

Abstract
Diatoms are phytoplankton eukaryotic microalgae that are widely distributed in the world’s oceans and are responsible for 20–25% of total carbon fixation on the planet. Using transcriptome sequencing here we show for the first time that the ubiquitous diatom Thalassiosira rotula expresses biosynthetic pathways that potentially lead to the synthesis of interesting secondary metabolites with pharmaceutical applications such as polyketides, prostaglandins and secologanin. We also show that these pathways are differentially expressed in conditions of silica depletion in comparison with standard growth conditions.
Subject terms: Gene ontology, Marine biology, Marine chemistry
Introduction
Diatoms are a major group of eukaryotic microalgae in the phytoplankton widely distributed in the world’s oceans, and capable through photosynthesis of fixing about 20–25% of the total carbon on the planet1. Their worldwide distribution is supported by a molecular tool-kit (M-T) that allows them to adapt to different conditions through the perception of environmental cues and the control of competitors and grazers2–5. Some of the metabolites which are part of this M-T have already been identified as having potential applications as pharmaceuticals or cosmeceuticals6. Nevertheless, the full potential of marine diatoms still needs to be unlocked. One way to unlock this potential is to expose diatoms to stressful conditions to force changes in their metabolism and activate biosynthetic pathways inducing the production of secondary metabolites. One of the stressful conditions to promote such changes is nutrient depletion during microalgae growth. For example, phosphorus (P) deficiency in Thalassiosira rotula induces a remodelling of the transcriptome including changes in cellular P allocation patterns, enzyme activity and lipid composition, whereas nitrogen (N) deprivation reduces primary carbon metabolism leading to accumulation of lipids7. In addition to P and N, also carbon deprivation, independently from the light intensity (30, 300 or 1000 μmol photon m−2 s−1), induces a rearrangement of the Carbon Concentration Mechanisms (CCM) toward the synthesis of pyruvate8,9 reorienting the carbon metabolism toward lipid accumulation. A similar remodelling of metabolic pathways occurs in conditions of silica depletion10. Diatoms require silica (Si) to generate the intricate frustules that surround the cell and allow the entrance of light to the chloroplasts and the transport of gases and solutes. The silica cell wall is believed to provide an ecological advantage over other phytoplankton groups due to its mechanical strength that protects diatoms against grazers. Deprivation of Si arrests progression of cell cycle events, such as cell division and DNA replication, attesting the importance of silica in the diatom life cycle11.
Here we analyse the transcriptome of the diatom Thalassiosira rotula, isolated from the Gulf of Naples in the Mediterranean Sea, comparing cultures grown in normal condition (CTR) versus cultures grown in conditions of stress such as silica depletion (STRESS). Our analysis shows that this diatom species has the potential to produce new metabolites, i.e. secologanin, polyketides and prostaglandins, the related pathways of which are differentially expressed under this stress condition.
We experimentally confirmed the expression of these pathways identified in the newly sequenced transcriptome of our clone and also compared their expression with those from other T. rotula clones, grown under different stress conditions and sequenced in the Marine Microbial Eukaryotic Transcriptome Sequencing Project (MMETSP)12 supported by the Moore Foundation. Our study also confirms that the identified pathways are actually present in the T. rotula genome and do not derive from bacteria commonly associated with diatom cells.
Results
De Novo transcriptome assembly and functional annotation
Total RNA from Thalassiosira rotula cultured in f/2 complete medium (control condition, CTR), and in f/2 silica depleted medium, (stress condition, STRESS), was obtained from cells collected at the beginning of the stationary phase of growth, when cells are more prone to produce secondary metabolites13. Table 1 shows the statistics for the total transcriptome assembly after filtering of unspecific sequences. The total assembly was composed by 142 Mbp in 107,289 transcripts grouped into 66,496 genes, with a N50 of 2,034 bp.
Table 1.
Transcriptome sequencing statistics.
| Assembly (Mbp) | 142 |
| Transcripts (n=) | 107,289 |
| Genes (n=) | 66,496 |
| Mean GC% | 47.53 |
| Average contig lenght (bp) | 1,332.76 |
| Median contig lenght (bp) | 970 |
| N50 (bp) | 2,034 |
| Proteins (n=) | 60,172 |
The final dataset translated into proteins (minimum length 50aa) includes 60,172 protein sequences, 28,128 (46.7%) of which were identified as complete, i.e. having a start methionine and a stop codon.
The sequences were also analysed for the presence of repetitive elements and more than 3 Mbp were identified as repetitive elements including, for example, DNA transposons, retro-elements, satellites, and rRNA.
Functional annotation of the combined transcriptomes was performed on proteome sequences, 89% of which showed homology with other diatoms, in particular T. pseudonana, T. oceanica and Phaeodactylum tricornutum (Supplementary Data 1 and 2).
Out of a total 60,172 peptide sequences, 63% had at least one blast hit, and almost half (35%) retrieved a blast hit against conserved domains (Fig. 1a).
Figure 1.
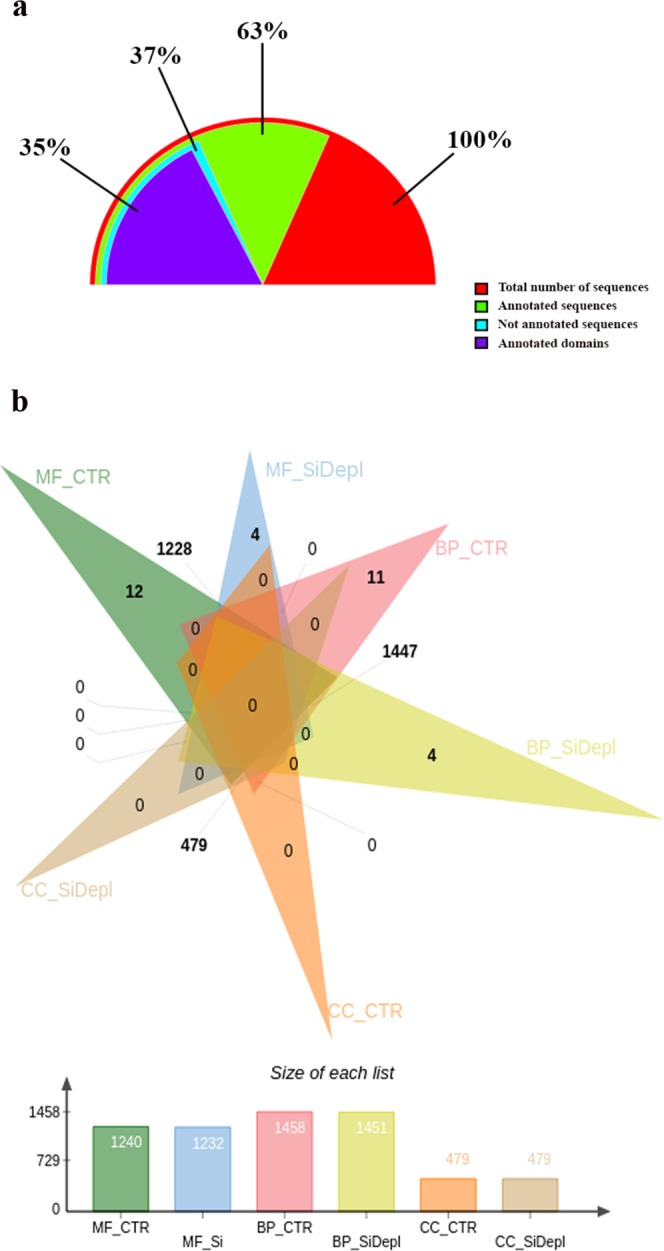
Thalassiosira rotula functional annotation. (a) Percentage of annotated sequences and domains of the total number of peptides in the diatom Thalassiosira rotula. (b) Venn diagram showing the number of common and unique GO terms between the two growth conditions for each category in the diatom Thalassiosira rotula. Abbreviations: MF: Molecular Functions; CTR: control condition; SiDepl: silica depleted medium corresponding to the STRESS condition; BP: Biological Processes; CC: CellularComponents.
The functional annotation of peptides assigned the majority of proteins and domains to the GO terms in the Molecular Functions (MF) category (Table 2). Except for the Cellular Components (CC) category, for both Biological Processes (BP) and MF categories, the number of associated domains was almost half the number of associated protein sequences.
Table 2.
Number of sequences associated to each GO term category.
| Number of sequences with at least one blast hit | 37999 |
| Number of peptides sequences associated to Molecular Functions GO terms | 26255 |
| Number of domains associated to Molecular Functions GO terms | 11622 |
| Number of peptides sequences associated to Biological Processes GO terms | 15459 |
| Number of domains associated to Biological Processes GO terms | 7809 |
| Number of peptides sequences associated to Cellular Components GO terms | 17794 |
| Number of domains associated to Cellular Components GO terms | 2694 |
Subdivision of the sequencing retrieving at least one blast hit and an association with a Gene Ontology number in the three functional categories: Molecular Functions (MF), Biological Processes (BP) and Cellular components (CC).
The “ATP binding” GO term was the most represented in the MF category while “transmembrane transport” was the most represented GO term in the BP category, and “integral to membrane” was the most represented GO term in the CC category (Supplementary Data 3 to 8, SI1: Supplementary Fig. 2a–f).
Functional annotation analysis: STRESS versus CTR
Principal Component Analysis showed that replicates 1 and 4, in the CTR and STRESS group, respectively, did not behave coherently within the group, so they were excluded from subsequent analysis (SI1: Supplementary Fig. 1).
Fisher test on the number of GO terms associated with each peptide sequence for each treatment did not reveal any significant difference. However, we were able to identify uniquely annotated GO terms. The Venn diagram in Fig. 1b shows the occurrence of 12 and 4 unique MF GO terms in CTR and STRESS, respectively. Similarly, 11 and 4 BP GO terms were uniquely annotated in CTR and STRESS, respectively. No unique GO term annotations were present in the CC category for both treatments (see Supplementary Data 9 for complete list of GO Terms). Table 3 lists the unique GO terms found.
Table 3.
Unique GO terms.
| Category | GO Number | GO Term | CTR | STRESS |
|---|---|---|---|---|
| MF | 0004062 | ATP-dependent helicase activity | + | − |
| 0008379 | L-leucine transaminase activity | |||
| 0008658 | Metalloaminopeptidase activity | |||
| 0015416 | Oxidoreductase activity | |||
| 0015430 | Oxidoreductase activity, acting on a sulfur group of donors, disulfide as acceptor | |||
| 0015604 | Oxidoreductase activity, acting on iron-sulfur proteins as donors | |||
| 0005391 | Hydro-lyase activity | |||
| 0008909 | NAPDH binding | |||
| 0034189 | Pyrophosphatase activity | |||
| 0043621 | RNA strand-exchange activity | |||
| 0015439 | snRNA stem-loop binding | |||
| 0015439 | Oxidoreductase activity, acting on a sulfur group of donors, NAD or NADP as acceptor | |||
| 0003983 | Anion transmembrane-transporting ATPase activity | − | + | |
| 0008320 | Lipid transporter activity | |||
| 0008761 | Methyltransferase activity | |||
| 0009002 | Nicotinate phosphoribosyltransferase activity | |||
| BP | 0001407 | Body fluid secretion | + | − |
| 0007281 | Interleukin-8 biosynthetic process | |||
| 0008354 | Lactate biosynthetic process | |||
| 001096 | Negative regulation by host of viral transcription | |||
| 0032802 | Post-chaperonin tubulin folding pathway | |||
| 0042744 | Purine ribonucleoside salvage | |||
| 004523 | Regulation of exit from mitosis | |||
| 0044458 | Regulation of protein phosphatase type 2A activity | |||
| 0050427 | RNA modification | |||
| 0051923 | Sterol biosynthetic process | |||
| 0006011 | Cellular amino acid metabolic process | − | + | |
| 001932 | Peptidyl-tyrosine dephosphorylation | |||
| 0043953 | Regulation of mRNA stability | |||
| 0000022 | (1-3)-beta_D-glucan biosynthetic process |
Unique GO terms for each growth condition for each category: MF: Molecular Function; BP: Biological Processes; CC: Cellular Components.
Except for ‘Interleukin-8 biosynthetic process’, ‘lactate biosynthetic process’, CTR specific, and ‘1-3-beta-D-glucan biosynthetic process’, STRESS specific in the BP category, all other terms referred to more broad categories (Table 3).
Differential expression analysis
In silico expression analysis on single transcripts identified 5,114 DET (Differential Expressed Transcripts) in STRESS vs CTR, with 2,949 upregulated and 2,165 downregulated transcripts (Supplementary Data 10). The GO terms associated to the DETs are reported in SI1: Supplementary Table 2 and Supplementary Fig. 3.
Enzymes in the chains for the sphingolipid, sterol, spermidine, secologanin, polyunsaturated fatty acids (PUFA), malonylCoA, lipotheicoic, L-ascorbate, L-cysteine biosynthesis and isoprenoid pathways were upregulated in STRESS condition.
Considering 2-fold change as the minimum value for significant results, the proportion of significant DET was 20% for downregulated and 34% for upregulated transcripts. Among these groups, 1,604 (54%) of the upregulated and 1,278 (60%) of the downregulated transcripts were not annotated but indicated as “protein”, or “predicted protein” or “hypothetical protein” or “NA” (not annotated at all). It is interesting to note that part of these “un-annotated” groups were considerably differentially expressed. Indeed, 21% of the “un-annotated”-upregulated transcripts had an expression fold change between +2.50 and +19.12, and 13% of the “un-annotated”-downregulated transcripts had an expression fold change between −2.50 and −20.03 indicating the presence in these groups of still unknown functions that are very important for the homeostasis of the cells (Supplementary Data 10).
Of the downregulated transcripts, it is interesting to note that the ‘ribosomal protein s6 kinase beta 2’ showed a fold change of −18, whereas the upregulated transcripts ‘cmp-sialic acid transporter’ and ‘peroxisomal trans-2-enoyl- reductase’ showed a fold change of +15 and +8, respectively.
In silico pathway expression analysis
Total number of general pathways, for both growth conditions, was 68 with all essential and secondary metabolic pathways present. The most represented pathways, based on the number of associated sequences, are those involved in carbohydrate, lipid and protein metabolism (Table 4). Analysis of general pathways expressed in FPKM (Fragments Per Kilobase of transcript per Million mapped reads) indicated some differences between the two conditions, CTR and STRESS (Table 5).
Table 4.
Top 10 most represented general pathways based on the number of associated sequences.
| Pathway name | Number of associated sequences |
|---|---|
| Protein modification | 456 |
| Amino-acid biosynthesis | 240 |
| Cofactor biosynthesis | 186 |
| Lipid metabolism | 175 |
| Carbohydrate degradation | 169 |
| Amino-acid degradation | 95 |
| Porphyrin containing compounds metabolism | 77 |
| Purine metabolism | 62 |
| Sulfur metabolism | 48 |
| Carbohydrate biosynthesis | 48 |
| Carbohydrate metabolism | 48 |
Table 5.
Top 10 most expressed pathways in FPKM, Fragments Per Kilobase of transcript per Million mapped reads. Differentially expressed pathways are reported in bold.
| Pathway Name | CTR_FPKM | STRESS_FPKM | Fold STRESS vs CTR |
|---|---|---|---|
| Protein biosynthesis | 88.73 | 245.92 | +2.8 |
| Carbohydrate degradation | 42.97 | 26.17 | −1.6 |
| Thermoadapter biosynthesis | 36,45 | 17.8 | −1.9 |
| Flavonoid metabolism | 27.73 | 47 | +1.7 |
| Organosulfur biosynthesis | 24.26 | 19.77 | |
| Carbohydrate biosynthesis | 20.89 | 16.51 | |
| Lipid metabolism | 19.65 | 21.85 | |
| Spore coat biogenesis | 18.6 | 28.11 | |
| Carbohydrate acid metabolism | 18.07 | 17.59 | |
| Nucleotide-sugar biosynthesis | 15.07 | 21.1 |
The ‘protein biosynthesis’ and ‘flavonoid metabolism’ pathways had almost three- and two-fold lower expression values in CTR with respect to STRESS, respectively. ‘Carbohydrate degradation’ and ‘thermoadapter’ biosynthesis on the contrary were almost two-fold higher in CTR with respect to STRESS.
Despite these differences, FPKM box plot expression analysis revealed no statistically significant differences between the two conditions (Supplementary Table 4 (in SI1), Supplementary Files 1 and 2).
The number of pathways identified in both conditions on the second level of annotation was 258 (Supplementary Data 11). The most represented pathways were ‘protein ubiquitination’ (293 sequences) followed by ‘protein glycosylation’ (127 sequences) and ‘glycolysis’ (105 sequences). Also within this level of pathway annotation there were no statistically significant differences in FPKM values between the two conditions (Supplementary Files 3 and 4).
The pipeline annotated also a third level with 327 pathways for CTR and 325 for STRESS (Supplementary Data 12). The best represented pathways were: ‘Pyruvate from D-glyceraldehyde 3-phosphate step 3/5’ with 26 associated sequences; ‘D-ribulose 5-phosphate from D-glucose 6-phosphate (oxidative stage) step 1/3’ with 25 associated sequences, ‘glutathione from L-cysteine and L-glutamate step 1/2’ with 21 associated sequences, ‘D-glyceraldehyde 3-phosphate and glycerone phosphate from D-glucose step 4/4’ with 20 associated sequences, ‘L-cysteine from L-serine’ step 2/2’ with 20 associated sequences. Again, no differences in FPKM values were appreciable (Supplementary File 5 and 6).
In silico expression analysis of selected pathways
Despite the fact that no significant differences were evident in the FPKM expression of the annotated pathways, we focused our attention on single transcripts associated with pathways leading to secondary metabolites that have been poorly studied or that were previously unknown for diatoms: secologanin, prostaglandin and polyketides biosynthesis (Supplementary File 4, highlighted pathways).
Secologanin biosynthesis
The ‘Secologanin synthase (SLS)’ annotation in the transcriptome of T. rotula was associated to four peptides that when aligned matched one another, with the peptide TR26037|c1_g1_i1 being the longest (SI1: Supplementary Fig. 4). Heat map in Fig. 2a shows expression levels of the corresponding transcript, calculated in FPKM, which was ten times higher in STRESS with respect to CTR (Fig. 2). In addition, the related transcript was also present in the DET table with a logFC of +3.1 (Supplementary Data 10).
Figure 2.

In silico expression analysis of selected pathways. (a) Heat map showing the presence and the expression levels, calculated in FPKM on the contig’s reads, of transcripts, belonging to our Thalassiosira rotula from the Mediterranean Sea, in the selected pathways for each growth condition considered. (b) Heat map showing the presence and the expression levels, calculated in FPKM on the contig’s reads of the transcripts in the selected pathways of the Thalassiosira rotula strains, grown in different conditions, from the MMETSP project in comparison with our clone from the Mediterranean Sea (Complete medium and SiOH4 limited).
Prostaglandin biosynthesis
Peptide TR754|c0_g1_i1 annotated as ‘Prostaglandin G/H synthase 2 (PGHS-2)’, TR25058|_c1_g1_i2 annotated as ‘Prostaglandin E synthase 2-like (mPGES-2)’ and TR11659|c0_g1_i1 annotated as ‘Prostaglandin F synthase (PGFS)’ were associated to the ‘Prostaglandin biosynthesis’ pathway (Supplementary Data 13).
In addition, three putative peptides and two transcripts were associated to the ‘Prostaglandin F synthase’ function. Table 6 shows the blast results of the five PGFS associated transcripts.
Table 6.
Blast results of the prostaglandin F synthase related transcript.
| Transcript | Annotation on: | First three BlastX results | Query cover (%) | Identity (%) |
|---|---|---|---|---|
| TR11659|c0_g1_i1 | Peptide | Aldo-keto oxidoreductase Thalassiosira pseudonana; | 76 | 63 |
| Hypothetical protein Thalassiosira ocenanica; | 74 | 58 | ||
| Prostaglandin F synthase Symbiodinium microadriaticum. | 70 | 45 | ||
| TR3003|c0_g1_i1 | Peptide | Hypothetical protein Thalassiosira pseudonana; | 72 | 66 |
| Reductase with NAD or NADP as acceptor Phaeodactylum tricornutum; | 76 | 48 | ||
| Aldo/keto reductase Fragilariopsis cylindrus. | 69 | 48 | ||
| TR17558|c0_g1_i1 | Peptide | Hypothetical protein Thalassiosira ocenanica; | 42 | 42 |
| Hypothetical protein Fistulifera solaris; | 41 | 44 | ||
| Acetyl-CoA synthetase Fistulifera solaris. | 41 | 43 | ||
| TR22640|c0_g1_i1 | Transcript | Predicted protein Thalassiosira pseudonana; | 72 | 75 |
| Hypothetical protein Thalassiosira oceanica; | 76 | 60 | ||
| Hypothetical protein Fistulifera solaris. | 58 | 59 | ||
| TR60026|c0_g1_i1 | Transcript | Predicted protein Thalassiosira pseudonana; | 67 | 82 |
| Hypothetical protein Thalassiosira oceanica; | 71 | 74 | ||
| Hypothetical protein Fistulifera solaris. | 55 | 54 |
TR11659|c0_g1_i1 was chosen for the following analysis since this transcript blasted to other Thalassiosira species and to a dinoflagellate PGFS function. Moreover, the match with T. pseudonana was a protein annotated with the more general function of aldo/keto oxidoreductase, which is a family of proteins that also include PGFS.
Furthermore, browsing the ‘Enzyme Description’ column of the annotation table (Supplementary Data 13) we found six peptides annotated as ‘Carbonyl reductase (NADPH)’ or ‘Prostaglandin E2 9-reductase’ that reduces prostaglandin E2 (PGE2) to prostaglandin F2α (PGF2α) (SI1: Supplementary Table 4) differently from PGFS that produces PGF2α starting from prostaglandin D2 (PGD2). However, we did not include this enzyme in our analysis as it was not annotated in the specific pathway.
Heat Map (Fig. 2a) shows no difference in mPGES-2 expression calculated in FPKM, between the two growth conditions, while PGFS and PGHS-2 expression were downregulated in STRESS by two- and three-fold, respectively. PGHS-2 was also present in the DET table with a LogFC of −1.57 (Supplementary Data 10).
Polyketides biosynthesis
No specific pathway annotation exists for polyketides biosynthesis as they are included in the general lipid metabolism pathway. Searching in the complete annotation table (Supplementary Data 13), we found polyketide-related functional annotations of at least 4 putative enzymes, i.e. ‘Polyketide cyclase dehydrase and lipid transport protein’ (PK_Cyc, TR3754|c0_g1_i2), ‘Polyketide enoylreductase’ (PK_ER, TR30039|c0_g1_i1), ‘Polyketide hydroxylase-like’ (PK_Hxl, TR21208|c0_g1_i1), ‘Phthiocerol synthesis polyketide synthase type I’ (ppsC, TR40329|c0_g1_i6). Except for ppsC synthase, whose related transcript was downregulated in the STRESS samples by two-fold (in FPKM value) and had a LogFC value equal to −0.99 in DET table (Supplementary Data 10), the other three related transcripts were almost equal in expression with low FPKM values in both growth conditions (Fig. 2a).
Finally, we also found annotations for single modules associated to both polyketide synthesis and fatty acid elongation, like acyltransferase, acyl carrier protein, β-ketoacyl reductase and ketosynthase, methyl transferases, thioesterases and dehydrogenase domains (Supplementary Datas 13 to 19). These transcripts were not considered for the expression analysis due to their high number of redundant related transcripts.
Comparative analysis of selected pathways with other strains of Thalassiosira
To understand if selected pathway expression in the transcriptome of T. rotula is a prerogative of the strain from the Mediterranean, we used the public available MMETSP transcriptomic dataset to verify their presence in other T. rotula strains, 3096 and GSO102, isolated from different geographical areas and grown in different conditions (Table 7 and Fig. 2b).
Table 7.
Presence of the selected pathways in different Thalassiosira rotula strains. N= not present; Y= present.
| T. rotula Strain | CCMP 3096 | CCMP 3096 | GSO 102 | GSO 102 | GSO 102 | GSO 102 | CCMP1647 | CCMP1647 |
|---|---|---|---|---|---|---|---|---|
| MMETSP code | 0403 | 0404 | 910 | 911 | 912 | 913 | — | — |
| Treatment | Light cycle | Dark Cycle | 0.4 um PO4 | 4 nM Fe, 12.5 um EDTA | 4 nM Fe, 12.5 um EDTA, 0.4 uM PO4 | replete condition | Complete Medium | SiOH4 Limited |
| Site Name | Pacific Ocean | Pacific Ocean | Pacific Ocean | Pacific Ocean | Pacific Ocean | Pacific Ocean | Mediterranean sea | Mediterranean sea |
| Prostaglandin G/H synthase (PGHS-2) | N | N | N | N | N | N | Y | Y |
| Prostaglandin E synthase (mPGES-2) | Y | N | N | N | N | N | Y | Y |
| Prostaglandin F synthase (PGFS) | Y | Y | Y | Y | Y | Y | Y | Y |
| Prostaglandin D synthase (PTGDS) | Y | Y | Y | N | Y | N | N | N |
| Phthiocerol synthesis polyketide synthase type I (ppsC) | N | Y | N | N | N | N | Y | Y |
| Polyketide cyclase dehydrase and lipid transport protein (PYL5) | N | N | N | N | N | N | Y | Y |
| Polyketide enoylreductase (PK_ER) | N | N | N | N | N | N | Y | Y |
| Polyketide hydroxylase-like (PK_HxL) | Y | Y | N | N | N | N | Y | Y |
| Secologanin synthase (SLS) | Y | N | N | N | Y | N | Y | Y |
The available conditions were: 3096LC = day portion of the day/night cycle; 3096DC = dark portion of the day/night cycle; GSO102Fe = iron depletion; GSO102P = phosphorus depletion; GSO102Fe2 = iron and phosphorus depletion; GSO102R = nutrient replete.
Secologanin synthase was present in the 3096LC and GSO102Fe2’s transcriptomes but not in those of 3096DC, GSO102Fe, GSO102P and GSO102R (Table 7, Fig. 2b). Of all the conditions considered in which we found SLS annotated, silica depletion and the light portion of the day/night cycle were similar and had the highest values (41.02 fpkm and 39.6 fpkm, respectively) compared to the other conditions.
Among the polyketide biosynthesis enzymes, PKS_ER annotation has not been reported for other strains. The synthase ppsC was present only in 3096DC and the best condition of expression was found in our clone in CTR. The hydrolase-lyase PK_HxL was present in both 3096LC and 3096DC. It was present with two isoforms, here named as PK_Hlx_1 and PK_Hlx_2, in 3096LC and with one isoform, PK_Hlx_1, in 3096DC and in our clone in CTR. The highest expression for PK_Hlx_1 was in the 3096DC condition. The PK-Hxl_2, present only in 3096LC, showed a higher FPKM value than PK_Hlx_1. The GSO102 strain, in all conditions tested, did not show any polyketide related enzyme annotation. Finally, all strains had annotations for the single modules associated to both polyketide synthesis and fatty acid elongation (Supplementary Data 13 to 19). Examining the prostaglandin pathway, we did not find any annotation for PGHS-2 in strains 3096 and GSO102. On the contrary, PGFS was present in all strains, together with a second isoform present in 3096LC, 3096DC, GSO102P and GSO102Fe2. The transcript for mPGES-2 was present only in the 3096LC transcriptome. Finally, annotation for prostaglandin D synthase (PTGDS) that was absent in our transcriptomes, was instead present in the 3096LC, 3096DC, GSO102P and GSOFe2 transcriptomes. The best condition of expression was 3096DC for PTGDS, 3096LC and CTR for mPGES-2, GSO102Fe and GSO102Fe2 for PGFS_1 and 3096DC for PGFS_2.
Experimental confirmation of selected pathways
In order to confirm in silico expression data, we performed quantitative Polymerase Chain Reaction (qPCR) amplification on coding DNA (cDNA) from the same sample used for RNA-seq and qPCR amplification on genomic DNA (gDNA) from an axenic T. rotula (Ax) and a non-axenic T. rotula (Mx) culture (Fig. 3) in which bacterial DNA was predominant (unpublished data).
Figure 3.

Experimental validation of the transcripts associated to the selected pathways in the Thalassiosira rotula from the Mediterranean Sea. (a–c) qPCR on cDNA (1), PCR on gDNA (2), qPCR on gDNA (3). (a) Secologanin synthase; (b) Prostaglandin biosynthesis; (c) Polyketides biosynthesis. Abbreviations: PGHS-2: Prostaglandin G/H synthase 2; mPGES-2: Prostaglandin E synthase 2-like; PGFS: Prostaglandin F synthase; ppsC: Phthiocerol synthesis polyketide synthase type I; PK_ER: Polyketide enoylreductase; PK_Cyc: Polyketide cyclase dehydrase and lipid transport protein; PK_Hxl: Polyketide hydroxylase-like; Ax: Axenic genomic DNA; Mx: mixed Bacterial-Algae genomic DNA; CT: cycle treshold. Original gel picture is shown in Supplementary Fig. 7.
Secologanin synthase
qPCR on cDNA confirmed the significant overexpression of Secologanin synthase (SLS) in STRESS with respect to CTR with a p-value of 0.0062 (Fig. 3a(1)). PCR on Ax- and Mx-gDNA confirmed the presence of the corresponding gene exclusively in the algal genome excluding a co-partnership with the associated bacteria for the possible production of secologanin (Fig. 3a(2)). qPCR amplification on Ax-gDNA had a cycle threshold (CT) value of 21,6 compared to 31,5 of the amplification on the Mx-gDNA, confirming the algal specific expression of SLS (Fig. 3a(3)). Melt curve analysis showed one peak for the Ax-gDNA overlapping with the cDNA peak, while the Mx-gDNA showed two separated peaks. Sequencing of the corresponding amplicon definitively demonstrated that the additional amplicon from the Mx culture was non-specific.
Prostaglandin biosynthesis
qPCR (Fig. 3b(1)) showed a significant downregulation of all three transcripts in STRESS with p-value = 0.2 for PGHS-2, 0.1 for mPGES-2 and 0.07 for PGFS. PGHS-2 had the highest level of expression in both conditions with respect to PGFS and mPGES-2 that had expression levels in the order of 10−2 and 10−3, respectively.
Amplifications on gDNAs (Fig. 3b(2)) and sequencing of the amplicons demonstrated the specific origin of the corresponding transcripts from the algae and not from the associated bacteria. All three transcript amplicons had a CT value equal to about 21 on the Ax-gDNA and equal to about 33 on the Mx-gDNA, confirming the specific derivation of the prostaglandin biosynthetic pathway transcripts from the alga and not from the associated bacteria (Fig. 3b(3A–C)). Differently from PGHS-2 and mPGES-2, PGFS melt curve on non-axenic Mx-gDNA showed one peak that did not overlap with that of the Ax-gDNA. Sequencing of the corresponding amplicon demonstrated its lack of specificity. The absence of a peak corresponding to the one present in the Ax-gDNA is probably due to its very low abundance also considering the low presence of eukaryotic DNA in the sample (unpublished data).
Polyketides biosynthesis
qPCR (Fig. 3c(1)) confirmed the results of the FPKM calculation showing no regulation of PKS_ER, and PK_Hxl but a significant downregulation of the ppsC transcript with a p-value = 0.03.
Classical PCR amplification (Fig. 3c(2)), amplicon sequencing and qPCR analysis on gDNA (amplification and melt curve, Fig. 3c (3A–D) of ppsC, PK_Hxl, PKS_ER, of the Ax-gDNA, demonstrated the specificity of this transcript to the eukaryotic alga.
Discussion
Expression analysis to identify transcripts and differentially expressed metabolic pathways between control and silica-limited growth conditions highlighted the presence of a large group of uncharacterized proteins in the diatom Thalassiosira rotula, which were differentially expressed between the two growth conditions, representing, as such, a large source of potentially still undiscovered new functions and correlated metabolites.
Among the differentially expressed annotated sequences, we found that Ribosomal protein s6 kinase beta 2 (S6K2) was the one with the highest downregulation (−18.03 fold change) in the silica-limited condition with respect to the control. S6K2 is an important cell growth regulator and metabolism and its downregulation is consistent with the status of starvation induced by silica limitation14. S6K2 is indeed activated in response to growth factors, cytokines and nutrients, together with other kinases of the same family. The activity of S6 kinases is linked to fundamental cellular processes, including protein synthesis, mRNA processing, glucose homeostasis, cell growth and survival. They also play an important role in several human pathologies, including obesity, diabetes, ageing and cancer15.
Upregulated functions in silica-limited conditions with respect to the control include upregulation of cmp-sialic acid transporter (+15.15) and peroxisomal trans-2-enoyl- reductase (+8), indicating that cells are coping with starvation by accumulating defensive molecules and lipids confirming again the results reported by Heydarizadeh in which light stress and carbon deprivation stress redirect the CCM toward lipids accumulation8,9. Cmp-sialic acid is one of a series of nucleotide sugar transporters, a class of transporters that have been associated with inflammation, tumours, virus infections, and other processes16. It transports CMP-sialic acid from the cytosol into Golgi vesicles where glycosyltransferases are activated and produce sialilated glycolipids that can act as defensive molecules. The presence of sialic acids has been ascertained only in animals, even though recently they have also been discovered in plants17,18 and in green microalgae19, but their role is still uncertain.
Peroxisomal trans-2-enoyl-reductase seems to be involved in the peroxisomal degradation of phytols, a product derived from chlorophyll degradation, and has also been proposed to participate in a peroxisomal fatty acid chain elongation system, indicating that cells are probably accumulating long fatty acids in response to starvation status20.
The CTR-specific uniquely annotated GO terms, ‘Interleukin-8 biosynthetic process’ and ‘lactate biosynthetic process’, and the STRESS specific ‘1-3-beta-D-glucan biosynthetic process’ are all involved in defensive systems and in pathogenic mechanisms in humans21,22. It would thus be very interesting to further investigate these processes to unveil their role in diatoms.
Worthy of mention is also the upregulation of the ‘flavonoid metabolism’ pathway in STRESS, which suggests that this condition could be useful to possibly increase the yield of this important class of molecules that are beneficial for human health23.
In addition to the above-mentioned differentially expressed transcripts, also enzymes in the chains for sphingolipid, sterol, spermidine, PUFA, malonylCoA, lipotheicoic, l-ascorbate, l-cysteine biosynthesis, and isoprenoid pathways were upregulated in STRESS confirming that the depletion of silica is a good condition to stimulate the synthesis of bioactive molecules.
Furthermore, with our study we show for the first time, that diatoms are capable of synthesising secondary metabolites such as polyketides and secologanin, the latter at increased levels under silica starvation. Moreover, we confirm the presence of genes that synthesize animal hormone-like molecules, such as prostaglandins also in this diatom species24, Thalassiosira rotula, other than Skeletonema marinoi already published in the 2017.
Secologanin is involved in a complex metabolic pathway leading to the synthesis of Monoterpenoid Indole Alkaloids (MIAs), a large class of chemically different bioactive molecules considered very attractive due to their activity against cancer and other severe diseases in humans25. Some of the most active anticancer drugs such as camptothecans and vinca alkaloids26 have this type skeleton. Secologanin is a precursor for many of these compounds that are present in low amounts in the Madagascar periwinkle Catharanthus roseus leaves27,28. The enzymes involved in its formation are target of increasing research efforts in the phytochemical area, but to date they have been identified only in a few plant species. Discovering secologanin synthase in the genome of a diatom species paves the way to a completely new research opportunity for the understanding of the synthetic pathway and the role of secologanin in eukaryotic unicellular photosynthetic organisms.
Surprisingly, we found the transcripts for enzymes involved in polyketide biosynthesis. Polyketides (PKs) are an important class of molecules with a broad range of biological and pharmacological activities29. They include toxins, siderophores, pigments, antibiotics, cytostatics, and immune-suppressants. PK Synthases (PKSs) are widely distributed in bacteria and sporadically in archaea and eukarya where they probably derive from bacteria via horizontal gene transfer30–32. There has been no report until now on the existence of polyketide enzymes in a diatom species even if this is not the first attempt to find them in diatoms. Kohli et al. (2016) made an evolutionary study on PKSs in marine microbial genera, including diatoms, utilizing the data in the MMETSP database. The species they considered also included the T. rotula strains we used in our in silico analysis. However, they were unable to find any PKS in their transcriptome, nor in all the Bacillariophytes they took into consideration29 whereas we found a low expression of PKS in the strain 3096 transcriptome, both in the dark (DC) and light portion (LC) of the day/night cycle. A possible reason for this discrepancy is the low sensitivity of the annotation pipeline used for data analysis in the MMETSP project, while for our analysis, we used Annocript33, an assembler pipeline specifically developed in our institute that differs from the ones used in the MMETSP project.
Prostaglandins are a very interesting class of PUFA-derived molecules acting as hormones that play a pivotal role in many physiological processes in animals34. They are present not only in mammals, but also in both marine and terrestrial invertebrates35 and only few studies report their occurrence in terrestrial plants36.
Some macroalgae also possess the prostaglandin pathway, but only a limited number of prostaglandin molecules have been described from this source37,38. Recently, we were able to demonstrate the presence of this pathway also in diatoms, showing that Skeletonema marinoi has the ability to synthesize the complete panel of three classes of prostaglandins24. With the present study, we demonstrate the expression of the prostaglandin pathway in another diatom species and the significant downregulation of the first enzyme, PGHS-2, induced by silica depletion, differently from S. marinoi in which we did not observe any regulation of the pathway by nutrient limitation24.
Overall, the results obtained from the transcriptomic analysis of T. rotula, provide evidence for the presence of biosynthetic pathways involved in the production of secondary metabolites with potentially interesting biotechnological applications and the possibility to modulate their expression by varying culturing conditions. Although the results do not definitely provide information on the conditions to stimulate secondary metabolite production, the use of different transcriptomes used together may facilitate the understanding of the most suitable conditions for the expression of the metabolites of interest. Another criticism to our work could be that our T. rotula strain from the Mediterranean Sea (Gulf of Naples, Italy) and T. rotula strains from other geographic regions do not represent the same species. However, a recent phylogenetic study by Whittaker et al.39 among different Thalassiosira species and strains, including a T. rotula strain from the Gulf of Naples, concluded that “T. rotula lineages should be considered as a single species”.
In conclusion, our results provide new stimuli to investigate the role and function of these newly discovered molecules in diatoms, especially as concerns their interactions with other phytoplankton species and with the surrounding environment.
Methods
Strain culturing and RNA extraction
T. rotula, strain CCMP1647, lab-name FE80, was isolated in 2011 in the Gulf of Naples (40°48.5′N, 14°15′E), Mediterranean Sea. Clonal cultures were established by isolating single cells from phytoplankton net samples collected from the surface layer of the water column. Cultures were grown in sterile filtered oligotrophic seawater amended with f/240 nutrients at a temperature of 18–20 °C, at 12:12 h light:dark cycle, with a photon flux of 100 μ mol photons m−2 s−1, under agitation true filtered air influx.
2 L cultures, containing 2,5 × 103 cells/mL, were grown in complete f/2 (CTR) and in silica depleted f/2 media with 36 μM Na2SiO3 (STRESS), in triplicates, until they reached the stationary growth phase (day 6 for CTR cultures and day 7 for the STRESS cultures). Cultures were harvested by filtration onto 1.2 μm pore size filters (RAWP04700 Millipore) and immediately frozen in liquid nitrogen.
10 Liter cultures of T. rotula, in triplicate, were used to follow their growth from day 3 to day 10. Every day, 250 mL of each culture was harvested by filtration onto 1.2 μm pore size filters (RAWP04700 Millipore) and immediately frozen in liquid nitrogen, and 100–200 mL of culture media recovered from the cell filtration was collected and stored at −80 °C until sample processing.
Total RNA was extracted with the RNeasy Micro Kit Quiagen according to the manufacturer’s instructions including a step with DNase digestion. RNA concentration was determined using a Qubit® 2.0 Fluorometer (Invitrogen) and a quality check was performed by gel electrophoresis (1% agarose w/v) and an Agilent2100 bioanalyzer.
Antibiotic Treatment to produce axenic cultures
1 mL of exponentially growing culture was inoculated in F/2 medium containing final concentrations of 0.1 mg/mL Streptomycin (PanReac Applichem, A1852), 0.06 mg/mL Penicillin (PanReac Applichem, A1837), 1 mg/mL Ampicillin (PanReac Applichem, A0839), 0.1 mg/mL kanamicine (PanReac Applichem, A1493) and 0.02 mg/mL cefotaxime (Sigma-Aldrich, C7039) and allowed to grow for 5–6 days under standard growth conditions. Bacterial contamination was checked in two ways (SI1: Supplementary Fig. 6): (i) by staining DNA with DAPI and examining cultures under the microscope to check for the presence/absence of bacterial nucleoids; (ii) by performing peptone tests. For DAPI staining, 1 µL of DAPI stock solution (4′,6-diamidino-2-phenylindole, 1 mg/mL, Roche, Basel, Switzerland) was added to 1 mL of formalin preserved culture, incubated for 10 minutes and observed under the epifluorescence microscope. For peptone tests, 1 mL of diatom culture was added to a tube containing a peptone solution (1 mg/mL), incubated in the dark and checked after 2–3 days and 1–2 weeks; growth of bacteria in the tubes indicated contamination. If bacterial contamination persisted, the treatment was repeated. Large volume cultures used for DNA extraction were grown with antibiotics and the contamination tests were always performed on an aliquot of the culture.
DNA extraction
Axenic T. rotula cells (strain FE80) were collected onto a 1.2 µm RAWP membrane filter (Millipore, Billerica, MA,US). The filter was rinsed with 1.5 mL seawater and cells were further collected into Eppendorf tubes and pelleted by centrifugation at 3,800 g at 4 °C for 5 minutes. The DNA was extracted following a Phenol-Chloroform extraction method41 with slight modifications that included cell disruption by adding 400 mg of 0.2–0.3 mm diameter silica beads and vortex mixing at 30 hertz for 85 seconds (3 times), and cooling the pellet on ice between the vortex mixings. The extracted DNA was ethanol precipitated, air dried, dissolved in 50 µL of sterile water and stored at −20 °C until sequencing.
RNA sequencing
Next generation sequencing experiments, including sample quality control, were performed by Genomix4life S.R.L. (Baronissi, Salerno, Italy). Indexed libraries were prepared from 2 µg/each purified RNA with TruSeq Stranded mRNA Sample Prep Kit (Illumina) according to the manufacturer’s instructions. Libraries were quantified using the Agilent 2100 Bioanalyzer (Agilent Technologies) and pooled such that each index-tagged sample was present in equimolar amounts, with a final concentration of the pooled samples of 2 nM. The pooled samples were subject to cluster generation and sequencing using an Illumina HiSeq 2500 System (Illumina) in a 2 × 100 paired-end format at a final concentration of 8 pmol.
Transcriptome assembly and annotation
Illumina paired-end 100 bp reads from 6 T. rotula samples were processed to produce the transcriptome assembly. Raw reads were trimmed and clipped with BBDuk (https://jgi.doe.gov/data-and-tools/bbtools/) setting a minimum Phred-like quality of 35 and a minimum length of 35 nucleotides. The quality of the reads before and after trimming was checked with the software FASTQC (http://www.bioi http://www.bioinformatics.babraham.ac.uk/projects/fastqc/nformatics.babraham.ac.uk/projects/fastqc/). Possible human contaminant reads were removed after mapping the high quality reads against the reference human genome (GRCh38) with STAR (v 2.4.0j)42. High quality reads were then normalized with Trinity43 using the options: –SS_lib_type RF–pairs_together–max_cov 50. De novo transcriptome assembly was then performed with Trinity using the options: –SS_lib_type RF–no_normalize_reads–min_kmer_cov 1–KMER_SIZE 32. Transcriptome redundancy was removed with CD-HIT-EST44 using the following options: -r 0 -g 1. A filter for contaminants was performed by BLASTing the transcripts against the NCBI nr database, discarding all the sequences having a significant hit (evalue < = 0.0001) against bacteria or metazoa.
The completeness of the assembly was checked against the Core Eukaryotic Genes database (http://korflab.ucdavis.edu/Datasets/genome_completeness/ and http://korflab.ucdavis.edu/datasets/cegma/).
In silico translation was performed with TransDecoder45 whereas Functional Annotation was performed with Blast2GO software46.
Transcriptome expression quantification and differential expression analysis
Transcript expression quantification was performed using Express (v 1.5.1)47 after mapping the reads against the assembly with STAR42. Posterior counts were used as input to perform transcript differential expression analysis with EBSeq48 transcripts with a probability of being differentially expressed higher than 0.95 were considered significant.
The contigs and peptide sequences for T. rotula FE80 (CCMP1647) can be found in Supplementary Files 7 and 8. Peptide sequences were used for the subsequent analyses. To annotate the translated transcriptome we used the custom pipeline Annocript33. We used the Swiss-Prot (SP) and UniRef9049 (version: August 2013) for the blastp against proteins with the following parameters: word_size = 4; e-value = 10−5; num_descriptions = 5; num_alignments = 5; threshold = 18.
For each sequence the best hit, if any, was chosen. Rpsblast parameters, to identify domains composition of putative proteins in the Conserved Domains Database, were: e-value = 10−5; num_descriptions = 20; num_alignments = 20.
The software returned GO functional classification50, the Enzyme Commission IDs51 and Pathways52 descriptions associated to the resulting best matches.
R scripts were used to perform further analyses and graphs (http://www.r-project.org/).
Venn diagram was done using the freely available software interactivenn (http://www.interactivenn.net/).
We used the t-test to compare the FPKM for groups of peptides associated to the same GO/pathway and the Fisher exact test to compare the corresponding proportions of transcripts.
Bioinformatic identification and selection of pathways of interest
Pathways annotated as ‘prostaglandin biosynthesis’ and ‘secologanin biosynthesis’ were found among the second level pathways list generated within the Annocript pipeline annotation of the proteome from the T. rotula CCMP 1647 RNA-seq. Transcripts associated to the above pathways were extrapolated from the total proteome annotation table (Supplementary Data 13). Transcript and enzymes involved in polyketide biosynthesis were found by searching in the transcriptome and proteome blast description table, using the term ‘polyketide’ as search term.
Comparative analysis
Fasta files and contig reads of T. rotula strains CCMP3096 and GSO102’s RNA-seq were retrieved from the public database CAMERA (http://camera.crbs.ucsd.edu/mmetsp/list.php) and iMicrobes interactive query tool for microbial data (http://data.imicrobe.us/sample/view/1867). Corresponding proteome were annotated with Annocript and the output files for pathway and global annotation were used to search for the selected pathways. For comparisons among the proteomes, we used FPKM. Paired aligned reads were used to calculate the Fragments Per Kilobase of transcript per Million fragments mapped (FPKM) of each sequence for each transcriptome as a measure of expression levels.
The FPKM were calculated as follows: FPKM = [mapped reads pairs]/([length of transcript]/1000)/([total reads pairs]/106).
Reverse transcription
1 μg of total RNA used for RNA-seq was retro-transcribed in the T100 Thermal cycler (Bio-Rad Laboratories, Hercules, CA, USA) following the manufacturer’s instructions of the sensiFAST™ cDNA synthesis kit (Bioline, Cat. No. BIO-65054).
Primer design and real time quantitative PCR
Candidate reference genes and genes of interest were selected considering the annotation of the peptides reported in the annotated transcriptome of T. rotula FE80 (CCMP1647) (Supplementary Data 13).
Primers for amplification of selected transcripts, designed using Primer3 program V. 0.453,54, are listed in SI1-Supplementary Table 5, together with their relative sequences and characteristics.
Each sequence was initially tested by standard PCR. Reactions were carried out in 25 μL volume with 2,5 μL of 10× PCR reaction buffer (Roche, Basel, Switzerland), 2,5 μL of 10 × 2 mM dNTP, 0.3 μL of 5 U/μL Taq (Roche, Basel, Switzerland), 1 μL 10 μΜ of each oligo, 1 μL of cDNA templates and nuclease-free water up to 25 μL. The PCR program consisted of a denaturation step at 95 °C for 3 min, 40 cycles at 95 °C for 30 s, 53 °C 30 s, 72 °C for 30 s, and a final extension step at 72 °C for 7 min. Amplified PCR products were analysed by agarose gel electrophoresis. The resulting bands were excised from the gel and extracted according to the GenElute Gel Extraction Kit protocol (Sigma-Aldrich, St. Louis, MO, USA). Sequences were obtained by BigDye Terminator Cycle Sequencing Technology (Applied Biosystems, Foster City, CA, USA) and purified using the Agencourt CleanSEQ Dye terminator removal Kit (Agencourt Bioscience Corporation, Beverly, MA, USA) in automated robotic station Biomek FX (Beckman Coulter, Pasadena, CA, USA). Products were analysed on the Automated Capillary Electrophoresis Sequencer 3730 DNA Analyser (Applied Biosystems, Foster City, CA, USA). Alignments were performed with BioEdit software V. 7.0.5.3 (http://www.mbio.ncsu.edu/BioEdit/bioedit.html).
Reverse transcription-quantitative PCR (rt-qPCR) experiments were performed in MicroAmp Optical 384-Well reaction plate (Applied Biosystems, Foster City, CA, USA) with Optical Adhesive Covers (Applied Biosystems, Foster City, CA, USA) in a Viia7 Real Time PCR System (Applied Biosystem, Foster City, CA, USA). Five serial dilutions of mixed cDNAs were used to determine primer reaction efficiency using the formula: E = 10−1/slope (SI1-Supplementary Fig. 5). The PCR volume for each sample was 10 μL, with 5 μl of SensiFASTTM SYBR® Lo-ROX Kit (BIO_94020, Bioline), 1 μL of cDNA template (1 to 5 dilution each template) and 4 μL of 0.7 μM oligo mix (forward and reverse). Program reaction used was: 95 °C for 20 s, 40 cycles of 95 °C for 1 s and 60 °C for 20 s. The program was set to reveal the melting curve of each amplicon from 60 °C to 95 °C, and read every 0.5 °C. Single peaks for all genes confirmed gene-specific amplification and the absence of primer-dimers. All RT-qPCR reactions were carried out in triplicate to capture intra-assay variability. Each assay included three no-template negative controls for each primer pair.
The normalized expression levels of each gene of interest relative to the most stable reference genes (see Supplementary Information 2 and 3 for reference genes analysis), actin and TBP, were calculated by using the Q-Gene tool55. Only TBP normalized values were reported in the main text and figure results.
Relative expression ratios above two fold were considered significant. Statistical analysis was performed using the unpaired t-test with Welch’s correction for comparison between conditions, using GraphPad Prim statistic software V. 6.01 (GraphPad Software Inc., San Diego, CA, USA).
Three different algorithms were utilized to identify the best reference genes in our experimental design: BestKeeper56; NormFinder57 and geNorm58.
Statistics
Using GraphPadPrism6 software t-test was performed between CTR and STRESS sample for qPCR amplification data to determine significant differences between treatments.
R software was used for bioinformatic analysis of the transcriptome performing the following tests: Fisher test to identify unique GO terms; t-test between the CTR and STRESS condition considering the FPKM expression value of each pathway; heatmaps in different conditions for the selected pathways; boxplot analysis of the expression in FPKM of each pathway in each level of annotation for each replicate and group.
Supplementary information
Acknowledgements
We thank the Molecular Biology and Sequencing Facility at the Stazione Zoologica Anton Dohrn Napoli (P. De Luca, E. Biffali, R. Pannone, E. Mauriello) for oligo supply service, qPCR support, PCR amplicons sanger sequencing service. We are grateful to Massimo Perna for his technical assistance for growth media preparation and Alberto Amato for useful discussions. This work was supported by grants from the Italian National Project ‘PON (Programma Operativo Nazionale) 01_00117-Antigeni e Adiuvanti per Vaccini e Immunoterapia’, the H2020-MSCA-RISE 2015: Ocean Medicines (Project ID: 690944) and from the Italy-South Africa joint Research Programme 2017: ‘Genomics for a blue economy’ funded by Ministero dell’Ambiente e della Tutela del Territorio e del Mare.
Author Contributions
V.D.D., A.I. and G.R., designed the experiments. V.D.D., F.D.C., R.B. and A.P., performed experiments. V.D.D. grew the cultures and performed the RNA extraction for the transcriptome sequencing, performed the bioinformatics analysis of the transcriptome and designed the molecular analysis by rt-PCR and rt-qPCR; F.D.C., R.B. and A.P. performed the rt-PCR and rt-qPCR runs; F.D.C. performed the gel extraction and sequencing verification of the gDNA PCR amplicons; V.D.D. wrote the paper with assistance from G.R. and A.I., V.D.D. made the figures with the assistance of F.D.C.
Data Availability
The sequencing data described in this article, i.e. the transcript and peptide sequences of T. rotula, strain CCMP1647, lab-name FE80, are in supplementary files 7 and 8 respectively. The sequencing data relative to the T. rotula strains CCMP3096 and GSO102 used for the comparative analysis are available at https://www.imicrobe.us/#/projects/104.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-46276-8.
References
- 1.Kroth PG, et al. A model for carbohydrate metabolism in the diatom Phaeodactylum tricornutum deduced from comparative whole genome analysis. PLoS One. 2008;3:e1426. doi: 10.1371/journal.pone.0001426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nisbet RER, Kilian O, McFadden GI. Diatom Genomics: Genetic Acquisitions and Mergers. Current Biology. 2004;14:R1048–R1050. doi: 10.1016/j.cub.2004.11.043. [DOI] [PubMed] [Google Scholar]
- 3.Armbrust E. Virginia. The life of diatoms in the world's oceans. Nature. 2009;459(7244):185–192. doi: 10.1038/nature08057. [DOI] [PubMed] [Google Scholar]
- 4.Lauritano C, Carotenuto Y, Miralto A, Procaccini G, Ianora A. Copepod Population-Specific Response to a Toxic Diatom Diet. PLOS One. 2012;7:e47262. doi: 10.1371/journal.pone.0047262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falciatore A, d’Alcalà MR, Croot P, Bowler C. Perception of environmental signals by a marine diatom. Science. 2000;288:2363–2366. doi: 10.1126/science.288.5475.2363. [DOI] [PubMed] [Google Scholar]
- 6.Romano G, et al. Marine microorganisms as a promising and sustainable source of bioactive molecules. Mar. Environ. Res. 2017;128:58–69. doi: 10.1016/j.marenvres.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 7.Muhseen ZT, Xiong Q, Chen Z, Ge F. Proteomics studies on stress responses in diatoms. Proteomics. 2015;15:3943–3953. doi: 10.1002/pmic.201500165. [DOI] [PubMed] [Google Scholar]
- 8.Heydarizadeh, P. et al. Carbon Orientation in the Diatom Phaeodactylum tricornutum: The Effects of Carbon Limitation and Photon Flux Density. Front Plant Sci10 (2019). [DOI] [PMC free article] [PubMed]
- 9.Heydarizadeh, P. et al. Response of CO2-starved diatom Phaeodactylum tricornutum to light intensity transition. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 372 (2017). [DOI] [PMC free article] [PubMed]
- 10.Shrestha RP, Hildebrand M. Evidence for a regulatory role of diatom silicon transporters in cellular silicon responses. Eukaryotic Cell. 2015;14:29–40. doi: 10.1128/EC.00209-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brzezinski M, Olson R, Chisholm S. Silicon availability and cell-cycle progression in marine diatoms. Marine Ecology Progress Series. 1990;67:83–96. doi: 10.3354/meps067083. [DOI] [Google Scholar]
- 12.Keeling PJ, et al. The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLoS Biol. 2014;12:e1001889. doi: 10.1371/journal.pbio.1001889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan Y, Bates SS, Cembella AD. Environmental stress and domoic acid production by Pseudo-nitzschia: a physiological perspective. Nat. Toxins. 1998;6:127–135. doi: 10.1002/(SICI)1522-7189(199805/08)6:3/4<127::AID-NT9>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 14.Shrestha RP, et al. Whole transcriptome analysis of the silicon response of the diatom Thalassiosira pseudonana. BMC Genomics. 2012;13:499. doi: 10.1186/1471-2164-13-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fenton TR, Gout IT. Functions and regulation of the 70 kDa ribosomal S6 kinases. Int. J. Biochem. Cell Biol. 2011;43:47–59. doi: 10.1016/j.biocel.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 16.Gerardy-Schahn R, Oelmann S, Bakker H. Nucleotide sugar transporters: biological and functional aspects. Biochimie. 2001;83:775–782. doi: 10.1016/S0300-9084(01)01322-0. [DOI] [PubMed] [Google Scholar]
- 17.Bakker H, et al. A CMP-sialic acid transporter cloned from Arabidopsis thaliana. Carbohydr. Res. 2008;343:2148–2152. doi: 10.1016/j.carres.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 18.Takashima S, et al. Analysis of CMP-sialic acid transporter-like proteins in plants. Phytochemistry. 2009;70:1973–1981. doi: 10.1016/j.phytochem.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 19.Mamedov T, Yusibov V. Green algae Chlamydomonas reinhardtii possess endogenous sialylated N-glycans. FEBS Open Bio. 2011;1:15–22. doi: 10.1016/j.fob.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das AK, Uhler MD, Hajra AK. Molecular cloning and expression of mammalian peroxisomal trans-2-enoyl-coenzyme A reductase cDNAs. J. Biol. Chem. 2000;275:24333–24340. doi: 10.1074/jbc.M001168200. [DOI] [PubMed] [Google Scholar]
- 21.Yuan A, Chen JJW, Yao P-L, Yang P-C. The role of interleukin-8 in cancer cells and microenvironment interaction. Front. Biosci. 2005;10:853–865. doi: 10.2741/1579. [DOI] [PubMed] [Google Scholar]
- 22.Ferguson BS, et al. Lactate metabolism: historical context, prior misinterpretations, and current understanding. Eur. J. Appl. Physiol. 2018;118:691–728. doi: 10.1007/s00421-017-3795-6. [DOI] [PubMed] [Google Scholar]
- 23.Tungmunnithum, D., Thongboonyou, A., Pholboon, A. & Yangsabai, A. Flavonoids and Other Phenolic Compounds from Medicinal Plants for Pharmaceutical and Medical Aspects: An Overview. Medicines (Basel)5 (2018). [DOI] [PMC free article] [PubMed]
- 24.Di Dato V, et al. Animal-like prostaglandins in marine microalgae. ISME J. 2017;11:1722–1726. doi: 10.1038/ismej.2017.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Connor SE, Maresh JJ. Chemistry and biology of monoterpene indole alkaloid biosynthesis. Nat Prod Rep. 2006;23:532–547. doi: 10.1039/b512615k. [DOI] [PubMed] [Google Scholar]
- 26.Venditto VJ, Simanek EE. Cancer Therapies Utilizing the Camptothecins: A Review of in Vivo Literature. Mol Pharm. 2010;7:307–349. doi: 10.1021/mp900243b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Irmler S, et al. Indole alkaloid biosynthesis in Catharanthus roseus: new enzyme activities and identification of cytochrome P450 CYP72A1 as secologanin synthase. Plant J. 2000;24:797–804. doi: 10.1046/j.1365-313x.2000.00922.x. [DOI] [PubMed] [Google Scholar]
- 28.English BJ, Williams RM. A divergent strategy for the synthesis of secologanin derived natural products. J. Org. Chem. 2010;75:7869–7876. doi: 10.1021/jo101775n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kohli GS, John U, Van Dolah FM, Murray SA. Evolutionary distinctiveness of fatty acid and polyketide synthesis in eukaryotes. ISME J. 2016;10:1877–1890. doi: 10.1038/ismej.2015.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H, Fewer DP, Holm L, Rouhiainen L, Sivonen K. Atlas of nonribosomal peptide and polyketide biosynthetic pathways reveals common occurrence of nonmodular enzymes. Proc. Natl. Acad. Sci. USA. 2014;111:9259–9264. doi: 10.1073/pnas.1401734111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stewart C, Vickery CR, Burkart MD, Noel JP. Confluence of structural and chemical biology: plant polyketide synthases as biocatalysts for a bio-based future. Curr. Opin. Plant Biol. 2013;16:365–372. doi: 10.1016/j.pbi.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 32.Wang H, Sivonen K, Fewer DP. Genomic insights into the distribution, genetic diversity and evolution of polyketide synthases and nonribosomal peptide synthetases. Curr. Opin. Genet. Dev. 2015;35:79–85. doi: 10.1016/j.gde.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 33.Musacchia F, Basu S, Petrosino G, Salvemini M, Sanges R. Annocript: a flexible pipeline for the annotation of transcriptomes able to identify putative long noncoding RNAs. Bioinformatics. 2015;31:2199–2201. doi: 10.1093/bioinformatics/btv106. [DOI] [PubMed] [Google Scholar]
- 34.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 35.Rowley AF, Vogan CL, Taylor GW, Clare AS. Prostaglandins in non-insectan invertebrates: recent insights and unsolved problems. J. Exp. Biol. 2005;208:3–14. doi: 10.1242/jeb.01275. [DOI] [PubMed] [Google Scholar]
- 36.Groenewald EG, van der Westhuizen AJ. Prostaglandins and related substances in plants. The Botanical Review. 1997;63:199–220. doi: 10.1007/BF02857948. [DOI] [Google Scholar]
- 37.Ritter A, et al. Copper stress induces biosynthesis of octadecanoid and eicosanoid oxygenated derivatives in the brown algal kelp Laminaria digitata. New Phytol. 2008;180:809–821. doi: 10.1111/j.1469-8137.2008.02626.x. [DOI] [PubMed] [Google Scholar]
- 38.Imbs AB, et al. Distribution of polyunsaturated fatty acids in red algae of the genus Gracilaria, a promising source of prostaglandins. Russ J Mar Biol. 2012;38:339–345. doi: 10.1134/S1063074012040049. [DOI] [Google Scholar]
- 39.Whittaker KA, Rignanese DR, Olson RJ, Rynearson TA. Molecular subdivision of the marine diatom Thalassiosira rotulain relation to geographic distribution, genome size, and physiology. BMC Evolutionary Biology. 2012;12:209. doi: 10.1186/1471-2148-12-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guillard Robert R. L. Culture of Marine Invertebrate Animals. Boston, MA: Springer US; 1975. Culture of Phytoplankton for Feeding Marine Invertebrates; pp. 29–60. [Google Scholar]
- 41.Sabatino Valeria, Russo Monia Teresa, Patil Shrikant, d’Ippolito Giuliana, Fontana Angelo, Ferrante Maria Immacolata. Establishment of Genetic Transformation in the Sexually Reproducing Diatoms Pseudo-nitzschia multistriata and Pseudo-nitzschia arenysensis and Inheritance of the Transgene. Marine Biotechnology. 2015;17(4):452–462. doi: 10.1007/s10126-015-9633-0. [DOI] [PubMed] [Google Scholar]
- 42.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 45.Haas BJ, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Conesa A, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 47.Roberts A, Pachter L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat. Methods. 2013;10:71–73. doi: 10.1038/nmeth.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leng N, et al. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013;29:1035–1043. doi: 10.1093/bioinformatics/btt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics. 2007;23:1282–1288. doi: 10.1093/bioinformatics/btm098. [DOI] [PubMed] [Google Scholar]
- 50.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bairoch A. The ENZYME database in 2000. Nucleic Acids Res. 2000;28:304–305. doi: 10.1093/nar/28.1.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morgat A, et al. UniPathway: a resource for the exploration and annotation of metabolic pathways. Nucleic Acids Res. 2012;40:D761–769. doi: 10.1093/nar/gkr1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Untergasser A, et al. Primer3–new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23:1289–1291. doi: 10.1093/bioinformatics/btm091. [DOI] [PubMed] [Google Scholar]
- 55.Simon P. Q-Gene: processing quantitative real-time RT-PCR data. Bioinformatics. 2003;19:1439–1440. doi: 10.1093/bioinformatics/btg157. [DOI] [PubMed] [Google Scholar]
- 56.Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004;26:509–515. doi: 10.1023/B:BILE.0000019559.84305.47. [DOI] [PubMed] [Google Scholar]
- 57.Andersen CL, Jensen JL, Ørntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64:5245–5250. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
- 58.Vandesompele J, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data described in this article, i.e. the transcript and peptide sequences of T. rotula, strain CCMP1647, lab-name FE80, are in supplementary files 7 and 8 respectively. The sequencing data relative to the T. rotula strains CCMP3096 and GSO102 used for the comparative analysis are available at https://www.imicrobe.us/#/projects/104.