

Abstract
This analysis aims to describe the outcomes of two nonambulatory patients with Duchenne muscular dystrophy (DMD) who participated in two clinical studies. The two consecutive trials of eteplirsen (studies 201 and 202) were conducted in patients with DMD (N = 12) and confirmed genetic mutations amenable to exon 51 skipping.
In study 201, 12 patients were randomized to receive once-weekly, double-blind intravenous infusions of eteplirsen 30 or 50 mg/kg or placebo for 24 weeks; patients then received open-label eteplirsen during weeks 25 through 28. All 12 patients continued onto open-label extension study 202 and received long-term treatment with eteplirsen. We compared cardiac, pulmonary, and upper limb function and dystrophin production in the nonambulatory twin patients versus the 10 ambulatory patients through 240 combined treatment weeks.
Ten study patients remained ambulatory through both studies, while the identical twin patients both experienced early, rapid loss of ambulation. The twin patients had greater disease severity at baseline (6-minute walk test [6MWT], 330 and 256 m) versus the other patients (n = 10; 6MWT range, 341–418 m). They maintained cardiac and upper limb function through combined week 240, with outcomes similar to those of the patients who remained ambulatory. Dystrophin production was confirmed following eteplirsen treatment.
Despite the loss of ambulation, other markers of disease progression remained relatively stable in the eteplirsen-treated twin patients and were similar to those of the ambulatory patients.
Keywords: dystrophin, echocardiography, left ventricular ejection fraction, upper extremity, ventricular dysfunction, vital capacity
1. Introduction
Duchenne muscular dystrophy (DMD) is a progressive, X-linked recessive, degenerative, universally fatal neuromuscular disease caused by lack of dystrophin that results in loss of ambulation and wheelchair dependency by the early teens.[1,2] A primary concern for nonambulatory patients and their caregivers is the maintenance of respiratory and cardiac function, as respiratory and cardiac deterioration are leading causes of death in patients with DMD.[3–6] In addition, preservation of upper extremity function[3] is important to maintain the ability to independently engage in activities of daily living, including self-care (e.g., feeding and grooming), computer use, writing, and control of manual or powered wheelchairs. Most clinical trials of disease-modifying agents for DMD focused on preservation of lower limb function.[7,8] However, as DMD-associated morbidity is not restricted to lower limb dysfunction, clinical assessments of disease-modifying treatments should also focus on preservation of pulmonary, cardiac, and upper limb function.
Eteplirsen (Exondys 51 [formerly AVI-4658]; Sarepta Therapeutics, Inc., Cambridge, MA) is the first DMD treatment approved by the US Food and Drug Administration, and is indicated for the treatment of DMD in patients with a genetically confirmed DMD gene mutation that is amenable to exon 51 skipping.[9] Patients with certain deletion mutations adjacent to exon 51 of the DMD gene produce an out-of-frame mRNA that results in the production of an unstable or nonfunctional protein product.[1] Eteplirsen targets exon 51 in dystrophin pre-mRNA to trigger skipping of exon 51,[1] resulting in restoration of the reading frame and allowing production of an internally truncated but functional dystrophin protein.[9–11]
Data from two consecutive studies of 12 patients treated with eteplirsen for up to 240 weeks at the time of this analysis were previously compared with data for untreated controls[10] or with natural history data.[12] These comparisons showed that long-term treatment with eteplirsen slowed disease progression, including measures of ambulatory and pulmonary function, and had no negative impact on cardiac function.[10,12,13] Two patients in the trial experienced early, rapid deterioration in ambulation. In this observational study, we compare long-term pulmonary, cardiac, and upper extremity function and dystrophin production in muscle biopsy samples obtained at week 180 in these two patients with that of 10 study patients who remained ambulatory throughout the trial.
2. Materials and methods
2.1. Study design
Details of the design of eteplirsen studies 201 and 202 have been described previously.[10] Briefly, study 201 (NCT01396239) was a 28-week trial conducted from July 2011 to February 2012 that comprised a 24-week double-blind phase and a 4-week open-label phase. Patients were randomized 1:1:1 to receive once-weekly, double-blind intravenous (IV) infusions of eteplirsen (30 or 50 mg/kg) or placebo for 24 weeks. Placebo patients were then randomized 1:1 to receive eteplirsen 30 or 50 mg/kg for weeks 25 through 28. During the last visit of study 201, eligible patients could be enrolled in study 202 (NCT01540409), an open-label extension study designed to assess the long-term efficacy and safety of eteplirsen, which initiated in February 2012 and ended in April 2016. A dose extension was completed and ended in August 2017. Patients continued on the same dose of eteplirsen through completion of study 202 (combined week 240 of studies 201 and 202). The studies were conducted in accordance with the principles of the Declaration of Helsinki and the International Council for Harmonisation Good Clinical Practice guidelines, and the ethics committee at Nationwide Children's Hospital approved the study protocol. Parents or legal guardians of all patients provided written informed consent before study participation and genetic testing.
2.2. Patients
Eligible patients for study 201 were aged 7 to 13 years with DMD and a genetically confirmed mutation amenable to exon 51 skipping, were able to walk 200 to 400 m (±10%) on the 6-minute walk test (6MWT), were receiving stable doses of oral corticosteroids for at least 24 weeks before study entry, and remained on stable corticosteroid therapy throughout the study.[2] Patients who completed study 201 were eligible to enroll in study 202, a long-term extension.
2.3. Functional efficacy
2.3.1. Ambulatory and pulmonary function assessments
The 6MWT and pulmonary function tests were performed at baseline, at least every 12 weeks through week 96, and every 24 weeks thereafter until week 240 and were described previously.[10]
2.3.2. Cardiac function assessment
As part of safety monitoring, standard 2-dimensional echocardiography (ECHO) of left ventricular ejection fraction (LVEF) was performed at the central site at baseline of study 201 and at prespecified time points every 10, 12, 14, or 24 weeks thereafter, through combined study week 240, to assess cardiac function. Medical personnel reviewed each ECHO, noting LVEF and designating findings as clinically or not clinically significant.
2.3.3. Upper limb functional assessments
The 9-Hole Peg Test was administered at least every 24 weeks using methods previously described.[14] The patient was timed on how quickly he could take 9 pegs from a shallow bowl indentation in the testing apparatus, place each peg into a hole one at a time, and put the pegs back, one at a time, in the shallow bowl indentation. Dominant and nondominant hands were tested twice, and the shorter time was used for analysis. Results of the dominant hand assessments are reported.
A maximum voluntary isometric contraction test (MVICT) was performed every 12 weeks through week 96 and every 24 weeks thereafter, using a Quantitative Movement Assessment system that utilized force transducers to assess elbow flexion and extension (kg) and hand grip (kg). For assessments of elbow muscle function, patients were placed supine with a strap attached to a cuff worn around the wrist; the opposite end of the strap was attached to the force transducer, which was secured to a stable frame. Hand grip function was evaluated while patients were sitting with their feet supported and the elbow positioned at a 90-degree angle. Patients completed a minimum of 3 trials. If the third trial produced the greatest value, up to 2 additional trials were then performed until either a maximum effort was achieved or 5 trials were completed. The maximum value for each muscle group was recorded.
2.4. Genetic diagnosis
Genetic mutation analysis was performed at Nationwide Children's Hospital as part of the study criteria. The presence of out-of-frame deletion(s) that could be corrected by skipping exon 51 (e.g., deletion mutations of exons 45–50, 47–50, 48–50, 49–50, 50, 52, or 52–63) were confirmed in a Clinical Laboratory Improvement Act–accredited laboratory by any peer-reviewed and published methodology that evaluates all exons. This methodology included, but was not limited to, multiplex ligation-dependent probe, comparative genomic hybridization, and single condition amplification/internal primer analysis.
2.5. Dystrophin assessments
2.5.1. Muscle biopsies
Details of muscle biopsy procedures conducted in studies 201 and 202 were described previously.[2,11] Eleven patients consented to an additional, optional fourth biopsy of the contralateral deltoid at week 180. A total of 9 baseline comparator muscle biopsies were also evaluated, including 3 baseline samples from studies 201 and 202 and 6 biceps biopsy samples from the eteplirsen PROMOVI clinical trial (NCT02255552), a study with comparable inclusion criteria to studies 201 and 202.
2.5.2. Western immunoblot for dystrophin
Muscle biopsy tissue sections were homogenized in buffer containing 4 M urea (Teknova; Hollister, California), 125-mM Tris pH 6.8 (Amresco; Solon, OH), 4% sodium dodecyl sulfate solution (Fisher; Loughborough, UK), and 1 complete mini protease inhibitor tablet/7-mL buffer (Roche; Basel, Switzerland). A total protein assay of the tissue extract was performed with the RC DC Protein Assay Kit II per the manufacturer's instructions (BioRad; Hercules, CA). For each patient sample, 50 μg total protein was loaded onto a NuPAGE Novex 10 well, 1-mm, mini 3% to 8% polyacrylamide Tris-acetate gel (Life Technologies; Carlsbad, CA), followed by transfer onto Invitrolon polyvinylidene difluoride (PVDF) membranes (Life Technologies; Carlsbad, CA). Dystrophin was detected by incubation PVDF membranes with 1:20 dilution of DYS1 (Leica; Wetzlar, Germany) followed by 1:40,000 anti-mouse IgG-conjugated horseradish peroxidase (GE Healthcare; Little Chalfont, UK). Signal from the membrane was developed using ECL Prime Buffer (GE Healthcare) followed by film exposure. Western blot films were analyzed using ImageQuant TL Plus software version 8.1 (GE Healthcare), and linear regression analysis was performed using GraphPad software. To determine the percent dystrophin in a sample, dystrophin band intensities as well as the input percent of normal control (NC) were log-transformed, and linear regression analysis was performed on the log-transformed data. The percent NC was then calculated using the equation: %NC = 10^((LOG(band intensity)-y intercept)/slope).
2.5.3. Percent dystrophin-positive fibers
The methodology for immunohistochemical staining was described previously.[2,11] Dystrophin-positive fibers were manually scored by independent, blinded analysts at Nationwide Children's Hospital and at Flagship Biosciences from randomized digital images of muscle biopsies stained using indirect immunofluorescence labeling with the primary antibody MANDYS106.
2.5.4. Bioquant analysis
Bioquant Life Sciences imaging software (Bioquant Image Analysis Corporation; Nashville, TN) was used to quantify relative dystrophin levels associated with muscle fiber membranes by measuring relative fluorescence levels from blinded, randomized digital images of muscle biopsies stained using indirect immunofluorescence labeling with the primary antibody MANDYS106 (Developmental Studies Hybridoma Bank; University of Iowa, Department of Biology, Iowa City, IA). Percent intensity was calculated for each test sample relative to the NC value, as previously described.[2,11]
2.6. Safety assessments
Safety assessments included the review and evaluation of treatment-emergent adverse events (TEAEs), physical examinations, vital signs, clinical laboratory testing, and the use of concomitant medications.
2.7. Statistical analysis
Baseline was defined as the last value prior to the first dose of eteplirsen. For the 6MWT, a distance of 0 m was used if the patient was unable to complete the 6MWT. Fold difference in PDPF and Bioquant intensity at week 180 was calculated as the mean in the eteplirsen group divided by the mean in the untreated control group. P values comparing eteplirsen patients versus untreated controls for dystrophin assessments (Western blot, PDPF, and fiber intensity) were calculated using a 2-sample t test, with statistical significance set at P < .05. For safety data, TEAEs were summarized using descriptive statistics.
3. Results
The patients who lost ambulation (patients A and B) were identical twin brothers, started ambulating independently at 16 months of age, were initially randomized to the eteplirsen 30-mg/kg treatment group, and remained on eteplirsen 30 mg/kg/week throughout the study. They were aged 9.9 years at study enrollment compared with a mean age for the remaining 10 patients (patients C–L) of 9.2 years (range, 7.4–11.0). Patient A had a body mass index (BMI) of 20.9 kg/m2 and weighed 39.8 kg; patient B had a BMI of 21.5 kg/m2 and weighed 39.7 kg. In comparison, the mean BMI of the remaining 10 patients was 20.3 kg/m2 (range, 16.4–25.6 kg/m2), and their mean weight was 29.9 kg (range, 22.1–38.3 kg). Baseline demographics and disease characteristics for all 12 male patients who completed the studies have been described previously.[2,10] Overall, the DMD gene mutations of all 12 patients represented 5 genotypes that caused out-of-frame deletions eligible for exon 51 skipping (deletion mutation of exons 45–50, 48–50, 49–50, 50, and 52)[2]; patients A and B had a deletion mutation of exons 45–50 as did 2 of the other 10 patients. Patients A and B lost ambulation at approximately week 36 while the other 10 patients remained ambulatory throughout the study.
As part of their preventive cardiovascular regimen, patients A and B were receiving losartan 25 mg once daily throughout the study, and carvedilol 3.125 mg twice daily until week 109, when they were switched to 6.25 mg twice daily and were maintained at that dose through the remainder of the study. Seven of the 10 patients who remained ambulatory were also taking concomitant medications as preventive cardiovascular regimens. Patients A and B were also taking albuterol as needed for mild asthma. The current daily medication regimen for patients A and B includes prednisone 40 mg, carvedilol 6.25 mg, eplerenone 0.5 mg, and losartan 25 mg.
3.1. Ambulation
The baseline 6MWT distances of patients A and B, 330 m and 256 m, respectively, were lower than the mean 6MWT distance of 376.4 m (range, 341–418 m) observed in the remaining 10 patients (Fig. 1). Longitudinally, patient A had a 6MWT distance of 50 m at week 24, 12 m at week 32, and 0 m at week 36; patient B had 6MWT distances of 17, 3, and 0 m at weeks 24, 32, and 36, respectively. The mean 6MWT distances achieved by the 10 ambulatory patients at weeks 24 and 36 were 381.8 m (range, 325–431 m) and 359.5 m (range, 302–458 m), respectively (Fig. 2).
Figure 1.
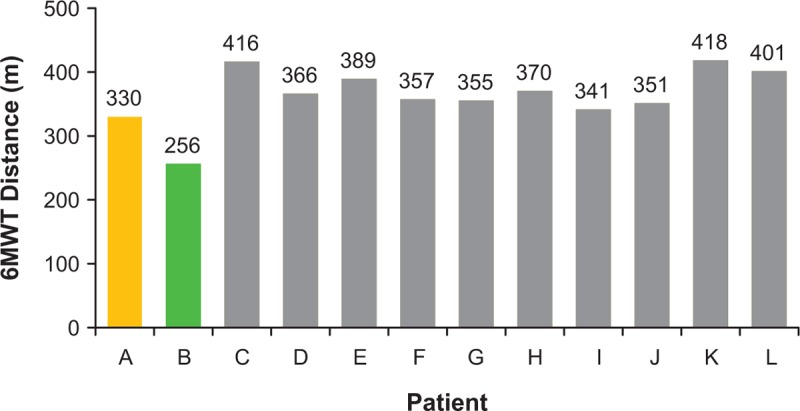
Six-minute walk test distance for all patients at baseline (day 1 visit). 6MWT = 6-minute walk test.
Figure 2.
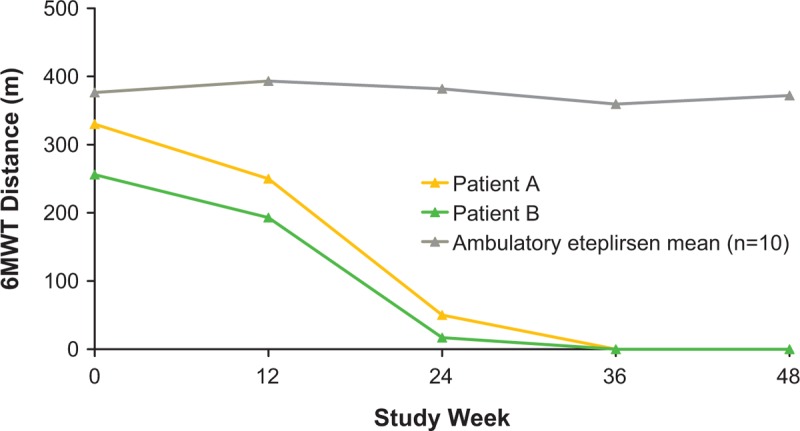
Rapid decline and loss of ambulation in the eteplirsen-treated twin patients. 6MWT = 6-minute walk test.
3.2. Pulmonary function
Patients A and B experienced an initial decline in percent predicted forced vital capacity (FVC%p), followed by a period of stability through week 96. Their FVC%p values subsequently declined between weeks 96 and 120, and again plateaued for the duration of the study (Fig. 3A). Values for FVC%p at week 240 in the nonambulatory twins were 80% for patient A and 72% for patient B. The study mean at week 240 for the ambulatory patients treated with eteplirsen was 92% (range, 61–112%). At the time of this writing, patients A and B were aged 16 years and had initiated intermittent, nocturnal ventilation at age 15.
Figure 3.
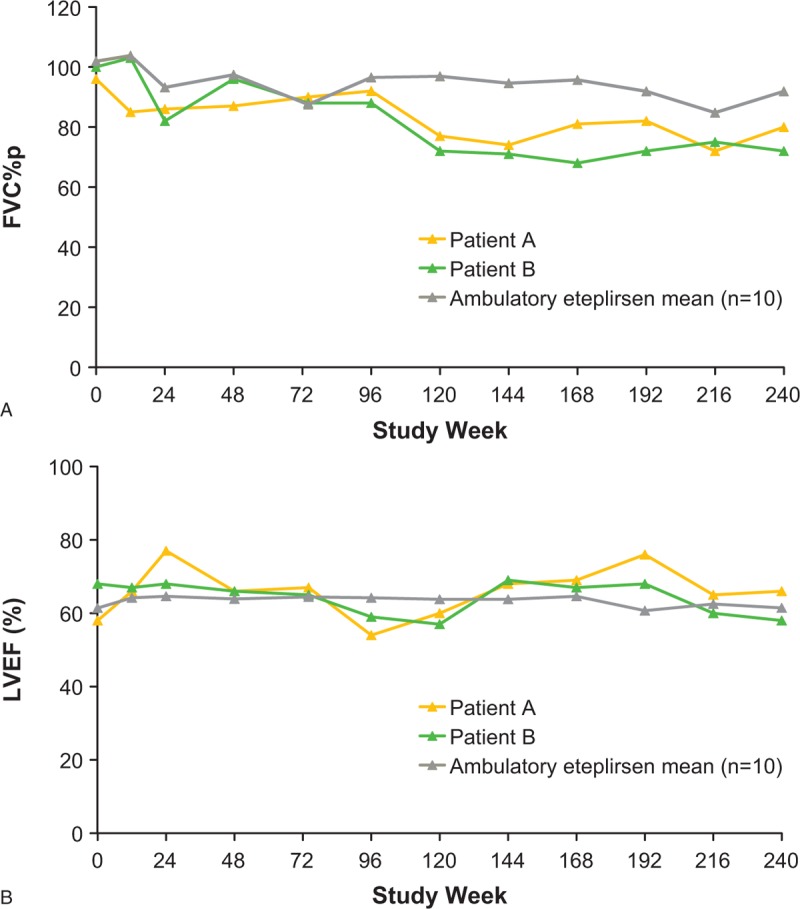
Measures of pulmonary and cardiac function in patients A and B versus study mean through 240 weeks of eteplirsen treatment: (A) FVC%p and (B) LVEF. FVC%p = percent predicted forced vital capacity, LVEF = left ventricular ejection fraction.
3.3. Cardiac function
Both twins maintained normal LVEF throughout the study (Fig. 3B), with values similar to study means through week 240. The LVEF values for patients A and B at week 240 were 66% and 58%, respectively. Similarly, the study mean at week 240 for the eteplirsen-treated ambulatory patients was 61% (range, 50–70%).
3.4. Upper limb function
Measures of upper limb function in the twins were reasonably stable (Fig. 4). For patient A, mean values at week 240 were 0:22 s for the 9-Hole Peg Test, 9.18 kg for dominant (right) hand grip strength, 0.81 kg for right elbow flexion, and 2.39 kg for right elbow extension at week 240. For patient B, mean values at week 240 were 0:31 s for the 9-Hole Peg Test, 9.60 kg for dominant hand grip strength, 1.44 kg for right elbow flexion, and 2.24 kg for right elbow extension. The 0:31 s 9-Hole Peg Test time for patient B may have resulted from a spinal fusion surgery conducted prior to week 240; prior to the surgery at week 216, patient B achieved a 9-Hole Peg Test completion time of 21 s. Taking this time point into consideration, overall, these findings were similar to study means for the 10 ambulatory patients through week 240, which were 0:19 s (9-Hole Peg Test), 7.5 kg (dominant hand grip strength), 1.77 kg (right elbow flexion), and 2.97 kg (right elbow extension). Currently, neither patient A nor B has developed hand contractures. In addition, both patients are generally able to feed themselves and to utilize computers or handheld devices to complete their schoolwork. They both require some assistance with hand transfer, particularly with heavy objects.
Figure 4.
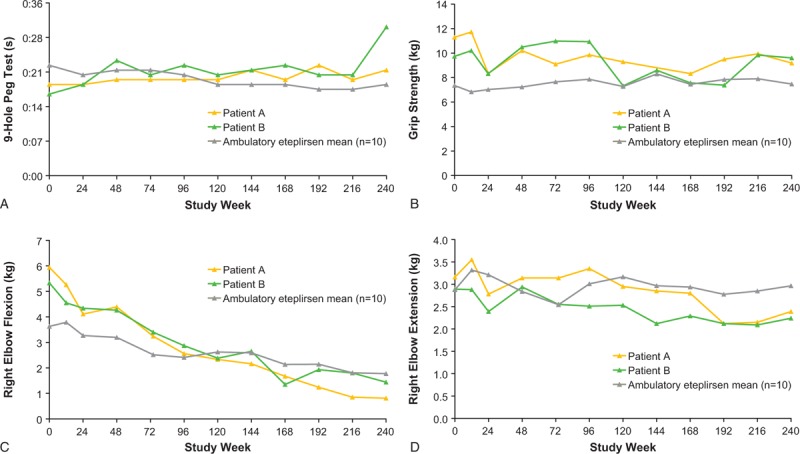
Measures of upper limb function in patients A and B versus study mean through 240 weeks of eteplirsen treatment: (A) 9-Hole Peg Test; (B) grip strength; (C) elbow flexion; and (D) elbow extension. All data represent the right hand, which was the dominant hand for all 12 patients.
3.5. Dystrophin levels
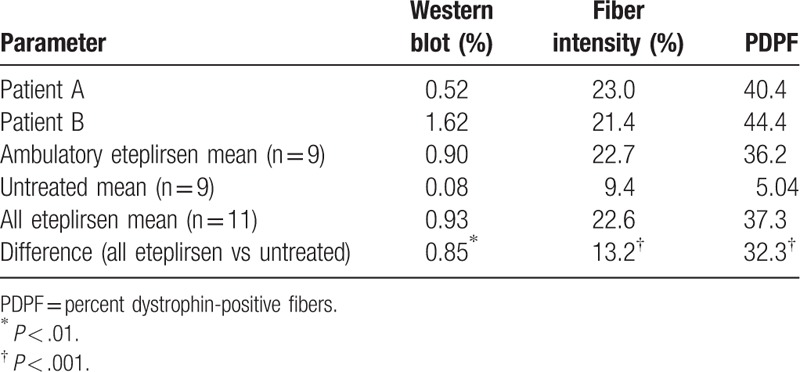
Dystrophin expression was assessed in 11 patients (patients A and B and 9 other patients) who provided consent for the week 180 biopsy, which was analyzed by 3 methods, including Western blot (% normal controls), percent dystrophin-positive fibers (PDPF), and fiber intensity (% intensity based on fluorescence; Fig. 5; Table 1). Patients A and B had higher dystrophin expression as measured by all 3 methods (patient A: 0.52%, 40.4%, and 23.0%, respectively; patient B: 1.62%, 44.4%, and 21.4%, respectively) relative to the untreated comparator group (0.08%, 5.04%, and 9.4%, respectively) and had comparable or greater dystrophin expression relative to the mean of the ambulatory eteplirsen-treated patients (0.90%, 36.2%, and 22.7%, respectively). The difference in dystrophin expression between the mean of the untreated controls and the mean of the 11 eteplirsen-treated patients was also evaluated and found to be statistically significant for all 3 measures (Western blot, P < .01; PDPF, P < .001; fiber intensity, P < .001; Table 1). As reported by Charleston et al,[11] these differences equated to an 11.6-fold difference by Western blot, a 7.4-fold difference by PDPF, and a 2.4-fold difference by Bioquant fluorescence intensity. The PDPF results were confirmed by 3 pathologists who independently scored the image sets and observed a 15.5-fold difference between the untreated controls and the treated patient population.
Figure 5.
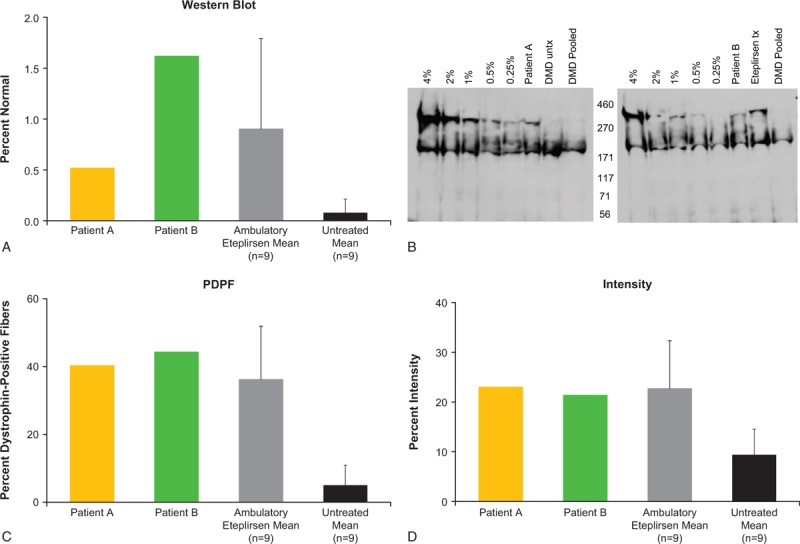
Dystrophin expression following 180 weeks of eteplirsen treatment, based on Western blot analysis and immunohistochemistry (A), Western blot image (B), PDPF (C), and the intensity of dystrophin fluorescent signal in muscle fibers (D). In panels A, C, and D, standard deviations are represented by error bars. In panel B, lanes in the Western blot image are identified left to right as the standard curve (4%, 2%, 1%, 0.5%, 0.25% of normal), patient samples (treated or untreated in blinded random order in next two lanes), and negative pooled DMD control sample. Patient samples representing patient A and patient B are identified in both figures. Note that either an unrelated patient sample representing an untreated (left figure) or treated (right figure) patient was run on the adjacent lane due to the random blinded nature that samples were evaluated. DMD = Duchenne muscular dystrophy, PDPF = percent dystrophin-positive fibers, tx = treated, untx = untreated.
Table 1.
Novel dystrophin production confirmed by Western blot, fiber intensity, and percent dystrophin-positive fibers[11].
3.6. Safety
All 12 patients experienced at least 1 TEAE. The most commonly reported TEAE, procedural pain, was reported by 11 of 12 patients. The TEAEs reported by at least 4 patients for up to 240 weeks of eteplirsen treatment in the combined studies 201 and 202 are shown in Table 2; TEAEs that occurred during 24 weeks of treatment with placebo in study 201 were previously reported.[10]
Table 2.
Most frequent treatment-emergent adverse events reported in at least 4 patients for up to 240 weeks of eteplirsen treatment in combined studies 201 and 202.
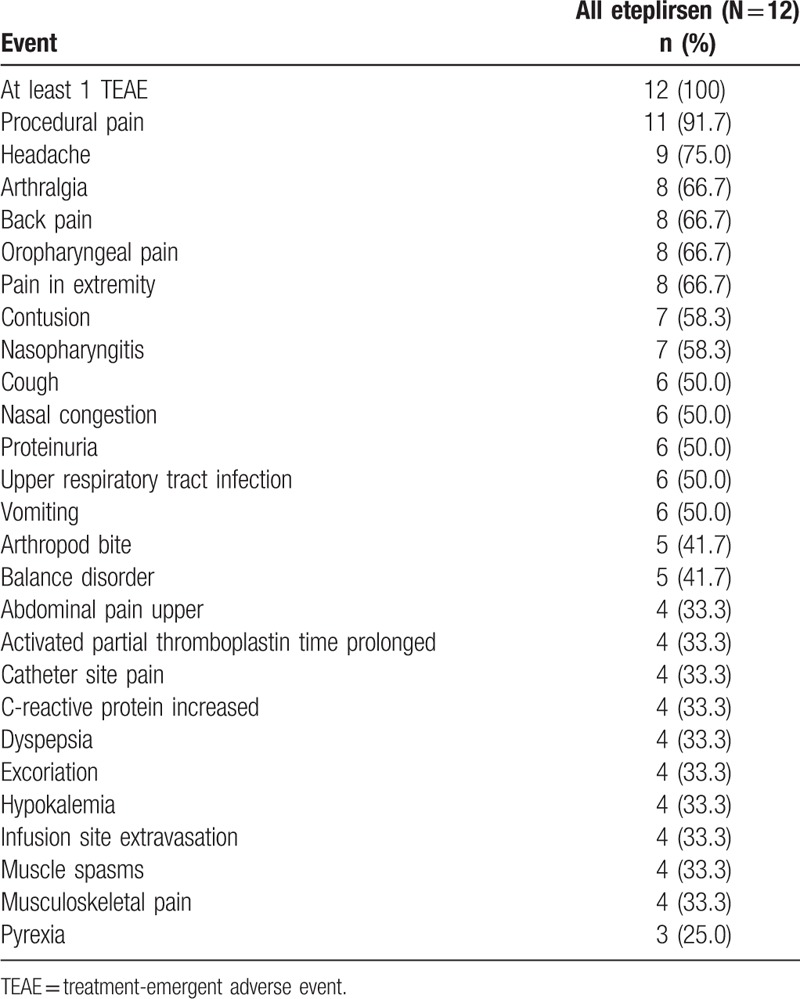
The 2 patients who lost ambulation experienced TEAEs that were similar in frequency and severity to those of the 10 patients who remained ambulatory throughout the study. A total of 34 and 38 TEAEs were reported for patients A and B, respectively, compared with a range of 36 to 81 TEAEs reported for the 10 patients who remained ambulatory. Six of 12 patients (50.0%) experienced TEAEs that were considered by the investigator to be related to eteplirsen. Patients A and B both experienced fibrin clots in a chest port placed for eteplirsen infusion intravenous access (week 136 in patient A, weeks 137 and 164 in patient B), which were considered to be non-serious TEAEs of moderate severity, possibly or probably related to the study drug.
Four of 12 patients (33.3%) experienced serious adverse events. Both patient A and patient B experienced serious adverse events that were considered by the investigator to be unrelated to eteplirsen treatment. At week 87, approximately 51 weeks after loss of ambulation, patient A sustained a severe, closed, stable fracture of the right distal femur after falling out of his wheelchair; he recovered 2 months later. During week 171, approximately 31 months after loss of ambulation, patient B experienced a non-serious event of moderate scoliosis related to DMD progression that was not considered treatment-related. During week 212, approximately 40 months after loss of ambulation, worsening of the scoliosis was noted and the patient underwent posterior spinal fusion surgery, which was considered serious. At the time of this writing, both patients A and B had undergone scoliosis surgery. Patient B experienced complications from this surgery, including postoperative anemia, cardiac function disturbance, and postoperative respiratory distress.
4. Discussion
Two patients with DMD who were enrolled in a clinical trial of eteplirsen lost ambulation but maintained relative stability on nonambulatory measures of disease progression. Through 240 weeks of eteplirsen treatment in combined studies 201 and 202, both patients demonstrated cardiac and upper limb function as well as dystrophin production similar to that of 10 ambulatory patients evaluated. Compared with the other study patients at initiation of eteplirsen treatment, the twin patients were further progressed in key measures of disease considered to be predictive of loss of ambulation. These measures included advanced age (>7 years) and shorter 6MWT distance (≤330 m).[2] Natural history studies show that age and 6MWT distance are predictive of disease progression.[15,16] A 3-year, prospective, longitudinal natural history study in ambulatory boys with DMD showed that the greatest decline in patients older than 7 years occurred in those who had 6MWT distances <350 m at baseline.[15]
Previous studies reported dystrophin expression at baseline and following 12 or 24 weeks of treatment with eteplirsen, with an increase in the PDPF at week 24 but not at week 12. These findings suggest that a period of treatment with eteplirsen is needed before an increase in dystrophin may be detected.[2] Therefore, given that the twins showed more advanced disease at trial initiation, it is not surprising that they lost ambulation early in the study. However, their stability on non-ambulatory outcome measures suggests a potential therapeutic benefit of eteplirsen. This is supported by the observation that both nonambulatory twin patients demonstrated a similar level of increased novel dystrophin compared with ambulatory patients following 180 weeks of treatment.[2] Dystrophin production was demonstrated by 3 independent measures—Western blot, PDPF, and fiber intensity—that effectively assess dystrophin quantity and correct localization of dystrophin to the sarcolemma membrane.[11]
Pulmonary function assessments showed that FVC%p declined initially and again between weeks 96 and 120, then remained stable for the duration of the study. Values for FVC%p at week 240 were 80% and 72% for patients A and B, respectively, and 92% for the ambulatory study mean. Pulmonary dysfunction is a key contributor to morbidity and mortality in patients with DMD.[17–19] FVC%p is an accurate spirometry measure that has been recommended as a reproducible, sensitive method for monitoring pulmonary function in DMD clinical trials.[20,21] Multiple publications investigating pulmonary function in DMD disease natural history report continual declines in FVC%p.[22–24] Interestingly, in a prospective follow-up of nonambulatory men and boys with DMD employing a natural history trial design, FVC%p remained stable over 12 months (61.0–61.8%), but declined significantly over 24 months from 60% to 54.4% (P = .02).[24]
With regard to cardiac function, LVEF values for the nonambulatory twin patients remained stable and within the normal range throughout 240 weeks of treatment with eteplirsen and were similar to the mean LVEF value of the 10 ambulatory patients. Decline in cardiac function, including LVEF decline by orthogonal measures, is reported in the natural history literature,[25–27] and cardiac failure is a leading cause of death in patients with DMD.[28] It should be noted that ECHO evaluation of LVEF is not as sensitive as orthogonal LVEF measurements by imaging modalities such as cardiac magnetic resonance imaging or radionuclide ventriculography. At a minimum, it can be stated that eteplirsen did not have an obvious negative impact on cardiac function during the duration of the trial.
Regarding the 9-Hole Peg Test, a separate study of healthy reference individuals reported mean completion times for the 9-Hole Peg Test of 19.9 s for boys aged 8 to 9 years that decreased steadily to 18.0 s for boys aged 14 to 15 years.[14] As expected, mean completion times are higher for patients with DMD; a prospective, natural history study of nonambulatory patients with DMD reported a mean 9-Hole Peg Test completion time at baseline of 27.2 s, which did not change significantly over 1 or 2 years of follow-up.[24] In the current report, mean 9-Hole-Peg Test completion times were similar for the nonambulatory twin patients compared with the ambulatory eteplirsen mean through week 216, followed by a slowing of completion time for patient B at week 240. This observation in patient B may have been attributable to a spinal fusion surgery conducted prior to week 240, as this surgical procedure often results in loss of function resulting from change in movement patterns. Notably, at week 216, patient B achieved a 9-Hole Peg Test completion time of 21 s.
Studies of upper limb function in nonambulatory patients have shown a decline in measures of grip strength and elbow flexion, over a period of 12 to 24 months.[24,29] In the present study, dominant-hand grip strength of nonambulatory patients A and B remained remarkably stable throughout the study, particularly following confirmation of dystrophin expression at week 24, and was similar to or greater than the mean grip strength of ambulatory patients. Right elbow flexion declined over time, and right elbow extension remained relatively stable and was similar to that of the mean of the other patients throughout the study. Neither patient A nor B developed hand contractures, a complication of DMD resulting from the absence of dystrophin that can substantially limit functioning and potentially limit the ability to administer and interpret commonly used tests of upper arm function and strength.[20,30] Importantly, both patients A and B retained anti-gravity arm function, which is vital to activities of daily living.
Weekly administration of eteplirsen was well tolerated through 240 weeks of treatment in the nonambulatory patients. The TEAEs experienced by these patients were similar in nature and frequency to those reported for ambulatory patients. Twin patients A and B each experienced 1 serious TEAE of fracture and scoliosis, respectively, which were considered unrelated to treatment with eteplirsen. Moreover, these TEAEs are not unexpected in patients diagnosed with DMD, as their fracture risk is substantially higher compared with age- and sex-matched individuals without DMD,[31] and the development of scoliosis is not uncommon in nonambulatory patients with DMD.[5,32]
Limitations to these studies include the observational nature of this analysis, the small number of patients evaluated, and the open-label nature of treatment in the extension study.
In conclusion, after loss of ambulation, disease progression was relatively stable in twin patients treated with eteplirsen and was similar to the mean of the study patients who remained ambulatory. The 2 patients who lost ambulation had the most progressed disease at initiation of treatment, experienced a rapid decline in ambulatory ability prior to confirmation of dystrophin production, and lost ambulation shortly thereafter. However, both patients experienced relative stability on nonambulatory outcome measures. Each twin patient experienced maintenance of normal cardiac function and stability of upper limb function after losing ambulation. Sustained dystrophin expression was confirmed in both patients following 180 weeks of eteplirsen treatment by 3 complementary dystrophin assays. This observational study highlights the potential benefit of eteplirsen in patients who are non-ambulatory or who lose ambulation during the course of treatment. As additional eteplirsen clinical trials progress, further evidence will become available to provide context for these clinical observations.
Acknowledgments
This study was sponsored by Sarepta Therapeutics, Inc. Medical writing and editorial support were provided by Callie Grimes, PhD, and Barbara Zeman, PhD, of Peloton Advantage, LLC, an OPEN Health company (Parsippany, NJ), and funded by Sarepta Therapeutics. The authors thank the team at Flagship Biosciences (Westminster, CO) who participated in the dystrophin analyses for this study, and John R. Kean, MD, and Jeffrey R. Leonard, MD, of Nationwide Children's Hospital, who performed the muscle biopsy procedures. The authors also gratefully acknowledge Dr. Steven A. Moore, the University of Iowa Hospitals and Clinics Histology Laboratory, and the Iowa Wellstone Muscular Dystrophy Cooperative Research Center (NIH grant U54, NS053672) for providing frozen sections of non-dystrophic control muscle biopsies, Becker muscular dystrophy muscle biopsies, and untreated DMD biopsy samples from the PROMOVI clinical study described in this manuscript. Dr. Moore also assisted in establishing the criteria for defining dystrophin-positive muscle fibers.
Author contributions
Conceptualization: Lindsay N. Alfano, Jerry R. Mendell, Linda Lowes.
Data curation: Lindsay N. Alfano, Jay S. Charleston, Anne M. Connolly, Linda Cripe, Cas Donoghue, Johannes Dworzak, Helen Eliopoulos, Diane E. Frank, Sarah Lewis, Jessie Lynch, A. J. Milici, Louise R. Rodino-Klapac, Zarife Sahenk, Frederick J. Schnell, G. David Young, Linda Lowes.
Formal analysis: Lindsay N. Alfano, Jay S. Charleston, Amy Flynt.
Investigation: Robert Dracker, Jerry R. Mendell.
Writing – review & editing: Lindsay N. Alfano, Jay S. Charleston, Anne M. Connolly, Linda Cripe, Cas Donoghue, Robert Dracker, Johannes Dworzak, Diane E. Frank, Sarah Lewis, Karin Lucas, Jessie Lynch, A. J. Milici, Amy Flynt, Emily Naughton, Louise R. Rodino-Klapac, Zarife Sahenk, Frederick J. Schnell, G. David Young, Jerry R. Mendell, Linda Lowes.
Footnotes
Abbreviations: 6MWT = 6-minute walk test, BMI = body mass index, DMD = Duchenne muscular dystrophy, IV = intravenous, LVEF = left ventricular ejection fraction, MVICT = maximum voluntary isometric contraction test, NC = normal control, PDPF = percent dystrophin-positive fibers, PDVF = polyvinylidene difluoride, TEAE = treatment-emergent adverse event.
Source of funding: This study was sponsored by Sarepta Therapeutics, Inc.
Role of funding source: This study and manuscript were funded by Sarepta Therapeutics, Inc. Sarepta employees were involved in the study design, the collection, analysis, and interpretation of data, the review of the manuscript, and the decision to submit for publication.
Conflicts of interest: Medical writing and editorial support were provided by Callie Grimes, PhD, and Barbara Zeman, PhD, of Peloton Advantage, LLC, an OPEN Health company (Parsippany, NJ), and funded by Sarepta Therapeutics, Inc. L. N. Alfano, L. Cripe, R. Dracker, Z. Sahenk, and L. Lowes have no conflicts to disclose. J. S. Charleston, J. Dworzak, H. Eliopoulos, D. E. Frank, K. Lucas, J. Lynch, E. Naughton, and F.J. Schnell are employees of Sarepta Therapeutics, Inc. and may own stock or options in the company. C. Donoghue was an employee at the time of the study. A. M. Connolly has served on advisory boards for AveXis, Inc., Cytokinetics, Inc., Sanofi Genzyme, and Sarepta Therapeutics, Inc. She serves on the data management safety board for Catabasis Pharmaceuticals, Inc., and as an investigator for AveXis, Inc., Biogen, Bristol-Myers Squibb, Cytokinetics, Inc., FibroGen, Italfarmeco SpA, NS Pharma, Inc., Pfizer Inc, PTC Therapeutics, Inc., and Sarepta Therapeutics, Inc. S. Lewis is an employee of Nationwide Children's Hospital, which received funding from Sarepta Therapeutics in the form of the study budget. A. J. Milici serves as a remunerated consultant for Sarepta Therapeutics, Inc. A. Flynt is an employee of PharPoint Research, which received funding from Sarepta Therapeutics for data analysis for the study. At the time of the study, L.R. Rodino-Klapac was an employee of Nationwide Children's Hospital, which received funding from Sarepta Therapeutics in the form of a research agreement; she is currently an employee of Sarepta Therapeutics, Inc., and may own stock/options in the company. She is the inventor of technology that has been exclusively optioned to Sarepta Therapeutics. G. D. Young serves as a remunerated consultant for Sarepta Therapeutics, Inc. J. R. Mendell serves on an advisory board for Sarepta Therapeutics, Inc.
References
- [1].Kole R, Krieg AM. Exon skipping therapy for Duchenne muscular dystrophy. Adv Drug Deliv Rev 2015;87:104–7. [DOI] [PubMed] [Google Scholar]
- [2].Mendell JR, Rodino-Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol 2013;74:637–47. [DOI] [PubMed] [Google Scholar]
- [3].McNeil DE, Davis C, Jillapalli D, et al. Duchenne muscular dystrophy: drug development and regulatory considerations. Muscle Nerve 2010;41:740–5. [DOI] [PubMed] [Google Scholar]
- [4].Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol 2010;9:177–89. [DOI] [PubMed] [Google Scholar]
- [5].Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010;9:77–93. [DOI] [PubMed] [Google Scholar]
- [6].Fayssoil A, Nardi O, Orlikowski D, et al. Cardiomyopathy in Duchenne muscular dystrophy: pathogenesis and therapeutics. Heart Fail Rev 2010;15:103–7. [DOI] [PubMed] [Google Scholar]
- [7].Voit T, Topaloglu H, Straub V, et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol 2014;13:987–96. [DOI] [PubMed] [Google Scholar]
- [8].Bushby K, Finkel R, Wong B, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014;50:477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Exondys 51 [package insert]. Cambridge, MA: Sarepta Therapeutics, Inc.; 2018. [Google Scholar]
- [10].Mendell JR, Goemans N, Lowes LP, et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol 2016;79:257–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Charleston JS, Schnell FJ, Dworzak J, et al. Eteplirsen treatment for Duchenne muscular dystrophy: exon skipping and dystrophin production. Neurology 2018;90:e2146–54. [DOI] [PubMed] [Google Scholar]
- [12].Kinane TB, Mayer OH, Mendell JR, et al. Effects of long-term eteplirsen treatment on pulmonary function in patients with Duchenne muscular dystrophy: findings of two phase 2 clinical trials [poster]. Presented at: Annual Scientific Conference of the Muscular Dystrophy Association; March 19–22, 2017; Arlington, VA. [Google Scholar]
- [13].Duda PW, Moody S, Colan S, et al. Effects of long-term treatment with eteplirsen on cardiac function in patients with DMD versus disease natural history [poster]. Presented at: Annual Scientific Conference of the Muscular Dystrophy Association; March 19–22, 2017; Arlington, VA. [Google Scholar]
- [14].Poole JL, Burtner PA, Torres TA, et al. Measuring dexterity in children using the Nine-hole Peg Test. J Hand Ther 2005;18:348–51. [DOI] [PubMed] [Google Scholar]
- [15].Pane M, Mazzone ES, Sivo S, et al. Long term natural history data in ambulant boys with Duchenne muscular dystrophy: 36-month changes. PLoS One 2014;9:e108205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mazzone E, Vasco G, Sormani MP, et al. Functional changes in Duchenne muscular dystrophy: a 12-month longitudinal cohort study. Neurology 2011;77:250–6. [DOI] [PubMed] [Google Scholar]
- [17].LoMauro A, D’Angelo MG, Aliverti A. Assessment and management of respiratory function in patients with Duchenne muscular dystrophy: current and emerging options. Ther Clin Risk Manag 2015;11:1475–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Roberto R, Fritz A, Hagar Y, et al. The natural history of cardiac and pulmonary function decline in patients with Duchenne muscular dystrophy. Spine 2011;36:E1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mayer OH, Finkel RS, Rummey C, et al. Characterization of pulmonary function in Duchenne muscular dystrophy. Pediatr Pulmonol 2015;50:487–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Connolly AM, Malkus EC, Mendell JR, et al. Outcome reliability in non-ambulatory boys/men with Duchenne muscular dystrophy. Muscle Nerve 2015;51:522–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mayhew JE, Florence JM, Mayhew TP, et al. Reliable surrogate outcome measures in multicenter clinical trials of Duchenne muscular dystrophy. Muscle Nerve 2007;35:36–42. [DOI] [PubMed] [Google Scholar]
- [22].Mayer OH, Henricson EK, McDonald CM, et al. Advances in pulmonary care in Duchenne muscular dystrophy. US Neurology 2017;13:35–41. [Google Scholar]
- [23].Finder J, Mayer OH, Sheehan D, et al. Pulmonary endpoints in Duchenne muscular dystrophy. A workshop summary. Am J Respir Crit Care Med 2017;196:512–9. [DOI] [PubMed] [Google Scholar]
- [24].Connolly AM, Florence JM, Zaidman CM, et al. Clinical trial readiness in non-ambulatory boys and men with Duchenne muscular dystrophy: MDA-DMD network follow-up. Muscle Nerve 2016;54:681–9. [DOI] [PubMed] [Google Scholar]
- [25].Duboc D, Meune C, Lerebours G, et al. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol 2005;45:855–7. [DOI] [PubMed] [Google Scholar]
- [26].Tandon A, Villa CR, Hor KN, et al. Myocardial fibrosis burden predicts left ventricular ejection fraction and is associated with age and steroid treatment duration in Duchenne muscular dystrophy. J Am Heart Assoc 2015;4:e001338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Schram G, Fournier A, Leduc H, et al. All-cause mortality and cardiovascular outcomes with prophylactic steroid therapy in Duchenne muscular dystrophy. J Am Coll Cardiol 2013;61:948–54. [DOI] [PubMed] [Google Scholar]
- [28].Birnkrant DJ, Ararat E, Mhanna MJ. Cardiac phenotype determines survival in Duchenne muscular dystrophy. Pediatr Pulmonol 2016;51:70–6. [DOI] [PubMed] [Google Scholar]
- [29].Ricotti V, Evans MR, Sinclair CD, et al. Upper limb evaluation in Duchenne muscular dystrophy: fat-water quantification by MRI, muscle force and function define endpoints for clinical trials. PLoS One 2016;11:e0162542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Henricson EK, Abresch RT, Cnaan A, et al. The Cooperative International Neuromuscular Research Group Duchenne Natural History Study: glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and other commonly used clinical trial outcome measures. Muscle Nerve 2013;48:55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Vestergaard P, Glerup H, Steffensen BF, et al. Fracture risk in patients with muscular dystrophy and spinal muscular atrophy. J Rehabil Med 2001;33:150–5. [PubMed] [Google Scholar]
- [32].Archer JE, Gardner AC, Roper HP, et al. Duchenne muscular dystrophy: the management of scoliosis. J Spine Surg 2016;2:185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]