

Supplemental Digital Content is available in the text
Keywords: HER2+ breast cancer, single-cell RNA-Seq, TCGA, transcriptome profile, trastuzumab, trastuzumab-mediated cardiotoxicity
Abstract
Human epidermal growth factor receptor 2-positive (HER2+) breast cancer accounts for ∼20% of invasive breast cancers and is associated with poor prognostics. The recent outcome of HER2+ breast cancer treatment has been vastly improved owing to the application of antibody-targeted therapies. Trastuzumab (Herceptin) is a monoclonal antibody designed to target HER2+ breast cancer cells. In addition to improved survival in the adjuvant treatment of HER2+ breast cancer, trastuzumab treatment has also been associated with cardiotoxicity side effect. However, the molecular mechanisms of trastuzumab action and trastuzumab-mediated cardiotoxicity are still not fully understood. Previous research utilized bulk transcriptomics analysis to study the underlining mechanisms, which relied on averaging molecular signals from bulk tumor samples and might have overlooked key expression features within breast cancer tumor. In contrast to previous research, we compared the single cancer cell level transcriptome profile between trastuzumab-treated and nontreated patients to reveal a more in-depth transcriptome profile. A total of 461 significantly differential expressed genes were identified, including previously defined and novel gene expression signatures. In addition, we found that trastuzumab-enhanced MGP gene expression could be used as prognostics marker for longer patient survival in breast invasive carcinoma patients, and validated our finding using TCGA (The Cancer Genome Atlas) breast cancer dataset. Moreover, our study revealed a 48-gene expression signature that is associated with cell death of cardiomyocytes, which could be used as early biomarkers for trastuzumab-mediated cardiotoxicity. This work is the first study to look at single cell level transcriptome profile of trastuzumab-treated patients, providing a new understanding of the molecular mechanism(s) of trastuzumab action and trastuzumab-induced cardiotoxicity side effects.
1. Introduction
Breast cancer is the most common cancer diagnosed in women worldwide.[1] The human epidermal growth factor receptor 2-positive (HER2+) breast cancer accounts for ∼20% of invasive breast cancers, which overexpresses the HER2 and is correlated with poor prognosis.[2] In the recent years, the outcome of HER2+ breast cancer treatment has been improved largely due to the application of targeted therapies.[3] Trastuzumab (Herceptin; F. Hoffmann-La Roche, Basel, Switzerland) is an anti-HER2 monoclonal antibody that explicitly directed against the HER2 extracellular domain.[4] Trastuzumab-based therapies have brought significant clinical benefits to HER2+ breast cancer patients including improved responsive rate, responsive duration, and survival rate.[5] Improved clinical outcomes have been associated with induction of cell cycle arrest and apoptosis, PIK3t pathway inhibition, and antigen-dependent cellular cytotoxicity, in addition to the direct inhibition of HER2.[4] Apart from its clinical benefits, trastuzumab can also induce cardiotoxicity,[6] which can cause “left ventricular dysfunction, arrhythmias, hypertension, disabling cardiac failure, cardiomyopathy, and cardiac death” according to the medical package insert. The cardiotoxicity-caused discontinuation rate of trastuzumab treatment ranges between 6% and 31% in multiple randomized adjuvant trials and observational studies.[7] Trastuzumab-induced cardiotoxicity is thought to be related with the neuregulin (NRG) ERBB pathway.[8] However, the complete mechanism of trastuzumab action and trastuzumab-mediated cardiotoxicity is still yet to be illuminated.
A detailed gene profile of trastuzumab is very essential to fully understand trastuzumab's mechanism of action and can provide new target/biomarkers for trastuzumab-mediated cardiotoxicity. Although a handful of studies have investigated the global gene expression pattern under trastuzumab treatment,[9,10] these studies applied conventional molecular profiling methods such as microarray or bulk RNA sequencing (bulk RNA-Seq), which relied on averaging molecular phenotypes from a large population of cells. Because of the growing evidence showing heterogeneity and infiltration within breast cancer tumor,[11] measuring the average values of gene expression might ignore the differences within breast cancer cell population that may be crucial for cancer progression and response to target therapies. Therefore, conventional molecular profiling methods can provide only partial information of the molecular state under trastuzumab treatment. The recent development of single-cell RNA sequencing (scRNA-Seq) technology has combined the single-cell isolation and RNA-sequencing technologies which enabled the global transcriptome profiling on single-cell level.[12] scRNA-Seq can provide a higher resolution of cancer cell transcriptome profile comparing with the conventional cell-averaging experiments. Hence, scRNA-Seq could reveal novel biomarkers that might have been overlooked in traditional bulk transcriptome analysis.
In this pilot study, we compared the single cancer cell expression profile between trastuzumab-treated and nontreated HER2+ breast cancer patients. To the best of our knowledge, this is the first research to exam trastuzumab action in single cell level, which could contribute to a better understanding of the trastuzumab-induced therapeutic and cardiotoxicity effects.
2. Materials and methods
2.1. Single-cell RNA-Seq dataset
The scRNA-seq dataset was downloaded from NCBI Gene Expression Omnibus database (accession number GSE75688). This dataset contains single-cell and bulk RNA-seq data from 3 HER2+ patients diagnosed with invasive ductal carcinoma. Among these 3 patients, 1 patient (T1) received adjuvant trastuzumab treatment and other 2 patients (C1, C2) did not receive any treatment before tumor removal. All 3 patients are Asian females diagnosed with T2 or T3 breast cancer. Primary tumor specimens were collected by breast-conserving surgery or total mastectomy and processed for single-cell and bulk RNA sequencing. A total of 74, 48, and 19 carcinoma scRNA-seq samples and 1 bulk RNA-seq samples were retrieved from T1, C1, and C2 patients, respectively. These carcinoma single cells were classified by Chung et al[11] using chromosomal gene expression patterns and validated by ESTIMATE algorithm.[13] The trastuzumab-treated patient T1 was excluded by Chung et al[11] for further transcriptome analysis because their research was focused on classifying nontreated cancer single cells. However, we felt this dataset provided a great opportunity to explore single cell transcriptome profile under trastuzumab treatment, and revisited this dataset. All data in this study were based on previously published studies, and thus, no ethical approval or patient consent was required.
2.2. TCGA dataset
A total of 1112 mRNA expression profiles along with their corresponding clinical data from patients identified with primary breast invasive carcinoma were downloaded from TCGA data portal.[14] After filtering out patients with follow-up shorter than 90 days, a total of 1078 patient profiles were used in this study.
2.3. Sequence analysis
The scRNA-Seq and bulk RNA-Seq sequences were aligned to human reference genome GRCh37 by bowtie2 program[15] using default parameters. Relative gene express as transcript per million (TPM) was qualified using RSEM v1.3.0 (Ensemble 92 gene model).[16]
2.4. Statistical analysis
Principal component analysis was performed in R using prcomp package.[17] Unsupervised hierarchical clustering based on heatmap was conducted using ComplexHeatmap package in R. t-distributed stochastic neighbor embedding analysis (tSNE) was performed by the Rtsne package in R.[18]
We applied the R Seurat package[19] to identify differential expressed genes (DEGs) in scRNA-Seq samples between treated and nontreated patients. The Seurat package implemented a likelihood ratio test model to identify DEGs between 2 cell groups. Raw P value output by Seurat package was adjusted by Bonferroni correction. Genes with adjusted P value <.05 and fold change >1.5 were considered as significantly differential expressed.
Survival analysis for TCGA dataset was performed using Kaplan-Meier method by survival package in R.[20] Patients were divided into high expression, low expression, and median expression groups according to MGP gene's expression value. Patients were categorized in high expression group if MGP expression was in the fourth quartile, low expression group if in the first quartile, and median expression group if in the second or third quartile. Differences between survival curves were measured by a log-rank test using survdiff package in R.
2.5. Enriched biological pathway analysis
We used Ingenuity Pathway Analysis software (IPA; Qiagen, Valencia, CA) to evaluate the most significant pathways and biological functions involved in trastuzumab treatment. DEGs identified from scRNA-Seq dataset and their expression values were uploaded to IPA for core analysis, which included canonical pathways, upstream regulators, disease and bio functions, and toxic functions analysis. IPA applied a Fisher exact test to identify significantly enriched canonical pathways, and only pathways with P value <.05 were further investigated. The IPA “Top Networks” analysis calculated a P score (-log10 [P value]) based on the fitting of uploaded genes to a list of known biological function. Networks with P score >3 were further investigated.
3. Results
3.1. Separation of treated and nontreated scRNA-seq samples
We applied 3 clustering methods to reveal the differences of transcriptome profiles between treated and nontreated scRNA-Seq samples. First, PCA analysis was applied to identify the potential clusters of single cells based on TPM values of detected genes. Our PCA result indicated that the whole variance of single-cell dataset could be well-explained by first 3 principle components (PCs), while PC1 explaining 10.96 of total variance (Fig. 1). The 3D PCA plot also showed that PC1-3 separated the treated versus nontreated single-cell samples into 2 distinguishing clusters. Secondly, we selected the top 1000 highly expressed genes and conducted the unsupervised hierarchical clustering based on heatmap. The separation of trastuzumab treated and nontreated single cells was also supported by the results of heatmap and dendrogram as the heatmap has been visually separated into 2 distinctive clusters (Fig. 2). Third, separation of single cells was also corroborated by our tSNE analysis as single cells from treated and nontreated patients were clustered into 2 distinguishing groups (Fig. 3A).
Figure 1.
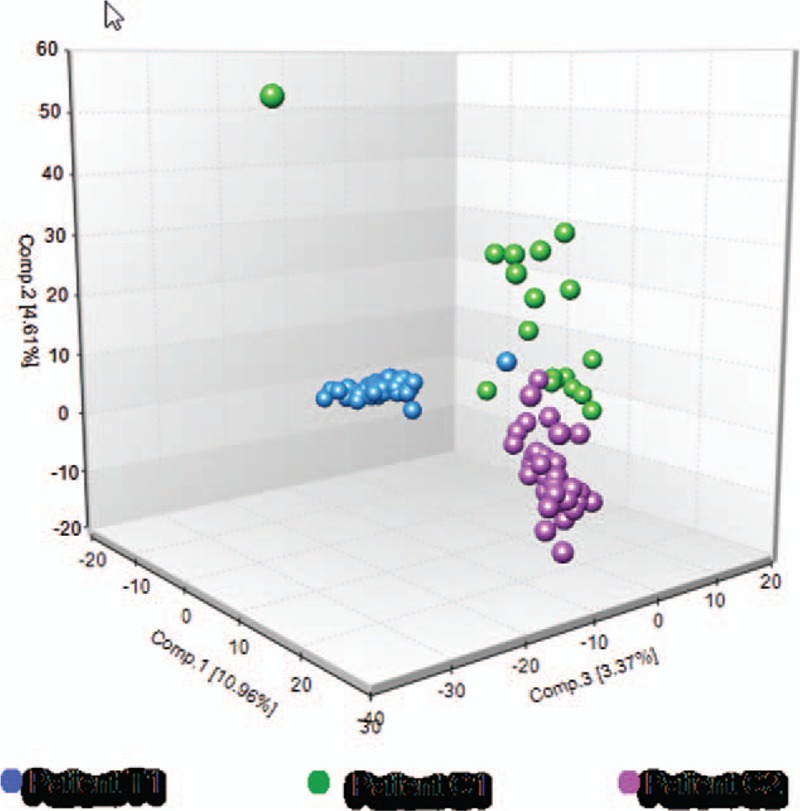
Principal component analysis (PCA) of trastuzumab-treated (T1) versus nontreated (C1, C2) single cancer cells. Unsupervised 3D PCA plot reveals 2 major groups of cells showing the separation of trastuzumab-treated and nontreated single cancer cells. Individual cells are colored blue for patient T1, green for patient C1, and purple for patient C2. The color scheme is maintained throughout the manuscript.
Figure 2.
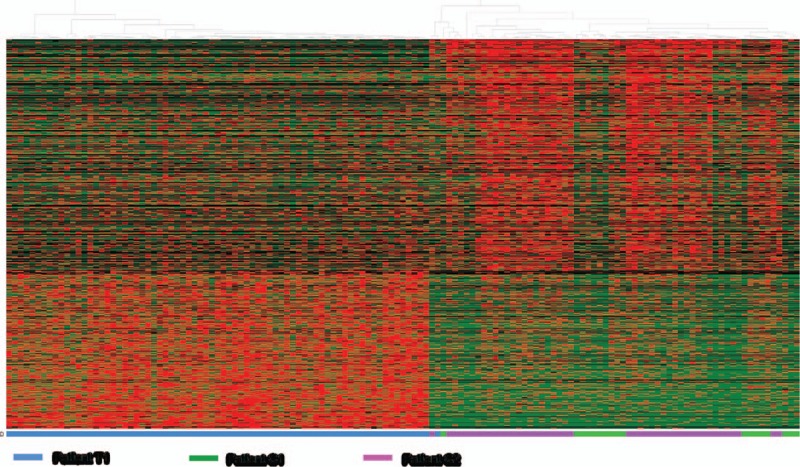
Heat map based hierarchical clustering separates trastuzumab-treated (T1) versus nontreated (C1, C2) single cancer cells. Individual cells are colored blue for patient T1, green for patient C1, and purple for patients C2 on color bar underneath hierarchical clustering heatmap.
Figure 3.
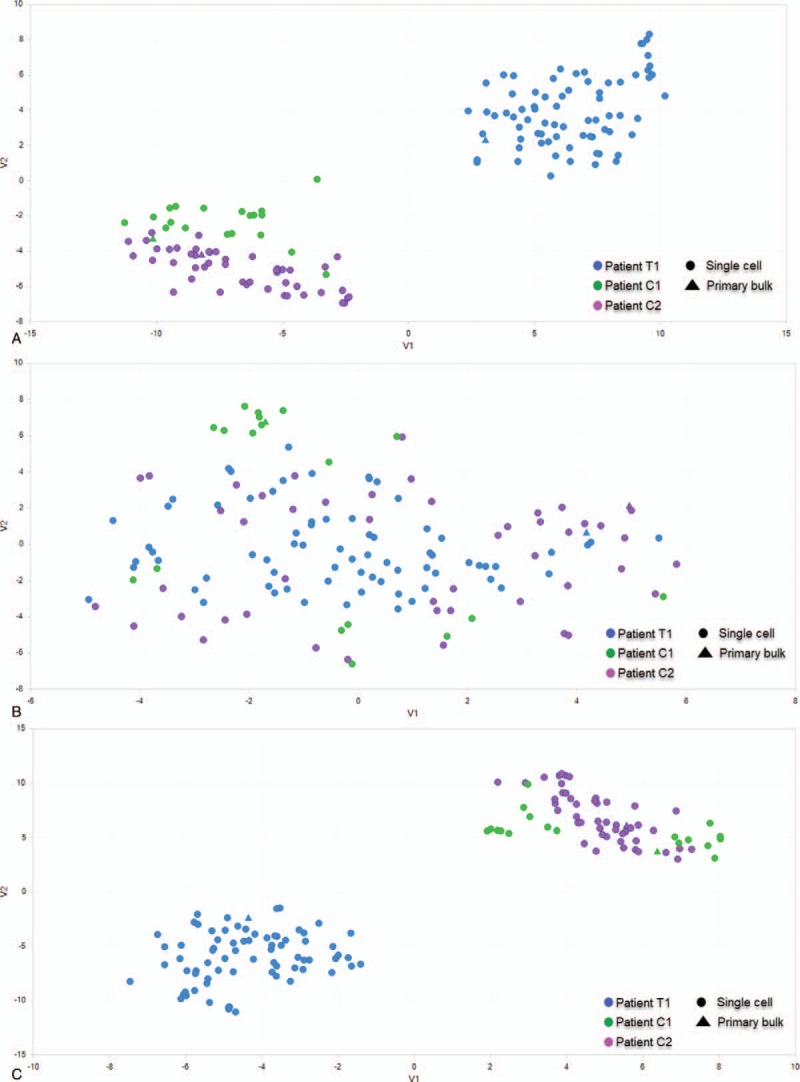
Tsne clustering of trastuzumab-treated (T1) versus nontreated (C1, C2) single cancer cells using different sets of genes. (A) Tsne separates single cancer cells into 2 major groups using top 2000 highly expressed genes. (B) Tsne is not able to separate single cancer cells using a list of cell cycle related genes. (C) Tsne separates single cancer cells into 2 major groups using a list of trastuzumab-responsive genes.
We hypothesized that the separation of 2 major single-cell clusters resulted from trastuzumab treatment. However, an alternative hypothesis was that cells were clustered by cell cycle phases. To test these 2 hypotheses, we applied tSNE analysis using a set of cell cycle-related genes[21] and a set of trastuzumab-responsive genes.[9] Our results showed that clustering using only cell cycle-related genes was not able to distinct treated or nontreated single cells (Fig. 3B). Clustering based on trastuzumab-responsive genes revealed a grouping that is highly like original grouping (Fig. 3C). This suggests the clustering of single cells was due to trastuzumab treatment, but not different cell cycle phases.
3.2. Identification of DEGs
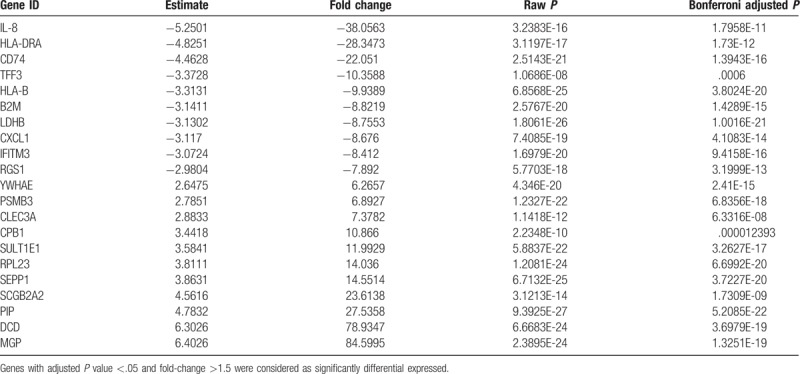
We identified 461 DEGs between treated and nontreated scRNA-Seq samples (Table 1 and Supplementary Table 1). Among these, a total of 303 genes were upregulated and 158 genes were downregulated. Top dysregulated genes were selected as candidate prognostic indicators for further validation using TCGA dataset.
Table 1.
Top 10 most significantly upregulated and downregulated genes between trastuzumab treated versus nontreated single cancer cells.
3.3. TCGA survival analysis
Using mRNA profiles and clinical data that retrieved from 1078 patients in TCGA database, we tested whether the top dysregulated genes in the single cell dataset could be used as prognostic indicators for breast cancer overall survival. We found the expression of MGP gene, 1 of the most significantly upregulated gene under trastuzumab treatment, was significantly associated with breast cancer patients survival (P = .03). Low MGP expression group has significant poorer survival rate comparing with high MGP expression or median MGP expression group (Fig. 4).
Figure 4.
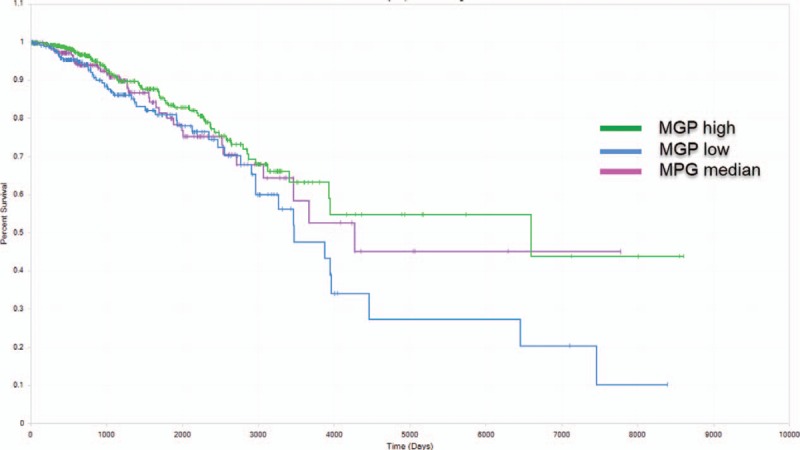
Overall survival curves based on MGP expression. Patients from TCGA breast cancer dataset were divided into high expression (green), average expression (purple), and low expression (blue) group according to MGP gene's expression value. MGP low expression group was associated with poor overall survival (P = .03).
3.4. IPA analysis
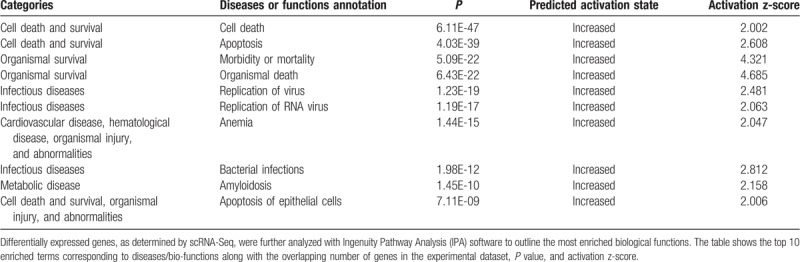
Our IPA analysis identified 33 significantly enriched canonical pathways under trastuzumab treatment (Table 2, Supplementary Table 2). IPA analysis revealed the top molecular and cellular functions related with DEGs are “cell death and survival,” “cellular movement,” “cellular development,” “cellular growth and proliferation,” and “protein synthesis.” In addition, IPA predicted increased cell death, apoptosis and decreased metabolism of protein, cell movement, and invasion in trastuzumab-treated cancer cells (Supplementary Table 2). Interestingly, our IPA analysis also identified 48 DEGs (Supplementary Table 3) that were associated with “cell death of cardiomyocytes” (P = .0003), which might explain the trastuzumab-induced cardiotoxicity.
Table 2.
Top 10 enriched diseases and biological functions identified by IPA.
4. Discussion
In this study, we investigated the expression pattern of HER2+ breast cancer cells in response to trastuzumab treatment. Using single-cell transcriptomics analysis, we could clearly separate the single cancer cells from trastuzumab-treated or nontreated patients. The separation of cancer cells was well-supported by our PCA, tSNE, and hierarchical clustering results. We further tested the hypothesis that separation of cancer single cells was due to trastuzumab treatment, but not different cell cycle phases. Our tSNE results supported this hypothesis because tSNE clustering using trastuzumab-responsive genes could clearly separate trastuzumab-treated and nontreated cancer cells, whereas cell cycle-related genes cannot.
Many previously defined expression signatures and pathways at bulk tumor level were also captured in our scRNA-Seq analysis. For instance, we found significantly upregulated CLU and SEPP1 gene in trastuzumab-treated patient (T1), which agreed with Necela et al[22] that CLU and SEPP1 were upregulated in trastuzumab-treated breast cancer cell lines. Interestingly, Necela et al[22] also observed upregulated CLU and SEPP1 expression in trastuzumab-treated cardiomyocytes. This suggests CLU and SEPP1 could be used as early indicator of trastuzumab-mediated cardiotoxicity. In addition, we found GATA3 gene, a top predictive gene for trastuzumab response,[23] was significantly upregulated in T1 patient. Consistently, our IPA analysis revealed enhanced antigen presentation pathway under trastuzumab treatment, which has also been well documented by Kono et al.[24] The identification of previously defined expression signatures and pathways indicates that traditional bulk transcriptome profiling can still be useful to reveal part of the global gene expression profile. Overlapping gene expression signatures between our study and previous research indicate that our identified DEGs were induced by trastuzumab treatment, and it further confirmed our hypothesis that separation of single cancer cells was due to trastuzumab treatment.
In addition to previously identified expression signatures, our scRNA-Seq analysis revealed novel expression signatures that might have been overlooked in previous bulk transcriptome analysis. For example, we found 2 chemokine (C-X-C motif) ligands CXCL1 and CXCL8 (also known as IL-8) among the most significantly downregulated genes under trastuzumab treatment. These 2 chemokines act as key cytokine to expand tumor neovasculature and promote tumor establishment. In lymph node-negative breast cancer, patients with enhanced CXCL8 expression were associated with poor prognosis of shorter survival time and higher chance of distant metastasis.[25] Therefore, decreased CXCL8 expression under trastuzumab treatment could contribute to a better clinical outcome. In addition, we found multiple tumor promoting genes, such as CIB1 and IFITM1, were significantly downregulated in trastuzumab-treated patients. There is increasing evidence showing CIB1 serve critical roles in cancer cell proliferation, apoptosis, and migration, and can promote tumor growth and angiogenesis.[26] Another tumor-promoting gene IFITM1 is required for the progression of colorectal cancer.[27] The downregulation of these 2 tumor-promoting genes could also explain the trastuzumab-mediated therapeutic effects. To the best of our knowledge, this is first time that the dysregulation of CXCL1, CXCL8, CIB1, and IFITM1 were associated with in vivo trastuzumab treatment. Our results highlighted new roles of these genes in trastuzumab treatment of HER2+ breast cancer. We expect these novel gene expression signature and the future follow-up studies could contribute to a better understanding of the molecular mechanisms of trastuzumab therapy.
Another important finding of this research is the upregulation of MGP expression under trastuzumab treatment and its potential role to serve as prognostic marker. We found MGP gene among 1 of the most significantly upregulated genes under trastuzumab treatment and hypothesized that upregulation of MGP could contribute to better clinical outcomes. Matrix Gla protein (MGP) is a vitamin K2-dependent, Gla-containing protein that acts as a multifunctional inhibitor of normal and abnormal angiogenesis, therefore acting as an endogenous inhibitor of tumor angiogenesis.[28]MGP downregulation was observed in lung cancer[29] and colorectal adenocarcinoma.[28] Repression of MGP gene was also shown to stimulate breast cancer cell proliferation and invasiveness.[30] Using TCGA breast cancer dataset, we confirmed our hypothesis that patients with enhanced MGP expression had significantly longer overall survival period than patients with lower MGP expression. Consistently, Tuo and Ye[31] also reported upregulation of MGP gene may predict better survival outcomes in ER+ breast cancer patients. This suggests enhanced MGP expression could contribute to better clinical outcome and could serve as prognostic marker for breast cancer overall survival.
Apart from our findings in trastuzumab's mechanism of action, we also identified a 48-gene signature that is related to cell death of cardiomyocytes. Molecular mechanisms that can potently suppress cancer cells growth, proliferation, and survival in both direct and indirect manners are considered as trastuzumab's mechanism of action. Molecular mechanisms that can directly and indirectly lead to cardiotoxicity are considered as trastuzumab-induced cardiotoxicity. Among these 48 genes, 14 genes were upregulated and 34 genes were downregulated under trastuzumab treatment. Several of the upregulated genes such as SPP1 and HMOX1 are known to increase the apoptosis of cardiac myocytes.[32,33] Further, many of the downregulated genes, such as TIMP1 and NAMPT, are known to decrease apoptosis of cardiomyocytes. Other downregulated genes, such as CXADR, are essential for cardiomyocyte development.[34] As predicted by IPA analysis, dysregulation of these genes could increase apoptosis of cardiomyocytes and disrupt the cardiomyocytes development, which could explain trastuzumab-mediated cardiotoxicity. Further validation of this 48-gene signature on cardiomyocytes is still required, which could discover novel therapeutic targets to prevent cardiac side effects or novel biomarkers to monitor trastuzumab-mediated cardiotoxicity.
5. Conclusions
To summarize, this is the first study exploring single cell level transcriptome profile of trastuzumab-treated HER2+ breast cancer patients. Our study offered new insights into the genes and potential biomarkers of trastuzumab-induced therapeutic effects and cardiotoxicity side effects. Gaining better understanding of these novel expression signatures remains a critical area to investigate in the future study. Limitation of this study includes the small number of patients and single cells. Due to the small sample size of this pilot study, analysis of more patient samples and more single cells per patient sample will be required.
Author contributions
Investigation: Jun Wang, Yunning Zhang.
Methodology: Jun Wang, Yunning Zhang.
Project administration: Jun Wang, Yunning Zhang.
Resources: Jun Wang, Haiyan Yuan.
Software: Jun Wang, Haiyan Yuan.
Supervision: Sean Cheng.
Validation: Rengen Xu.
Visualization: Rengen Xu, Haiyan Yuan, Yunning Zhang.
Writing – original draft: Jun Wang, Rengen Xu, Yunning Zhang, Sean Cheng.
Writing – review & editing: Jun Wang, Sean Cheng.
Supplementary Material
Footnotes
Abbreviations: DEGs =differential expressed genes, HER2 = human epidermal growth factor 2, RNA-Seq = RNA sequencing, scRNA-Seq = single-cell RNA sequencing, TPM = transcript per million, Tsne = t-distributed stochastic neighbor embedding analysis.
The authors declare that they have no conflicts of interest.
Supplemental Digital Content is available for this article.
References
- [1].Forouzanfar MH, Foreman KJ, Delossantos AM, et al. Breast and cervical cancer in 187 countries between 1980 and 2010: a systematic analysis. Lancet 2011;378:1461–84. [DOI] [PubMed] [Google Scholar]
- [2].Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature 2000;406:747–52. [DOI] [PubMed] [Google Scholar]
- [3].Dean-Colomb W, Esteva FJ. Her2-positive breast cancer: herceptin and beyond. Eur J Cancer 2008;44:2806–12. [DOI] [PubMed] [Google Scholar]
- [4].Hudis CA. Trastuzumab: mechanism of action and use in clinical practice. N Engl J Med 2007;357:39–51. [DOI] [PubMed] [Google Scholar]
- [5].Chung A, Cui X, Audeh W, Giuliano A. Current status of anti-human epidermal growth factor receptor 2 therapies: predicting and overcoming herceptin resistance. Clin Breast Cancer 2013;13:223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ewer SM, Ewer MS. Cardiotoxicity profile of trastuzumab. Drug Saf 2008;31:459–67. [DOI] [PubMed] [Google Scholar]
- [7].Bonifazi M, Franchi M, Rossi M, et al. Trastuzumab-related cardiotoxicity in early breast cancer: a cohort study. Oncologist 2013;18:795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sengupta PP, Northfelt DW, Gentile F, Zamorano JL, Khandheria BK. Trastuzumab-induced cardiotoxicity: heart failure at the crossroads. Mayo Clin Proc 2008;83:197–203. doi:10.4065/83.2.197. [DOI] [PubMed] [Google Scholar]
- [9].Kauraniemi P, Hautaniemi S, Autio R, et al. Effects of Herceptin treatment on global gene expression patterns in HER2-amplified and nonamplified breast cancer cell lines. Oncogene 2004;23:1010–3. [DOI] [PubMed] [Google Scholar]
- [10].Végran F, Boidot R, Coudert B, et al. Gene expression profile and response to trastuzumab-docetaxel-based treatment in breast carcinoma. Br J Cancer 2009;101:1357–64. doi:10.1038/sj.bjc.6605310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chung W, Eum HH, Lee H-O, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun 2017;8:15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hedlund E, Deng Q. Single-cell RNA sequencing: technical advancements and biological applications. Mol Aspects Med 2018;59:36–46. [DOI] [PubMed] [Google Scholar]
- [13].Yoshihara K, Shahmoradgoli M, Martínez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hutter C, Zenklusen JC. The cancer genome atlas: creating lasting value beyond its data. Cell 2018;173:283–5. [DOI] [PubMed] [Google Scholar]
- [15].Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].R: The R Project for Statistical Computing. Available at: https://www.r-project.org/ Accessed January 8, 2019. [Google Scholar]
- [18].Justin Donaldson M. T-Distributed Stochastic Neighbor Embedding for R (t-SNE); 2016. Available at: https://cran.r-project.org/web/packages/tsne/tsne.pdf Accessed January 8, 2019. [Google Scholar]
- [19].Stuart T, Butler A, Hoffman P, et al. Comprehensive integration of single cell data. bioRxiv 2018;460147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jager KJ, van Dijk PC, Zoccali C, et al. The analysis of survival data: the Kaplan–Meier method. Kidney Int 2008;74:560–5. [DOI] [PubMed] [Google Scholar]
- [21].Whitfield ML, Sherlock G, Saldanha AJ, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell 2002;13:1977–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Necela BM, Axenfeld BC, Serie DJ, et al. The antineoplastic drug, trastuzumab, dysregulates metabolism in iPSC-derived cardiomyocytes. Clin Transl Med 2017;6:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lu X-F, Zeng D, Liang W-Q, Chen CF, Sun SM, Lin HY. FoxM1 is a promising candidate target in the treatment of breast cancer. Oncotarget 2018;9:842–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kono K, Sato E, Naganuma H, et al. Trastuzumab (Herceptin) enhances class I-restricted antigen presentation recognized by HER-2/neu-specific T cytotoxic lymphocytes. Clin Cancer Res 2004;10:2538–44. [DOI] [PubMed] [Google Scholar]
- [25].Milovanovic J, Todorovic-Rakovic N, Abu Rabi Z. The prognostic role of interleukin-8 (IL-8) and matrix metalloproteinases -2 and -9 in lymph node-negative untreated breast cancer patients. J BUON 2019;18:866–73. [PubMed] [Google Scholar]
- [26].Wang X, Peng X, Zhang X, et al. The emerging roles of CIB1 in cancer. Cell Physiol Biochem 2017;43:1413–24. [DOI] [PubMed] [Google Scholar]
- [27].Sari IN, Yang Y-G, Phi LTH, et al. Interferon-induced transmembrane protein 1 (IFITM1) is required for the progression of colorectal cancer. Oncotarget 2016;7:86039–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mertsch S, Schurgers LJ, Weber K, Paulus W, Senner V. Matrix gla protein (MGP): an overexpressed and migration-promoting mesenchymal component in glioblastoma. BMC Cancer 2009;9:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bianchi F, Hu J, Pelosi G, et al. Lung cancers detected by screening with spiral computed tomography have a malignant phenotype when analyzed by cDNA microarray. Clin Cancer Res 2004;10:6023–8. [DOI] [PubMed] [Google Scholar]
- [30].Tiago DM, Conceição N, Caiado H, Laizé V, Cancela ML. Matrix Gla protein repression by miR-155 promotes oncogenic signals in breast cancer MCF-7 cells. FEBS Lett 2016;590:1234–41. [DOI] [PubMed] [Google Scholar]
- [31].Tuo Y-L, Ye Y-F. MGP is downregulated due to promoter methylation in chemoresistant ER+ breast cancer and high MGP expression predicts better survival outcomes. Eur Rev Med Pharmacol Sci 2017;21:3871–8. [PubMed] [Google Scholar]
- [32].Yet S-F, Perrella MA, Layne MD, et al. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J Clin Invest 1999;103:R23–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dalal S, Zha Q, Daniels CR, et al. Osteopontin stimulates apoptosis in adult cardiac myocytes via the involvement of CD44 receptors, mitochondrial death pathway, and endoplasmic reticulum stress. Am J Physiol Heart Circ Physiol 2014;306:H1182–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].2005;Asher DR, Cerny AM, Weiler SR, et al. Coxsackievirus and adenovirus receptor is essential for cardiomyocyte development. genesis. 42:77–85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.