

Abstract
We previously identified that ngr1 allele deletion limits the severity of experimental autoimmune encephalomyelitis (EAE) by preserving axonal integrity. However, whether this favorable outcome observed in EAE is a consequence of an abrogated neuronal-specific pathophysiological mechanism, is yet to be defined. Here we show that, Cre-loxP-mediated neuron-specific deletion of ngr1 preserved axonal integrity, whereas its re-expression in ngr1−/− female mice potentiated EAE-axonopathy. As a corollary, myelin integrity was preserved under Cre deletion in ngr1flx/flx, retinal ganglion cell axons whereas, significant demyelination occurred in the ngr1−/− optic nerves following the re-introduction of NgR1. Moreover, Cre-loxP-mediated axon-specific deletion of ngr1 in ngr1flx/flx mice also demonstrated efficient anterograde transport of fluorescently-labeled ChTxβ in the optic nerves of EAE-induced mice. However, the anterograde transport of ChTxβ displayed accumulation in optic nerve degenerative axons of EAE-induced ngr1−/− mice, when NgR1 was reintroduced but was shown to be transported efficiently in the contralateral non- recombinant adeno-associated virus serotype 2-transduced optic nerves of these mutant mice. We further identified that the interaction between the axonal motor protein, Kinesin-1 and collapsin response mediator protein 2 (CRMP2) was unchanged upon Cre deletion of ngr1. Whereas, this Kinesin-1/CRMP2 association was reduced when NgR1 was re-expressed in the ngr1−/− optic nerves. Our data suggest that NgR1 governs axonal degeneration in the context of inflammatory-mediated demyelination through the phosphorylation of CRMP2 by stalling axonal vesicular transport. Moreover, axon-specific deletion of ngr1 preserves axonal transport mechanisms, blunting the induction of inflammatory demyelination and limiting the severity of EAE.
SIGNIFICANCE STATEMENT Multiple sclerosis (MS) is commonly induced by aberrant immune-mediated destruction of the protective sheath of nerve fibers (known as myelin). However, it has been shown that MS lesions do not only consist of this disease pattern, exhibiting heterogeneity with continual destruction of axons. Here we investigate how neuronal NgR1 can drive inflammatory-mediated axonal degeneration and demyelination within the optic nerve by analyzing its downstream signaling events that govern axonal vesicular transport. We identify that abrogating the NgR1/pCRMP2 signaling cascade can maintain Kinesin-1-dependent anterograde axonal transport to limit inflammatory-mediated axonopathy and demyelination. The ability to differentiate between primary and secondary mechanisms of axonal degeneration may uncover therapeutic strategies to limit axonal damage and progressive MS.
Keywords: axonal degeneration, collapsin response mediator protein 2, demyelination, experimental autoimmune encephalomyelitis, Kinesin-1, Nogo receptor 1
Introduction
In contrast to the original research focus defining the autoimmune mechanisms that are operative in multiple sclerosis (MS) and the therapeutic developments that have achieved substantive reductions in relapse rates, axonal degeneration and neural tissue injury still remain the arbiter of profound neurological deficit in individuals living with this severe disease (for review, see Lee et al., 2014). Two models of axonopathy/demyelination have been suggested in MS pathology; primary axonal degeneration or oligodendrocyte dystrophy followed by demyelination (inside-out), or demyelination leading to secondary axonal degeneration (outside-in; Trapp and Nave, 2008). Although there is no clear indication of exact cause and effect in MS lesions, the validity of models precipitating axonal degeneration can be argued.
Nogo-A, an integral protein of oligodendrocytes and myelin, with potent neurite outgrowth inhibitory effects has been an attractive research target for the development of MS therapeutics, (for review, see Lee and Petratos, 2013). We have previously demonstrated that antagonizing this myelin-associated inhibitory factors (MAIFs) in a mouse model of MS [experimental autoimmune encephalomyelitis (EAE)] can reduce the incidence, severity, and pathological sequelae of EAE by limiting axonal damage (Karnezis et al., 2004; Petratos et al., 2012). Moreover, our recent finding that mice deficient for the Nogo-66 receptor (ngr1−/−) display a reduced severity of EAE, emphasizes the importance of investigating the molecular mechanism(s) underscoring this result (Petratos et al., 2012). However, as NgR may be expressed on other cell types, particularly those of immune cell lineage (Fry et al., 2007; David et al., 2008), there is a unique requirement to dissect out whether the neuroprotective effects exhibited by ngr1−/− mice are a consequence of inhibited axonal-specific mechanisms or abrogated inflammatory sequelae. Furthermore, as there are now documented ultrastructural anatomical differences exhibited in ngr1−/− mice that include axodendritic synaptic stability (Wills et al., 2012; Akbik et al., 2013) and recently documented white matter axo-glial units (Lee et al., 2017), it is important to delineate whether the axonal preservation germane to EAE-induced ngr1−/− mice (Petratos et al., 2012), is based on direct NgR1-dependent signaling or indirect ultrastructural differences, whereby the plasticity-dependent responsiveness of neural cells may limit degeneration.
In this study, we addressed the axon-specific role of NgR1 during neuroinflammation through recombinant adeno-associated virus serotype 2 (rAAV2) transduction of retinal ganglion cells (RGCs) by intraocular delivery of either, Cre recombinase to adult ngr1flx/flx or, full-length NgR1 to ngr1−/− mice, before EAE-induction. We report that axon-specific deletion of ngr1 preserves axons and conversely, the re-expression of NgR1 in ngr1−/− optic nerves exacerbates axonopathy during neuroinflammatory insult. Anterograde transport of vesicles labeled by AlexaFluor 555 Cholera toxin β (ChTxβ), showed accumulation in the proximal optic nerve segments of EAE-induced wild-type, or ngr1−/− mice following the re-expression of NgR1. In cortical neuronal cultures we identified that stimulation of NgR1 with Nogo-66 can slow or stall the anterograde trafficking of AlexaFluor 555 ChTxβ and pDendra-labeled amyloid precursor protein (APP). These data which demonstrate the NgR1-dependent reduction of anterograde transport, were further substantiated in the spinal cords and optic nerves of EAE-induced mice, where we identified reduced interactions between the Kinesin-1 motor protein and collapsin response mediator protein 2 (CRMP2), a result of increased phosphorylation of CRMP2 (Petratos et al., 2012). Moreover, the preservation of axons upon deletion of axonal ngr1 also preserved the integrity of myelin, whereas re-expression of NgR1 caused demyelination (inside-out).
Our results support the neurobiological role of NgR1 driving axonopathy during EAE-induced neuroinflammation and that the preservation of axons observed in ngr1−/− or ngr1-Cre-deleted mice is a consequence of preserved anterograde axonal transport. Furthermore, to our knowledge we provide the first experimental evidence that the direct inhibition of NgR1-dependent axonopathy also prevents demyelination in EAE with the working hypothesis that the axonal status plays an active role in the maintenance of CNS myelin under inflammatory conditions.
Materials and Methods
Animals.
C57BL/6 ngr1+/+, ngr1−/− (Kim et al., 2004), and ngr1flx/flx (Wang et al., 2011) mice were bred and maintained at the Alfred Medical Research and Education Precinct animal facility. The AMREP Animal Ethics Committee (AEC nos. E/1532/2015/M and E/1602/2015/M) approved the use of these animals for experimentation in accordance with the guidelines and regulations set out by the National Health and Medical Research Council of Australia. All animal experiments are governed by the Australian Code for the care and use of animals for scientific purposes (2013) and comply with the Victorian Cruelty to Animals Act 1986. Both, age- and sex-matched (female) ngr1+/+, ngr1flx/flx, and ngr1−/− mice were used in the experimental paradigms outlined in this study.
AAV2 construction and production.
The construction of recombinant AAV2 carrying enhanced green fluorescent protein (eGFP) reporter (rAAV2-eGFP), eGFP reporter and improved Cre (rAAV2-eGFP-iCre), eGFP reporter and full-length mouse NgR1 (rAAV2-eGFP-mNgR1) were generated through Vector BioLabs. Generation of rAAV2 carrying phospho-mutant CRMP2 and eGFP reporter (rAAV2-Flag-CRMP2T555A-eGFP) was previously described (Petratos et al., 2012). Full vector sequences are available on request.
Intraocular injections and EAE induction.
Intraocular injections of rAAV2 vectors were performed as previously described (Petratos et al., 2012). Animals were anesthetized by intraperitoneal injection of 100 μl per 10 g of body weight consisting: 2% xylazine (7 mg/kg body weight) and 10% ketamine (95 mg/kg body weight). Subsequently, local eye anesthetic was applied at the site of injection. Using a finely pulled micro-syringe, 1.5 μl of rAAV2-eGFP, rAAV2-eGFP-iCre, rAAV2-eGFP-NgR1, or rAAV2-Flag-CRMP2T555A-eGFP (0.75 × 1010 genomic copies per eye) was injected directly into the vitreous of the right eye. Following injection, the animals were monitored for 7 d then immunized with MOG35–55 as described previously (Petratos et al., 2012). All animal groups were randomized by coding individual cages to limit experimenter bias during EAE clinical score assessment. The codes and data were stored electronically and as hard copies by the senior investigator (S.P.) and two other co-authors (P.M.A. and S.T.). The mice were then left to progress to the peak stage of disease where ongoing spinal cord axonal degeneration is frequently observed in and around axonal inflammatory lesions (18–21 d post-EAE induction; Petratos et al., 2012). The day before killing the animals, they were anesthetized by ketamine/xylazine and 2 μl of AlexaFluor 555 ChTxβ (C34776, Life Technologies) was injected directly into the vitreous of the left and right eye. The animals were then monitored for 1 h post-injection and experimental mouse groups decoded only following the collection of neural tissues.
Retinal whole-mounts.
Retinal whole-mounts were prepared as previously described (Petratos et al., 2012). They were postfixed with 4% paraformaldehyde (PFA) for 20 min and blocked with blocking buffer consisting of 10% normal goat serum and 0.3% Triton X-100 in PBS for 2 h at RT. Sections were subsequently immunostained with primary antibodies (Table 1) diluted in blocking buffer overnight at 4°C. Secondary antibodies (Table 2) were diluted in blocking buffer and applied for 2 h at RT. The sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (D1306, Life Technologies; 1:2000) for 15 min at RT. To analyze myelin integrity, sections were counterstained with FluoroMyelin Red (F34652, Life Technologies; 1:300) for 1 h at RT. The sections were then mounted and coverslipped using fluorescent mounting medium (Dako).
Table 1.
Primary antibodies used in study
| Antibody | Dilution | Catalog | Supplier | Application |
|---|---|---|---|---|
| Rabbit anti-GFP | 1:500 | Ab290 | Abcam | Retina whole-mounts, Optic nerve immunofluorescence |
| Mouse anti-βIII-tubulin | 1:2000 | MMS-435P | Covance | Retina whole-mounts |
| Mouse anti-AT8 | 1:40 | MN1020 | ThermoFisher Scientific | Optic nerve immunofluorescence |
| Mouse anti-βAPP clone 3E9 | 1:100 | MA1-25489 | ThermoFisher Scientific | Optic nerve and Human post-mortem MS brain immunofluorescence |
| Mouse anti-SMI32 | 1:1000 | SMI-32R | Covance | Optic nerve and Human post-mortem MS brain immunofluorescence |
| Rat anti-NgR | 1:100 | MAB1659 | R&D Systems | Optic nerve immunofluorescence |
| Mouse anti-NF-200 | 1:400 | N0142 | Sigma-Aldrich | Optic nerve immunofluorescence |
| Rabbit anti-pCRMP2T555 | 1:200 | (Petratos et al., 2012; Mokhtar et al., 2018) | Optic nerve immunofluorescence | |
| Mouse anti-MBP | 1:1000 | NE1018 | Millipore | Human post-mortem brain and EAE-induced spinal cord tissues. |
| Mouse anti-NgR | 1:100 | MAB1208 | R&D Systems | Human post-mortem MS brain immunofluorescence |
| Rabbit anti-proteolipid (PLP) | 1:500 | Ab28486 | Abcam | Human post-mortem MS brain immunofluorescence |
| Rabbit anti-pCRMP2-T555 | 1:40,000 | (Petratos et al., 2012; Mokhtar et al., 2018) | Western blot | |
| Mouse anti-CRMP2 | 1:40,000 | 11096 | IBL | Western blot |
| Rabbit anti-KIF5c | 1:4000 | LS-C81914 | LSBio | Western blot |
| Mouse anti-β-actin | 1:50,000 | MAB1501 | Millipore | Western blot |
| Mouse anti-Kinesin light chain-1 (KLC-1) | 1:2000 | MAB1617 | Millipore | Western blot |
| Mouse anti-α-tubulin | 1:10,000 | 05-829 | Millipore | Western blot |
| Biotinylated anti-GFP | 1:200 | Ab6658 | Abcam | Oligodendrocyte apoptosis assay |
| Mouse anti-CC1 | 1:50 | M11217 | Calbiochem | Oligodendrocyte apoptosis assay |
| Rabbit anti-cleaved caspase-3 | 1:400 | 9661 | Cell Signaling Technology | Oligodendrocyte apoptosis assay |
Table 2.
Secondary antibodies used in study
| Antibody | Dilution | Catalogue | Supplier | Application |
|---|---|---|---|---|
| AlexaFluor 488 conjugated goat anti-rabbit IgG (H + L) | 1:500 | A-11008 | Life Technologies | Retina whole-mount, Optic nerve immunofluorescence |
| AlexaFluor 546 conjugated goat anti-mouse IgG (H + L) | 1:500 | A-11030 | Life Technologies | Retina whole-mount, Optic nerve immunofluorescence, Oligodendrocyte apoptosis assay |
| AlexaFluor 647 conjugated goat anti-rat IgG (H + L) | 1:500 | A-21247 | Life Technologies | Retina whole-mount, Optic nerve immunofluorescence, Oligodendrocyte apoptosis assay |
| HRP-conjugated goat anti-mouse IgG (H + L) | 1:20,000 | AB308P | Millipore | Western blot |
| HRP-conjugated goat anti-rabbit IgG (H + L) | 1:20,000 | AB307P | Millipore | Western blot |
| Streptavidin, AlexaFluor 488 conjugated | 1:500 | S11223 | Life Technologies | Oligodendrocyte apoptosis assay |
Quantitative PCR after reverse-transcription.
mRNA was isolated from rAAV2-transduced retinae, and nontransduced retinae of naive ngr1+/+, ngr1−/−, and ngr1flx/flx mice 7 d post-intraocular injection by using RNeasy Mini kit (Qiagen) with DNase-I to remove genomic DNA. From the isolated mRNA, cDNA was synthesized using a high-capacity cDNA reverse-transcription kit following the manufacturer's instructions (Applied Biosystems). Quantitative PCR after reverse-transcription (qRT-PCR) was performed with 50 ng of cDNA using TaqMan probes (Applied Biosystems): mouse Reticulon-4 receptor (rtn4r) (Mm0045228_m1) mouse gapdh (Mm99999915_g1) on ABI Prism 7900HT sequence detection system (Applied Biosystems). The CT value was measured in triplicate and normalized to the gapdh endogenous control. These ΔCT values of samples were then standardized against the ΔCT values of non-rAAV2-transduced retinae of ngr1+/+ mice, derived as relative mRNA expression (ΔΔCT).
Preparation of tissues.
Animals (uninjected naive control and EAE-induced mice) were killed by CO2 inhalation then transcardially perfused with PBS. For mRNA and protein extraction, the optic nerves, retinae, and lumbosacral spinal cords were collected and immediately snap frozen and stored at −80°C. For histological experiments, following PBS perfusion, mice were perfused with 4% PFA. The optic nerves and retinae were postfixed with 4% PFA overnight at 4°C for immunofluorescence analysis or immunogold-labeled electron microscopy (EM; see Fig. 5-1).
Optic nerve immunofluorescence.
Optic nerves from naive and EAE-induced mice were prepared for immunofluorescence as previously described (Petratos et al., 2012). Optic nerves were embedded in OCT and 10 μm serial longitudinal sections were cut on a cryostat. Before sectioning, all embedded tissue was coded by a different investigator (S.T.) listed in the authorship and electronically categorized, then stored by the senior investigator. The subsequent analysis was thereby performed blinded by the dual first authors, to exclude bias. Three sections per optic nerve, at least 50 μm apart, were selected to perform all quantification following immunohistochemistry. Briefly, sections were postfixed with 4% PFA for 20 min and blocked with blocking buffer (10% normal goat serum and 0.3% Triton X-100 in PBS) for 2 h at RT. Sections were subsequently immunostained with primary antibodies (Table 1) diluted in blocking buffer overnight at 4°C. Secondary antibodies (Table 2) were diluted in blocking buffer and applied for 2 h at RT. The sections were counterstained with DAPI (D1306, Life Technologies; 1:2000) for 15 min at RT. To analyze myelin integrity, sections were counterstained with FluoroMyelin Red (F34652, Life Technologies; 1:300) for 1 h at RT. The sections were then mounted and coverslipped using fluorescent mounting medium (Dako). The fluorescence intensity of FluoroMyelin was measured surrounding eGFP+ axons by identifying the Integrated density (IntDen) on ImageJ. All measurements performed were normalized against background intensity.
Quantification of axonal degeneration.
Briefly, at least three sections (> 50 μm apart) from each mouse optic nerve (minimum of n = 3 mice per experimental group) were used to analyze the number of degenerative axons. Axonal degeneration was defined according to the number of eGFP+ (i.e., rAAV2-transduced axons) that exhibited the presence of swelling retraction bulbs/spheroids at the distal end of the axon transection with fragmentation (Nikić et al., 2011). This number of eGFP+ swollen axons at the distal end was then divided by the total number of eGFP+ (rAAV2-transduced axons) and represented as a percentage. Validation of these morphological outcomes was performed by quantifying the number of colabeled eGFP+ axons with either SMI32, βAPP, or AT8 divided by the total number of eGFP+ axons.
Quantification of the fluorescent intensity of ChTxβ.
The fluorescent intensity of ChTxβ was measured exclusively in the eGFP+ axons as IntDen on a separate window on ImageJ. The transport of ChTxβ- on rAAV2-injected and uninjected optic nerves was quantified by measuring the intensity at the proximal, intermediate, and distal parts, all separated by a 400 μm length of nerve. All the measurements were normalized against the background intensity.
Preparation of primary cortical neuron cultures.
E15–E16 C56Bl/6 wild-type and ngr1−/− pregnant mice were killed by cervical dislocation. The cortical neuron cultures were prepared as described by Reddrop et al. (2005) and Samson et al. (2008). Briefly, the isolated cortices pellet was trypsinized for 5 min at 37°C with gentle agitation and followed by addition of trypsin inhibitor. The pellet was triturated through an 18-gauge blunt-ended needle. The resultant cell pellet resuspended in neurobasal medium, supplemented with 1 × B27 supplement, 10% dialyzed fetal calf serum, 1% Glutamax (Invitrogen), and 1% penicillin/streptomycin (P/S; Invitrogen). The cell suspension was seeded onto poly-d-lysine (0.05 mg/ml; BD Bioscience) precoated 6-well plates at 150,000 cells/cm2. Twenty-four hours after seeding [1 d in vitro (DIV)], the serum-containing medium was aspirated and replaced with serum-free neurobasal medium supplemented with 1× B27 supplement, 1% Glutamax and 1% P/S. The culture was monitored every day for neurite outgrowth and replaced with 1:1 fresh medium every 3 d.
Nogo-66 ligand stimulation of Nogo receptor in primary cortical neurons.
At 7 DIV, a scratch injury was performed to the culture by aspirating the middle of the plate with a glass pasture pipette, followed by the addition of Nogo-66 ligand to stimulate Nogo-66 receptor (NgR1). Briefly, 330 ng/cm2 of Nogo-66 (488230, Calbiochem) was added to each well in a 6-well plate (Fournier et al., 2003). For control experiments, 3 μg of Nogo-66 ligand was pre-absorbed with 0.3 μg full-length NgR-Fc peptide (1440-NG, R&D systems) by incubating in the medium for 30 min at 37°C and centrifuged at 15,000 × g for 10 min at 37°C, then adding the supernatant to the culture within the 6-well plate, separately. The cell lysates were collected 8 h after the stimulation.
Primary cortical neuron culture in the microfluidic chamber and live cell imaging.
Primary cortical neuron culture prepared as described in Preparation of primary cortical neuron cultures. Cells (100,000) were seeded in each chamber. At 7 DIV, AlexaFluor 555 ChTxβ subunit (1 μl) was added to the soma side of the microfluidic chamber (RD150, Xona Microfluidics), 1 d before live cell imaging. The transection injury was performed on the axonal side by aspirating the chamber then immediately 0.3 μg Nogo-66 peptide was added. Three hours after the stimulation, live cell imaging was performed using 40× objective lens on a Nikon A1r confocal microscope. KymoAnalyzer (Neumann et al., 2017) was performed to generate kymographs and determine the velocity of the axonal transport.
Transient transfection of primary cortical neurons with pDendra-Fag-βAPP.
At 7 DIV, the culture was transfected with 5 μg of pDendra-Flag-βAPP cDNA using Lipofectamine 3000 (Invitrogen). The scratch injury was performed by aspirating the middle of the plate with a glass pasture pipette, then adding 3 μg Nogo-66 within the aspirated groove. Three hours after the stimulation, live cell imaging was performed using a 40× objective lens on a Nikon A1r confocal microscope and time-lapse videos captured over a 15 min interval. The cell lysates were then collected for immunoprecipitation and Western blotting, which were performed as described in the sections Immunoprecipitation and Western immunoblotting.
Human MS tissue histology and immunofluorescence.
Human MS tissues were embedded in OCT compound and 10 μm sections were prepared then stained using Luxol fast blue and periodic acid Schiff (LFB-PAS) to demonstrate demyelinated plaques and inflammatory cell infiltration as described previously (Petratos et al., 2012). Immunofluorescence was performed according to standard procedures (see Optic nerve immunofluorescence).
Human tissue selection.
All frozen human deep-cortical white matter tissue was acquired from the Victorian Brain Bank Network (VBBN) under the National Health and Medical Research Council guidelines and the Monash University Human Research Ethics Committee approval number CF13/1646–2013000831. Human brain tissue included five subjects with non-neurological disease control (NNDC), other neurological disease controls including four subjects with Alzheimer's disease (AD), four subjects with frontotemporal dementia (FTD), four subjects with Huntington's disease (HD), and five subjects with progressive MS with active lesions (Table 3). Postmortem interval did not exceed 56 h. All specimens were frozen under liquid nitrogen and then stored in the Brain Biobank (−80°C until required). Tissue sections were coded and stored electronically and as hard copies by the senior investigator (S.P.) and two other co-authors (P.M.A. and S.T.). Isolated proteins underwent either immunoprecipitation and/or Western immunoblotting analyses, performed according to standard procedures for all tissues (see the sections Western immunoblotting and Immunoprecipitation).
Table 3.
Details of human tissues
| Patient No. | Details | Age | Sex | Postmortem intervals, h |
|---|---|---|---|---|
| 1 | MS/AD | 65.1 | Female | 12 |
| 2 | MS | 38.5 | Male | 25 |
| 3 | MS | 49.9 | Male | 43 |
| 4 | MS | 62.7 | Female | 56 |
| 5 | MS | 49.9 | Male | 56.5 |
| 6 | AD | 71.4 | Male | 9.5 |
| 7 | AD | 73 | Male | 9.5 |
| 8 | AD | 83.8 | Male | 10 |
| 9 | AD | 88.4 | Female | 11 |
| 10 | FTD | 60.6 | Male | 12 |
| 11 | FTD | 61.4 | Female | 20.5 |
| 12 | FTD | 77 | Male | 6 |
| 13 | FTD | 66.8 | Male | 37 |
| 14 | HD | 61.1 | Male | 17 |
| 15 | HD | 66.7 | Female | 18.5 |
| 16 | HD | 57.2 | Female | 22 |
| 17 | HD | 72.2 | Female | 22 |
| 18 | NNDC | 52.1 | Male | 33 |
| 19 | NNDC | 82.7 | Male | 27 |
| 20 | NNDC | 63.4 | Female | 30.5 |
| 21 | NNDC | 63.9 | Male | 31.5 |
Histological analysis (human postmortem brain).
Images immunostained with anti-PLP/βAPP and anti-MBP/pCRMP2-T555 antibodies (Table 1) were analyzed through ImageJ for quantitation. To estimate the level of axonal degeneration, the number of βAPP+ axons were manually counted in regions predefined as normal-appearing white matter (NAWM), periplaque white matter (PPWM) + lesion border, and NAWM from LFB-PAS-stained sections. The degree of axonal degeneration was represented as the number of βAPP+ axons within the analyzed area per square millimeter of tissue area. For quantitation of demyelinated regions around pCRMP2-T555-positive axons was assessed by measuring MBP+ immunostained area surrounding pCRMP2-T555+ axons, counted within the NAWM (n = 47 axons measured, across 4 separate tissue samples) or PPWM (n = 33 axons measured, across 4 separate tissue samples). More specifically, using the Imaris software, the intensity of MBP immunostaining that was <1 (A.U.). Subsequently the degree of demyelination was represented as the area under the curve (MBP fluorescence intensity <1).
Western immunoblotting.
Fresh-frozen mouse lumbosacral spinal cords, optic nerves, and human brain tissues were ground into a fine powder in liquid N2 and then lysed in 1× RIPA buffer (Cell Signaling Technology) supplemented with protease and phosphatase inhibitor cocktails (Millipore). Homogenates were centrifuged at 13,000 × g for 20 min and the supernatants were collected. Protein concentrations were determined using BCA assay (ThermoFisher Scientific). For each Western immunoblot, 5 μg of protein was denatured and differentiated on a 4–12% Bis-Tris gel (Invitrogen). The gels were then electrophoretically transferred onto PVDF membranes (Millipore). The membranes were blocked with TBS containing 5% nonfat dried milk powder including and 0.1% Tween 20. Primary antibodies (Table 1) were incubated overnight at 4°C, then secondary antibodies (Table 2) were incubated for 1 h at RT. Immunoreactive proteins were detected using an ECL Prime kit (GE Healthcare). Densitometric analysis was performed using ImageJ and the level of immunoreactive signal was normalized against the ubiquitous actin loading-control band.
Immunogold labeled EM.
Tokuyasu sample preparation was performed as previously described (Slot and Geuze, 2007). Optic nerves were fixed overnight at 4°C with 0.1 m phosphate buffered 2% (w/v) EM-graded PFA and 0.2% (w/v) glutaraldehyde. The fixed samples were embedded in 12% (w/v) gelatin in 0.1 m phosphate buffer at 37°C, which was allowed to set at 4°C before being cut into small cubes measuring ∼0.5 mm on each edge. The gelatin embedded tissues were infiltrated with 2.3 m sucrose in 0.1 m phosphate buffer at 4°C overnight on a rocker. The sucrose infiltrated gelatin blocks were mounted on cryo-pins then frozen in liquid nitrogen for cryo-ultramicrotomy. Frozen samples were trimmed at −100°C and sectioned at −120°C using a Cryo-EM UC7 ultra microtome (Leica) equipped with a 45° diamond cryo-trimming knife (Diatome) and a 35° diamond cryo-immuno knife (Diatome). Cryosections were retrieved by pick-up loop with a droplet of phosphate buffered 1% (w/v) methyl cellulose and 1.15 m sucrose, then deposited on carbon-coated Formvar grids for immunolabeling. The grids were prepared for immunolabeling by melting up-side-down on PBS at 37°C, then rinsing the grids on droplets of 0.02 m glycine in PBS. The grids were blocked with 1% (w/v) bovine serum albumin (BSA) in PBS, incubated with goat anti-GFP-Biotin (600–106-215, 1:600, Rockland) in 1% (w/v) BSA in PBS at RT, for 1 h and incubated with a bridging rabbit anti-biotin (100–4198, 1:10,000; Rockland) then rinsed with 0.1% (w/v) BSA in PBS. The grids were then incubated with protein-A-conjugated 10 nm gold particles diluted (1:50; Cell Biology, Utrecht University, Utrecht, The Netherlands) in 1% (w/v) BSA in PBS for 20 min at RT, rinsed with PBS. After stabilization of the reaction by 1% (w/v) glutaraldehyde in PBS for 5 min, grids were rinsed in distilled water. Finally, the grids were stained with 2% (w/v) uranyloxaalacetate (pH 7) for 5 min at RT and 0.4% (w/v) uranyl acetate in 1.8% (w/v) aqueous methyl cellulose (pH 4) for 5 min at 4°C and dried in a thin film of the final stain in the center of a wire loop. EM imaging was done on JEM-1400Flash (Jeol).
Immunoprecipitation.
One hundred micrograms of either mouse or human brain tissue lysate was incubated overnight at 4°C with 1 μg of mouse anti-CRMP2 (Table 1) capture antibody, under constant agitation. These samples were then incubated with 50 μl of protein G magnetic beads (Millipore) to capture the antibody-antigen complex. Samples were then denatured, and Western immunoblotting was performed (see Western immunoblotting).
Analysis of oligodendrocyte apoptosis.
Optic nerve immunofluorescence was performed according to standard procedures as defined (see Optic nerve immunofluorescence). Apoptotic oligodendrocytes were defined as CC-1-immunolabeled cells (CC-1+) with cleaved caspase-3-immunolabeled (caspase-3+) nuclei that appeared condensed and fragmented, as assessed by DAPI counterstaining. The data were plotted as the number of cleaved caspase-3/CC-1+ cells divided by the total number of CC-1+ oligodendrocytes.
Statistical analyses.
Statistical analyses were conducted on GraphPad Prism v7. For Densitometric analysis and stereological counts, a Student's t test or one-way analysis of variance (ANOVA) with Tukey's post hoc test was used. The data is presented as mean ± standard error of the mean (SEM). To analyze the differences between genotypes during EAE progression, a two-way ANOVA was performed. A p value <0.05 was considered statistically significant.
Results
RGC-specific deletion or overexpression of NgR1 regulates axonal degeneration during EAE
We previously reported that NgR1-dependent phosphorylation of CRMP2 may potentiate axonal degeneration during neuroinflammation (Petratos et al., 2012). Here, we investigated how selective deletion of NgR1 in neurons, may prevent this signal transduction pathway from executing axonal degeneration during EAE. As a corollary, we also posited the question: how would the re-expression of full-length NgR1 in ngr1−/− CNS neurons modify axonal integrity during EAE?
For axon-specific manipulation of the NgR1, we used an intraocular delivery system of the rAAV2 as previously described (Petratos et al., 2012). Recombinant AAV2-eGFP, rAAV2-eGFP expressing the improved Cre (rAAV2-eGFP-iCre), and rAAV2-eGFP expressing mouse full-length NgR1 were generated (Fig. 1A–C). One week before EAE induction, direct intraocular injections into the vitreous cavity of right mouse eyes were performed using these vectors, to transduce RGCs and their axons. Retinal whole mount immunofluorescence revealed ∼40% of βIII-tubulin-labeled RGCs to be consistently transduced by all three rAAV2 vectors (Fig. 1G). qRT-PCR analysis of mRNA collected from the retinae revealed a ∼twofold downregulation of ngr1 upon rAAV2-eGFP-iCre transduction in ngr1flx/flx mice, and a ∼twofold upregulation upon rAAV2-eGFP-mNgR1 ngr1−/− mice compared with the uninjected left eye (Fig. 1H). Therefore, we verified that this rAAV2-mediated gene delivery system efficiently transduces RGCs to modulate the expression of NgR1.
Figure 1.
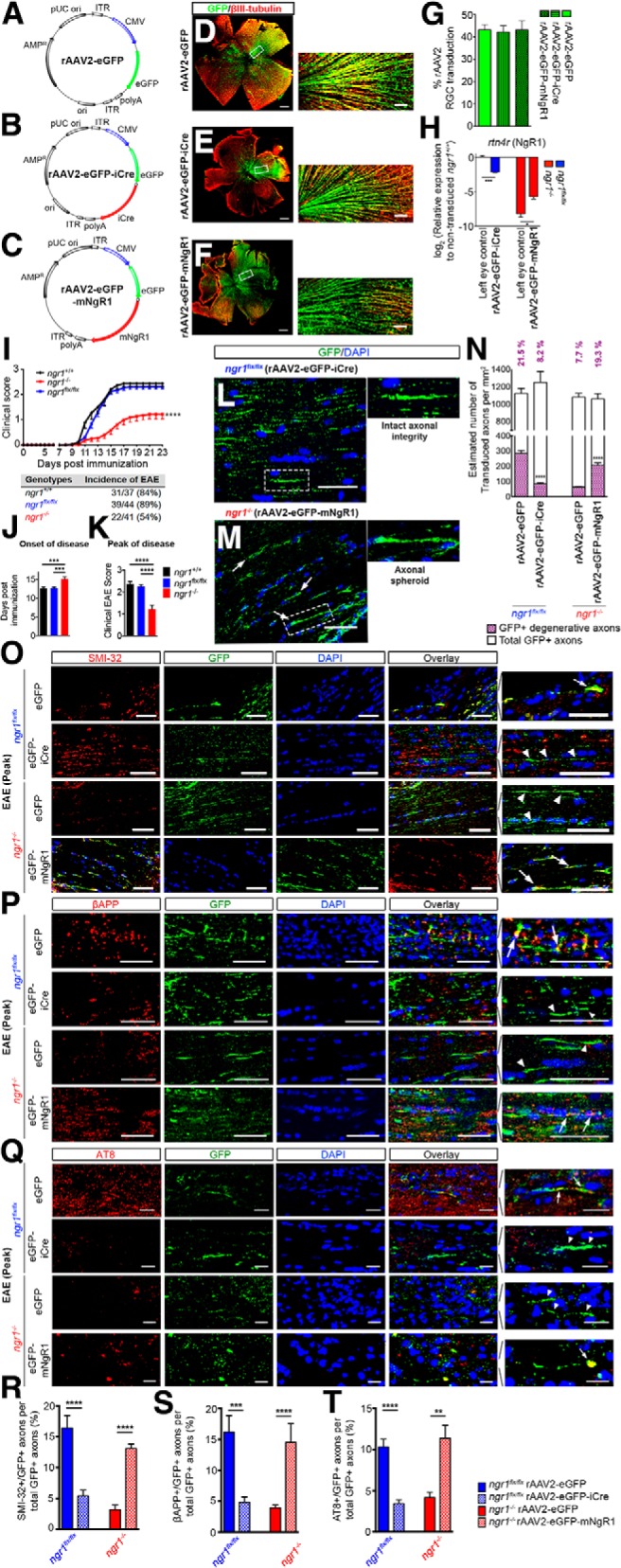
Axon-specific ablation of ngr1 preserves axonal integrity during EAE whereas its re-expression potentiates axonal degeneration. A–C, rAAV2 vector maps that used in this study, (A) rAAV2-eGFP, (B) rAAV2-eGFP-iCre, and (C) rAAV2-eGFP-mNgR1. D–F, Representative retinal whole-mounts immunolabeled for eGFP and βIII-tubulin from (D) rAAV2-eGFP-, (E) rAAV2-eGFP-iCre-, and (F) rAAV2-eGFP-mNgR1-injected retinae. Scale bar, 500 μm. Magnified images showing colocalization of eGFP with βIII-tubulin are shown at the right-hand side. Scale bar, 50 μm. G, The percentage of rAAV2 transduction in RGCs (n = 22–29). H, qRT-PCR analysis of rAAV2-eGFP-iCre transduced retinae of ngr1flx/flx and rAAV2-eGFP-mNgR1 transduced retinae of ngr1−/− mice and their left retinae controls using rtn4r (encoding NgR1) probe (n = 3, mean ± SEM, one-way ANOVA with Tukey's post test; *p < 0.05, ***p < 0.001). I, Clinical progression of EAE upon immunization with CFA and MOG35–55 in female ngr1+/+ (n = 37), ngr1flx/flx (n = 44), and ngr1−/− (n = 41) mice (mean ± SEM, repeated-measures ANOVA; ****p < 0.0001). The incidence of EAE for each genotype is shown underneath the graph. J, Average onset of EAE, defined as the day of the first clinical score reached in each mouse. K, Average peak score of disease (mean ± SEM, one-way ANOVA with Tukey's post test; ***p < 0.001, ****p < 0.0001). L, M, Representative images immunolabeled for eGFP and DAPI from (L) rAAV2-eGFP-iCre-injected right optic nerve of ngr1flx/flx mice and (M) rAAV2-eGFP-mNgR1-injected right optic nerve of ngr1−/− mice at the peak stage of EAE. Magnified images of single eGFP+ transduced axons are shown underneath the larger scaled images. N, Estimated numbers of transduced axons and morphologically degenerative axons per square millimeter of right optic nerve sections in either rAAV2-eGFP- or rAAV2-eGFP-iCre-injected ngr1flx/flx and either rAAV2-eGFP- or rAAV2-eGFP-mNgR1-injected ngr1−/− mice at the peak stage of EAE. (n = 10, Student's t test; ****p < 0.0001). O–Q, Representative images from either rAAV2-eGFP- or rAAV2-eGFP-iCre-injected optic nerves of ngr1flx/flx mice and either rAAV2-eGFP- or rAAV2-eGFP-mNgR1-injected optic nerves of ngr1−/− mice coimmunolabeled for either (O) SMI-32, (P) βAPP, or (Q) AT8 and eGFP, followed by DAPI counterstaining. Scale bar, 50 μm. Magnified images of intact (in rAAV2-eGFP-iCre-injected ngr1flx/flx and rAAV2-eGFP-injected ngr1−/− mice optic nerves) or degenerative axons (in rAAV2-eGFP-injected ngr1flx/flx and rAAV2-eGFP-mNgR1-injected ngr1−/− optic nerves) are shown on the right (arrow indicates degenerative axons whereas arrowhead indicates intact axons). Scale bar, 10 μm. R–T, The percentage of colocalization of eGFP+ transduced axons with (R) SMI-32, (S) βAPP, and (T) AT8 (n = 10, mean ± SEM, Student's t test; **p < 0.01, ***p < 0.001, ****p < 0.0001). For axon-specific overexpression of NgR1 in optic nerves of naive ngr1−/− mice (Figure 1-1).
Axon-specific overexpression of NgR1 in optic nerves of naïve ngr1-/- mice did not lead into axonal degeneration (A) Immunolabeling of rAAV2-eGFP-mNgR1 injected optic nerve sections of naïve ngr1-/- mice for NF-200, GFP, NgR1, and DAPI counterstaining. (B) Immunolabeling of serial section for βAPP, GFP, and DAPI counterstaining. (C) The percentage of degenerative GFP+ axons over total number of GFP+ transduced axons (n = 4, mean ± SEM). Download Figure 1-1, TIF file (18.7MB, tif)
We first analyzed rAAV2-eGFP-mNgR1-injected optic nerves of naive (non-EAE) ngr1−/− mice to investigate possible axonal changes, which may occur without inflammation (basal changes as a consequence of NgR1 re-expression). We verified the expression of axonal NgR1. However, we could not detect axonal abnormalities, morphologically or by demonstrating βAPP accumulation in the transduced optic nerves (eGFP+/NgR1+; Fig. 1-1). This indicates that re-introduction of NgR1 to RGC axons, does not cause damage, without inflammatory infiltrates being present.
In line with our previous data generated for ngr1−/− mice (Petratos et al., 2012), we demonstrated a significant delay in the onset of EAE and reduction in the peak clinical score compared with ngr1+/+ or ngr1flx/flx mice (Fig. 1I–K). Furthermore, by Day 18 post-EAE induction, only 54% of ngr1−/− mice exhibited any form of clinical symptoms, whereas 84% of ngr1+/+ and 89% of ngr1flx/flx mice displayed EAE onset and progression (Fig. 1I). Immunofluorescence studies performed on transduced optic nerves at Days 18–21 post-EAE induction (corresponding to peak stage of EAE) revealed a significant reduction in morphologically identifiable degenerative axons of ngr1flx/flx mice transduced with rAAV2-eGFP-iCre (mean ± SEM: 8.220 ± 0.4239%) compared with the optic nerves transduced with rAAV2-eGFP transduction (controls; mean ± SEM: 21.53 ± 0.7954%). Furthermore, a significant increase in morphologically identifiable degenerative axons in the optic nerves of ngr1−/− mice transduced with rAAV2-eGFP-mNgR1 (mean ± SEM: 19.25 ± 1.298%) was identified compared with rAAV2-eGFP transduction in the same genotype (controls; mean ± SEM: 7.734 ± 0.3381%; Fig. 1L–N).
To further demonstrate axonal degeneration, we immunostained serial sections of rAAV2-transduced optic nerves localizing SMI-32, βAPP, and hyperphosphorylated tau (AT8). Consistent with the observed morphological changes of axonal degeneration observed within the eGFP+ transduced optic nerve axons following intraocular injection with rAAV2-eGFP-iCre, we identified significant reductions in the colocalization of SMI-32, βAPP, and AT8 (compared with the rAAV2-eGFP-injected optic nerves of ngr1flx/flx mice; Fig. 1O–T). Conversely, we demonstrated significant increases in the colocalization of all axonal dystrophy markers with GFP+ expression in the rAAV2-eGFP-mNgR1-injected optic nerves compared with rAAV2-eGFP-injected control optic nerves from ngr1−/− mice (Fig. 1O–T). These results suggest that axonal NgR1 drives axonopathy during neuroinflammation.
Inhibiting axon-specific NgR1-dependent signaling preserves axonal transport in the optic nerves during EAE
To assess the importance of neuronal NgR1-dependent signaling during neuroinflammation in the optic nerves of EAE-induced mice, we used the vesicular axonal transport qualities of ChTxβ subunit conjugated to the AlexaFluor 555 (ChTxβ-555) label. We identified stalling of ChTxβ-555-labeled vesicles as accumulation of immunofluorescence within dystrophic axons in the proximal ends of optic nerves that were either transduced with rAAV2-eGFP-mNgR1 in ngr1−/− mice (right optic nerves) following EAE induction, or in the nontransduced optic nerves (left optic nerves) of EAE-induced ngr1+/+ or ngr1flx/flx mice (Fig. 2B,C). The intensity of ChTxβ-555 fluorescence was minimal in the proximal ends of rAAV2-eGFP-iCre transduced optic nerves of ngr1flx/flx mice (Fig. 2C). This reduction was also observed within the rAAV2-Flag-CRMP2T555A-eGFP transduced optic nerves (phospho-mutant CRMP2 construct, Rho-associated kinase II phosphorylation site) of ngr1+/+ mice (Fig. 2B).
Figure 2.

Anterograde axonal transport is stalled when NgR1-dependent phosphorylation of CRMP2 is potentiated in dystrophic axons during EAE progression. A, Schematic diagram of proximal, intermediate, and distal segments (400 μm apart) of optic nerves. Representative images from right (rAAV2 transduced) and left (rAAV2 uninjected) optic nerve of (B) rAAV2-CRMP2T555A-eGFP-injected ngr1+/+ mice, (C) rAAV2-eGFP-iCre-injected ngr1flx/flx mice, and (D) rAAV2-eGFP-mNgR1-injected ngr1−/− mice. Scale bar, 70 μm. E, Number of eGFP+ rAAV2-transduced axons and accumulation of intense Alexa Fluor 555 ChTxβ in eGFP+ degenerative axons per square millimeter of sections from optic nerve, at the peak stage of EAE (n = 3, mean ± SEM, one-way ANOVA with Tukey's post test; ****p < 0.0001). F, Percentage of rAAV2-transduced axons that are dystrophic (n = 3, mean ± SEM, one-way ANOVA with Tukey's post test; *p < 0.05). G, The intensity of ChTxβ+ dystrophic axons in the area of rAAV2-transduced axons (n = 3, mean ± SEM, one-way ANOVA with Tukey's post test; **p < 0.01, ***p < 0.001). H, I, Anterograde ChTxβ transport at peak stage of EAE in (H) rAAV2-transduced right optic nerve, and (I) rAAV2 uninjected contralateral optic nerve (n = 3, mean ± SEM, one-way ANOVA with Tukey's post test; **p < 0.01, ***p < 0.001). For the net velocity of in vitro axonal transport with and without stimulation of Nogo-66 (Figure 2-1).
The net velocity of axonal transport with and without stimulation of Nogo66 after transection injury. (A) Schematic diagram of the compartmentalized culture in the microfluidic chamber where the transection injury and subsequent Nogo66 stimulation were performed. (B) The net velocities of the tracks in each group. Representative time-lapse of ChTxβ added cortical neurons from ngr1+/+ mice (C) untreated and (D) treated with 0.3 μg of Nogo66 peptide after transection injury. These time-lapse videos are available as Movies 1 and 2, respectively. Representative time-lapse of ChTxβ added cortical neurons from ngr1-/- mice (E) untreated and (F) treated with 0.3 μg of Nogo66 peptide after transection injury. These time-lapse videos are available as Movies 3 and 4, respectively. (G-K) Representative kymographs generated from time-lapse videos of each group. Colored lines represent the tracks that were measured for net velocities. (n = 80 tracks, mean ± SEM, one-way ANOVA, ****P<0.0001). Scale bars = 20 μm. Download Figure 2-1, TIF file (14.4MB, tif)
These observations were further validated when we measured the ChTxβ-555 fluorescence intensity along the distance of the optic nerve, from the proximal optic disc to the chiasm. We identified that rAAV2-eGFP-mNgR1 transduced axons of ngr1−/− mice following EAE induction, displayed increased accumulation of ChTxβ-555 fluorescence intensity in the proximal segment of the right optic nerve compared with the contralateral nontransduced left optic nerve (Fig. 2B–D). We calculated the amount of ChTxβ-555 fluorescence accumulation in the rAAV2-eGFP-mNgR1 transduced axons of ngr1−/− mice following EAE induction, to be 126 ± 10 axons/mm2 of right optic nerve tissue, a threefold increase in the level of accumulation compared with rAAV2-CRMP2T555A-eGFP transduced optic nerves of ngr1+/+ mice (43 ± 8 axons/mm2); or twofold increase compared with rAAV2-eGFP-iCre transduced optic nerves of ngr1flx/flx mice (57 ± 8 axons/mm2; Fig. 2E). The overall percentage of dystrophic axons in the rAAV2-eGFP-mNgR1 transduced optic nerves of ngr1−/− mice, was 28.6 ± 3.7% of all transduced (eGFP+) axons (Fig. 2F,G).
We further interrogated the ChTxβ-555 fluorescence accumulation in the transduced or non-transduced optic nerve at the proximal, intermediate and distal segments, to assess the level of vesicular transport stalling in the axons of EAE-induced mice at the peak stage of disease (Fig. 2H,I). We demonstrated that in the proximal and intermediate segments of the rAAV2-eGFP-mNgR1 transduced right optic nerves of EAE induced ngr1−/− mice, the intensity of ChTxβ-555 fluorescence was 50.3 ± 6.6 and 53.3 ± 7.3 (A.U.) in these segments, respectively (Fig. 2H). This compared with 34.6 ± 2.8 and 13.3 ± 2.5 (A.U.) in the proximal and intermediate segments, respectively, as exhibited in the rAAV2-eGFP-iCre transduced optic nerves of ngr1flx/flx mice (Fig. 2H). Similarly, comparison levels of ChTxβ-555 fluorescence within the proximal and intermediate segments of rAAV2-CRMP2T555A-eGFP transduced optic nerves of ngr1+/+ mice were 28.3 ± 2.1 or 24.2 ± 4.3 (A.U.), respectively (Fig. 2H).
Conversely, the accumulation of ChTxβ-555 fluorescence in the uninjected (nontransduced) optic nerves of EAE-induced ngr1+/+ and ngr1flx/flx mice at the peak stage of disease, illustrated the existence of abrogated transport at the proximal and intermediate segments of the optic nerve, resembling the NgR1-overexpression studies performed in the ngr1−/− rAAV2-eGFP-mNgR1 transduced optic nerve (Fig. 2I). Because there was a greater intensity of ChTxβ-555 fluorescence detected at the distal ends in the uninjected (nontransduced) optic nerves of EAE-induced ngr1−/− mice, without accumulation in the proximal and intermediate segments, we showed that the transport of ChTxβ was unimpeded in these mice. These data suggest that stalling of anterograde vesicular transport exists in the proximal through to intermediate segments of optic nerves when NgR1 is expressed in neurons during neuroinflammation. However, does this transport deficit occur because of Nogo-A (a potent myelin inhibitory factor) ligation through NgR1?
To answer this question, we isolated cortical neurons from wild-type and ngr1−/− mice allowing them to grow in microfluidic chambers. The axons of these neurons where allowed to grow freely into the grooves of the chambers, isolating the cortical neuron somas from their distal axonal terminals that had grown into the neighboring chamber (Fig. 2-1; also time-lapse videos are available from Movies 1, 2, 3, 4). After transecting the distal axonal terminals by aspirating the axonal chamber, we added 0.3 μg of Nogo-66 peptide, thereby stimulating NgR1 present on the axolemmal growth cone (if expressed). In the ngr1+/+ cortical neuronal cultures, we identified that Nogo-66 stimulation slows and stalls anterograde axonal transport of ChTxβ+ vesicles (∼0.0019 ± 0.0015 SEM μm/s) compared with transected but unstimulated ngr1+/+ axons (∼0.038 ± 0.015 SEM μm/s; Fig. 2-1). In fact, these anterograde vesicular transport velocity measurements were significantly different to those obtained from the ngr1−/− axons that showed (∼0.232 ± 0.028 SEM μm/s without Nogo-66 and ∼0.045 ± 0.018 SEM μm/s with Nogo-66 stimulation, respectively; Fig. 2-1). These data identify the stalling of anterograde axonal transport following stimulation with Nogo-66, a mechanism that we then set out to interrogate.
Real-time imaging of axonal transport of ChTxβ-positive vesicles from ngr1+/+ cortical neurons. The movie shows simultaneous imaging of individual ChTxβ-positive (red) vesicles along the axons.

Real-time imaging of axonal transport of ChTxβ-positive vesicles from ngr1+/+ cortical neurons 3 h after treated with 0.3 μg of Nogo-66 peptide after transection injury. The movie shows simultaneous imaging of individual ChTxβ-positive (red) vesicles along the axons.

Real-time imaging of axonal transport of ChTxβ-positive vesicles from ngr1−/− cortical neurons. The movie shows simultaneous imaging of individual ChTxβ-positive (red) vesicles along the axons.

Real-time imaging of axonal transport of ChTxβ-positive vesicles from ngr1−/− cortical neurons 3 h after treated with 0.3 μg of Nogo-66 peptide after transection injury. The movie shows simultaneous imaging of individual ChTxβ-positive (red) vesicles along the axons.

Axon-specific deletion of ngr1 prevents dissociation of Kinesin-1 from CRMP2 during EAE
The vesicular cargo protein CRMP2, has been previously shown to associate with the Kinesin-1 motor protein to transport tubulin heterodimers in an anterograde manner (Kimura et al., 2005). Because NgR1-dependent phospho-CRMP2-Thr555 levels are increased within dystrophic axons during EAE (Petratos et al., 2012) and moreover, this specific phosphorylation can dissociate it from tubulin (Petratos et al., 2008, 2012), we further investigated whether this signaling can modulate the interaction of CRMP2 and Kinesin-1 [the main anterograde motor protein that transports α and β tubulin heterodimers to the “plus” ends of axonal microtubules (Yoshimura et al., 2005)], thereby impairing axonal transport during EAE and by extension MS.
Initially, we stimulated ngr1+/+ and ngr1−/− cortical neurons that were transiently transfected with the pDendra2 construct encoding a flag-tagged mouse APP, with Nogo-66 to demonstrate the association of APP with Kinesin-1 and CRMP2. Upon Nogo-66 stimulation of ngr1+/+ cultures following a scratch injury, we showed accumulation of pDendra2+ APP to the proximal end of the axon (Fig. 3B, arrowheads; also time-lapse videos are available from Movies 5, 6) compared with the more punctate and distributed vesicles exhibited without Nogo-66 stimulation or the profile observed in the ngr1−/− cortical axons (Fig. 3C; also time-lapse videos are available from Movies 7, 8). Moreover, we collected the protein lysates from the un-stimulated and stimulated cultures from this experiment and identified the association of APP with KIF5c through immunoprecipitation and Western blotting (Fig. 3E). We identified increased association of APP with Kinesin-1 in ngr1−/− compared with the ngr1+/+ cortical neuron cultures (Fig. 3E). We next investigated how CRMP2 in ngr1+/+ and ngr1−/− cortical neuron cultures can regulate the association of Kinesin-1 in the presence or absence of neurite inhibitory factor, Nogo-66. By immunoprecipitation of CRMP2 from lysates of these stimulated neurons, we were able to identify that Nogo-66 inhibited the association with CRMP2 with a ∼sixfold reduction in the level of KIF5c observed in the stimulated ngr1+/+ and ngr1−/− cortical neuron cultures (Fig. 3F,G). This association was partially recovered following the blockade of Nogo-66 by pre-incubating this ligand with the soluble NgR(310)-Fc (∼3-fold increase from Nogo-66 stimulation alone; Fig. 3F,G).
Figure 3.

Reduced interaction between CRMP2 and KIF5c within cortical neuron lysates and spinal cord lysates of ngr1+/+ compared with lysates from ngr1−/− mice during the peak stage of EAE. Representative time-lapse axonal transport of pDendra-Flag-βAPP transfected ngr1+/+ cortical neurons (A) untreated and (B) treated with 0.3 μg of Nogo-66 peptide after transection injury. These time-lapse videos are available as Movies 5 and 6, respectively. Representative time-lapse axonal transport of pDendra-Flag-βAPP transfected ngr1−/− cortical neurons (C) untreated and (D) treated with 0.3 μg of Nogo-66 peptide after transection injury. These time-lapse videos are available as Movies 7 and 8, respectively. E, Immunoprecipitation of Flag-tagged βAPP from transfected ngr1+/+ and ngr1−/− cortical neuron lysates, following a scratch injury and with or without Nogo-66 ligand treatment. Membranes were re-probed using an anti-KIF5c antibody. Pre-immunoprecipitation (5% input of protein) from spinal cord lysates are shown under each immunoprecipitation panel. F, Immunoprecipitation of CRMP2 performed on lysates of transfected ngr1+/+ and ngr1−/− cortical neuron lysates after scratch injury, with or without Nogo-66 ligand treatment. Membranes were re-probed using an anti-KIF5c antibody. Pre-immunoprecipitation (5% input of protein) from spinal cords lysates are shown under each immunoprecipitation panel. G, H, Densitometric quantification of the levels of (D) CRMP2 associated KIF5c, and (E) total KIF5c normalized to actin. I, Immunoprecipitation of CRMP2 or pCRMP2-T555 from lumbo-sacral spinal cord lysates from naive mice (controls), EAE-induced ngr1+/+ and ngr1−/− mice at the peak of EAE, followed by re-probing of these membranes using either anti-KIF5c or anti-CRMP2 antibodies. Pre-immunoprecipitation (5% input of protein) from spinal cords lysates are shown under each immunoprecipitation panel. actin used as a loading control. J–M, Densitometric quantification of the levels of (J) CRMP2 associated KIF5c, (K) total KIF5c normalized to actin, (L) pCRMP2-T555 normalized to total CRMP2, and (M) total CRMP2B normalized to actin (n = 4, mean ± SEM, one-way ANOVA with post Tukey's test; *p < 0.05, **p < 0.01, ****p < 0.0001).
Real-time imaging of axonal transport of pDendra-Flag-βAPP transfected ngr1+/+ cortical neurons. The movie shows simultaneous imaging of individual βAPP-positive (green) vesicles along the axons.
Real-time imaging of axonal transport of pDendra-Flag-βAPP transfected ngr1+/+ cortical neurons 3 h after treated with 0.3 μg of Nogo-66 peptide after transection injury. The movie shows simultaneous imaging of individual βAPP-positive (green) vesicles along the axons.

Real-time imaging of axonal transport of pDendra-Flag-βAPP transfected ngr1−/− cortical neurons. The movie shows simultaneous imaging of individual βAPP-positive (green) vesicles along the axons.

Real-time imaging of axonal transport of pDendra-Flag-βAPP transfected ngr1−/− cortical neurons 3 h after treated with 0.3 μg of Nogo-66 peptide after transection injury. The movie shows simultaneous imaging of individual βAPP-positive (green) vesicles along the axons.
We then collected protein lysates from inflamed lumbosacral spinal cords of ngr1+/+ and ngr1−/− mice at the peak stage of EAE and performed immunoprecipitation using a monoclonal anti-CRMP2 antibody. Western blotting using an anti-KIF5c (Kinesin family member 5c) antibody showed a significant reduction in the level of association with CRMP2 in spinal cord lysates from EAE-induced (peak stage) ngr1+/+ mice compared with naive ngr1+/+ controls (Fig. 3I–K). Whereas, we did not observe changes in the levels of these proteins following coimmunoprecipitation, within the spinal cords of EAE-induced (peak stage) ngr1−/− mice compared with naive ngr1−/− controls (Fig. 3I–K). We also demonstrated that the level of phospho-CRMP2-Thr555 was increased in the spinal cords of ngr1+/+ mice at the peak stage of EAE. Whereas, no modulation of pCRMP2-T555 levels was seen for spinal cord lysates obtained from ngr1−/− mice, at the peak stage of EAE (Fig. 3I,L,M). Therefore, the dissociation of KIF5c from CRMP2 in the spinal cords of ngr1+/+ mice during EAE, may be a result of NgR1-dependent signaling.
Because NgR1-dependent signaling may abrogate the interaction between CRMP2 and KIF5c, we then asked whether time-dependent and axon-specific deletion of NgR1 preserves this interaction. We collected protein lysates from rAAV2-transduced right optic nerves and their left optic nerves from uninjected eyes during EAE. We then performed immunoprecipitation and Western blotting. As expected, the left uninjected optic nerves of ngr1+/+ and ngr1flx/flx mice during EAE, showed a reduction in the levels of CRMP2 bound KIF5c compared with the optic nerves of naive ngr1+/+ mice (Fig. 4A,B). However, no difference in this interaction could be found in the left uninjected optic nerves of ngr1−/− mice during EAE (Fig. 4A,B). Importantly, both rAAV2-CRMP2T555A-eGFP-injected optic nerves of ngr1+/+ and rAAV2-eGFP-iCre-injected optic nerves of ngr1flx/flx mice showed a significant increase in the level of CRMP2-bound KIF5c compared with the left optic nerves lacking rAAV2-transduction, at the peak stage of EAE (Fig. 4A,B). Whereas, we found a reduction in the levels of CRMP2-bound KIF5c within rAAV2-eGFP-mNgR1-injected optic nerves of ngr1−/− mice compared with the left nontransduced nerves at the peak stage of EAE, although these did not reach statistical significance (Fig. 4A,B).
Figure 4.

Axon-specific deletion of ngr1 maintains the protein interactions between CRMP2 and KIF5c in the optic nerve during EAE. A, First row, Immunoprecipitation of CRMP2 of optic nerve lysates from naive ngr1+/+ and ngr1−/−; EAE-induced optic nerve lysates (at peak stage of disease) from either: ngr1+/+ transduced with rAAV2-CRMP2T555A-eGFP, ngr1flx/flx transduced with rAAV2-eGFP-iCre, ngr1−/− mice transduced with rAAV2-eGFP-mNgR1 and their left (contralateral) uninjected optic nerve controls. The membranes were then re-probed using anti-KIF5c antibody. Second row, A pre-immunoprecipitation 5% input of total protein. Third row, Western immunoblot detection of the phosphorylation of CRMP2 at the threonine 555 site (pCRMP2-T555), (fourth row) total CRMP2, and (fifth row) actin loading control. B, Densitometric quantification of total KIF5c and CRMP2 bound KIF5c (after immunoprecipitation) from optic nerve lysates. C, Densitometric quantification of level of phosphorylation of CRMP2B at T555 over total CRMP2B (n = 3, mean ± SEM, one-way ANOVA with post Tukey's test; *p < 0.05, **p < 0.01).
The dissociation of KIF5c from CRMP2 during EAE in the optic nerves of ngr1+/+ mice may be a result of direct axonal NgR1-signaling as the levels of pCRMP2-T555 were found to be reduced both in the rAAV2-CRMP2T555A-eGFP-injected optic nerves of ngr1+/+ and rAAV2-eGFP-iCre-injected optic nerves of ngr1flx/flx mice (Fig. 4A,C). Although not statistically significant, an increase in the level of phospho-CRMP2-Thr555 was observed in the rAAV2-eGFP-mNgR1-injected optic nerves of ngr1−/− mice compared with left nontransduced nerves, at the peak stage of EAE (Fig. 4A,C). These results suggest that axonal NgR1-signaling during EAE impairs Kinesin-1 association with CRMP2, a result of phosphorylation, and may govern the anterograde axonal transport blockade observed during axonal degeneration, although this hypothesis awaits further validation.
Axon-specific deletion of ngr1 preserves myelin integrity in the optic nerves during EAE
Given that axonal degeneration during EAE is potentiated by neuronal NgR1 expression, we sought to determine whether the preservation of axons by conditionally deleting neuronal NgR1 could protect the myelin sheath from degeneration, following inflammatory infiltration. FluoroMyelin immunostained sections of optic nerves from all rAAV2-injected EAE-induced mice demonstrated reductions in demyelination surrounding rAAV2-CRMP2T555A-eGFP transduced axons in ngr1+/+ mice (∼1.5-fold) and restoration in FluoroMyelin intensity compared with optic nerves of ngr1+/+ mice transduced with rAAV2-eGFP during the peak stage of EAE (Fig. 5A,B). Optic nerve myelin integrity was also preserved in EAE-induced ngr1flx/flx mice following axonal transduction with rAAV2-eGFP-iCre compared with the optic nerves of rAAV2-eGFP injected EAE-induced mice at peak stage of disease (∼1.6-fold restoration in FluoroMyelin intensity; Fig. 5A,B). On the other hand, extensive demyelination was demonstrated surrounding rAAV2-eGFP-mNgR1 transduced axons in ngr1−/− mice following EAE induction compared with rAAV2-eGFP transduced optic nerves following EAE at peak stage of disease (∼1.3-fold reduction in FluoroMyelin intensity). We observed an overall reduction of 1.8-fold FluoroMyelin intensity of optic nerves compared with naive ngr1−/− mice (Fig. 5A,B). Further observation of enhanced axo-myelin integrity was achieved at the ultrastructure level whereby immunogold-labeled rAAV2 particles carrying the iCre construct (Fig. 5-1, arrowheads) or the CRMP2T555A mutant construct (Fig. 5-1, arrowheads), localized in optic nerve axons, show lamellated intact myelin sheaths (Fig. 5-1). However, nontransduced axons within the same optic nerve segments demonstrated Wallerian degeneration with macrophage phagocytic activity (Fig. 5-1, arrows and asterisks). These results indicate that preserving axons during neuroinflammation via conditional deletion of ngr1 or, antagonizing NgR1-dependent signaling downstream to CRMP2, can also preserve myelin integrity.
Figure 5.

Axon-specific deletion of ngr1 preserves myelin integrity during EAE. A, Representative images from either rAAV2-eGFP- or rAAV2-CRMP2T555A-eGFP-injected optic nerves of ngr1+/+; rAAV2-eGFP- or rAAV2-eGFP-iCre-injected optic nerves of ngr1flx/flx, and rAAV2-eGFP- or rAAV2-eGFP-mNgR1-injected optic nerves of ngr1−/− mice; immunolabeled for FluoroMyelin, eGFP, and DAPI (arrow indicates degenerative axons whereas arrowhead indicates intact axons). Scale bar, 50 μm. B, Quantification of demyelination near eGFP+ transduced axons as measured by FluoroMyelin staining intensity per area (mm2) of optic nerve (n = 10, mean ± SEM, one-way ANOVA with post Tukey's test; *p < 0.05, *** p < 0.001, **** p < 0.0001). For immunogold-labeled electron microscopic images of the transduced axons (Figure 5-1).
Intact axonal structure with compact myelin in rAAV2-transduced axons Immunogold-labeled electron microscopic analysis of axons in (A) rAAV2-eGFP-iCre, (B) rAAV2-CRMP2T555A-GFP transduced right optic nerves, and (C) non-transduced control nerve. Arrowheads indicate the immunogold-labeled GFP+ rAAV2 particles. Asterisks indicate degenerative axons where rAAV2 was not transduced. Arrows indicate macrophages engulfing myelin debris in non-transduced control optic nerve. Scale bar = 500 nm. Download Figure 5-1, TIF file (28MB, tif)
Axon-specific deletion of ngr1 limits oligodendrocyte dystrophy in the optic nerve during EAE
Having demonstrated that rAAV2-eGFP-iCre transduction within the optic nerves of EAE-induced ngr1flx/flx mice preserves myelin integrity, but extensive demyelination exhibited in rAAV2-eGFP-mNgR1 transduced axons of ngr1−/− mice, we next investigated whether axonal degeneration and demyelination could potentiate oligodendrocyte dystrophy. We identified significant numbers of apoptotic oligodendrocytes in the rAAV2-eGFP-mNgR1 transduced optic nerves within EAE-induced ngr1−/− mice at peak stage of disease compared with rAAV2-eGFP-iCre transduced and rAAV2-CRMP2T555A-eGFP-injected optic nerves of ngr1flx/flx and ngr1+/+ mice, respectively (Fig. 6A,B; see Fig. 6-1) for the number of CC-1+ mature oligodendrocytes in optic nerves of naïve ngr1+/+ and ngr1−/− mice). In particular, we identified that the numbers of cleaved caspase-3-immunopositive, mature oligodendrocytes (CC-1+) were ∼2.5-fold greater in the rAAV2-eGFP-mNgR1 transduced axons of ngr1−/− mice (149.5 ± 7.31 per mm2) compared with the contralateral uninjected left optic nerves from the same mice (31.0 ± 5.6 per mm2; Fig. 6B,D). However, the numbers of cleaved caspase-3-immunopositive, mature oligodendrocytes (CC-1+) in rAAV2-eGFP-iCre transduced optic nerves of ngr1flx/flx mice (99.05 ± 7.67 per mm2), were reduced by ∼1.5-fold compared with the rAAV2-eGFP-mNgR1 transduced axons in ngr1−/− mice during the peak stage of EAE (Fig. 6B). A similar reduction (∼1.5-fold) was calculated for the rAAV2-CRMP2T555A-eGFP transduced axons in ngr1+/+ mice, at the peak stage of EAE (79.07 ± 10.09 per mm2; Fig. 6B). These data demonstrate that NgR1-dependent axonal expression drives oligodendrocyte dystrophy during inflammation of the optic nerve.
Figure 6.

Axon-specific deletion of ngr1 protects oligodendrocytes during EAE. Representative images of apoptotic CC-1-positive mature oligodendrocytes, expressing cleaved caspase-3 in (A) rAAV2-injected right optic nerves, and (C) rAAV2-uninjected left optic nerves. Scale bar, 50 μm. High-power images show anti-activated caspase 3 staining and the presence of pyknotic nuclei (from yellow dotted box). Number of CC-1-positive mature oligodendrocytes and cleaved caspase-3-immunopositive oligodendrocytes per square millimeter of (B) right and (D) left optic nerves at the peak stage of EAE (n = 4, mean ± SEM, one-way ANOVA with post Tukey's test; *p < 0.05, ****p < 0.0001). Arrowheads indicate apoptotic mature oligodendrocytes. For the number of CC-1+ mature oligodendrocytes in optic nerves of naive ngr1+/+ and ngr1−/− mice, see Figure 6-1.
Number of CC1+ mature oligodendrocytes in optic nerves of naïve ngr1+/+ and ngr1-/- mice. Representative images of CC1+ mature oligodendrocytes in naïve ngr1+/+ and ngr1-/- mice. Greater number of CC1+ mature oligodendrocytes in optic nerves of naïve ngr1+/+ and ngr1-/- mice per mm2 (n = 3, mean ± SEM, student's t-test, *P<0.05). Scale bars = 50μm. Download Figure 6-1, TIF file (6.5MB, tif)
NgR1 is coexpressed with phosphorylated CRMP2 in degenerating neurons of MS brain and EAE spinal cord lesions
Elevated levels of phosphorylated CRMP2 at the threonine 555 site (pCRMP2-T555) occur during the pathological sequelae governing acute spinal cord injury (Mimura et al., 2006), chronic MS lesions and in the spinal cord following EAE induction (Petratos et al., 2012). These findings have set an important precedent for investigation of the molecular mechanisms underscoring CNS axon damage during neuroinflammation. We have previously shown that in the MOG35–55 model of EAE, elevated levels of pCRMP2-T555 occur in the optic nerves of mice that express NgR1 during the peak stage of disease and are associated with significant axonopathy (Petratos et al., 2012). In this study, the optic nerves of EAE-induced mice that were transduced with rAAV2-CRMP2T555A-eGFP inhibited profound axonopathy, demyelination, and oligodendrocyte dystrophy. This suggests that by abrogating CRMP2 phosphorylation, we cannot only limit axonal damage but also preserve myelin integrity and oligodendrocyte viability. However, the direct importance of NgR1-dependent CRMP2 phosphorylation localized in MS lesions and how this relates to disruption of the axonal transport machinery is an important question that needs to be addressed. We therefore first investigated the localization of pCRMP2-T555 expressed within deep cortical white-matter lesions of postmortem frontal lobe frozen tissue samples from secondary progressive MS (Fig. 7A,B).
Figure 7.

pCRMP2-T555 localization in degenerating neuronal somata and axons in chronic-active lesions of MS brains. A, LFB-PAS immunohistochemistry showing representative chronic demyelinated lesion from secondary progressive MS frontal cortex. B, Immunostaining against anti-proteolipid protein (PLP; green) and anti-βAPP (red) on serial section showing significant numbers of βAPP-positive axons in a representative image of an active demyelinating lesion (active border) and a few βAPP-positive axons in an inactive demyelinated plaque. Scale bar, 100 μm. C, βAPP expression in MS lesions of different demyelinating activity [NAWM, periplaque along with the active lesion border (active demyelinating lesion), plaque (inactive demyelinated lesion); n = 4, mean ± SEM, one-way ANOVA with post Tukey's test; *p < 0.05, ****p < 0.0001]. Expression of pCRMP2-T555 alongside common neurodegenerative markers: (D) pCRMP2-T555 expression in the distal segment of an βAPP-positive degenerating axon, and (E) colocalization with non-phosphorylated SMI-32-positive neurofilament. F, Degenerative ovoid formation and swelling in pCRMP2-T555-positive axons in human deep cortical white matter. Arrowheads indicate extracellular localization of NgR and downstream intracellular phosphorylation of CRMP2 in a degenerative axon. G, Immunohistochemistry showing pCRMP2-T555 (red), MBP (green) in NAWM and PPWM. H, The degree of demyelination around pCRMP2-T555-positive axons is significantly higher within active demyelination lesions compared with NAWM (n = 4, mean ± SEM, Student's t test; ****p < 0.0001). Scale bar, 10 μm.
In line with our previous study (Petratos et al., 2012), pCRMP2-T555 was shown to be elevated in dystrophic axons localized largely in chronic demyelinated lesions of brain tissue from individuals with secondary progressive MS (Table 3; Fig. 7D–G). In fact, colocalization of pCRMP2-T555 with the axonal degenerative markers βAPP (Fig. 7D, arrowhead), and non-phosphorylated neurofilament, SMI-32 (Fig. 7E, arrowhead), were observed. Moreover, axolemmal NgR1 immunostaining demonstrated colabeling of axonal spheroids with intra-axonal pCRMP2-T555 (Fig. 7F, arrowhead). In areas of apparent demyelination (represented by diffuse and discontinuous MBP-immunopositive myelin sheath staining), pCRMP2-T555 immunofluorescence was of greatest intensity within the PPWM lesions (n = 33 axons measured, across 4 separate tissue samples) compared with NAWM (n = 47 axons measured, across 4 separate tissue samples; Fig. 7G,H). These results suggest that pCRMP2-T555 is involved in axonal degeneration in MS and contiguous with myelin disruption.
Elevated NgR1 occurs with disassociated CRMP2-bound Kinesin and tubulin in brain lysates from individuals with MS
We finally correlated the levels of NgR1 expressed within postmortem tissue from individuals with MS and other neurological disorders, to the association of CRMP2/tubulin and Kinesin-1 motor protein complexes, in an attempt to identify the possible molecular events governing NgR1-dependent axonal dysfunction linked with anterograde transport. We found that only the lysates from individuals with secondary progressive MS, demonstrated elevated levels of NgR1 (Fig. 8A,B). We identified that there were ∼90, 85, 80, and 70% reductions in the percentage levels of Kinesin-bound to CRMP2 in AD, MS, HD, and FTD samples, respectively, compared with brain lysates from non-neurological control tissue (Table 3; Fig. 8C,D). We further identified that ∼80, 50, 60, and 40% reductions of tubulin-bound CRMP2 occurred in AD, MS, FTD, and HD brain lysates, respectively, compared with the non-neurological controls (Fig. 8C,E). These data suggest that tubulin and Kinesin dissociation from CRMP2 could be central to neurodegeneration although a dominant molecular mechanism governing neuroinflammatory axonopathy may be related to the expression of NgR1.
Figure 8.

Increased NgR1 and putatively impaired CRMP2-depdent axonal transport in brain lysates of patients with progressive MS. A, Western immunoblot detection for (first row) NgR1 and (second row) actin loading control of brain lysates from NNDC, AD, FTD, HD, and secondary progressive MS. B, Densitometric quantification of the levels of NgR1 normalized to actin loading control (n = 4, mean ± SEM, one-way ANOVA with post Tukey's test; *p < 0.05). C, Immunoprecipitation of pCRMP2-T555 using polyclonal anti-pCRMP2-T555 antibody and probed with Kinesin light chain 1 (KLC-1) and α-tubulin. D, AD (n = 7), FTD (n = 9), HD (n = 4), and MS (n = 5) lysates have ∼90, 85, 80, and 70% decrease in the percentage of Kinesin-bound to CRMP2 compared with non-neurological disease control lysates, respectively. E, AD (n = 8), FTD (n = 9), HD (n = 4), and MS (n = 5) lysates exhibit an approximate 80, 60, 50, and 40% reduction in the percentage of α-tubulin-bound to CRMP2 compared with non-neurological disease control lysates, respectively (mean ± SEM, one-way ANOVA with post Tukey's test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Discussion
In the current study, we have defined that axon-specific NgR1 can drive inflammatory-dependent degeneration in the CNS by stalling the Kinesin-dependent anterograde transport mechanism, a direct consequence of the downstream phosphorylation of CRMP2. By using the highly efficient rAAV2 transduction system to either conditionally delete or overexpress neuronal NgR1, we identified that transduced RGCs were either salvaged from or driven toward degeneration, respectively. Importantly, we identified that by ablating NgR1 expression during inflammatory challenge in the optic nerve, not only could we preserve axonal integrity but simultaneously preserve myelin. Reintroduction of NgR1 in the ngr1−/− mice illustrated its central role in driving axonal damage and demyelination as a consequence of neuroinflammation. Moreover, the overexpression of a phospho-mutant T555A CRMP2 construct was able to structurally protect axonal and myelin integrity along with providing sustained axonal transport under neuroinflammatory conditions within the ngr1+/+ mouse optic nerve. Our data suggest that NgR1-dependent axonal damage and demyelination is potentiated under inflammatory condition, and this degeneration may be dependent upon the downstream molecular association of KIF5c with CRMP2. This molecular motor dissociation may lead to slowing or stalling anterograde vesicular axonal trafficking, leading to synaptic dysfunction.
A functional measure of CRMP2 phosphorylation is the degree to which it associates with tubulin heterodimers (Arimura et al., 2005; Kimura et al., 2005). Previous reports have implicated this mechanism during developmentally regulated axonal growth, albeit through the phosphorylation of CRMP2 at the glycogen synthase kinase-3β Thr514 site (Yoshimura et al., 2005, 2006). However, phosphomimetic mutants display a weakened ability to drive axon growth during development and following acute spinal cord injury (Nagai et al., 2016). The importance of Rho kinase-mediated Thr555 phosphorylation of CRMP2 in potentiating acute axonal damage, was demonstrated through MAIF-dependent signaling, in an acute spinal cord injury model (Mimura et al., 2006). Indeed, blocking this signaling pathway can facilitate microtubule re-association with CRMP2, directly benefiting axon growth in this injury paradigm (Mimura et al., 2006). However, there remains a knowledge gap in the molecular drivers of axonal degeneration during inflammatory demyelination. In particular, the mechanism governing frank axonal dystrophy readily observed in NAWM within the CNS of individuals with MS, is yet to be elucidated (for review, see Lassmann, 2010).
The physiological importance of Kinesin/CRMP2 association is that it can be a marker for the fidelity of anterograde axonal transport and axon growth (Kimura et al., 2005), possibly through the Kinesin molecular motor protein associating with key developmental and functionally dynamic molecular cargo such as, βAPP, Wiskott–Aldrich syndrome protein family verprolin-homologous protein (WAVE), SRA1/WAVE1 (specifically Rac1-associated protein-1/WASP family verprolin-homologous protein-1) complex/WAVE1 (Kamal et al., 2001; Kawano et al., 2005; Takata et al., 2009; Hensley et al., 2011). Stalling of Kinesin motor proteins during fast axonal transport has been shown to accumulate βAPP within swollen axons (Stokin et al., 2005). Indeed, Kinesin blockade and dystrophic axons have been reported in the pathogenesis of AD and other neurodegenerative disorders (for review, see Maday et al., 2014). Therefore, maintaining this anterograde transport is integral to the fidelity of axons and it is known that both are severely compromised during EAE and by extension, MS (Sorbara et al., 2014; Hares et al., 2017). We identified that the anterograde transport of βAPP by Kinesin-1 can be maintained if NgR1-dependent activation is abrogated.
The phosphorylation of CRMP2 can limit Kinesin anterograde motor proteins from binding and carrying vital molecular cargo for axonal growth and maintenance (Arimura et al., 2005). Our studies have identified the biochemical dissociation of Kinesin-1 from CRMP2 in chronic-active lesions of autopsy tissue obtained from secondary progressive MS patients and this was directly related to the intense βAPP localization in dystrophic axons. Therefore, the question was posed that if NgR1 signaling regulates the phosphorylation of CRMP2 during MS and EAE (Petratos et al., 2012) can this cause the stalling of Kinesin motor proteins within these axons potentiating their degeneration? Moreover, are these molecular events antecedent to myelin degeneration?
We initially asked whether the overexpression of full-length NgR1 in the RGCs from ngr1−/− mice promotes axonal damage during EAE, as a model of inside-out axonopathy. Overexpression was performed by transduction of RGCs with a rAAV2-eGFP-mNgR1, we were then able to demonstrate how NgR1 expression (within the ngr1−/− optic nerve) may promote degeneration of axons during inflammatory demyelination (assessed at peak stage of EAE) compared with those unlabeled (nontransduced axons–contralateral optic nerve) within the ngr1−/− mice, or following intraocular injection with rAAV2-eGFP in the control experiment. Our data implicate NgR1 as a driver of axonal damage in the context of CNS inflammatory challenge. Because the overexpression of NgR1 in naive ngr1−/− optic nerves did not exhibit axonal damage and there was a disconnect between the preservation of axons in the ngr1flx/flx optic nerve following Cre-mediated deletion, with axonal damage exhibited in the uninjected contralateral nerve of the same mice. This suggests that inflammatory-mediated axonal damage can be driven through NgR1 signaling.
It is then plausible that either: (1) the limitation of myelin debris enriched with Nogo-A, initiating signaling through the NgR1-dependent phosphorylation of CRMP2 (inflammatory-mediated demyelination and secondary axonal damage; outside-in axonopathy), preserves axons or; (2) limiting the phosphorylation of CRMP2 within the axon prevents myelin from degenerating (inside-out axonal damage; secondary demyelination) and abrogates the cycle of inflammatory cell infiltration within the CNS. These outside-in versus inside-out mechanisms of axonopathy still require investigation, particularly because Nogo-A is also highly expressed in axons and may play an active role in driving degeneration during neuroinflammation (Ineichen et al., 2017; Zemmar et al., 2018). Although neuronal Nogo-A-dependent signaling downstream to CRMP2 has not been well defined but has been identified in the regulation of axonal transport (Joset et al., 2010).
It was previously demonstrated that axonal transport deficits precede structural/morphological alterations of myelin during EAE (Sorbara et al., 2014). If indeed it holds true that the axon deficit can propagate the demyelinating pathology because of the blockade of vesicular transport, then it would seem that the physiological status of CRMP2 is fundamental for the preservation of an integrated CNS. Indeed recent protein interactome data have identified a further 78 protein partners for CRMP2 in the brain, vital for healthy neuronal function and plasticity (Martins-de-Souza et al., 2015). Importantly, the direct relevance of CRMP2 post-translational modifications in neurodegenerative pathologies have been highlighted through the targeting of proteins that may interact with CRMP2 (for review, see Taghian et al., 2012) or small molecules such as lanthionine ketimine-5-ethyl ester (LKE), that can facilitate the binding of CRMP2 to other proteins (Dupree et al., 2015). Importantly, the later small molecule has recently been trialed in EAE experiments whereby the investigators demonstrated significant protection of axonal caliber and myelin sheaths (Dupree et al., 2015), highlighting the translational potential for this cell-permeable analog of lanthionine ketamine. Whether LKE can in fact potentiate the association of CRMP2 with Kinesin and re-establish fast axonal transport remains to be elucidated.
It is now evident that the morphological hallmarks of axonal injury can precede histologically detectable demyelination (Onuki et al., 2001; Wang et al., 2005), and correlate strongly with the progression of EAE in mice (Ayers et al., 2004; Nikić et al., 2011). NAWM histologically resembles appropriately myelinated axonal fibers situated at least 1 cm from the edges of MS plaques (Filippi et al., 2012). However, NAWM from the CNS of MS patients demonstrates diffuse pathology, which includes inflammatory infiltrates, microglial activation and demyelination but of note is the loss of axons with ∼12–42% reduction in axonal density within the spinal cord and corpus callosum (Evangelou et al., 2000; Bjartmar et al., 2001; Moll et al., 2011). Despite its histopathological classification, the mechanism governing axonal degeneration in NAWM remains elusive. Recently, an elegant study identified that transport deficits actually precede structural alterations of axons and their myelin (Sorbara et al., 2014). These investigators argue that under the breach of CNS inflammation, axonal transport is disrupted before demyelination or damage to organelles and that this can be reversed through therapeutically administering redox scavenging agents (Nitric oxide scavenger, 2-(4-Carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide [cPTIO]) and anti-inflammatory methylprednisolone. The data highlight that inflammation is fundamentally linked to axonal transport deficits that can lead to progressive neurodegeneration. Our work suggests that NgR1 expression in axons under inflammatory conditions can propagate transport deficits in secondary progressive MS and support the recent identification synaptic dysfunction in experimental glaucoma models (Fu et al., 2011; Wang et al., 2015).
The hypothesis for induction of axonal degeneration manifest in MS, may suggest that the inflammatory attack of axons (outside-in) or primary neuronal and/or oligodendrocyte (inside-out) degeneration can both ultimately result in a similar neurological outcome, despite the possibility of differing pathogenesis. Here we show that the Nogo-A/NgR1/pCRMP2-T555 signal cascade drives axonal transport deficits that can lead to the degeneration of axons before robust demyelination and disease severity in EAE. Importantly, this mechanism may be operative during secondary progressive MS. Directly targeting axon-specific signaling may be a plausible therapeutic approach to limit neurological decline in MS patients.
Footnotes
This work was supported by a Multiple Sclerosis Research Australia and Trish Multiple Sclerosis Research Foundation Postgraduate Scholarship to J.Y.L.; by National Multiple Sclerosis Society Project Grant RG4398A1/1, International Progressive Multiple Sclerosis Alliance Challenge Award PA0065, Multiple Sclerosis Research Australia and Trish Multiple Sclerosis Research Foundation 15-022, and Bethlehem Griffiths Research Foundation BGRF1706 to S.P.; and by the NIH to S.M.S. We thank Stephen Cody from Monash Micro Imaging, Central Clinical School, Monash University, Melbourne, Australia.
The authors declare that S.M.S. is a co-founder and consultant for ReNetX Bio, which seeks to develop NgR1-based therapies for neural repair. The remaining authors declare no competing financial interests.
References
- Akbik FV, Bhagat SM, Patel PR, Cafferty WB, Strittmatter SM (2013) Anatomical plasticity of adult brain is titrated by nogo receptor 1. Neuron 77:859–866. 10.1016/j.neuron.2012.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimura N, Ménager C, Kawano Y, Yoshimura T, Kawabata S, Hattori A, Fukata Y, Amano M, Goshima Y, Inagaki M, Morone N, Usukura J, Kaibuchi K (2005) Phosphorylation by rho kinase regulates CRMP-2 activity in growth cones. Mol Cell Biol 25:9973–9984. 10.1128/MCB.25.22.9973-9984.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers MM, Hazelwood LJ, Catmull DV, Wang D, McKormack Q, Bernard CC, Orian JM (2004) Early glial responses in murine models of multiple sclerosis. Neurochem Int 45:409–419. 10.1016/j.neuint.2003.08.018 [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Kinkel RP, Kidd G, Rudick RA, Trapp BD (2001) Axonal loss in normal-appearing white matter in a patient with acute MS. Neurology 57:1248–1252. 10.1212/WNL.57.7.1248 [DOI] [PubMed] [Google Scholar]
- David S, Fry EJ, López-Vales R (2008) Novel roles for nogo receptor in inflammation and disease. Trends Neurosci 31:221–226. 10.1016/j.tins.2008.02.002 [DOI] [PubMed] [Google Scholar]
- Dupree JL, Polak PE, Hensley K, Pelligrino D, Feinstein DL (2015) Lanthionine ketimine ester provides benefit in a mouse model of multiple sclerosis. J Neurochem 134:302–314. 10.1111/jnc.13114 [DOI] [PubMed] [Google Scholar]
- Evangelou N, Konz D, Esiri MM, Smith S, Palace J, Matthews PM (2000) Regional axonal loss in the corpus callosum correlates with cerebral white matter lesion volume and distribution in multiple sclerosis. Brain 123:1845–1849. 10.1093/brain/123.9.1845 [DOI] [PubMed] [Google Scholar]
- Filippi M, Riccitelli G, Mattioli F, Capra R, Stampatori C, Pagani E, Valsasina P, Copetti M, Falini A, Comi G, Rocca MA (2012) Multiple sclerosis: effects of cognitive rehabilitation on structural and functional MR imaging measures: an explorative study. Radiology 262:932–940. 10.1148/radiol.11111299 [DOI] [PubMed] [Google Scholar]
- Fournier AE, Takizawa BT, Strittmatter SM (2003) Rho kinase inhibition enhances axonal regeneration in the injured CNS. J Neurosci 23:1416–1423. 10.1523/JNEUROSCI.23-04-01416.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry EJ, Ho C, David S (2007) A role for nogo receptor in macrophage clearance from injured peripheral nerve. Neuron 53:649–662. 10.1016/j.neuron.2007.02.009 [DOI] [PubMed] [Google Scholar]
- Fu QL, Liao XX, Li X, Chen D, Shi J, Wen W, Lee DH, So KF (2011) Soluble nogo-66 receptor prevents synaptic dysfunction and rescues retinal ganglion cell loss in chronic glaucoma. Invest Ophthalmol Vis Sci 52:8374–8380. 10.1167/iovs.11-7667 [DOI] [PubMed] [Google Scholar]
- Hares K, Redondo J, Kemp K, Rice C, Scolding N, Wilkins A (2017) Axonal motor protein KIF5A and associated cargo deficits in multiple sclerosis lesional and normal-appearing white matter. Neuropathol Appl Neurobiol 43:227–241. 10.1111/nan.12305 [DOI] [PubMed] [Google Scholar]
- Hensley K, Venkova K, Christov A, Gunning W, Park J (2011) Collapsin response mediator protein-2: an emerging pathologic feature and therapeutic target for neurodisease indications. Mol Neurobiol 43:180–191. 10.1007/s12035-011-8166-4 [DOI] [PubMed] [Google Scholar]
- Ineichen BV, Kapitza S, Bleul C, Good N, Plattner PS, Seyedsadr MS, Kaiser J, Schneider MP, Zörner B, Martin R, Linnebank M, Schwab ME (2017) Nogo-A antibodies enhance axonal repair and remyelination in neuro-inflammatory and demyelinating pathology. Acta Neuropathol 134:423–440. 10.1007/s00401-017-1745-3 [DOI] [PubMed] [Google Scholar]
- Joset A, Dodd DA, Halegoua S, Schwab ME (2010) Pincher-generated nogo-A endosomes mediate growth cone collapse and retrograde signaling. J Cell Biol 188:271–285. 10.1083/jcb.200906089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS (2001) Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature 414:643–648. 10.1038/414643a [DOI] [PubMed] [Google Scholar]
- Karnezis T, Mandemakers W, McQualter JL, Zheng B, Ho PP, Jordan KA, Murray BM, Barres B, Tessier-Lavigne M, Bernard CC (2004) The neurite outgrowth inhibitor nogo A is involved in autoimmune-mediated demyelination. Nat Neurosci 7:736–744. 10.1038/nn1261 [DOI] [PubMed] [Google Scholar]
- Kawano Y, Yoshimura T, Tsuboi D, Kawabata S, Kaneko-Kawano T, Shirataki H, Takenawa T, Kaibuchi K (2005) CRMP-2 is involved in kinesin-1-dependent transport of the sra-1/WAVE1 complex and axon formation. Mol Cell Biol 25:9920–9935. 10.1128/MCB.25.22.9920-9935.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JE, Liu BP, Park JH, Strittmatter SM (2004) Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron 44:439–451. 10.1016/j.neuron.2004.10.015 [DOI] [PubMed] [Google Scholar]
- Kimura T, Watanabe H, Iwamatsu A, Kaibuchi K (2005) Tubulin and CRMP-2 complex is transported via kinesin-1. J Neurochem 93:1371–1382. 10.1111/j.1471-4159.2005.03063.x [DOI] [PubMed] [Google Scholar]
- Lassmann H. (2010) Axonal and neuronal pathology in multiple sclerosis: what have we learnt from animal models. Exp Neurol 225:2–8. 10.1016/j.expneurol.2009.10.009 [DOI] [PubMed] [Google Scholar]
- Lee JY, Petratos S (2013) Multiple sclerosis: does Nogo play a role? Neuroscientist 19:394–408. 10.1177/1073858413477207 [DOI] [PubMed] [Google Scholar]
- Lee JY, Taghian K, Petratos S (2014) Axonal degeneration in multiple sclerosis: can we predict and prevent permanent disability? Acta neuropathologica communications 2:97. 10.1186/s40478-014-0097-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Kim MJ, Li L, Velumian AA, Aui PM, Fehlings MG, Petratos S (2017) Nogo receptor 1 regulates caspr distribution at axo-glial units in the central nervous system. Sci Rep 7:8958. 10.1038/s41598-017-09405-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Twelvetrees AE, Moughamian AJ, Holzbaur EL (2014) Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron 84:292–309. 10.1016/j.neuron.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins-de-Souza D, Cassoli JS, Nascimento JM, Hensley K, Guest PC, Pinzon-Velasco AM, Turck CW (2015) The protein interactome of collapsin response mediator protein-2 (CRMP2/DPYSL2) reveals novel partner proteins in brain tissue. Proteomics Clinical applications 9:817–831. 10.1002/prca.201500004 [DOI] [PubMed] [Google Scholar]
- Mimura F, Yamagishi S, Arimura N, Fujitani M, Kubo T, Kaibuchi K, Yamashita T (2006) Myelin-associated glycoprotein inhibits microtubule assembly by a rho-kinase-dependent mechanism. J Biol Chem 281:15970–15979. 10.1074/jbc.M510934200 [DOI] [PubMed] [Google Scholar]
- Mokhtar SH, Kim MJ, Magee KA, Aui PM, Thomas S, Bakhuraysah MM, Alrehaili AA, Lee JY, Steer DL, Kenny R, McLean C, Azari MF, Birpanagos A, Lipiec E, Heraud P, Wood B, Petratos S (2018) Amyloid-beta-dependent phosphorylation of collapsin response mediator protein-2 dissociates kinesin in Alzheimer's disease. Neural Regen Res 13:1066–1080. 10.4103/1673-5374.233451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll NM, Rietsch AM, Thomas S, Ransohoff AJ, Lee JC, Fox R, Chang A, Ransohoff RM, Fisher E (2011) Multiple sclerosis normal-appearing white matter: pathology-imaging correlations. Ann Neurol 70:764–773. 10.1002/ana.22521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai J, Owada K, Kitamura Y, Goshima Y, Ohshima T (2016) Inhibition of CRMP2 phosphorylation repairs CNS by regulating neurotrophic and inhibitory responses. Exp Neurol 277:283–295. 10.1016/j.expneurol.2016.01.015 [DOI] [PubMed] [Google Scholar]
- Neumann S, Chassefeyre R, Campbell GE, Encalada SE (2017) KymoAnalyzer: a software tool for the quantitative analysis of intracellular transport in neurons. Traffic 18:71–88. 10.1111/tra.12456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikić I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, Bareyre FM, Bruck W, Bishop D, Misgeld T, Kerschensteiner M (2011) A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med 17:495–499. 10.1038/nm.2324 [DOI] [PubMed] [Google Scholar]
- Onuki M, Ayers MM, Bernard CC, Orian JM (2001) Axonal degeneration is an early pathological feature in autoimmune-mediated demyelination in mice. Microsc Res Tech 52:731–739. 10.1002/jemt.1057 [DOI] [PubMed] [Google Scholar]
- Petratos S, Li QX, George AJ, Hou X, Kerr ML, Unabia SE, Hatzinisiriou I, Maksel D, Aguilar MI, Small DH (2008) The β-amyloid protein of Alzheimer's disease increases neuronal CRMP-2 phosphorylation by a rho-GTP mechanism. Brain 131:90–108. 10.1093/brain/awm260 [DOI] [PubMed] [Google Scholar]
- Petratos S, Ozturk E, Azari MF, Kenny R, Lee JY, Magee KA, Harvey AR, McDonald C, Taghian K, Moussa L, Mun Aui P, Siatskas C, Litwak S, Fehlings MG, Strittmatter SM, Bernard CC (2012) Limiting multiple sclerosis related axonopathy by blocking nogo receptor and CRMP-2 phosphorylation. Brain 135:1794–1818. 10.1093/brain/aws100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddrop C, Moldrich RX, Beart PM, Farso M, Liberatore GT, Howells DW, Petersen KU, Schleuning WD, Medcalf RL (2005) Vampire bat salivary plasminogen activator (desmoteplase) inhibits tissue-type plasminogen activator-induced potentiation of excitotoxic injury. Stroke 36:1241–1246. 10.1161/01.STR.0000166050.84056.48 [DOI] [PubMed] [Google Scholar]
- Samson AL, Nevin ST, Croucher D, Niego B, Daniel PB, Weiss TW, Moreno E, Monard D, Lawrence DA, Medcalf RL (2008) Tissue-type plasminogen activator requires a co-receptor to enhance NMDA receptor function. J Neurochem 107:1091–1101. 10.1111/j.1471-4159.2008.05687.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot JW, Geuze HJ (2007) Cryosectioning and immunolabeling. Nat Protoc 2:2480–2491. 10.1038/nprot.2007.365 [DOI] [PubMed] [Google Scholar]
- Sorbara CD, Wagner NE, Ladwig A, Nikić I, Merkler D, Kleele T, Marinković P, Naumann R, Godinho L, Bareyre FM, Bishop D, Misgeld T, Kerschensteiner M (2014) Pervasive axonal transport deficits in multiple sclerosis models. Neuron 84:1183–1190. 10.1016/j.neuron.2014.11.006 [DOI] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 307:1282–1288. 10.1126/science.1105681 [DOI] [PubMed] [Google Scholar]
- Taghian K, Lee JY, Petratos S (2012) Phosphorylation and cleavage of the family of collapsin response mediator proteins may play a central role in neurodegeneration after CNS trauma. J Neurotrauma 29:1728–1735. 10.1089/neu.2011.2063 [DOI] [PubMed] [Google Scholar]
- Takata K, Kitamura Y, Nakata Y, Matsuoka Y, Tomimoto H, Taniguchi T, Shimohama S (2009) Involvement of WAVE accumulation in Aβ/APP pathology-dependent tangle modification in Alzheimer's disease. Am J Pathol 175:17–24. 10.2353/ajpath.2009.080908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Nave KA (2008) Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci 31:247–269. 10.1146/annurev.neuro.30.051606.094313 [DOI] [PubMed] [Google Scholar]
- Wang D, Ayers MM, Catmull DV, Hazelwood LJ, Bernard CC, Orian JM (2005) Astrocyte-associated axonal damage in pre-onset stages of experimental autoimmune encephalomyelitis. Glia 51:235–240. 10.1002/glia.20199 [DOI] [PubMed] [Google Scholar]
- Wang X, Duffy P, McGee AW, Hasan O, Gould G, Tu N, Harel NY, Huang Y, Carson RE, Weinzimmer D, Ropchan J, Benowitz LI, Cafferty WB, Strittmatter SM (2011) Recovery from chronic spinal cord contusion after nogo receptor intervention. Ann Neurol 70:805–821. 10.1002/ana.22527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lin J, Arzeno A, Choi JY, Boccio J, Frieden E, Bhargava A, Maynard G, Tsai JC, Strittmatter SM (2015) Intravitreal delivery of human NgR-fc decoy protein regenerates axons after optic nerve crush and protects ganglion cells in glaucoma models. Invest Ophthalmol Vis Sci 56:1357–1366. 10.1167/iovs.14-15472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills ZP, Mandel-Brehm C, Mardinly AR, McCord AE, Giger RJ, Greenberg ME (2012) The nogo receptor family restricts synapse number in the developing hippocampus. Neuron 73:466–481. 10.1016/j.neuron.2011.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K (2005) GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120:137–149. 10.1016/j.cell.2004.11.012 [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Arimura N, Kawano Y, Kawabata S, Wang S, Kaibuchi K (2006) Ras regulates neuronal polarity via the PI3-kinase/Akt/GSK-3beta/CRMP-2 pathway. Biochem Biophys Res Commun 340:62–68. 10.1016/j.bbrc.2005.11.147 [DOI] [PubMed] [Google Scholar]
- Zemmar A, Chen CC, Weinmann O, Kast B, Vajda F, Bozeman J, Isaad N, Zuo Y, Schwab ME (2018) Oligodendrocyte- and neuron-specific nogo-A restrict dendritic branching and spine density in the adult mouse motor cortex. Cereb Cortex 28:2109–2117. 10.1093/cercor/bhx116 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Axon-specific overexpression of NgR1 in optic nerves of naïve ngr1-/- mice did not lead into axonal degeneration (A) Immunolabeling of rAAV2-eGFP-mNgR1 injected optic nerve sections of naïve ngr1-/- mice for NF-200, GFP, NgR1, and DAPI counterstaining. (B) Immunolabeling of serial section for βAPP, GFP, and DAPI counterstaining. (C) The percentage of degenerative GFP+ axons over total number of GFP+ transduced axons (n = 4, mean ± SEM). Download Figure 1-1, TIF file (18.7MB, tif)
The net velocity of axonal transport with and without stimulation of Nogo66 after transection injury. (A) Schematic diagram of the compartmentalized culture in the microfluidic chamber where the transection injury and subsequent Nogo66 stimulation were performed. (B) The net velocities of the tracks in each group. Representative time-lapse of ChTxβ added cortical neurons from ngr1+/+ mice (C) untreated and (D) treated with 0.3 μg of Nogo66 peptide after transection injury. These time-lapse videos are available as Movies 1 and 2, respectively. Representative time-lapse of ChTxβ added cortical neurons from ngr1-/- mice (E) untreated and (F) treated with 0.3 μg of Nogo66 peptide after transection injury. These time-lapse videos are available as Movies 3 and 4, respectively. (G-K) Representative kymographs generated from time-lapse videos of each group. Colored lines represent the tracks that were measured for net velocities. (n = 80 tracks, mean ± SEM, one-way ANOVA, ****P<0.0001). Scale bars = 20 μm. Download Figure 2-1, TIF file (14.4MB, tif)
Real-time imaging of axonal transport of ChTxβ-positive vesicles from ngr1+/+ cortical neurons. The movie shows simultaneous imaging of individual ChTxβ-positive (red) vesicles along the axons.
Real-time imaging of axonal transport of ChTxβ-positive vesicles from ngr1+/+ cortical neurons 3 h after treated with 0.3 μg of Nogo-66 peptide after transection injury. The movie shows simultaneous imaging of individual ChTxβ-positive (red) vesicles along the axons.
Real-time imaging of axonal transport of ChTxβ-positive vesicles from ngr1−/− cortical neurons. The movie shows simultaneous imaging of individual ChTxβ-positive (red) vesicles along the axons.
Real-time imaging of axonal transport of ChTxβ-positive vesicles from ngr1−/− cortical neurons 3 h after treated with 0.3 μg of Nogo-66 peptide after transection injury. The movie shows simultaneous imaging of individual ChTxβ-positive (red) vesicles along the axons.
Real-time imaging of axonal transport of pDendra-Flag-βAPP transfected ngr1+/+ cortical neurons. The movie shows simultaneous imaging of individual βAPP-positive (green) vesicles along the axons.
Real-time imaging of axonal transport of pDendra-Flag-βAPP transfected ngr1+/+ cortical neurons 3 h after treated with 0.3 μg of Nogo-66 peptide after transection injury. The movie shows simultaneous imaging of individual βAPP-positive (green) vesicles along the axons.
Real-time imaging of axonal transport of pDendra-Flag-βAPP transfected ngr1−/− cortical neurons. The movie shows simultaneous imaging of individual βAPP-positive (green) vesicles along the axons.
Real-time imaging of axonal transport of pDendra-Flag-βAPP transfected ngr1−/− cortical neurons 3 h after treated with 0.3 μg of Nogo-66 peptide after transection injury. The movie shows simultaneous imaging of individual βAPP-positive (green) vesicles along the axons.
Intact axonal structure with compact myelin in rAAV2-transduced axons Immunogold-labeled electron microscopic analysis of axons in (A) rAAV2-eGFP-iCre, (B) rAAV2-CRMP2T555A-GFP transduced right optic nerves, and (C) non-transduced control nerve. Arrowheads indicate the immunogold-labeled GFP+ rAAV2 particles. Asterisks indicate degenerative axons where rAAV2 was not transduced. Arrows indicate macrophages engulfing myelin debris in non-transduced control optic nerve. Scale bar = 500 nm. Download Figure 5-1, TIF file (28MB, tif)
Number of CC1+ mature oligodendrocytes in optic nerves of naïve ngr1+/+ and ngr1-/- mice. Representative images of CC1+ mature oligodendrocytes in naïve ngr1+/+ and ngr1-/- mice. Greater number of CC1+ mature oligodendrocytes in optic nerves of naïve ngr1+/+ and ngr1-/- mice per mm2 (n = 3, mean ± SEM, student's t-test, *P<0.05). Scale bars = 50μm. Download Figure 6-1, TIF file (6.5MB, tif)