

Abstract
Essentials.
The role of formyl peptide receptor 1 (FPR1) and its ligand, fMLF, in the regulation of platelet function, hemostasis, and thrombosis is largely unknown.
Fpr1‐deficient mice and selective inhibitors for FPR1 were used to investigate the function of fMLF and FPR1 in platelets.
N‐formyl‐methionyl‐leucyl‐phenylalanine primes platelet activation and augments thrombus formation, mainly through FPR1 in platelets.
Formyl peptide receptor 1 plays a pivotal role in the regulation of platelet function.
Background
Formyl peptide receptors (FPRs) play pivotal roles in the regulation of innate immunity and host defense. The FPRs include three family members: FPR1, FPR2/ALX, and FPR3. The activation of FPR1 by its high‐affinity ligand, N‐formyl‐methionyl‐leucyl‐phenylalanine (fMLF) (a bacterial chemoattractant peptide), triggers intracellular signaling in immune cells such as neutrophils and exacerbates inflammatory responses to accelerate the clearance of microbial infection. Notably, fMLF has been demonstrated to induce intracellular calcium mobilization and chemotaxis in platelets that are known to play significant roles in the regulation of innate immunity and inflammatory responses. Despite a plethora of research focused on the roles of FPR1 and its ligands such as fMLF on the modulation of immune responses, their impact on the regulation of hemostasis and thrombosis remains unexplored.
Objective
To determine the effects of fMLF on the modulation of platelet reactivity, hemostasis, and thrombus formation.
Methods
Selective inhibitors for FPR1 and Fpr1‐deficient mice were used to determine the effects of fMLF and FPR1 on platelets using various platelet functional assays.
Results
N‐formyl‐methionyl‐leucyl‐phenylalanine primes platelet activation through inducing distinctive functions and enhances thrombus formation under arterial flow conditions. Moreover, FPR1 regulates normal platelet function as its deficiency in mouse or blockade in human platelets using a pharmacological inhibitor resulted in diminished agonist‐induced platelet activation.
Conclusion
Since FPR1 plays critical roles in numerous disease conditions, its influence on the modulation of platelet activation and thrombus formation may provide insights into the mechanisms that control platelet‐mediated complications under diverse pathological settings.
Keywords: fMLF, formyl peptide receptors, hemostasis, platelets, thrombosis
1. INTRODUCTION
Platelets are small circulating blood cells that play indispensable roles in the regulation of hemostasis to prevent excessive bleeding upon vascular injury. However, their unwarranted activation under pathological conditions leads to the formation of blood clots (thrombi) within the circulation.1 This results in reduced/retarded blood supply to vital organs including the heart and brain, which leads to heart attacks or strokes, respectively.2 Moreover, platelet activation during microbial infection results in their aggregation, thrombus formation in the microvasculature, and, in later stages, sequestration of platelets in organs such as the lungs, instigating thrombocytopenia and bleeding complications.3 In addition to their prominent roles in hemostasis and thrombosis, platelets play a crucial role in the regulation of innate immunity, inflammatory responses, and clearance of microbial infection.4, 5, 6 Platelets contain a broad spectrum of receptors that induce inflammatory responses during microbial infection and other pathological conditions. In addition, platelets secrete various inflammatory and immunomodulatory molecules from their granules upon activation. They also possess antimicrobial proteins including thrombocidins, cathelicidins, and human β‐defensins that trigger direct microbicidal activities.4 Furthermore, platelets can directly bind and internalize invading microbes.7 The presence of major inflammatory molecules such as formyl peptide receptors (FPRs) and toll‐like receptors facilitates platelets to recognize a diverse array of endogenous damage‐associated molecular patterns and exogenous pathogen‐associated molecular patterns. Collectively, these properties render platelets effector and sentinel cells in primary host defense against invading pathogenic microbes.7, 8
The FPRs belong to a family of G protein‐coupled receptors and are predominantly expressed in immune cells, where they play a prominent role in the regulation of inflammatory responses and host defense. In humans, three FPR family members have been identified: FPR1, FPR2/ALX, and FPR3.9 Although they were originally identified by their capability to recognize N‐formyl peptides produced from bacteria or mitochondria of damaged cells, FPRs can bind a wide variety of structurally and functionally diverse ligands. These include bacterial and mitochondrial formyl peptides, nonformylated peptides/proteins, and small lipid molecules.10 While FPR1 binds to bacterial‐derived N‐formyl peptides with high affinity, FPR2/ALX largely binds to mitochondrial formyl peptides.9, 11 The ligation of N‐formyl peptides to FPRs in immune cells triggers a range of signaling cascades resulting in numerous biological activities. For example, the stimulation of FPR1 by fMLF in neutrophils induces degranulation, chemotaxis, production of superoxide anions, calcium mobilization, cytokine release, and expression of various surface markers.12, 13 During microbial infection, invading bacteria release N‐formyl peptides that facilitate the recruitment of immune cells to the site of infection and accelerate the clearance of microbial infection and repair of tissue damage.14
Czapiga et al15 reported the presence of FPR1 in platelets and its ability to induce chemotactic and migratory responses upon ligation with N‐formyl peptides, which emphasize a crucial role for FPR1 in platelet‐mediated immune responses. Notably, bacterial or synthetic fMLF has been shown to act as a potent chemotactic agent through FPR1 in platelets. Despite numerous reports on the immune functions of FPR1, its impact upon ligation with fMLF on the modulation of hemostasis and thrombosis remains uncharacterized. Recently, we have reported the presence of FPR2/ALX in human platelets and its significance in the regulation of LL37‐induced platelet activation and thrombus formation.16 Here, we report the ability of fMLF to prime platelets and augment thrombus formation, and the significance of FPR1 in the regulation of platelet function in the presence and absence of fMLF.
2. METHODS
2.1. Preparation of human platelet‐rich plasma and isolated platelets
The University of Reading Research Ethics Committee approved all the experimental procedures using human blood from healthy volunteers. The blood samples were collected from healthy, aspirin‐free volunteers after obtaining written informed consent. The blood was collected into VACUETTE blood collection tubes containing 3.2% (w/v) sodium citrate. The blood samples were then centrifuged at 102 g for 20 minutes at room temperature to obtain platelet‐rich plasma (PRP). The PRP was rested at 30°C for 30 minutes prior to use. For the preparation of isolated platelets, the blood was mixed with ACD [2.5% (w/v) sodium citrate, 2% (w/v) D‐glucose, and 1.5% (w/v) citric acid] at 1 (ACD): 9 (blood) ratio and centrifuged at 102 g for 20 minutes. The PRP was collected, mixed with appropriate volume of ACD and prostaglandin I2 (PGI2), and centrifuged at 1413 g for 10 minutes at room temperature. The resultant platelet pellet was resuspended in modified Tyrodes‐HEPES buffer (134 mmol/L NaCl, 2.9 mmol/L KCl, 0.34 mmol/L Na2HPO4.12H2O, 12 mmol/L NaHCO3, 20 mmol/L HEPES, and 1 mmol/L MgCl2, pH 7.3) with appropriate volume of ACD and PGI2 and centrifuged once again at 1413 g for 10 minutes at room temperature. The resultant platelet pellet was resuspended to a final density of 4 × 108 cells/mL in modified Tyrode's‐HEPES buffer and allowed to rest for 30 minutes at 30°C prior to use.
2.2. Mouse blood collection and platelet preparation
The mouse strains of Fpr1 −/− 17 and Fpr2/3 −/− 18 on a C57BL/6 background obtained from William Harvey Research Institute, London, UK, and wild‐type C57BL/6 mice from Envigo, UK, were used in this study. The mice were sacrificed with CO2 and the blood was directly collected by cardiac puncture into a syringe containing 3.2% (w/v) sodium citrate at 1 (citrate):9 (blood) ratio. The blood was then centrifuged at 203 g for 8 minutes at room temperature and the PRP was collected. The remaining blood was resuspended in 500 μL of modified Tyrode's‐HEPES buffer and centrifuged once again at 203 g for 5 minutes. The resultant PRP with PGI2 then centrifuged at 1028 g for 5 minutes. The resultant platelet pellet was resuspended in modified Tyrode's‐HEPES buffer at a density of 2 × 108 cells/mL and rested for 30 minutes at 30°C prior to use.
2.3. In vitro thrombus formation assay
The in vitro thrombus formation assay under arterial flow conditions was performed as described previously.19, 20 Briefly, human DiOC6‐labeled (Sigma Aldrich) human whole blood was preincubated with a vehicle, MLF or fMLF (5 μmol/L) or cyclosporin H (CsH) (10 μmol/L), for 10 minutes before perfusion over collagen (Nycomed) (400 μg/mL)‐coated Vena8™ Biochips (Cellix Ltd) at a shear rate of 20 dynes/cm2. Z‐stack fluorescence images of thrombi were obtained every 30 s for up to 10 minutes using a Nikon eclipse (TE2000‐U) microscope (Nikon Instruments). The fluorescence intensity was calculated by analyzing the data using ImageJ software (National Institutes of Health).
2.4. Platelet adhesion assay
Platelet adhesion was measured by detecting the level of acid phosphatase in immobilized platelet lysates. One hundred microliters of collagen (10 μg/mL in 0.01 M acetic acid) were added to 96‐well plates and incubated overnight at 4°C. The unbound collagen was discarded, and the wells were blocked with 175 μL of 5% (w/v) bovine serum albumin in modified Tyrode's‐HEPES for 1 h. Plates were then washed three times with 175 μL per well of 0.1% bovine serum albumin in modifiedTyrode's‐HEPES buffer. Platelets (1 × 108 cells/mL, 50 μL per well) were then added to wells and incubated at room temperature for 1 h. Nonadhered platelets were discarded, and the wells were washed three times with modified Tyrode's‐HEPES buffer. One hundred and fifty microliters of citrate lysis buffer (3.53 mmol/L p‐nitrophenyl phosphate, 71.4 mmol/L trisodium citrate, 28.55 mmol/L citric acid, 0.1% [v/v] Triton X‐100; pH 5.4) were added and the plate was incubated for 1 h at room temperature. The reaction was stopped by adding 2 M NaOH and the absorbance was measured at 405 nm using a Fluostar Optima spectrofluorimeter.
2.5. Tail bleeding assay
The tail bleeding assay was performed as described previously.21 The British Home Office has approved the experimental procedures. In brief, C57BL/6 (10‐12 weeks old; Envigo, UK) or Fpr1 −/− mice were anesthetized using ketamine (80 mg/kg) and xylazine (5 mg/kg) administered via the intraperitoneal route and placed on a heated mat (37°C). The tail tip (3 mm) was dissected and immersed in sterile saline. The time to cessation of bleeding was measured and the assay was terminated at 20 minutes.
2.6. Platelet aggregation assay
The platelet aggregation assays were performed by optical aggregometry using a two‐channel platelet aggregometer (Chrono‐Log). Human isolated platelets or PRP (270 μL) were added into a siliconized cuvette and prewarmed at 37°C for 90 s. Upon addition of an agonist, the platelets were allowed to aggregate under continuous stirring at 1200 rpm for 5 minutes at 37ºC and the level of aggregation was monitored. The platelets were pretreated with different concentrations of fMLF (1, 5, 10, and 20 μmol/L) for 5 minutes before the addition of CRP‐XL (0.25 μg/mL), collagen (0.5 μg/mL), or thrombin (0.01 U/mL) and the level of aggregation was monitored. Data were analyzed by calculating the percentage of maximum platelet aggregation obtained at 5 minutes.
2.7. Adenosine triphosphate secretion assay
To assess the level of dense granule secretion in platelets, adenosine triphosphate (ATP) secretion was measured using a luciferin–luciferase luminescence substrate (Chrono‐Log) by lumiaggregometry (Chrono‐log). The level of ATP released from platelets upon stimulation with a platelet agonist, CRP‐XL (0.25 μg/mL), in the presence and absence of different concentrations of Boc‐MLF was measured by observing the level of luminescence released.
2.8. Statistical analysis
The data obtained in this study are represented as mean ± SEM. The statistical significance was analyzed using two‐tailed unpaired Student t test for two‐sample comparisons for the data obtained from the flow cytometric assay for FPR1 expression and platelet receptor characterization and cyclic adenosine monophosphate (cAMP) assay. For multiple comparisons such as for data obtained from in vitro thrombus formation, fMLF binding, ATP release, platelet aggregation, and activation, the statistical significance was established using 1‐way or 2‐way ANOVA followed by Bonferroni's correction. Data obtained from the tail bleeding assay were analyzed using a nonparametric Mann‐Whitney test. All statistical analyses were performed using Graphpad Prism 7 software (GraphPad Software Inc.).
3. RESULTS
3.1. Platelets express FPR1
Recently, we have reported the presence of FPR2/ALX in human and its orthologue, Fpr2/3 in human platelets.16 Similarly, the expression of FPR1 in human platelets at protein level, and in megakaryocytes at transcript level, has been previously reported.15 Furthermore, the presence of FPR1 transcripts in human and mouse platelets was demonstrated.22 In line with these findings, here we confirmed the presence of FPR1 on the surface of human isolated platelets by flow cytometry and in isolated platelet lysates using immunoblot analysis. Notably, the activation of platelets using 1 μg/mL cross‐linked collagen‐related peptide (CRP‐XL) increased the level of FPR1 on the surface as determined by flow cytometry (Figure 1AI), while the level of proteins identified by immunoblots remained unchanged (Figure 1AII). These data confirm the presence of FPR1 in platelets, possibly in granules or the open canalicular system or both, and their increase on the surface upon activation similar to FPR2/ALX.16
Figure 1.
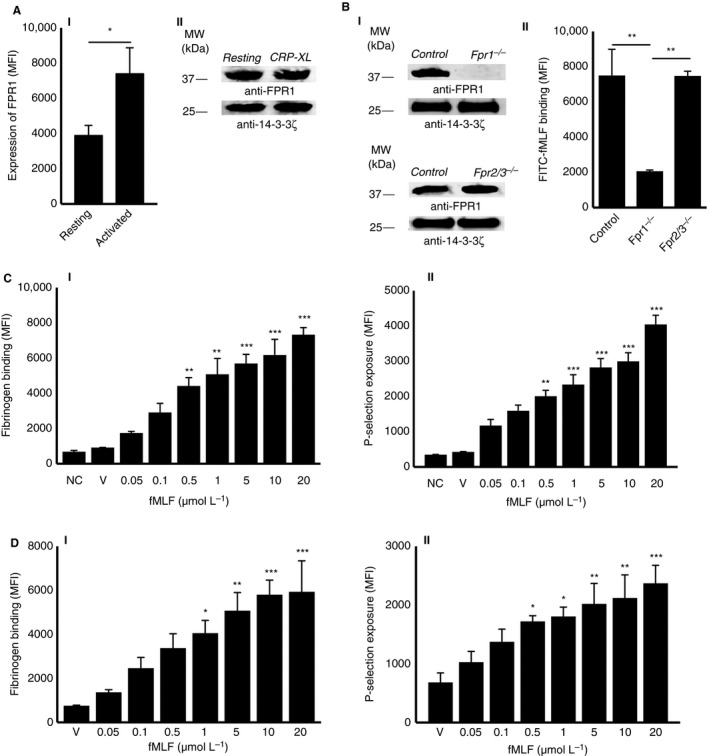
Expression of FPR1 in platelets and the impact of fMLF on platelet activation. A, the expression of FPR1 on the surface of resting or 1 μg/mL CRP‐XL‐ activated human isolated platelets was analyzed using FPR1‐selective and fluorescent‐labeled secondary antibodies by flow cytometry (AI). Data represent mean ± SEM (n = 8). Similarly, the presence of FPR1 in human isolated platelet lysates was confirmed by immunoblot analysis using selective antibodies (AII). B, the absence of Fpr1 was confirmed in isolated platelet lysates obtained from Fpr1 −/− in comparison to the control and Fpr2/3 −/− mice by immunoblot analysis using selective antibodies (BI). Isolated platelets obtained from control, Fpr1 −/−, and Fpr2/3 −/− mice were preincubated with 10 μmol/L FITC‐conjugated fMLF and their level of binding to the platelet surface was measured by flow cytometry (BII). Data represent mean ± SEM (n = 5). C, different concentrations of fMLF were used to determine their impact on platelet activation in human isolated platelets by quantifying the level of fibrinogen binding (CI) and P‐selectin exposure (CII) using flow cytometry. Data represent mean ± SEM (n = 10). D, different concentrations of fMLF were used to determine their effects on platelet activation in human platelet‐rich plasma (PRP) by quantifying the level of fibrinogen binding (DI) and P‐selectin exposure (DII) using flow cytometry. Data represent mean ± SEM (n = 10). The blots shown are representative of three separate experiments. Protein 14‐3‐3ζ was detected as a loading control in the immunoblots. The statistical significance was calculated by 1‐way ANOVA followed by Bonferroni's correction in most of the experiments except the data shown in panel A, which were analyzed by 2‐tailed unpaired Student t test (*P < 0.01, **P < 0.001, and ***P < 0.0001). FITC, fluorescently labeled; fMLF, N‐formyl‐methionyl‐leucyl‐phenylalanine; FPR1, N‐formyl peptide receptor‐1
3.2. N‐formyl‐methionyl‐leucyl‐phenylalanine selectively binds to FPR1 on the platelet surface
A fluorescently‐labeled fMLF was used to investigate its binding to FPR1 on the surface of platelets by flow cytometry. To ascertain the selective binding of fMLF to FPR1, platelets obtained from Fpr1 −/− and Fpr2/3 −/− (an orthologue of human FPR2/ALX) mice along with their controls were used in this assay. The absence of Fpr1 in platelets obtained from Fpr1 −/− mice was confirmed by immunoblot analysis (Figure 1BI). The level of Fpr1 identified in Fpr2/3 −/− mouse platelets was found to be same as the controls. Notably, the absence of Fpr2/3 protein in platelets obtained from Fpr2/3 −/− mice was confirmed previously.16 The binding of FITC‐fMLF (5 μmol/L) was significantly reduced in Fpr1 −/− mouse isolated platelets compared to the control and Fpr2/3 −/− mouse platelets (Figure 1BII). These data confirm the selective binding of fMLF to Fpr1 in mouse platelets.
3.3. N‐formyl‐methionyl‐leucyl‐phenylalanine stimulates platelet activation
To determine whether fMLF is able to stimulate platelet activation upon binding to FPR1, a range of platelet functional assays were performed. Platelet activation triggers inside‐out signaling to integrin αIIbβ3 on the platelet surface and converts it to a high‐affinity state to allow fibrinogen binding and subsequent platelet aggregation.23 To examine whether fMLF influences the inside‐out signaling to integrin αIIbβ3 in platelets, the level of fibrinogen binding on the platelet surface was measured as a marker for inside‐out signaling to integrin αIIbβ3. Indeed, fMLF has increased the level of fibrinogen binding in human isolated platelets in a concentration‐dependent manner (Figure 1CI). A minimum concentration of 0.5 μmol/L fMLF has shown significant increase in fibrinogen binding compared to the resting platelets. Similarly, the level of P‐selectin exposure on the platelet surface was measured as a marker for α‐granule secretion by flow cytometry. The results indicate that fMLF has induced α‐granule secretion in human isolated platelets in a concentration‐dependent manner (Figure 1CII). Similar to the isolated platelets, fMLF has increased the level of fibrinogen binding and P‐selectin exposure in human platelets when PRP was used in a concentration‐dependent manner (Figure 1D). In addition, fMLF has increased the level of platelet adhesion to immobilized collagen under static conditions (Figure 2A). Together, these data confirm that the fMLF significantly primes platelet activation and adhesion.
Figure 2.
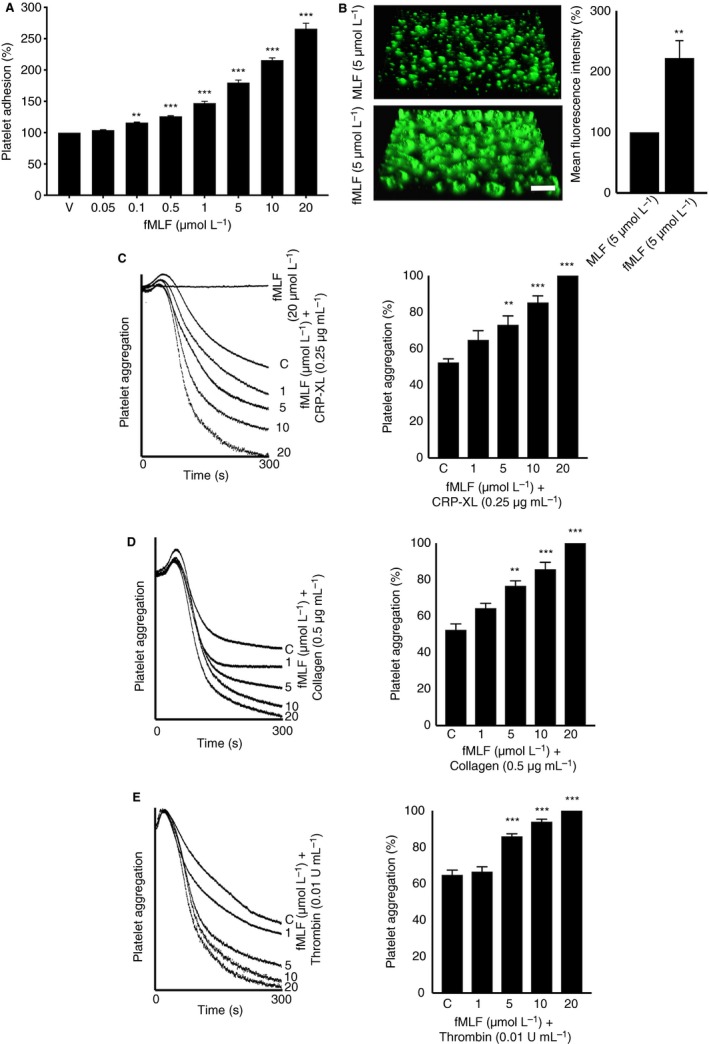
The impact of fMLF on platelet adhesion and aggregation. A, different concentrations of fMLF were used to analyze their influence on human platelet adhesion on immobilized collagen under static conditions. Data represent mean ± SEM (n = 10). B, the effect of fMLF on the modulation of thrombus formation was analyzed by using human DiOC6‐labeled whole blood that was preincubated with nonformylated MLF or fMLF (5 μmol/L) for 10 min prior to perfusion over collagen‐coated (400 μg/mL) Vena8™ Biochips. Images shown are representative of three separate experiments (10× magnification; scale bar ‐ 10 μm). Likewise, the effects of fMLF on C, cross‐linked collagen‐related peptide (CRP‐XL)‐induced, , D, collagen‐induced, or E, thrombin‐induced platelet activation were measured using isolated human platelets by optical aggregometry. Data represent mean ± SEM (n = 5). The statistical significance was calculated by 1‐way ANOVA followed by Bonferroni's correction in most of the experiments except the data shown in panels B, which were analyzed by 2‐tailed unpaired Student t test (*P <0.01, **P <0.001, and ***P <0.0001). fMLF, N‐formyl‐methionyl‐leucyl‐phenylalanine; MLF, methionyl‐leucyl‐phenylalanine
3.4. N‐formyl‐methionyl‐leucyl‐phenylalanine augments thrombus formation
Microbial infection and various inflammatory diseases including sepsis are associated with the risk of disseminated intravascular coagulation or thrombosis in the microvasculature.24 To investigate whether fMLF has a direct impact on thrombosis, its effect on thrombus formation under arterial flow conditions was analyzed. Human DiOC6‐labeled whole blood was preincubated with a control (nonformylated peptide, MLF) or fMLF (5 μM) for 10 minutes prior to perfusion over collagen‐coated Vena8™ biochips. Thrombus formation was monitored for 10 minutes by acquiring fluorescent images at every 30 s . Indeed, fMLF has increased the mean fluorescence intensity of thrombi by approximately 60% compared to the controls (Figure 2B). These data demonstrate the direct impact of fMLF on augmenting the thrombus formation under arterial flow conditions in human whole blood. The effect of fMLF on other blood cells, mainly leukocytes, and their subsequent influence on thrombus formation cannot be ruled out under these circumstances.
3.5. Agonist‐induced platelet aggregation is amplified by fMLF
Following the determination of the effects of fMLF on thrombus formation and platelet activation, aggregation assays were performed to establish its effects on isolated platelets. Human isolated platelets were incubated with various concentrations of fMLF (1, 5, 10, and 20 μmol/L) prior to stimulation with subthreshold concentrations of different platelet agonists such as CRP‐XL (0.25 μg/mL), collagen (0.5 μg/mL), and thrombin (0.01 U/mL), and the level of aggregation was monitored for 5 minutes by optical aggregometry. Notably, fMLF has failed to induce a noticeable platelet aggregation on its own (Figure 2C) although its preincubation with platelets markedly enhanced agonist‐induced platelet aggregation. Maximum aggregation (100%) was obtained in human isolated platelets that were treated with 20 μmol/L fMLF and 0.25 μg/mL CRP‐XL for 5 minutes (Figure 2C). Similar results were obtained with collagen‐induced (Figure 2D) and thrombin‐induced (Figure 2E) platelet aggregation. These data confirm the ability of fMLF to prime platelets and amplify their aggregation upon stimulation with different agonists although it was unable to aggregate platelets on its own under the current settings.
3.6. N‐formyl‐methionyl‐leucyl‐phenylalanine selectively acts through FPR1 in platelets
A large number of studies indicate that fMLF binds primarily to FPR1 and exerts its effects in immune cells.25, 26, 27 The functional dependence of fMLF on FPR1 in platelets was determined using a pharmacological inhibitor for FPR1, Boc‐MLF in human platelets, and platelets obtained from Fpr1 −/− mice through measuring the levels of fibrinogen binding and P‐selectin exposure by flow cytometry. Similar to human platelets, fMLF increased the level of fibrinogen binding [Figure 3AI (isolated platelets) and AII (whole blood)] and P‐selectin exposure [Figure 3BI (isolated platelets) and BII (whole blood)] on platelets obtained from control mice. However, the level of platelet activation by fMLF was significantly reduced in Fpr1 −/− mouse platelets, which demonstrates its functional dependence on FPR1. Here, the impact of leukocytes upon binding to fMLF on platelet activation cannot be ruled out in whole blood obtained from control mice. Notably, the characterization of platelets obtained from Fpr1 −/− mice failed to display any defects in the size and number of platelets or the levels of major platelet receptors such as GPVI (Figure S1i), GPIbα (Figure S1ii), αIIbβ3 (Figure S1iii), and ɑ2β1 (Figure S1iv) in comparison to the control mouse platelets. To establish the functional dependence of fMLF on FPR1 in human platelets, similar assays were performed in the presence or absence of Boc‐MLF. The preincubation of human isolated platelets with different concentrations of Boc‐MLF diminished fMLF (5 μmol/L)‐induced platelet activation as measured by the levels of fibrinogen binding (Figure 3CI) and P‐selectin exposure (Figure 3CII). To determine whether fMLF also acts through FPR2/ALX, the platelet activation was measured in the presence or absence of an FPR2/ALX‐selective inhibitor, WRW4. The preincubation of human isolated platelets with different concentrations of WRW4 did not affect the level of fMLF‐induced (5 μmol/L) platelet activation as measured by the levels of fibrinogen binding (Figure 3DI) and P‐selectin exposure (Figure 3DII). Together, these data emphasize the involvement of FPR1 in the regulation of fMLF‐mediated effects in platelets.
Figure 3.
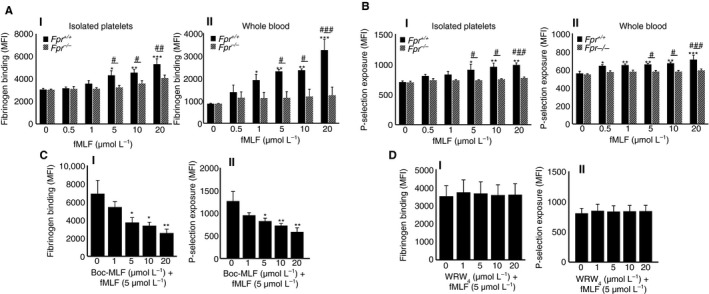
The effects of fMLF on platelet activation are largely mediated via FPR1. The platelet activation upon stimulation with various concentrations of fMLF was quantified by measuring A, the level of fibrinogen binding using FITC‐conjugated fibrinogen antibodies and P‐selectin exposure (B), using PECy5‐conjugated P‐selectin antibodies in isolated platelets (I) or whole blood (II) obtained from control and Fpr1 −/− mice by flow cytometry. Data represent mean ± SEM (n = 6 for isolated platelets and n = 5 for whole blood). C, human isolated platelets were stimulated with fMLF (5 μmol/L) in the presence or absence of different concentrations of Boc‐MLF (1, 5, 10, and 20 μmol/L), and the levels of fibrinogen binding (CI) and P‐selectin exposure (CII) were analyzed by flow cytometry. Data represent mean ± SEM (n = 5). D, similarly, human isolated platelets were stimulated with fMLF (5 μmol/L) in the presence or absence of various concentrations of WRW4 (1, 5, 10, and 20 μmol/L), and the levels of fibrinogen binding (DI) and P‐selectin exposure (DII) were analyzed by flow cytometry. Data represent mean ± SEM (n = 5). *Represents the significant difference between the various concentrations of fMLF within the Fpr1 +/+ group. #Represents the significant difference between Fpr1 +/+ and Fpr1 −/− groups. The statistical significance was calculated by 2‐way ANOVA followed by Bonferroni's correction in most of the experiments except the data shown in panel C and D, which were analyzed by 1‐way ANOVA followed by Bonferroni's correction (*P < 0.01. **P <0.001, and ***P < 0.0001). FITC, Fluorescein isothiocyanate; fMLF, N‐formyl‐methionyl‐leucyl‐phenylalanine; FPR1, N‐formyl peptide receptor‐1
3.7. Inhibition of FPR1 reduces the agonist‐induced platelet activation
In order to study the importance of FPR1 in the regulation of normal platelet activation, further experiments were performed using human isolated platelets in the presence or absence of Boc‐MLF. The CRP‐XL (0.25 μg/mL)‐induced platelet aggregation was significantly reduced in the presence of different concentrations of Boc‐MLF (1, 5, 10, and 20 μmol/L). For example, the addition of Boc‐MLF (20 μmol/L) reduced the platelet aggregation by around 89% (Figure 4A). Similar results were obtained with ADP‐induced platelet aggregation, wherein Boc‐MLF (20 μmol/L) reduced aggregation by approximately 82% (Figure 4B). Moreover, CRP‐XL (0.25 μg/mL)‐induced dense granule secretion as evidenced by the ATP release was significantly reduced by Boc‐MLF (Figure 4C). Similarly, to determine whether FPR1 has any influence on the outside‐in signaling triggered by integrin αIIbβ3, platelet spreading assay was performed on fibrinogen‐coated glass surfaces and analyzed using confocal microscopy. The preincubation of platelets with Boc‐MLF (1, 5, 10, and 20 μmol/L) significantly decreased the number of adhered (Figure 5AI) and spread (Figure 5AII) platelets, and the relative surface area of spreading on fibrinogen‐coated surfaces (Figure 5AIII) indicating a role for FPR1 in the modulation of integrin αIIbβ3‐mediated outside‐in signaling in platelets. In order to corroborate the involvement of FPR1 in the regulation of platelet function, CsH, an inverse agonist for FPR1, was employed. The CsH inhibited CRP‐XL (0.5 μg/mL)‐induced human isolated platelet activation as measured by the levels of fibrinogen binding (Figure 5BI) and P‐selectin exposure (Figure 5BII). Furthermore, CsH decreased the mean fluorescence intensity of thrombi in human whole blood under arterial flow conditions by approximately 60% compared to the controls (Figure 5C). These results highlight the prominent roles of FPR1 in the regulation of normal platelet function.
Figure 4.
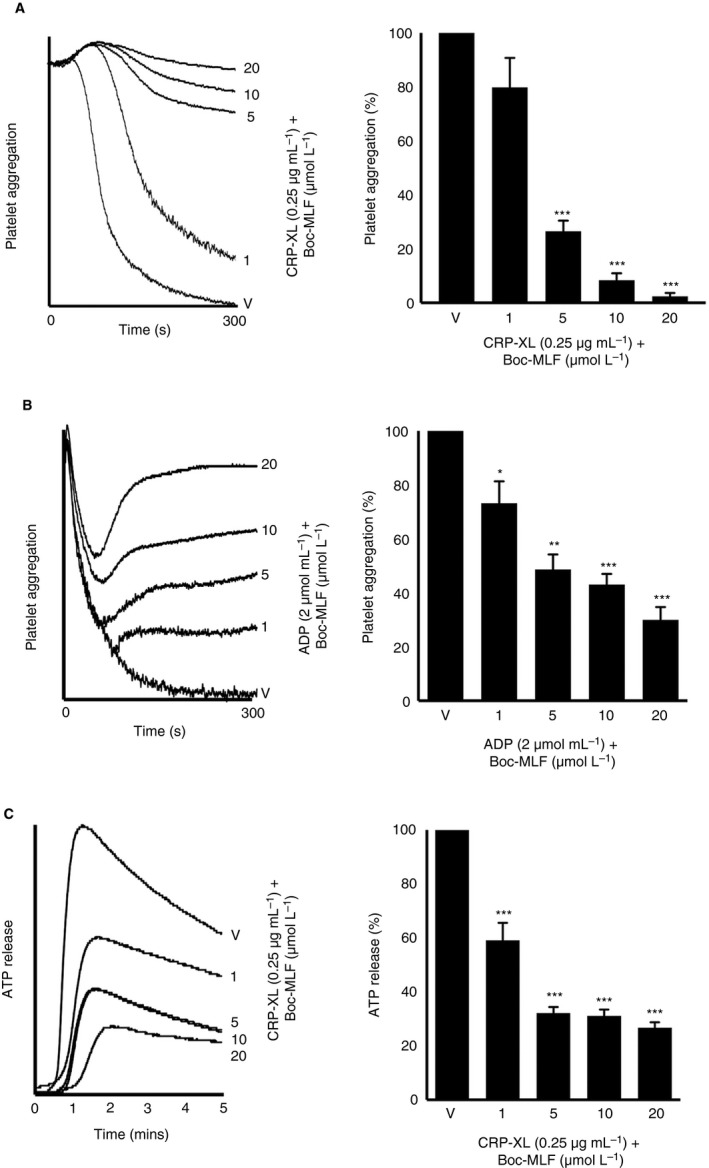
Blockade of FPR1 using a pharmacological inhibitor reduces agonist‐induced platelet aggregation. The effect of different concentrations of Boc‐MLF on CRP‐XL (0.25 μg/mL)‐induced aggregation using human isolated platelets (A) or ADP (2 μmol/L)‐induced aggregation using human platelet‐rich plasma (B) was analyzed by optical aggregometry. The level of aggregation obtained with the vehicle control was taken as 100% to calculate the extent of inhibition with Boc‐MLF‐treated samples. Data represent mean ± SEM (n = 3). Similarly, the level of ATP secretion in human isolated platelets upon activation with CRP‐XL (0.25 μg/mL) in the presence and absence of different concentrations of Boc‐MLF was measured by lumiaggregometry (C). Data represent mean ± SEM (n = 3). P values shown are as calculated by 1‐way ANOVA followed by Bonferroni's correction. (*P < 0.01, **P < 0.001, and ***P <0.0001). ATP, adenosine triphosphate; CRP‐XL, cross‐linked collagen‐related peptide; FPR1, N‐formyl peptide receptor‐1; MLF, methionyl‐leucyl‐phenylalanine
Figure 5.
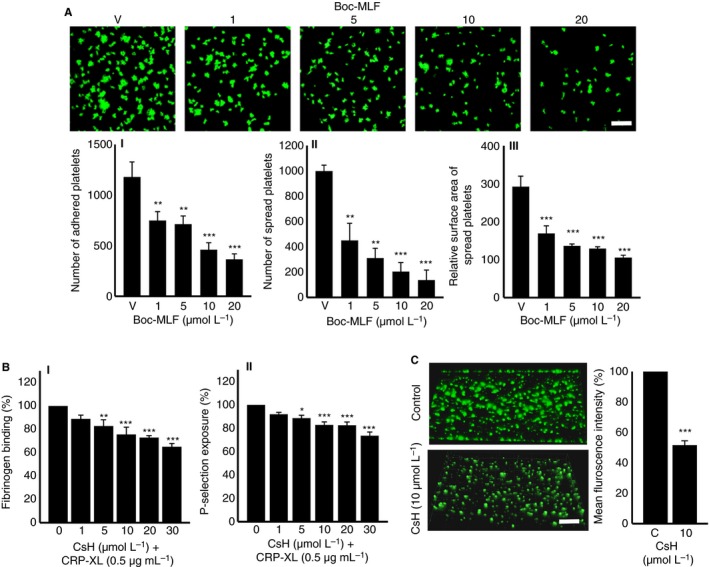
Blockade of FPR1 using a pharmacological inhibitor reduces platelet spreading, activation and thrombus formation. A, platelet adhesion and spreading on fibrinogen‐coated glass surface was analyzed in the absence or presence of Boc‐MLF (1, 5, 10, and 20 μmol/L) by confocal microscopy (60× magnification; scale bar ‐ 10 μm). The number of adhered (AI) and spread platelets (AII) and the relative surface area of spread platelets (AIII) were determined by analyzing the images using ImageJ. Ten random fields of view were recorded for each sample. Data represent mean ± SEM (n = 3). B, the levels of fibrinogen binding (BI) and P‐selectin exposure (BII) were analyzed in human platelet‐rich plasma (PRP) by flow cytometry upon stimulation with CRP‐XL (0.5 μg/mL), in the presence and absence of different concentrations of CsH (1, 5, 10, 20, and 30 μmol/L). Data represent mean ± SEM (n = 3). C, the impact of CsH on the modulation of thrombus formation was analyzed using human DiOC6‐labeled whole blood that was preincubated with a vehicle control or 10 μmol/L CsH for 10 min prior to perfusion over collagen‐coated (400 μg/mL) Vena8™ Biochips. Images shown are representative of three separate experiments (10× magnification; scale bar ‐ 10 μm). Data represent mean ± SEM (n = 3). P values shown are as calculated by 1‐way ANOVA followed by Bonferroni's correction except for data shown in C, which were analyzed by 2‐tailed unpaired Student t test, respectively. (*P < 0.01, **P < 0.001, and ***P < 0.0001) CRP‐XL, cross‐linked collagen‐related peptide; CsH, cyclosporin H; MLF, methionyl‐leucyl‐phenylalanine
3.8. Deletion of FPR1 affects mouse platelet activation
To examine further the impact of FPR1 in platelets, the whole blood obtained from control and Fpr1 −/− mice was used to assess the platelet activation upon stimulation with a range of conventional platelet agonists by measuring the levels of fibrinogen binding and P‐selectin exposure using flow cytometry. Similar to the results obtained with human platelets (Figure 4), the level of (I) fibrinogen binding and (II) P‐selectin exposure in platelets obtained from Fpr1 −/− mice upon stimulation with CRP‐XL (Figure 6A), ADP (Figure 6B), AY‐NH2 (a PAR4 agonist) (Figure 6C), and U46619, an analog of TXA2 (Figure 6D) was largely reduced compared to their controls. To determine the influence of FPR1 on the modulation of hemostasis, tail bleeding assay was performed in control and Fpr1‐deficient mice. A mean bleeding time of 429 s was observed in the control group; however, Fpr1 −/− mice had a significantly increased bleeding time to a mean of 1128 s (Figure 6E). These data indicate the importance of FPR1 in the modulation of platelet function and the maintenance of hemostasis under physiological conditions. Moreover, these results are similar to the data that were reported for Fpr2/3 −/− mice16 emphasizing the physiological significance of FPRs in the modulation of platelet function and hemostasis.
Figure 6.
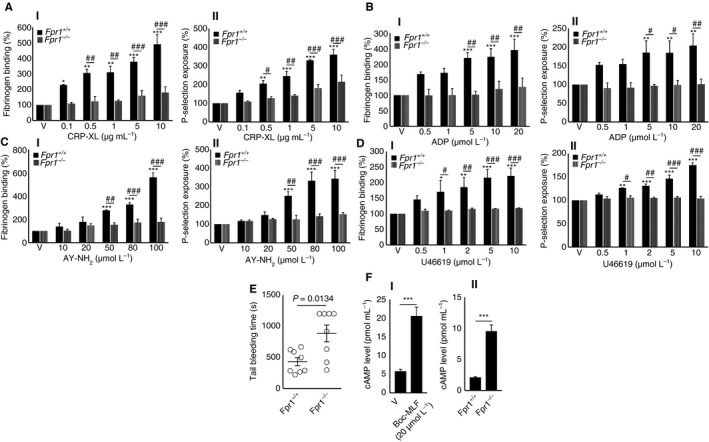
Deletion of Fpr1 in mice reduces the agonists‐induced platelet activation and affects hemostasis. The levels of fibrinogen binding (I) and P‐selectin exposure (II) were analyzed in isolated platelets obtained from control or Fpr1 −/− mice upon stimulation with various concentrations of agonists such as CRP‐XL (A), ADP (B), AY‐NH2 (C), or U46691 (D), by flow cytometry. Data represent mean ± SEM (n = 4). E, the impact of FPR1 on the modulation of hemostasis was analyzed by tail bleeding assay in control or Fpr1 −/− mice. Data represent mean ± SEM (n = 8). F, the level of cAMP in human isolated platelets in the presence or absence of Boc‐MLF (I), and in control and Fpr1 −/− mouse isolated platelets (II), was analyzed using a cAMP assay kit. *Represents the significant difference between the various concentrations of agonists within the Fpr1 +/+ group. #Represents the significant difference between Fpr1 +/+ and Fpr1 −/− groups. Data represent mean ± SEM (n = 4). The statistical significance was calculated by 2‐way ANOVA followed by Bonferroni's correction in most of the experiments except for the data shown in panels E and F, which were calculated by non parametric Mann‐Whitney test, and 2‐tailed unpaired Student t test, respectively (*P <0.01, **P <0.001, and ***P <0.0001). ADP, adenosine diphosphate; cAMP, cyclic adenosine monophosphate; CRP‐XL, cross‐linked collagen‐related peptide; FPR1, N‐formyl peptide receptor‐1
3.9. The inhibition or deletion of FPR1 increases the level of cyclic AMP
Cyclic AMP (cAMP) is a potent inhibitor of platelet function and its level is generally reduced upon platelet activation. Stimulants of cAMP generation are known to inhibit platelet activation.28 The FPRs are Gi protein‐coupled receptors,29 which are known to inhibit adenylate cyclase and thus, lead to a reduction in cAMP levels. Therefore, the deletion of genes for Gi‐coupled receptors in mice generally increases the basal levels of cAMP in target cells.30, 31 To investigate whether the effects of FPR1 in platelets are driven through cAMP‐dependent signaling, the level of cAMP was quantified in platelets using a cAMP assay kit. The inhibition of FPR1 in human isolated platelets with Boc‐MLF (20 μmol/L) significantly elevated the level of cAMP compared to the controls (Figure 6FI). Similarly, Fpr1 −/− mouse isolated platelets exhibited elevated basal levels of cAMP compared to the control mouse platelets at resting conditions (Figure 6FII). These data illustrate that the level of cAMP may play a key role in the regulation of FPR1‐mediated function in platelets.
4. DISCUSSION
N‐formyl peptides are released from bacteria or mitochondria of damaged cells.32, 33 They have been demonstrated to play substantial roles in the initiation of chemotaxis and subsequent inflammatory responses in immune cells including monocytes, mast cells, eosinophils, and neutrophils via FPRs.9 Despite their ability to mediate innate immune responses, they have been associated with the pathogenesis of microbial infection and inflammatory diseases. Notably, E coli‐derived fMLF34 has been implicated in bacterial cystitis,35 pneumococcal pneumonia,36 inflammatory bowel disease,37 pouchitis, colitis38 and juvenile peridontitis.39 It has been shown that inhalation or injection of fMLF can cause bronchial inflammation40 and induce rapid neutropenia, thereby increasing susceptibility to infection.41 The plasma levels of fMLF were increased in these conditions, and also in high‐fat‐diet‐treated mice due to altered microbiome where it impaired the glucose tolerance and insulin secretion.42 Many of these infectious and inflammatory conditions are associated with a risk for thrombosis and other platelet‐mediated complications.43, 44, 45 The concentrations of fMLF in the intestinal milieu have been reported to be at least in micromolar ranges.46 In this study, we demonstrate that fMLF is able to prime platelets and augment thrombus formation in micromolar concentrations. Therefore, the increased levels of fMLF under the aforementioned pathological conditions may lead to platelet activation and contribute to thrombotic complications.
The activation of platelets facilitates their adhesion to leukocytes and leads to the formation of platelet‐leukocyte aggregates (PLAs).47 P‐selectin on the surface of activated platelets drives the formation of PLAs via binding to P‐selectin glycoprotein ligand‐1 on the surface of leukocytes.48 In addition, the fibrinogen binding to integrin αIIbβ3 in platelets and Mac‐1 in neutrophils was suggested to play an important role in the formation of PLAs.49 The PLAs are known to amplify thrombotic and proinflammatory responses in diverse inflammatory settings.50 Indeed, fMLF has been reported to induce PLA formation51 through fibrinogen binding in platelets and neutrophils.49 Moreover, previous studies have reported the aggregation of platelets in response to fMLF‐induced neutrophil stimulation51, 52, 53 through release of platelet‐activating factor54 and cathepsin G.55, 56 In line with these, we demonstrate that fMLF has failed to induce aggregation of isolated platelets although the pretreatment of platelets with fMLF augmented agonist‐induced aggregation. Furthermore, fMLF augmented thrombus formation under arterial flow conditions in whole blood. Given that fMLF is able to upregulate the expression of adhesion molecules57, 58 and aggregate neutrophils,49, 53 its effects on thrombus formation may be partly attributed to its interactions with leukocytes. Therefore, fMLF‐induced fibrinogen binding and P‐selectin exposure in platelets may directly trigger PLA formation as detailed previously.59 Although in this study the direct impact of fMLF on platelet‐mediated inflammatory responses was not analyzed, the role of fMLF cannot be excluded in such responses. Together, these data demonstrate a prominent priming role for fMLF in platelets, which may augment thrombotic and proinflammatory responses through interactions with leukocytes in pathological settings.
Formyl peptide receptor 1 is a chemoattractant receptor that is widely expressed in various cell types including neutrophils, macrophages, and platelets.15 Despite its well‐characterized role in the modulation of inflammatory responses, the role of FPR1 in the regulation of platelet function is poorly studied. Here, we report a crucial role for FPR1 on the regulation of platelet activation, hemostasis, and thrombosis. In line with a previous study,15 we confirm the presence of this receptor in human platelets by immunoblot analysis, and its upregulation upon activation of platelets with an agonist. By using selective pharmacological inhibitor, Boc‐MLF and an inverse agonist, cyclosporin H (CsH), the significance of FPR1 in the regulation of fMLF‐induced and agonist‐induced (such as CRP‐XL and ADP) platelet activation was established. The blockade of FPR1 resulted in reduced ATP release upon activation with CRP‐XL, which not only affects secondary platelet activation but may also influence the modulation of inflammatory responses.60 Similarly, the inhibition of FPR1 impaired the ability of platelet spreading on fibrinogen, which is essential for thrombosis and subsequent wound repair.61 The specificity of inhibitors to FPR1 may be limited by their ability to bind FPR2/ALX nonspecifically at higher concentrations. Therefore, the prominent role of FPR1 on the regulation of platelet activation was also corroborated using platelets obtained from Fpr1‐deficient mice, wherein the effects of fMLF and platelet agonists were largely diminished. The selective binding of fMLF to Fpr1 was confirmed using platelets obtained from Fpr1‐deficient mice. In addition, the hemostasis in Fpr1‐deficient mice was affected further emphasizing its significance in the regulation of platelet function. Fpr1‐deficient mice displayed severe inflammation, higher mortality during pneumococcal meningitis,62 increased susceptibility to Listeria monocytogenes infection, and impaired antibacterial host defense.17, 63 In addition, Fpr1‐deficient mice demonstrate major roles in sterile inflammation triggered by mitochondrial N‐formyl peptides as demonstrated by the attenuation of inflammation in response to sterile acute lung injury in these mice.64 In line with these studies, here we demonstrate the dysfunction of platelets and affected hemostasis in Fpr1‐deficient mice, which may also substantiate the reduced inflammatory responses due to significant contribution of platelets in inflammation and innate immunity.
As a Gi protein‐coupled receptor, FPR1 triggers downstream signaling via various molecules such as phospholipase C, PI3K/AKT, and MAPK, and modulates calcium mobilization in neutrophils.13 The ability of fMLF to induce calcium mobilization in platelets has been previously reported.15 Here, we report the impact of FPR1 on cAMP‐mediated signaling in platelets using Boc‐MLF and platelets obtained from Fpr1‐deficient mice. Indeed, the inhibition of FPR1 in human platelets or its deletion in mouse platelets resulted in elevated levels of cAMP, which is a potent inhibitor for platelet activation. This confirmed the involvement of cAMP‐dependent signaling in the regulation of FPR1‐mediated effects in platelets.
Given the significance of FPRs and their ligands under various clinical settings, the therapeutic potential of these are being widely investigated. Notably, honokiol, a plant‐derived bioactive agent, has recently been demonstrated to reduce the proinflammatory responses induced by fMLF in neutrophils through inhibiting FPR1.65 Recently, we have reported the presence of FPR2/ALX in platelets, and its significance in the activation of platelets and thrombus formation upon ligation with an antimicrobial cathelicidin, LL37.16 In conclusion, similar to LL37, this study demonstrates a prominent role for fMLF for priming platelet activation and augmenting thrombus formation under arterial flow conditions. Using an FPR1 selective inhibitor and Fpr1‐deficient mice, the functional dependence of fMLF on this receptor was established. Therefore, the priming effects of fMLF on platelets may significantly contribute to the development of thrombotic and proinflammatory diatheses in pathological settings where the level of fMLF is elevated. Hence, FPR1 may act as a potential therapeutic target and its ligands may provide a powerful platform to develop novel therapeutic agents in order to treat and prevent thrombotic and inflammatory complications in diverse pathophysiological settings.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
AUTHOR CONTRIBUTIONS
Maryam F. Salamah and Sakthivel Vaiyapuri designed the study, performed experiments, analyzed data, and wrote the paper; Divyashree Ravishankar, Rajendran Vaiyapuri, and Leonardo A. Moraes performed experiments and analyzed data; Ketan Patel, Mauro Perretti, and Jonathan M. Gibbins provided guidance and support for the design of experiments and analysis of data.
Supporting information
ACKNOWLEDGMENTS
We would like to thank the British Heart Foundation (Grant number: PG/16/64/32311), the Wellcome Trust (204389/Z/16/Z), the Royal Society, the Physiological Society, and the Saudi Arabian Ministry of Higher Education for their funding support for this research.
Salamah MF, Ravishankar D, Vaiyapuri R, et al. The formyl peptide fMLF primes platelet activation and augments thrombus formation. J Thromb Haemost. 2019;17:1120–1133. 10.1111/jth.14466
Funding information
We would like to thank the British Heart Foundation (Grant number: PG/16/64/32311), The Wellcome Trust (204389/Z/16/Z), The Royal Society, The Physiological Society, and the Saudi Arabian Ministry of Higher Education for their funding support for this research.
Manuscript handled by: Diego Mezzano
Final decision: Pieter Reitsma, 18 April 2019
REFERENCES
- 1. Duhamel TA, Xu YJ, Arneja AS, Dhalla NS. Targeting platelets for prevention and treatment of cardiovascular disease. Expert Opin Ther Targets. 2007;11:1523–33. [DOI] [PubMed] [Google Scholar]
- 2. Michelson AD. Antiplatelet therapies for the treatment of cardiovascular disease. Nat Rev Drug Discovery. 2010;9:154–69. [DOI] [PubMed] [Google Scholar]
- 3. Claushuis TA, van Vught LA, Scicluna BP, Wiewel MA, Klein Klouwenberg PM, Hoogendijk AJ, et al. Thrombocytopenia is associated with a dysregulated host response in critically ill sepsis patients. Blood. 2016; 127:3062‐72. [DOI] [PubMed] [Google Scholar]
- 4. Hamzeh‐Cognasse H, Damien P, Chabert A, Pozzetto B, Cognasse F, Garraud O. Platelets and infections ‐ complex interactions with bacteria. Front Immunol. 2015;6:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rondina MT, Garraud O. Emerging evidence for platelets as immune and inflammatory effector cells. Front Immunol. 2014;5:653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nording HM, Seizer P, Langer HF. Platelets in inflammation and atherogenesis. Front Immunol. 2015;6:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yeaman MR. Platelets in defense against bacterial pathogens. Cell Mol Life Sci. 2010;67:525–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Speth C, Loffler J, Krappmann S, Lass‐Florl C, Rambach G. Platelets as immune cells in infectious diseases. Future Microbiol. 2013;8:1431–51. [DOI] [PubMed] [Google Scholar]
- 9. Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, et al. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev. 2009;61:119–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. He HQ, Ye RD. The formyl peptide receptors: diversity of ligands and mechanism for recognition. Molecules. 2017;22:E455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rabiet MJ, Huet E, Boulay F. Human mitochondria‐derived N‐formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes‐derived peptides preferentially activate FPR. Eur J Immunol. 2005;35:2486–95. [DOI] [PubMed] [Google Scholar]
- 12. Sklar LA, Jesaitis AJ, Painter RG. The neutrophil N‐formyl peptide receptor: dynamics of ligand‐receptor interactions and their relationship to cellular responses. Contemp Top Immunobiol. 1984;14:29–82. [DOI] [PubMed] [Google Scholar]
- 13. Dorward DA, Lucas CD, Chapman GB, Haslett C, Dhaliwal K, Rossi AG. The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation. Am J Pathol. 2015;185:1172–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dahlgren C, Gabl M, Holdfeldt A, Winther M, Forsman H. Basic characteristics of the neutrophil receptors that recognize formylated peptides, a danger‐associated molecular pattern generated by bacteria and mitochondria. Biochem Pharmacol. 2016;114:22–39. [DOI] [PubMed] [Google Scholar]
- 15. Czapiga M, Gao JL, Kirk A, Lekstrom‐Himes J. Human platelets exhibit chemotaxis using functional N‐formyl peptide receptors. Exp Hematol. 2005;33:73–84. [DOI] [PubMed] [Google Scholar]
- 16. Salamah MF, Ravishankar D, Kodji X, Moraes LA, Williams HF, Vallance TM, et al. The endogenous antimicrobial cathelicidin LL37 induces platelet activation and augments thrombus formation. Blood Adv. 2018;2:2973–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gao JL, Lee EJ, Murphy PM. Impaired antibacterial host defense in mice lacking the N‐formylpeptide receptor. J Exp Med. 1999;189:657–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dufton N, Hannon R, Brancaleone V, Dalli J, Patel HB, Gray M, et al. Anti‐inflammatory role of the murine formyl‐peptide receptor 2: ligand‐specific effects on leukocyte responses and experimental inflammation. J Immunol. 2010;184:2611–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Spyridon M, Moraes LA, Jones CI, Sage T, Sasikumar P, Bucci G, et al. LXR as a novel antithrombotic target. Blood. 2011;117:5751–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vaiyapuri S, Jones CI, Sasikumar P, Moraes LA, Munger SJ, Wright JR, et al. Junctions and connexin hemichannels underpin hemostasis and thrombosis. Circulation. 2012;125:2479–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vaiyapuri S, Moraes LA, Sage T, Ali MS, Lewis KR, Mahaut‐Smith MP, et al. Connexin40 regulates platelet function. Nat Commun. 2013;4:2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rowley JW, Oler AJ, Tolley ND, Hunter BN, Low EN, Nix DA, et al. Genome‐wide RNA‐seq analysis of human and mouse platelet transcriptomes. Blood. 2011;118:e101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yun SH, Sim EH, Goh RY, Park JI, Han JY. Platelet activation: the mechanisms and potential biomarkers. Biomed Res Int. 2016;2016:9060143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Semeraro N, Ammollo CT, Semeraro F, Colucci M. Sepsis‐associated disseminated intravascular coagulation and thromboembolic disease. Mediterr J Hematol Infect Dis. 2010;2:e2010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kirpotina LN, Schepetkin IA, Khlebnikov AI, Ruban OI, Ge Y, Ye RD, et al. 4‐Aroyl‐3‐hydroxy‐5‐phenyl‐1H‐pyrrol‐2(5H)‐ones as N‐formyl peptide receptor 1 (FPR1) antagonists. Biochem Pharmacol. 2017;142:120–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Honda M, Takeichi T, Hashimoto S, Yoshii D, Isono K, Hayashida S, et al. Intravital imaging of neutrophil recruitment reveals the efficacy of FPR1 blockade in hepatic ischemia‐reperfusion injury. J Immunol. 2017;198:1718–28. [DOI] [PubMed] [Google Scholar]
- 27. Maaty WS, Lord CI, Gripentrog JM, Riesselman M, Keren‐Aviram G, Liu T, et al. Identification of C‐terminal phosphorylation sites of N‐formyl peptide receptor‐1 (FPR1) in human blood neutrophils. J Biol Chem. 2013;288:27042–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Smolenski A. Novel roles of cAMP/cGMP‐dependent signaling in platelets. J Thromb Haemost. 2012;10:167–76. [DOI] [PubMed] [Google Scholar]
- 29. Wenzel‐Seifert K, Arthur JM, Liu HY, Seifert R. Quantitative analysis of formyl peptide receptor coupling to g(i)alpha(1), g(i)alpha(2), and g(i)alpha(3). J Biol Chem. 1999;274:33259–66. [DOI] [PubMed] [Google Scholar]
- 30. Yang J, Wu J, Jiang H, Mortensen R, Austin S, Manning DR, et al. Signaling through G(i) family members in platelets ‐ Redundancy and specificity in the regulation of adenylyl cyclase and other effectors. J Biol Chem. 2002;277:46035–42. [DOI] [PubMed] [Google Scholar]
- 31. Liu YF, Ghahremani MH, Rasenick MM, Jakobs KH, Albert PR. Stimulation of cAMP synthesis by G(i)‐coupled receptors upon ablation of distinct G alpha(i) protein expression ‐ G(i) subtype specificity of the 5‐HT1A receptor. J Biol Chem. 1999;274:16444–50. [DOI] [PubMed] [Google Scholar]
- 32. Schiffmann E, Corcoran BA, Wahl SM. N‐formylmethionyl peptides as chemoattractants for leukocytes. P Natl Acad Sci USA. 1975;72:1059–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chadwick VS, Mellor DM, Myers DB, Selden AC, Keshavarzian A, Broom MF, et al. Production of peptides inducing chemotaxis and lysosomal enzyme release in human neutrophils by intestinal bacteria in vitro and in vivo. Scand J Gastroenterol. 1988;23:121–8. [DOI] [PubMed] [Google Scholar]
- 34. Marasco WA, Phan SH, Krutzsch H, Showell HJ, Feltner DE, Nairn R, et al. Purification and identification of formyl‐methionyl‐leucyl‐phenylalanine as the major peptide neutrophil chemotactic factor produced by Escherichia‐Coli. J Biol Chem. 1984;259:5430–9. [PubMed] [Google Scholar]
- 35. Schwarz NT, Jung SY, Kalff JC, Chancellor M, Bauer AJ. Bacterial toxin N‐formyl‐methionyl‐leucyl‐phenylalanine acutely contracts human and rabbit detrusor through the release of eicosanoids. J Urology. 2002;167:2603–12. [PubMed] [Google Scholar]
- 36. Gauthier JF, Fortin A, Bergeron Y, Dumas MC, Champagne ME, Bergeron MG. Differential contribution of bacterial N‐formyl‐methionyl‐leucyl‐phenylalanine and host‐derived CXC chemokines to neutrophil infiltration into pulmonary alveoli during murine pneumococcal pneumonia. Infect Immun. 2007;75:5361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Anton PA, Targan SR, Shanahan F. Increased neutrophil receptors for and response to the proinflammatory bacterial peptide formyl‐methionyl‐leucyl‐phenylalanine in Crohn's disease. Gastroenterology. 1989;97:20–8. [DOI] [PubMed] [Google Scholar]
- 38. Leduc LE, Nast CC. Chemotactic peptide‐induced acute colitis in rabbits. Gastroenterology. 1990;98:929–35. [DOI] [PubMed] [Google Scholar]
- 39. Perez HD, Kelly E, Elfman F, Armitage G, Winkler J. Defective polymorphonuclear leukocyte formyl peptide receptor(s) in juvenile periodontitis. J Clin Invest. 1991;87:971–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Peters MJ, Breslin AB, Kemp AS, Chu J, Berend N. Haematological effects of inhalation of N‐formyl‐methionyl‐leucyl‐phenylalanine in man. Thorax. 1992;47:284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yurt RW, Shires GT. Increased susceptibility to infection due to infusion of exogenous chemotaxin. Arch Surg‐Chicago. 1987;122:111–6. [DOI] [PubMed] [Google Scholar]
- 42. Wollam J, Johnson AM, Xu YJ, Jain M, Olefsky JM. Gut microbiome produced N‐formyl peptides act through the receptor FPR1 to modulate insulin secretion and metabolism. Diabetes. 2016;65:A453. [Google Scholar]
- 43. Danese S, de la Motte C, Fiocchi C. Platelets in inflammatory bowel disease: clinical, pathogenic, and therapeutic implications. Am J Gastroenterol. 2004;99:938–45. [DOI] [PubMed] [Google Scholar]
- 44. Tunjungputri RN, van de Heijden W, Urbanus RT, de Groot PG, van der Ven A, de Mast Q. Higher platelet reactivity and platelet‐monocyte complex formation in Gram‐positive sepsis compared to Gram‐negative sepsis. Platelets. 2017;28:595–601. [DOI] [PubMed] [Google Scholar]
- 45. Munoz‐Esquerre M, Ferreiro JL, Huertas D, Marcano AL, Lopez‐Sanchez M, Roura G, et al. Impact of acute exacerbations on platelet reactivity in chronic obstructive pulmonary disease patients. Int J Chronic Obstr. 2018;13:141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Roberts EC, Hobson CH, Anderson RP, Chadwick VS. Radioimmunoassay for formyl methionyl leucyl phenylalanine. 2. Demonstration of an enterohepatic circulation of immunoreactive bacterial chemotactic peptides in man. J Gastroen Hepatol. 1990;5:38–43. [DOI] [PubMed] [Google Scholar]
- 47. Zarbock A, Polanowska‐Grabowska RK, Ley K. Platelet‐neutrophil‐interactions: linking hemostasis and inflammation. Blood Rev. 2007;21:99–111. [DOI] [PubMed] [Google Scholar]
- 48. Ostrovsky L, King AJ, Bond S, Mitchell D, Lorant DE, Zimmerman GA, et al. A juxtacrine mechanism for neutrophil adhesion on platelets involves platelet‐activating factor and a selectin‐dependent activation process. Blood. 1998;91:3028–36. [PubMed] [Google Scholar]
- 49. Li NL, Hu H, Lindqvist M, Johnsson EW, Goodall AH, Hjemdahl P. Platelet‐leukocyte cross‐talk in whole blood: inhibition by GPIIb/IIIa and P‐selectin antagonists. Circulation. 2000;102:34. [DOI] [PubMed] [Google Scholar]
- 50. Esmon CT. The interactions between inflammation and coagulation. Brit J Haematol. 2005;131:417–30. [DOI] [PubMed] [Google Scholar]
- 51. Coeffier E, Joseph D, Prevost MC, Vargaftig BB. Platelet‐leukocyte interaction ‐ Activation of rabbit platelets by Fmlp‐stimulated neutrophils. Brit J Pharmacol. 1987;92:393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rossaint J, Kuhne K, Skupski J, Van Aken H, Looney MR, Hidalgo A, et al. Directed transport of neutrophil‐derived extracellular vesicles enables platelet‐mediated innate immune response. Nat Commun. 2016;7:13464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Oda M, Satouchi K, Yasunaga K, Saito K. Polymorphonuclear leukocyte‐platelet interactions: acetylglyceryl ether phosphocholine‐induced platelet activation under stimulation with chemotactic peptide. J Biochem. 1986;100:1117–23. [DOI] [PubMed] [Google Scholar]
- 54. Stewart AG, Harris T. Involvement of platelet‐activating factor in endotoxin‐induced priming of rabbit polymorphonuclear leukocytes. J Lipid Mediat. 1991;3:125–38. [PubMed] [Google Scholar]
- 55. Sambrano GR, Huang W, Faruqi T, Mahrus S, Craik C, Coughlin SR. Cathepsin G activates protease‐activated receptor‐4 in human platelets. J Biol Chem. 2000;275:6819–23. [DOI] [PubMed] [Google Scholar]
- 56. Evangelista V, Rajtar G, de Gaetano G, White JG, Cerletti C. Platelet activation by fMLP‐stimulated polymorphonuclear leukocytes: the activity of cathepsin G is not prevented by antiproteinases. Blood. 1991;77:2379–88. [PubMed] [Google Scholar]
- 57. Fan ST, Edgington TS. Integrin regulation of leukocyte inflammatory functions. CD11b/CD18 enhancement of the tumor necrosis factor‐alpha responses of monocytes. J Immunol. 1993;150:2972–80. [PubMed] [Google Scholar]
- 58. Fearon DT, Collins LA. Increased expression of C3b receptors on polymorphonuclear leukocytes induced by chemotactic factors and by purification procedures. J Immunol. 1983;130:370–5. [PubMed] [Google Scholar]
- 59. Evangelista V, Piccardoni P, White JG, Degaetano G, Cerletti C. Cathepsin G‐dependent platelet stimulation by activated polymorphonuclear leukocytes and its inhibition by antiproteinases ‐ role of P‐selectin mediated cell‐cell adhesion. Blood. 1993;81:2947–57. [PubMed] [Google Scholar]
- 60. Dosch M, Gerber J, Jebbawi F, Beldi G. Mechanisms of ATP release by inflammatory cells. Int J Mol Sci. 2018;19:E1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lee D, Fong KP, King MR, Brass LF, Hammer DA. Differential dynamics of platelet contact and spreading. Biophys J . 2012;102:472–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Oldekamp S, Pscheidl S, Kress E, Soehnlein O, Jansen S, Pufe T, et al. Lack of formyl peptide receptor 1 and 2 leads to more severe inflammation and higher mortality in mice with of pneumococcal meningitis. Immunology. 2014;143:447–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu MY, Chen KQ, Yoshimura T, Liu Y, Gong WH, Wang AM, et al. Formylpeptide receptors are critical for rapid neutrophil mobilization in host defense against Listeria monocytogenes. Sci Rep‐UK. 2012;2:786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dorward DA, Lucas CD, Doherty MK, Chapman GB, Scholefield EJ, Morris AC, et al. Novel role for endogenous mitochondrial formylated peptide‐driven formyl peptide receptor 1 signalling in acute respiratory distress syndrome. Thorax. 2017;72:928–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Liu FC, Yu HP, Syu YT, Fang JY, Lin CF, Chang SH, et al. Honokiol suppresses formyl peptide‐induced human neutrophil activation by blocking formyl peptide receptor 1. Sci Rep‐UK. 2017;7:6718 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials