60 ± 13.24%, Wilcox test, p = 0.03; U test, p = 0.002; Fig. 3E). Undisturbed p75NTR KO mice displayed unaltered long-lasting potentiation during the entire recording period of 4 h (158.00 ± 1.63%, Wilcox test, p = 0.03; U test, p = 0.008; Fig. 3F). In p75NTR KO mice subjected to gentle handling, the late-LTP was maintained at significant levels throughout the entire recording period of 4 h (156.80 ± 6.11%, Wilcox test, p = 0.03; U test, p = 0.008; Fig. 3F). The control synaptic input S2 (closed and open blue circles for undisturbed and mice subjected to gentle handling, respectively) in both WT and p75NTR KO mice (Fig. 3E,F) remained stable at baseline levels throughout the entire recording period. These results show that the effects of SD in this study were not related to the amount of stimulation the mice received from the SD method. This observation is consistent with a previous study which demonstrated that applying the same amount of stimulation (gentle handling) in the waking phase (i.e., the dark phase) does not lead to memory impairments (Hagewoud et al., 2010).
Deletion of p75NTR restores associative memory in SD mice
We investigated whether the effects of SD and p75NTR KO on synaptic plasticity were reflected in behavior by testing associative memory. To this end, we used a behavioral tagging paradigm (Moncada and Viola, 2007), in which mice are exposed to a novel environment or OF before the application of a weak foot shock (Fig. 4A). Previous studies have demonstrated that weak memory (i.e., weak foot shock) can be consolidated into a strong memory if it was coupled with strong stimulation (i.e., OF environment; Moncada and Viola, 2007). Associative memory strength was measured as the latency to step down to bars that were associated with a weak foot shock at various time points (1 h, 24 h, and 7 d). A longer step-down latency indicates a stronger associative memory as the mice would stay on the platform for a longer time before stepping down to the bars. The pre-shock (training) step-down latency was 11.61 ± 1.365% and 9.363 ± 1.077% for WT and p75NTR KO mice, respectively (Fig. 4B,C). In agreement with a previous study (Moncada and Viola, 2007), the step-down latency at all three different time points measured was significantly higher (p = 0.0006 for 1 h, p = 0.0029 for 24 h, and p = 0.0029 for 7 d) in mice that received the weak foot shock after OF spatial exploration, compared with mice that were not exposed to the OF (Fig. 4B,C). Both WT and p75NTR KO mice responded in a similar way. However, after SD, WT mice that experienced a foot shock after OF exploration had a significantly lower step-down latency compared with the control NSD WT mice at all three different time points (p = 0.007 for 1 h, p = 0.007 for 24 h, and p = 0.0227 for 7 d; Fig. 4B). In general, SD reduced the step-down latency in WT mice to a level similar to that shown by NSD mice that did not experience OF exploration. We could not observe any difference in step-down latency between non-behavioral tagging NSD group (Fig. 4B, blue bars) and behavioral tagging SD group (Fig. 4B, green bars), suggesting impaired memory acquisition and retention in WT mice subjected to SD. In contrast, both SD and NSD p75NTR KO mice exhibited normal memory that was maintained for up to 7 d (Fig. 4C). These results show that p75NTR KO mice are resistant to the detrimental effects of SD on associative memory.
Figure 4.
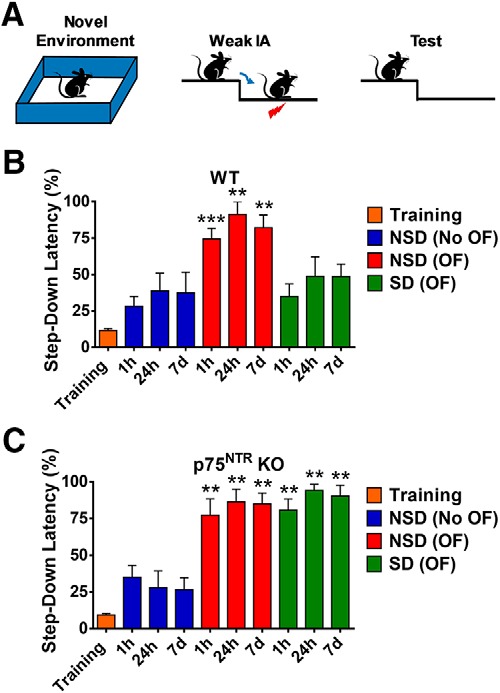
p75NTR KO displays normal associative memory regardless of SD. A, Schematic diagram of behavioral tagging experiment protocol. A mouse with or without being placed in a novel environment/OF for 10 min within 1 h before weak IA. Step-down latency was tested at 1 h, 24 h, and 7 d post-IA. B, Associative memory was impaired in SD WT mice. C, Associative memory was normal in SD p75NTR KO mice. Asterisks indicate significant differences between groups (unpaired t test, **p < 0.01, ***p < 0.001). Error bars indicate ±SEM.
Deletion of p75NTR prevents SD-mediated changes in PDE4A5-cAMP–CREB-BDNF pathway
Next, we investigated the possible mechanisms by which p75NTR prevents SD-mediated functional and structural deficits. Hippocampal phosphodiesterase PDE4A5 plays an important role in mediating the electrophysiological and behavioral effects of SD (Vecsey et al., 2009; Havekes et al., 2016a). Specifically, SD was shown to have resulted in increased PDE4A5 levels in the hippocampus (Vecsey et al., 2009). Consistent with earlier findings, we confirmed that SD significantly increased (p = 0.0079) the levels of hippocampal PDE4A5 in WT but not p75NTR KO mice compared with NSD mice (Fig. 5A,B). A previous study has demonstrated that PDE4A5 interacts with p75NTR (Sachs et al., 2007), an observation that we also confirmed by co-immunoprecipitation from mouse hippocampal extracts (Fig. 5C). PDE4A5 hydrolyzes cAMP and, in agreement with this, hippocampal cAMP levels were found to be significantly decreased (p = 0.0043) in SD WT mice compared with NSD WT mice (Fig. 5D). However, SD did not affect hippocampal cAMP levels in p75NTR KO mice (Fig. 5D). These results show that the deletion of p75NTR preserves cAMP levels in hippocampus from SD mice by affecting PDE4A5 function.
Figure 5.

Effects of SD on PDE4A5, ERK, CREB, and cAMP levels in the hippocampus. A, Western blot analysis of hippocampal PDE4A5, ERK, and CREB levels between NSD and SD from WT and p75NTR KO mice. B, Ratio of fold-change from Western blot. The PDE4A5 protein level was significantly increased (p = 0.0079) in SD WT mice compared with NSD WT, NSD p75NTR KO, and SD p75NTR KO mice; N = 4 for each group. The phospho/total ERK ratio was significantly decreased in SD WT mice compared with NSD WT (p = 0.0022), NSD p75NTR KO (p = 0.0022), and SD p75NTR KO (p = 0.0043) mice; N = 6 for each group. The phospho/total CREB ratio was significantly decreased (p = 0.0079) in SD WT mice compared with NSD WT, NSD p75NTR KO, and SD p75NTR KO mice; N = 5 for each group. The values of the individual groups were calculated in relation to the control group while tubulin serves as a loading control. Asterisks indicate significant differences between groups (one-way ANOVA, **p < 0.01). Error bars indicate ±SEM. C, Immunoprecipitation of hippocampal tissues reveals an interaction between p75NTR and PDE4A5 in both NSD and SD conditions. D, ELISA analysis of hippocampal cAMP level between NSD and SD mice. The cAMP level was significantly decreased in SD WT mice compared with NSD WT (p = 0.0043), NSD p75NTR KO (p = 0.0216), and SD p75NTR KO (p = 0.0087) mice; N = 6 for each group. Asterisks indicate significant differences between groups (one-way ANOVA, *p < 0.05, **p < 0.01). Error bars indicate ±SEM.
The extracellular signal-regulated kinases (ERK1/2) are also key players in the maintenance of long-term synaptic plasticity (Impey et al., 1999; Roth and Sweatt, 2008). In the hippocampus, the phosphorylation of ERK1/2, a key event in ERK1/2 activation, was significantly reduced (p = 0.0022) in WT mice after SD (Fig. 5A,B), consistent with previous findings (Guan et al., 2004; Ravassard et al., 2009). However, SD did not affect the levels of phosphorylated ERK1/2 in SD p75NTR KO mice (Fig. 5A,B). Similar effects were observed in the phosphorylation levels of cAMP responsive element-binding protein (CREB), a transcription factor important for synaptic plasticity and memory (Vecsey et al., 2009). A significant decrease (p = 0.0079) in phosphorylated CREB was observed after SD in WT but not p75NTR KO mice compared with NSD WT mice (Fig. 5A,B). Together, ERK1/2 and CREB signaling pathways were found to be preserved in p75NTR KO mice subjected to SD. This is in line with the resistance to SD-induced synaptic plasticity and memory deficits observed in p75NTR KO mice.
Given the fact that CREB regulates BDNF expression, ELISA and Western blot analysis were performed to detect BDNF expression in hippocampal extracts. The ELISA experiments revealed that both mature- and pro-BDNF levels were significantly decreased (p = 0.0128 for mature BDNF and p = 0.0012 for pro-BDNF) in SD WT mice compared with NSD WT mice (Fig. 6A), but p75NTR KO mice did not show these changes (Fig. 6A). Western blot analysis further confirmed that SD significantly reduced the expression of mature BDNF (p = 0.0138) and pro-BDNF (p = 0.0286) in WT mice but not p75NTR KO mice (Fig. 6B,C). To further elucidate the role of BDNF in SD, TrkB/Fc chimera recombinant protein (1 μg/ml) was bath applied to the hippocampal slices from SD p75NTR KO mice 30 min before and 30 min after the first STET (Fig. 6D,E). In the presence of TrkB/Fc, STET led to an immediate increase in fEPSP in S1, 151.40 ± 4.30% (Wilcox test, p = 0.02), but the potentiation was statistically significant only up to 75 min (Wilcox test, p = 0.04) or 140 min (U test, p = 0.04), after which the fEPSP remained at baseline until the end of the recording period of 4 h (108.40 ± 6.81%, Wilcox test, p = 0.68). Similarly, in the STC experiment, with TrkB/Fc bath application, potentiation in S1 was statistically significant (179.70 ± 8.28%, Wilcox test, p = 0.02) immediately after STET but remained only for 150 min (Wilcox test, p = 0.03) and gradually decayed to the baseline values toward the end of recording (108.20 ± 8.18%, Wilcox test, p = 0.38). The fEPSP in S2 showed statistically significant potentiation until 110 min (Wilcox test, p = 0.04) after which the potentiation gradually decayed to the baseline (104.60 ± 12.27%, Wilcox test, p = 0.4688). These results further support a significant role of BDNF in maintaining late-LTP and STC in p75NTR KO mice after SD.
Figure 6.

Effects of SD on BDNF levels in the hippocampus. A, ELISA analysis of hippocampal mature- and pro-BDNF level between NSD and SD mice. The mature BDNF level was significantly decreased (p = 0.0128) in SD WT mice compared with NSD WT mice; N = 6 for each group. The mature BDNF level was significantly decreased (p = 0.0221) in SD WT mice compared with both NSD and SD p75NTR KO mice; N = 6 for each group. The pro-BDNF level was significantly decreased (p = 0.0012) in SD WT mice compared with NSD WT, NSD p75NTR KO, and SD p75NTR KO mice; N = 6 for each group. Asterisks indicate significant differences between groups (one-way ANOVA, *p < 0.05, **p < 0.01). Error bars indicate ±SEM. B, Western blot analysis of hippocampal mature- and pro-BDNF levels between NSD and SD from WT and p75NTR KO mice. C, Ratio of fold-change from Western blot. The pro-BDNF level was significantly decreased (p = 0.0286) in SD WT mice compared with NSD WT, NSD p75NTR KO, and SD p75NTR KO mice; N = 4 for each group. The mature-BDNF level was significantly decreased in SD WT mice compared with NSD WT (p = 0.0138), NSD p75NTR KO (p = 0.0218), and SD p75NTR KO (p = 0.0286) mice; N = 4 for each group. Asterisk indicates significant differences between groups (one-way ANOVA, *p < 0.05). Error bars indicate ±SEM. D, Induction of late-LTP by STET in S1 (open red circles) in the presence of TrkB/Fc (1 μg/ml) resulted in a decremental LTP in SD p75NTR KO mice (N = 7). Control potentials from S2 (open blue circles) remained stable during the recording period. E, Both STET in S1 (open red circles) and WTET in S2 (open blue circle) in the presence of TrkB/Fc (1 μg/ml) resulted in early-LTP that was not reinforced into late-LTP in SD p75NTR KO mice (N = 7). Scale bars: vertical, 2 mV; horizontal, 3 ms. Error bars indicate ±SEM. Symbols and analog traces as in Figure 1.
Deletion of p75NTR prevents SD-mediated changes in RhoA-ROCK2 and cAMP-PKA-LIMK1-cofilin pathway
RhoA-ROCK and cAMP-PKA-LIMK-cofilin pathways are well known signaling cascades in the regulation of actin dynamics (Ohashi et al., 2000; Hsieh et al., 2006; Nadella et al., 2009). It has been previously reported that p75NTR signaling can modify the activity of the Ras homolog gene family member A (RhoA; Yamashita et al., 1999). We found that SD significantly increased (p = 0.03) RhoA protein levels in SD WT but not SD p75NTR KO mice compared with NSD control WT mice (Fig. 7A,B), indicating a role of p75NTR in the upregulation of RhoA protein after SD. Moreover, the level of Rho-associated coiled-coil containing protein kinase 2 (ROCK2) was also significantly increased (p = 0.0123) in WT but not p75NTR KO mice after SD (Fig. 7A,B). In addition, SD significantly decreased (p = 0.0016) the phosphorylation of LIMK1 at threonine 508 in WT mice after SD (Fig. 7A,B). No significant change (p = 0.8826) was observed in SD p75NTR KO mice (Fig. 7A,B). In turn, LIMK has been implicated in the regulation of cofilin activity (Endo et al., 2003). We found that cofilin phosphorylation at serine 3 was significantly lower (p = 0.0495) in SD WT mice compared with control WT mice (Fig. 7A,B). However, phosphorylated cofilin level was unchanged in p75NTR KO after SD (Fig. 7A,B). A previous study has demonstrated that SD decreases LIMK serine 596 phosphorylation and cofilin phosphorylation at serine 3 as a result of decreased cAMP-PKA activity (Havekes et al., 2016b). Spine counts from Golgi staining in CA1 pyramidal neurons of the hippocampus (Fig. 8A) showed that SD significantly decreased (p < 0.0001) spine density in WT mice compared with NSD WT, NSD p75NTR KO, and SD p75NTR KO mice (Fig. 8B). SD significantly reduced the number of apical dendrites (p < 0.0001; Fig. 8C) and basal dendrites (p = 0.0003; Fig. 8D) of CA1 neurons in WT but not p75NTR KO mice. Furthermore, SD significantly reduced (p = 0.0286) the spine density of apical/basal dendrites located 60–150 μm away from the soma of CA1 neurons in WT but not p75NTR KO mice (Fig. 8E). Immunostaining of the proteins RhoA, ROCK2, LIMK1 and cofilin (Fig. 7C) showed that these proteins were distributed in all subregions of the hippocampus although the staining of RhoA is weak. SD has been shown to lead to the loss of dendritic spines in CA1 and dentate gyrus but not CA3 region, although the underlying mechanisms that account for these region-specific changes remain to be investigated (Havekes et al., 2016b; Raven et al., 2019). Thus, we postulated that the RhoA-ROCK2 and cAMP-PKA-LIMK1-cofilin pathways in the CA1 and dentate gyrus regions may be more vulnerable to the negative consequences of SD. To further elucidate the role of p75NTR in SD, the cell-permeable p75NTR signaling inhibitor TAT-pep5 (0.1 μm) was bath applied to the hippocampal slices from SD WT mice throughout the LTP and STC experiments (Fig. 9A,B). In late-LTP experiment, the mean fEPSP slope value immediately after first tetanization was 155.30 ± 7.28% (Wilcox test, p = 0.03). The potentiation in S1 remained stable up to 4 h (158.7 ± 7.56%, Wilcox test, p = 0.03; U test, p = 0.002; Fig. 9A). In STC experiment, the increase in post-tetanization potential in S1 was statistically significant (156.30 ± 8.63%, Wilcox test, p = 0.04) immediately after STET and the mean fEPSP stayed up until the end of recording (141.70 ± 13.40%, Wilcox test, p = 0.04). The post-tetanization potentials in S2 also showed statistically significant potentiation until the end of recording (144.30 ± 2.65%, Wilcox test, p = 0.04). These results further support a significant role of p75NTR in mediating plasticity changes in SD. Together, these data suggest that SD modulates the RhoA-ROCK2 and cAMP-PKA-LIMK1-cofilin pathways via p75NTR. This in turn impairs actin dynamics, which is important for synaptic function and efficacy.
Figure 7.
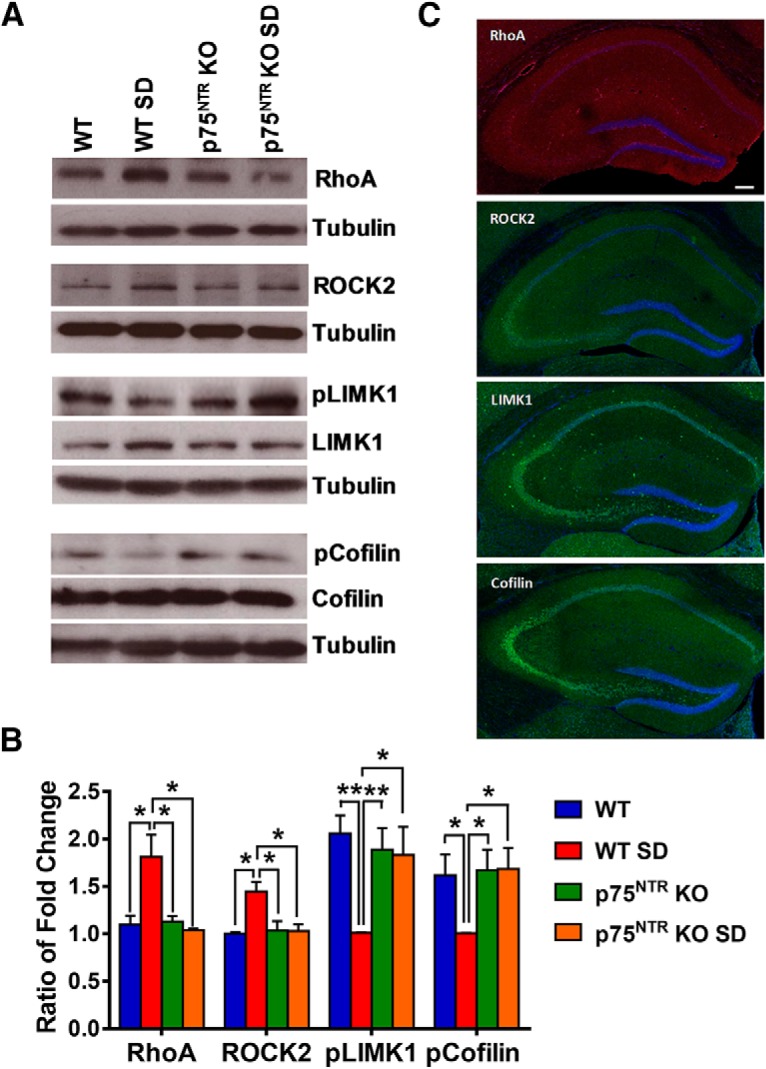
Effects of SD on RhoA, ROCK2, LIMK1, and cofilin levels in the hippocampus. A, Western blot analysis of hippocampal RhoA, ROCK2, LIMK1, and cofilin levels between NSD and SD mice. B, Ratio of fold-change from Western blot. The RhoA level was significantly increased in SD WT mice compared with NSD WT (p = 0.0300), NSD p75NTR KO (p = 0.0298), and SD p75NTR KO (p = 0.0164) mice; N = 4 for each group. The ROCK2 level was significantly increased in SD WT mice compared with NSD WT (p = 0.0123), NSD p75NTR KO (p = 0.0435), and SD p75NTR KO (p = 0.0282) mice; N = 3 for each group. The phosphorylated LIMK1 protein level was significantly decreased in SD WT mice compared with NSD WT (p = 0.0016), NSD p75NTR KO (p = 0.0084), and SD p75NTR KO (p = 0.0329) mice; N = 4 for each group. The phosphorylated cofilin level was significantly decreased in SD WT mice compared with NSD WT (p = 0.0495), NSD p75NTR KO (p = 0.0387), and SD p75NTR KO (p = 0.0388) mice; N = 3 for each group. The values of the individual groups were calculated in relation to the control group while tubulin serves as a loading control. Asterisks indicate significant differences between groups (one-way ANOVA, *p < 0.05, **p < 0.01). Error bars indicate ±SEM. C, Representative images of stained WT mice hippocampus with RhoA (red), ROCK2 (green), LIMK1 (green), and cofilin (green), and counterstained with DAPI (blue). Scale bars, 200 μm.
Figure 8.
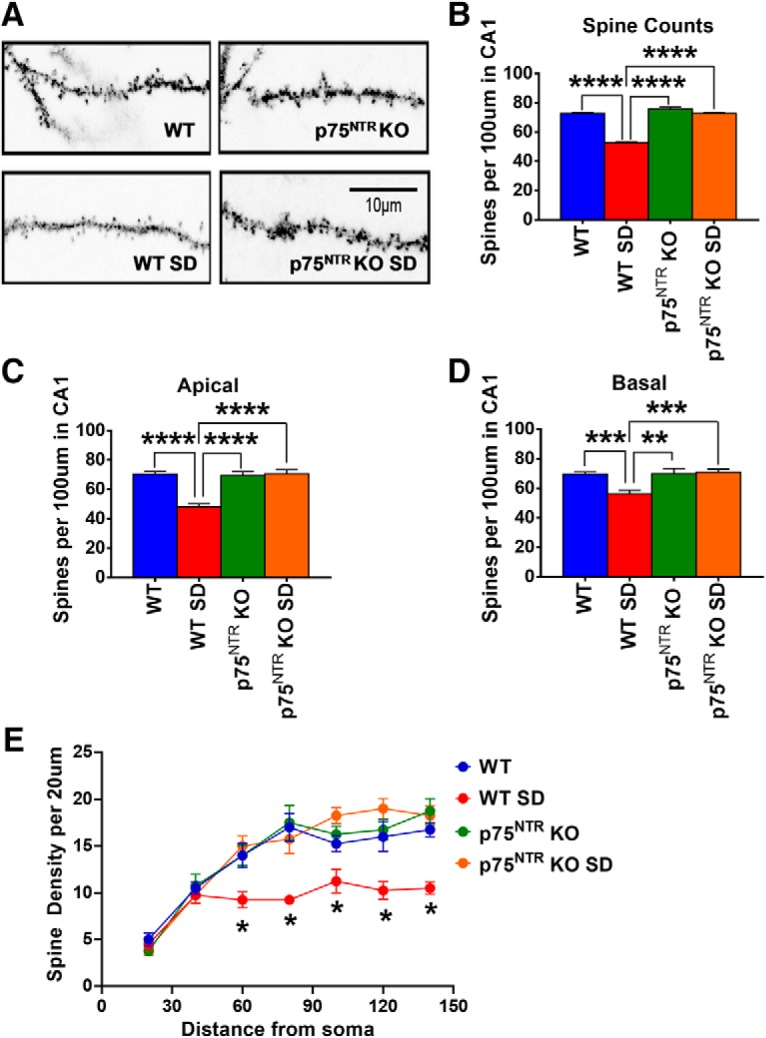
Effects of SD on spine density in hippocampus. A, Representative image of Golgi-Cox stained dendritic spines of CA1 pyramidal neurons from WT and p75NTR KO mice in both NSD and SD conditions. B, WT mice had significant reductions (p < 0.0001) in spine density post-SD compared with NSD WT and both NSD and SD p75NTR KO mice; N = 20 sections per group. C, WT mice had significant decrease (p < 0.0001) in the number of apical dendrites of CA1 neurons post-SD compared with NSD WT and both NSD and SD p75NTR KO mice; N = 10 sections per group. D, WT mice had significant decrease in the number of basal dendrites of CA1 neurons post-SD compared with NSD WT (p = 0.0003), NSD p75NTR KO (p = 0.007) and SD p75NTR KO (p = 0.0005) mice; N = 10 sections per group. E, WT mice had significant reductions (p = 0.0286) in spine density of apical/basal dendrites between 60 and 150 μm away from the soma of CA1 neurons post-SD compared with NSD WT and both NSD and SD p75NTR KO mice; N = 4 per each group. Asterisks indicate significant differences between groups (one-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Error bars indicate ±SEM.
Figure 9.

Effect of TAT-pep5 on synaptic plasticity associated with SD. A, The STET in S1 (open red circles) in the presence of TAT-pep5 (0.1 μm) resulted in late-LTP that was maintained for up to 4 h in SD WT mice (N = 6). Control potentials from S2 (open blue circles) remained stable during the recording period. B, Both STET in S1 (open red circles) and WTET in S2 (open blue circles) resulted in a significant potentiation that was maintained until the end of recording in SD WT mice (N = 5). Scale bars: vertical, 2 mV; horizontal, 3 ms. Error bars indicate ±SEM. Symbols and analog traces as in Figure 1.
Discussion
In the present study, we show that 5 h of SD in mice affected late-LTP but not the short-lasting form of LTP (early-LTP) in the hippocampal CA1 region. It has been reported earlier that prolonged SD (24–72 h) inhibits the induction of LTP in the hippocampus of rodents (Campbell et al., 2002; Ravassard et al., 2009). Consistent with earlier findings, our study indicates that the degree of impact of SD on hippocampal function correlates with the length of SD (Campbell et al., 2002; Ravassard et al., 2009). In general, acute SD is sufficient to disrupt the signaling pathways underlying hippocampal-dependent synaptic plasticity and efficacy.
Synaptic associativity transforms short-term plasticity to long-term plasticity, enabling the neural networks to encode stable long-term memories. One characteristic consequence of SD is a deficit in associative plasticity or memory consolidation evident predominantly in behavioral studies (Graves et al., 2003; Prince and Abel, 2013; Havekes et al., 2016b). Using the STC paradigm, our study provides evidence of disrupted associative plasticity after SD, which corroborates behavioral tagging experiments in which SD impaired associative memory. It has been postulated that during behavioral tagging, PRPs synthesized under the influence of novelty, could transform transient forms of plasticity into long-lasting forms (Moncada and Viola, 2007). Interestingly, evidence from rodent experiments has suggested that hippocampus replays recent awake experiences during sleep, which in turn can influence later memory performance. This demonstrates the important role of hippocampal replay in memory consolidation (Ji and Wilson, 2007; Bendor and Wilson, 2012). In humans, the consolidation of hippocampal-dependent memories during sleep is causally related to the replay of hippocampal neural activities during sleep (Hanert et al., 2017; Schapiro et al., 2018). Overall, the findings from our electrophysiological and behavioral tagging studies further support the importance of sleep in regulating hippocampal-dependent long-term memory and associative memory.
In agreement with previous studies (Rösch et al., 2005; Woo et al., 2005), our data show that p75NTR KO mice exhibit normal late-LTP and STC. Amazingly, the deletion of p75NTR prevented the deficits typically observed after SD, suggesting that the cellular and synaptic alterations that arise during SD do not take place in cells lacking this receptor. Together, our data suggest that p75NTR contributes to the inhibitory influence on hippocampal plasticity, associative plasticity, and associative memory that follows sleep loss.
What could be the possible mechanism for the preservation of associative plasticity and memory in SD mice lacking p75NTR? We found that SD-mediated increase in PDE4A5 levels did not occur in p75NTR mutant mice. Increased PDE4A5 levels have been shown to impair a long-lasting form of hippocampal synaptic plasticity and attenuate hippocampal-dependent memory consolidation by interfering with PKA activation (Havekes et al., 2016b). In fact, blockade of PDE4A5 signaling has been proven to be an effective mechanism to prevent SD-induced changes in synaptic plasticity (Vecsey et al., 2009). Our data show that SD increases p75NTR protein expression. Induction of p75NTR expression has been observed upon nervous system injury and in a number of neurodegenerative diseases, contributing to neuronal death (Dechant and Barde, 2002). A previous study demonstrated that p75NTR interacts directly with PDE4A5, thereby regulating the PDE4A5-cAMP-PKA pathway, leading to decreased cAMP levels (Sachs et al., 2007). We also provide evidence that PDE4A5 interacts with p75NTR in the hippocampus. Thus, our results support the idea that p75NTR contributes to enhanced PDE4A5 signaling during SD, thereby limiting cAMP-dependent plasticity and memory processes.
Similar to cAMP-PKA signaling (Vecsey et al., 2009), a decrease in ERK1/2 activation, visualized as ERK phosphorylation, has been reported in SD (Guan et al., 2004; Ravassard et al., 2009). Crosstalk between the ERK pathway and cAMP-PKA pathway, through the exchange protein activated by cAMP (Epac) and Ras (Roht and Sweatt, 2008), has also been described (Sindreu et al., 2007; Havekes et al., 2012). Hence, changes in cAMP-PKA activity could also influence ERK activity in SD. Both pathways are important for regulating CREB-mediated gene expression at the transcriptional level, which promotes the expression of proteins that will consolidate labile memories into long-term memories (Roberson et al., 1999). Interestingly, BDNF plays a critical role in maintaining late-LTP and STC in p75NTR KO mice after SD, which is consistent with earlier findings in which we and others have showed the critical role of BDNF as a plasticity protein in maintaining long-lasting plasticity and STC (Barco et al., 2005; Sajikumar et al., 2009).
Sleep loss has been shown to reduce spine density and attenuate synaptic efficacy in the hippocampus (Raven et al., 2017). The size and shape of spines are dependent on actin protein and its dynamics is strongly associated with activity-dependent long-term plasticity (Rudy, 2015). RhoA-ROCK and cAMP-PKA-LIMK-cofilin pathways have been implicated in regulating actin dynamics (Nadella et al., 2009; Tönges et al., 2011). p75NTR can activate RhoA in response to myelin-derived ligands and activated RhoA in turn causes ROCK2 activation (Maekawa et al., 1999; Wang et al., 2002; Wong et al., 2002). In our study, we found that the levels of hippocampal RhoA and ROCK2 increased after SD. RhoA-ROCK activation can activate downstream effectors, which regulate cytoskeleton reorganization such as growth cone collapse and neurite outgrowth inhibition (Lingor et al., 2007; Wu et al., 2013). LIMK1 activation can result in growth cone collapse (Maekawa et al., 1999) and its activity is regulated by the cAMP-PKA pathway (Nadella et al., 2009). Downregulation of LIMK1 was shown to have a critical role in BDNF/CaMKIIβ-mediated neurite outgrowth (Saito et al., 2013). Our data show that SD decreases phosphorylation of LIMK1 in the hippocampus. Actin depolymerizing protein cofilin is a downstream target of LIMK1. Modulation of cofilin activity has been shown to be essential for the reorganization of the actin cytoskeleton (Bamburg and Bernstein, 2010). An earlier study has demonstrated that SD reduced cofilin phosphorylation, reflecting higher activity of cofilin in the hippocampus (Havekes et al., 2016b). Indeed, our study shows a decrease in cofilin phosphorylation and spine density after SD in WT mice. Surprisingly, deletion of p75NTR prevents the SD-mediated modulation of RhoA-ROCK2 and cAMP-PKA-LIMK1-cofilin pathways in the hippocampus and hence, preserves spine density. Furthermore, p75NTR signaling inhibitor TAT-pep5 is able to restore late-LTP and STC deficits in the hippocampus after SD, revealing that the role of p75NTR in SD is not developmental, as it can be induced acutely in adult animals.
It has been reported that the setting of synaptic tags and stabilization of STC is governed by actin polymerization (Ramachandran and Frey, 2009) and cofilin is a widely distributed actin-modulating protein. On the other hand, BDNF/TrkB signaling, which is downregulated in SD, is important for regulating LTP maintenance in the hippocampus by promoting actin polymerization within dendritic spines (Rex et al., 2007). Moreover, cofilin also regulates the trafficking and insertion of AMPAR at postsynaptic sites upon LTP induction (Gu et al., 2010). Thus, we postulate that SD may interfere with the tagging process during LTP and STC, thus preventing their maintenance by interfering with the setting of synaptic tags and the distribution of PRPs. Inhibition of p75NTR could, however, hinder cofilin activity by preventing modulation of the cAMP-PKA-LIMK1 pathway while promoting actin dynamics. The exon III p75NTR KO mice used in this study may express an alternatively spliced isoform (von Schack et al., 2001), which is able to support some signaling activity although the expression level is expected to be low. Alternatively, there exists a conditional complete p75NTR KO mouse strain (Bogenmann et al., 2011), which carries a complete deletion of the transmembrane and cytoplasmic domains of the receptor. In the future, it will be intriguing to study the effects of SD using this conditional complete p75NTR KO mouse as it will not be hampered by complexities associated using the p75NTR KO mice with only exon III deletion. The conditional complete p75NTR KO mice are expected to produce a similar result or even better outcome in restoring plasticity deficits associated with sleep loss.
In conclusion, our findings demonstrate that p75NTR is an important mediator of the alterations in hippocampal-dependent synaptic plasticity and memory caused by SD. The study elucidates the signaling pathways activated by p75NTR to regulate both functional and structural plasticity changes associated with SD and contributes to the understanding of how deletion of p75NTR restores hippocampal plasticity changes after sleep loss. In general, deletion of p75NTR prevents SD-mediated changes of the following pathways: (1) PDE4A5-cAMP–CREB-BDNF, (2) cAMP-PKA-LIMK1-cofilin, and (3) RhoA-ROCK2 (Fig. 10). Alterations in these pathways underlie the disruption of functional and structural plasticity in hippocampus, which eventually contributes to the failure of STC and hence associative memory after SD. Thus, targeting p75NTR could be an attractive strategy to counteract the impairments in cognitive function that result from sleep loss.
Figure 10.
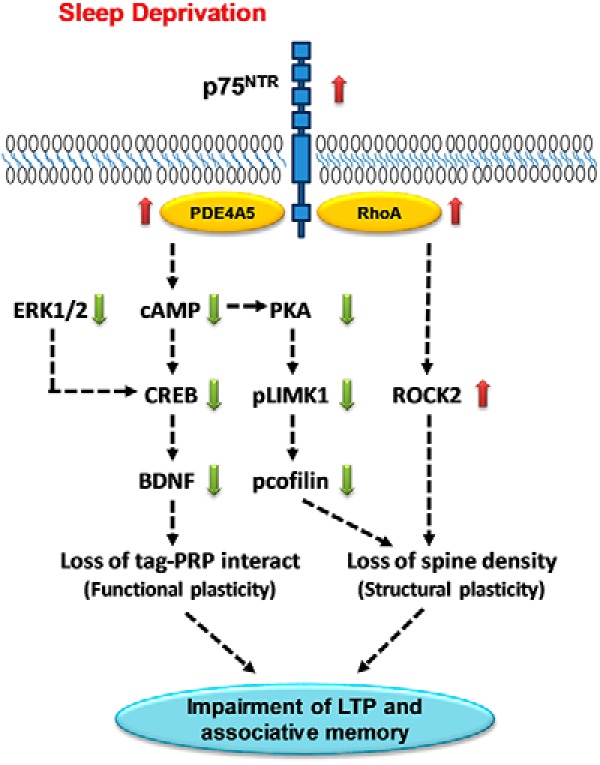
A proposed mechanism of the p75NTR as a mediator of synaptic changes during SD. This diagram depicts the signaling pathway by p75NTR in mediating LTP and associative memory impairment after sleep loss. SD increases PDE4A5 level and this protein can interact directly with p75NTR, which leads to a reduced level of cAMP. Moreover, SD also decreases ERK1/2 activity. Both cAMP and ERK pathways are important in regulating gene expression at the transcriptional level via CREB, which promotes upregulation of expression of proteins, for example BDNF that will consolidate labile memories into long-term memories. Both CREB and BDNF levels are decreased in SD. The BDNF and TrkB receptor have been shown to be involved in STC process, which is critical for memory consolidation, are impaired during SD. In addition, SD modulates the cAMP-PKA-LIMK1 pathway. Modulation of this pathway increases the activity of cofilin, which leads to loss of spine density. SD also increases RhoA level leading to an increase in ROCK2 activity, which has been implicated in regulating spine density. As a whole, p75NTR mediates PDE4A5-cAMP–CREB-BDNF, cAMP-PKA-LIMK1-cofilin, and RhoA-ROCK2 pathways, which eventually lead to changes in both functional and structural plasticity. As a consequence, LTP and associative memory are impaired after sleep loss. Red arrow indicates increases. Green arrow indicates decreases.
Footnotes
This work was supported by NUS-Strategic and Aspiration Research Funds to S.S., L.-W.W., and C.F.I., by a Fellowship from NUS Graduate School to J.Y.T., and by National Medical Research Council Collaborative Research Grants (NMRC-CBRG-0099-2015 and NMRC/OFIRG/0037/2017) and Ministry of Education Academic Research Fund Tier 3 (MOE2017-T3-1-002) to S.S. The behavioral experiments were performed at the Neuroscience Phenotyping Core Facility, which is supported by the NMRC NUHS Centre Grant–Neuroscience Phenotyping Core (NMRC/CG/M009/2017_NUH/NUHS). We thank Liu Chang and Eunice Sim for their help with immunostaining, and Sheila Ang, Karen Pang, and Amrita Benoy for their critical comments and help with editing the paper.
The authors declare no competing financial interests.
References
- Abel T, Havekes R, Saletin JM, Walker MP (2013) Sleep, plasticity and memory from molecules to whole-brain networks. Curr Biol 23:R774–R788. 10.1016/j.cub.2013.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeza-Raja B, Eckel-Mahan K, Zhang L, Vagena E, Tsigelny IF, Sassone-Corsi P, Ptáček LJ, Akassoglou K (2013) p75 neurotrophin receptor is a clock gene that regulates oscillatory components of circadian and metabolic networks. J Neurosci 33:10221–10234. 10.1523/JNEUROSCI.2757-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeza-Raja B, Li P, Le Moan N, Sachs BD, Schachtrup C, Davalos D, Vagena E, Bridges D, Kim C, Saltiel AR, Olefsky JM, Akassoglou K (2012) p75 neurotrophin receptor regulates glucose homeostasis and insulin sensitivity. Proc Natl Acad Sci U S A 109:5838–5843. 10.1073/pnas.1103638109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballarini F, Moncada D, Martinez MC, Alen N, Viola H, Izquierdo IA (2009) Behavioral tagging is a general mechanism of long-term memory formation. Proc Natl Acad Sci U S A 106:14599–14604. 10.1073/pnas.0907078106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamburg JR, Bernstein BW (2010) Roles of ADF/cofilin in actin polymerization and beyond. F1000 Biol Rep 2:62. 10.3410/B2-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks S, Dinges DF (2007) Behavioral and physiological consequences of sleep restriction. J Clin Sleep Med 3:519–528. [PMC free article] [PubMed] [Google Scholar]
- Barco A, Patterson SL, Alarcon JM, Gromova P, Mata-Roig M, Morozov A, Kandel ER (2005) Gene expression profiling of facilitated L-LTP in VP16-CREB mice reveals that BDNF is critical for the maintenance of LTP and its synaptic capture. Neuron 48:123–137. 10.1016/j.neuron.2005.09.005 [DOI] [PubMed] [Google Scholar]
- Bendor D, Wilson MA (2012) Biasing the content of hippocampal replay during sleep. Nat Neurosci 15:1439–1444. 10.1038/nn.3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Cooke SF (2011) Long-term potentiation and long-term depression: a clinical perspective. Clinics 66:3–17. 10.1590/S1807-59322011001300002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Lømo T (1973) Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol 232:331–356. 10.1113/jphysiol.1973.sp010273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogenmann E, Thomas PS, Li Q, Kim J, Yang LT, Pierchala B, Kaartinen V (2011) Generation of mice with a conditional allele for the p75NTR neurotrophin receptor gene. Genesis 49:862–869. 10.1002/dvg.20747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IG, Guinan MJ, Horowitz JM (2002) Sleep deprivation impairs long-term potentiation in rat hippocampal slices. J Neurophysiol 88:1073–1076. 10.1152/jn.2002.88.2.1073 [DOI] [PubMed] [Google Scholar]
- Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhäuser N, Böhm-Matthaei R, Baeuerle PA, Barde YA (1996) Selective activation of NF-κB by nerve growth factor through the neurotrophin receptor p75. Science 272:542–545. 10.1126/science.272.5261.542 [DOI] [PubMed] [Google Scholar]
- Chao MV. (2003) Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci 4:299–309. 10.1038/nrn1078 [DOI] [PubMed] [Google Scholar]
- Dechant G, Barde YA (2002) The neurotrophin receptor p75 NTR: novel functions and implications for diseases of the nervous system. Nat Neurosci 5:1131–1136. 10.1038/nn1102-1131 [DOI] [PubMed] [Google Scholar]
- Endo M, Ohashi K, Sasaki Y, Goshima Y, Niwa R, Uemura T, Mizuno K (2003) Control of growth cone motility and morphology by LIM kinase and slingshot via phosphorylation and dephosphorylation of cofilin. J Neurosci 23:2527–2537. 10.1523/JNEUROSCI.23-07-02527.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey U, Morris RG (1997) Synaptic tagging and long-term potentiation. Nature 385:533–536. 10.1038/385533a0 [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG (1998) Synaptic tagging: implications for late maintenance of hippocampal long-term potentiation. Trends Neurosci 21:181–188. 10.1016/S0166-2236(97)01189-2 [DOI] [PubMed] [Google Scholar]
- Friedman WJ. (2000) Neurotrophins induce death of hippocampal neurons via the p75 receptor. J Neurosci 20:6340–6346. 10.1523/JNEUROSCI.20-17-06340.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry JJ, Barker PA, Carter BD (2004) The p75 neurotrophin receptor: multiple interactors and numerous functions. Prog Brain Res 146:25–39. 10.1016/S0079-6123(03)46002-0 [DOI] [PubMed] [Google Scholar]
- Graves LA, Heller EA, Pack AI, Abel T (2003) Sleep deprivation selectively impairs memory consolidation for contextual fear conditioning. Learn Mem 10:168–176. 10.1101/lm.48803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunwald T, Lehnertz K, Heinze HJ, Helmstaedter C, Elger CE (1998) Verbal novelty detection within the human hippocampus proper. Proc Natl Acad Sci U S A 95:3193–3197. 10.1073/pnas.95.6.3193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Lee CW, Fan Y, Komlos D, Tang X, Sun C, Yu K, Hartzell HC, Chen G, Bamburg JR, Zheng JQ (2010) ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nature Neurosci 13:1208–1215. 10.1038/nn.2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Peng X, Fang J (2004) Sleep deprivation impairs spatial memory and decreases extracellular signal-regulated kinase phosphorylation in the hippocampus. Brain Res 1018:38–47. 10.1016/j.brainres.2004.05.032 [DOI] [PubMed] [Google Scholar]
- Hagewoud R, Whitcomb SN, Heeringa AN, Havekes R, Koolhaas JM, Meerlo P (2010) A time for learning and a time for sleep: the effect of sleep deprivation on contextual fear conditioning at different times of the day. Sleep 33:1315–1322. 10.1093/sleep/33.10.1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanert A, Weber FD, Pedersen A, Born J, Bartsch T (2017) Sleep in humans stabilizes pattern separation performance. J Neurosci 37:12238–12246. 10.1523/JNEUROSCI.1189-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havekes R, Park AJ, Tolentino RE, Bruinenberg VM, Tudor JC, Lee Y, Hansen RT, Guercio LA, Linton E, Neves-Zaph SR, Meerlo P, Baillie GS, Houslay MD, Abel T (2016a) Compartmentalized PDE4A5 signaling impairs hippocampal synaptic plasticity and long-term memory. J Neurosci 36:8936–8946. 10.1523/JNEUROSCI.0248-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havekes R, Park AJ, Tudor JC, Luczak VG, Hansen RT, Ferri SL, Bruinenberg VM, Poplawski SG, Day JP, Aton SJ, Radwańska K, Meerlo P, Houslay MD, Baillie GS, Abel T (2016b) Sleep deprivation causes memory deficits by negatively impacting neuronal connectivity in hippocampal area CA1. eLife 5:e13424. 10.7554/eLife.13424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havekes R, Vecsey CG, Abel T (2012) The impact of sleep deprivation on neuronal and glial signaling pathways important for memory and synaptic plasticity. Cell Signal 24:1251–1260. 10.1016/j.cellsig.2012.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh SH, Ferraro GB, Fournier AE (2006) Myelin-associated inhibitors regulate cofilin phosphorylation and neuronal inhibition through LIM kinase and slingshot phosphatase. J Neurosci 26:1006–1015. 10.1523/JNEUROSCI.2806-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibáñez CF, Simi A (2012) p75 neurotrophin receptor signaling in nervous system injury and degeneration: paradox and opportunity. Trends Neurosci 35:431–440. 10.1016/j.tins.2012.03.007 [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Storm DR (1999) Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron 23:11–14. 10.1016/S0896-6273(00)80747-3 [DOI] [PubMed] [Google Scholar]
- Ji D, Wilson MA (2007) Coordinated memory replay in the visual cortex and hippocampus during sleep. Nat Neurosci 10:100–107. 10.1038/nn1825 [DOI] [PubMed] [Google Scholar]
- Knight R. (1996) Contribution of human hippocampal region to novelty detection. Nature 383:256–259. 10.1038/383256a0 [DOI] [PubMed] [Google Scholar]
- Kopp C, Longordo F, Nicholson JR, Lüthi A (2006) Insufficient sleep reversibly alters bidirectional synaptic plasticity and NMDA receptor function. J Neurosci 26:12456–12465. 10.1523/JNEUROSCI.2702-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause AJ, Simon EB, Mander BA, Greer SM, Saletin JM, Goldstein-Piekarski AN, Walker MP (2017) The sleep-deprived human brain. Nat Rev Neurosci 18:404–418. 10.1038/nrn.2017.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledoux L, Sastre JP, Buda C, Luppi PH, Jouvet M (1996) Alterations in c-fos expression after different experimental procedures of sleep deprivation in the cat. Brain Res 735:108–118. 10.1016/0006-8993(96)00599-9 [DOI] [PubMed] [Google Scholar]
- Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV, Jaenisch R (1992) Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell 69:737–749. 10.1016/0092-8674(92)90286-L [DOI] [PubMed] [Google Scholar]
- Li Q, Rothkegel M, Xiao ZC, Abraham WC, Korte M, Sajikumar S (2014) Making synapses strong: metaplasticity prolongs associativity of long-term memory by switching synaptic tag mechanisms. Cereb Cortex 24:353–363. 10.1093/cercor/bhs315 [DOI] [PubMed] [Google Scholar]
- Lingor P, Teusch N, Schwarz K, Mueller R, Mack H, Bähr M, Mueller BK (2007) Inhibition of rho kinase (ROCK) increases neurite outgrowth on chondroitin sulphate proteoglycan in vitro and axonal regeneration in the adult optic nerve in vivo. J Neurochem 103:181–189. 10.1111/j.1471-4159.2007.04756.x [DOI] [PubMed] [Google Scholar]
- Lu B, Pang PT, Woo NH (2005) The yin and yang of neurotrophin action. Nat Rev Neurosci 6:603–614. 10.1038/nrn1726 [DOI] [PubMed] [Google Scholar]
- Lu Y, Ji Y, Ganesan S, Schloesser R, Martinowich K, Sun M, Mei F, Chao MV, Lu B (2011) TrkB as a potential synaptic and behavioral tag. J Neurosci 31:11762–11771. 10.1523/JNEUROSCI.2707-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S (1999) Signaling from rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285:895–898. 10.1126/science.285.5429.895 [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG (2000) Synaptic plasticity and memory: an evaluation of the hypothesis. Ann Rev Neurosci 23:649–711. 10.1146/annurev.neuro.23.1.649 [DOI] [PubMed] [Google Scholar]
- McDermott CM, Hardy MN, Bazan NG, Magee JC (2006) Sleep deprivation-induced alterations in excitatory synaptic transmission in the CA1 region of the rat hippocampus. J Physiol 570:553–565. 10.1113/jphysiol.2005.093781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott CM, LaHoste GJ, Chen C, Musto A, Bazan NG, Magee JC (2003) Sleep deprivation causes behavioral, synaptic, and membrane excitability alterations in hippocampal neurons. J Neurosci 23:9687–9695. 10.1523/JNEUROSCI.23-29-09687.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada D, Ballarini F, Viola H (2015) Behavioral tagging: a translation of the synaptic tagging and capture hypothesis. Neural Plast 2015:650780. 10.1155/2015/650780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada D, Viola H (2006) Phosphorylation state of CREB in the rat hippocampus: a molecular switch between spatial novelty and spatial familiarity? Neurobiol Learn Mem 86:9–18. 10.1016/j.nlm.2005.12.002 [DOI] [PubMed] [Google Scholar]
- Moncada D, Viola H (2007) Induction of long-term memory by exposure to novelty requires protein synthesis: evidence for a behavioral tagging. J Neurosci 27:7476–7481. 10.1523/JNEUROSCI.1083-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadella KS, Saji M, Jacob NK, Pavel E, Ringel MD, Kirschner LS (2009) Regulation of actin function by protein kinase A-mediated phosphorylation of Limk1. EMBO Rep 10:599–605. 10.1038/embor.2009.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niklison-Chirou MV, Steinert JR, Agostini M, Knight RA, Dinsdale D, Cattaneo A, Mak TW, Melino G (2013) TAp73 knockout mice show morphological and functional nervous system defects associated with loss of p75 neurotrophin receptor. Proc Natl Acad Sci U S A 110:18952–18957. 10.1073/pnas.1221172110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S, Mizuno K (2000) Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem 275:3577–3582. 10.1074/jbc.275.5.3577 [DOI] [PubMed] [Google Scholar]
- Prince TM, Abel T (2013) The impact of sleep loss on hippocampal function. Learn Mem 20:558–569. 10.1101/lm.031674.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran B, Frey JU (2009) Interfering with the actin network and its effect on long-term potentiation and synaptic tagging in hippocampal CA1 neurons in slices in vitro. J Neurosci 29:12167–12173. 10.1523/JNEUROSCI.2045-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasch B, Born J (2013) About sleep's role in memory. Physiol Rev 93:681–766. 10.1152/physrev.00032.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravassard P, Pachoud B, Comte JC, Mejia-Perez C, Scoté-Blachon C, Gay N, Claustrat B, Touret M, Luppi PH, Salin PA (2009) Paradoxical (REM) sleep deprivation causes a large and rapidly reversible decrease in long-term potentiation, synaptic transmission, glutamate receptor protein levels, and ERK/MAPK activation in the dorsal hippocampus. Sleep 32:227–240. 10.1093/sleep/32.2.227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raven F, Meerlo P, Van der Zee EA, Abel T, Havekes R (2019) A brief period of sleep deprivation causes spine loss in the dentate gyrus of mice. Neurobiol Learn Mem 160:83–90. 10.1016/j.nlm.2018.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raven F, Van Der Zee EA, Meerlo P, Havekes R (2017) The role of sleep in regulating structural plasticity and synaptic strength: implications for memory and cognitive function. Sleep Med Rev 39:3–11. 10.1016/j.smrv.2017.05.002 [DOI] [PubMed] [Google Scholar]
- Redondo RL, Morris RG (2011) Making memories last: the synaptic tagging and capture hypothesis. Nat Rev Neurosci 12:17–30. 10.1038/nrn2963 [DOI] [PubMed] [Google Scholar]
- Rex CS, Lin CY, Kramár EA, Chen LY, Gall CM, Lynch G (2007) Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J Neurosci 27:3017–3029. 10.1523/JNEUROSCI.4037-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD (1999) The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci 19:4337–4348. 10.1523/JNEUROSCI.19-11-04337.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rösch H, Schweigreiter R, Bonhoeffer T, Barde YA, Korte M (2005) The neurotrophin receptor p75NTR modulates long-term depression and regulates the expression of AMPA receptor subunits in the hippocampus. Proc Natl Acad Sci U S A 102:7362–7367. 10.1073/pnas.0502460102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth TL, Sweatt JD (2008) Rhythms of memory. Nat Neurosci 11:993–994. 10.1038/nn0908-993 [DOI] [PubMed] [Google Scholar]
- Roux PP, Barker PA (2002) Neurotrophin signaling through the p75 neurotrophin receptor. Prog Neurobiol 67:203–233. 10.1016/S0301-0082(02)00016-3 [DOI] [PubMed] [Google Scholar]
- Rudy JW. (2015) Actin dynamics and the evolution of the memory trace. Brain Res 1621:17–28. 10.1016/j.brainres.2014.12.007 [DOI] [PubMed] [Google Scholar]
- Sachs BD, Baillie GS, McCall JR, Passino MA, Schachtrup C, Wallace DA, Dunlop AJ, MacKenzie KF, Klussmann E, Lynch MJ, Sikorski SL, Nuriel T, Tsigelny I, Zhang J, Houslay MD, Chao MV, Akassoglou K (2007) p75 neurotrophin receptor regulates tissue fibrosis through inhibition of plasminogen activation via a PDE4/cAMP/PKA pathway. J Cell Biol 177:1119–1132. 10.1083/jcb.200701040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A, Miyajima K, Akatsuka J, Kondo H, Mashiko T, Kiuchi T, Ohashi K, Mizuno K (2013) Ca MKII β-mediated LIM-kinase activation plays a crucial role in BDNF-induced neuritogenesis. Genes Cells 18:533–543. 10.1111/gtc.12054 [DOI] [PubMed] [Google Scholar]
- Sajikumar S, Korte M (2011) Metaplasticity governs compartmentalization of synaptic tagging and capture through brain-derived neurotrophic factor (BDNF) and protein kinase mzeta (PKMzeta). Proc Natl Acad Sci U S A 108:2551–2556. 10.1073/pnas.1016849108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajikumar S, Li Q, Abraham WC, Xiao ZC (2009) Priming of short-term potentiation and synaptic tagging/capture mechanisms by ryanodine receptor activation in rat hippocampal CA1. Learn Mem 16:178–186. 10.1101/lm.1255909 [DOI] [PubMed] [Google Scholar]
- Schapiro AC, McDevitt EA, Rogers TT, Mednick SC, Norman KA (2018) Human hippocampal replay during rest prioritizes weakly learned information and predicts memory performance. Nat Commun 9:3920. 10.1038/s41467-018-06213-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty MS, Sharma M, Hui NS, Dasgupta A, Gopinadhan S, Sajikumar S (2015) Investigation of synaptic tagging/capture and cross-capture using acute hippocampal slices from rodents. J Vis Exp 103:e53008. 10.3791/53008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokri-Kojori E, Wang GJ, Wiers CE, Demiral SB, Guo M, Kim SW, Lindgren E, Ramirez V, Zehra A, Freeman C, Miller G, Manza P, Srivastava T, De Santi S, Tomasi D, Benveniste H, Volkow ND (2018) β-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc Natl Acad Sci U S A 115:4483–4488. 10.1073/pnas.1721694115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindreu CB, Scheiner ZS, Storm DR (2007) Ca2+ -stimulated adenylyl cyclases regulate ERK-dependent activation of MSK1 during fear conditioning. Neuron 53:79–89. 10.1016/j.neuron.2006.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson RE, Li Z, Li Z, Minichiello L, Riddle RC, Venkatesan A, Clemens TL (2017) NGF-TrkA signaling in sensory nerves is required for skeletal adaptation to mechanical loads in mice. Proc Natl Acad Sci U S A 114:E3632–E3641. 10.1073/pnas.1701054114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tönges L, Koch JC, Bähr M, Lingor P (2011) ROCKing regeneration: rho kinase inhibition as molecular target for neurorestoration. Front Mol Neurosci 4:39. 10.3389/fnmol.2011.00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy CM, Friedman JE, Friedman WJ (2002) Mechanisms of p75-mediated death of hippocampal neurons role of caspases. J Biol Chem 277:34295–34302. 10.1074/jbc.M205167200 [DOI] [PubMed] [Google Scholar]
- Underwood CK, Coulson EJ (2008) The p75 neurotrophin receptor. Int J Biochem Cell Biol 40:1664–1668. 10.1016/j.biocel.2007.06.010 [DOI] [PubMed] [Google Scholar]
- van der Borght K, Ferrari F, Klauke K, Roman V, Havekes R, Sgoifo A, van der Zee EA, Meerlo P (2006) Hippocampal cell proliferation across the day: increase by running wheel activity, but no effect of sleep and wakefulness. Behav Brain Res 167:36–41. 10.1016/j.bbr.2005.08.012 [DOI] [PubMed] [Google Scholar]
- Vecsey CG, Baillie GS, Jaganath D, Havekes R, Daniels A, Wimmer M, Huang T, Brown KM, Li XY, Descalzi G, Kim SS, Chen T, Shang YZ, Zhuo M, Houslay MD, Abel T (2009) Sleep deprivation impairs cAMP signalling in the hippocampus. Nature 461:1122–1125. 10.1038/nature08488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schack D, Casademunt E, Schweigreiter R, Meyer M, Bibel M, Dechant G (2001) Complete ablation of the neurotrophin receptor p75 NTR causes defects both in the nervous and the vascular system. Nat Neurosci 4:977–978. 10.1038/nn730 [DOI] [PubMed] [Google Scholar]
- Wang KC, Kim JA, Sivasankaran R, Segal R, He Z (2002) P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 420:74–78. 10.1038/nature01176 [DOI] [PubMed] [Google Scholar]
- Wong ST, Henley JR, Kanning KC, Huang KH, Bothwell M, Poo MM (2002) A p75(NTR) and nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat Neurosci 5:1302–1308. 10.1038/nn975 [DOI] [PubMed] [Google Scholar]
- Woo NH, Teng HK, Siao CJ, Chiaruttini C, Pang PT, Milner TA, Hempstead BL, Lu B (2005) Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat Neurosci 8:1069–1077. 10.1038/nn1510 [DOI] [PubMed] [Google Scholar]
- Wu BQ, Bi ZG, Qi Q (2013) Inactivation of the rho-ROCK signaling pathway to promote neurologic recovery after spinal cord injuries in rats. Chin Med J 126:3723–3727. 10.3760/cma.j.issn.0366-6999.20111068 [DOI] [PubMed] [Google Scholar]
- Wulff K, Gatti S, Foster RG, Wettstein JG (2010) Sleep and circadian rhythm disruption in psychiatric and neurodegenerative disease. Nat Rev Neurosci 11:589–599. 10.1038/nrn2868 [DOI] [PubMed] [Google Scholar]
- Yamashita T, Tucker KL, Barde YA (1999) Neurotrophin binding to the p75 receptor modulates rho activity and axonal outgrowth. Neuron 24:585–593. 10.1016/S0896-6273(00)81114-9 [DOI] [PubMed] [Google Scholar]
- Yamuy J, Morales FR, Chase MH (1995) Induction of rapid eye movement sleep by the microinjection of nerve growth factor into the pontine reticular formation of the cat. Neuroscience 66:9–13. 10.1016/0306-4522(95)00052-K [DOI] [PubMed] [Google Scholar]
- Yamuy J, Sampogna S, Chase MH (2000) Neurotrophin-receptor immunoreactive neurons in mesopontine regions involved in the control of behavioral states. Brain Res 866:1–14. 10.1016/S0006-8993(00)02204-6 [DOI] [PubMed] [Google Scholar]
- Yeo TT, Chua-Couzens J, Butcher LL, Bredesen DE, Cooper JD, Valletta JS, Mobley WC, Longo FM (1997) Absence of p75NTR causes increased basal forebrain cholinergic neuron size, choline acetyltransferase activity, and target innervation. J Neurosci 17:7594–7605. 10.1523/JNEUROSCI.17-20-07594.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SO, Casaccia-Bonnefil P, Carter B, Chao MV (1998) Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J Neurosci 18:3273–3281. 10.1523/JNEUROSCI.18-09-03273.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]