

Abstract
Metal nanoclusters (NCs) are considered ideal nanomaterials for biological applications owing to their strong photoluminescence (PL), excellent photostability, and good biocompatibility. This study presents a simple and versatile strategy to design proteins, via incorporation of a di‐histidine cluster coordination site, for the sustainable synthesis and stabilization of metal NCs with different metal composition. The resulting protein‐stabilized metal NCs (Prot‐NCs) of gold, silver, and copper are highly photoluminescent and photostable, have a long shelf life, and are stable under physiological conditions. The biocompatibility of the clusters was demonstrated in cell cultures in which Prot‐NCs showed efficient cell internalization without affecting cell viability or losing luminescence. Moreover, the approach is translatable to other proteins to obtain Prot‐NCs for various biomedical applications such as cell imaging or labeling.
Keywords: bioimaging, bionanohybrids, metal nanoclusters, photoluminescence, protein design
Novel metal nanomaterials with interesting size‐dependent properties have been intensely pursued not only for their fundamental scientific interest, but also for their many technological applications.1 In particular, metal nanoclusters (NCs) have attracted special attention in recent years owing to their size‐dependent electronic transitions, strong fluorescence, photostability, and biocompatibility. These assemblies of a few to several atoms, which are smaller than 2 nm in size,2 have discrete electron energy levels, resulting in highly distinct properties from bulk metals or classical nanoparticles.3 Such properties make metal NCs promising in biological analysis and imaging, environmental monitoring, industrial catalysis, and electronic devices.4
Metal NCs need to be stabilized by different molecules, such as dendrimers,5 small molecules,6 DNA,7 peptides,8 and proteins.9 NCs stabilized by peptides and proteins can be produced under mild conditions, and the obtained structures are stable under a wide range of pH values and ionic forces, making them ideal for biological applications. Natural and designed histidine‐rich and cysteine‐rich peptides have been used for the synthesis and stabilization of metal NCs.8, 10 Thiol, amine, and carboxylic acid side‐chain functionalities seem to promote metal binding.11 Different commercially available proteins, such as bovine serum albumin (BSA),12 papain,13 human transferrin,14 lysozyme,15 trypsin,16 pepsin,17 insulin,18 and peroxidase,19 have been employed in the preparation of metal NCs. Major efforts have been made to understand the role of protein characteristics, including protein size and amino acid content, in the formation and properties of metal NCs; the metal coordination environments that promote biomineralization; and the NCs structure and formation mechanisms as well as interactions of the metal core with the protein.20 However, a simple approach to design artificial and/or natural proteins with an incorporated predefined metal binding site for the synthesis of metal NCs is still not available. To date, there are only two examples reported that explored the rational introduction of amino acids for cluster coordination: the insertion of a single cysteine (Cys) for the of metal NCs,21 and the incorporation of two additional Cys residues in the ferritin cage to enhance the uptake of Au ions.20c
The current study explores the design of a cluster coordination site into a protein structure as a simple and versatile approach for the sustainable synthesis and stabilization of metal NCs into selected proteins. In particular, we used repeat proteins as an artificial protein system but we believe that this method can be used as a general approach to incorporate cluster coordination sites into functional proteins to expand their applications. The resulting designed repeat proteins were evaluated as templates for the synthesis and stabilization of NCs with different metal composition (Figure 1). Designed repeat proteins are an interesting template for the synthesis and stabilization of metal NCs owing to their modular structure22 and function in which specific ligand recognition activities can be encoded, opening the door to multiple applications in biosensing and molecular imaging.23 Specifically, the consensus tetratricopeptide repeat (CTPR) module, a 34 amino acids helix‐turn‐helix motif, was used as a study case.24 Previously, CTPR protein‐mediated synthesis of AuNCs and Au nanoparticles have been reported.21a, 25 In the present study, the capability of the designed CTPR proteins to act as templates for the green synthesis and stabilization of NCs composed of Au, Ag, or Cu is explored, by the incorporation of a designed cluster coordination site based on histidines (His) (Figure 1). The potential of these Prot‐NCs in the field of cellular imaging or labeling was shown.
Figure 1.
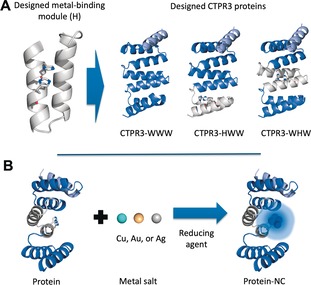
Design of CTPR proteins for the incorporation of NCs stabilization sites. A) Modified CTPR unit in which two His have been introduced at positions 5 and 9 (H module). Representation of the structure of the designed CTPR3 proteins constructed by combination of H (gray) and WT (blue) modules: WWW, HWW, and WHW, based on the structure of CTPR (PDB ID: 2HYZ). The solvating helix is shown in light blue. B) The general strategy for the synthesis and stabilization of fluorescent metal NCs by CTPR proteins.
Initially, based on the crystal structure of CTPR protein, (2HYZ),26 a metal coordination site based on two His was modeled on the protein concave surface, since His are known to coordinate noble metals, such as gold (0 and I state).20c Two His were introduced within a CTPR unit at positions 5 and 9 (H‐module; Figure 1 A), since an i, i+4 bis‐His motif on α‐helices is a high‐affinity bidentate motif frequently utilized in natural metalloproteins.27 The side chain conformations and backbone geometry of the designed His were compatible with the CTPR fold and with the metal coordination distances observed in protein coordination sites.27, 28 The potential of the designed site to coordinate metals was validated computationally using the metal ion‐binding site prediction and docking server.29 Arrays of three CTPR modules were constructed by combination of H‐modules and WT CTPR units (W‐modules), to obtain CTPR3‐WHW and CTPR3‐HWW proteins, with one H module localized at the central repeat or at the N‐terminal repeat, respectively. These proteins were compared to a CTPR3‐WWW protein without metal coordination site. The three proteins present an additional helix at the C‐terminal for improved solubility (Figure 1 A). Designed CTPR proteins were tested for the generation of Prot‐NCs (composed of Au, Ag, or Cu) using green biocompatible chemistry (Figure 1 B), to evaluate the role of the metal coordination site and its position within the protein scaffold. The proteins at 10 μm were incubated with 5 equiv of metal salts and after 30 min incubation the reduction of the metal was achieved with 10 equiv of sodium ascorbate with respect to metal ions. The Prot‐NCs were purified by gel filtration and the NCs formation was evaluated by the appearance of characteristic fluorescence features. The synthesis with CTPR3‐WWW showed no fluorescence, whereas the synthesis using CTPR3‐WHW showed the largest PL with all metals. This scaffold was selected for the optimization of synthetic parameters. PL from Prot‐NCs increased with the reaction time from 24 h to 72 h. Additional PL improvement was observed upon increasing the metal to protein molar ratio, and the reducing‐agent to metal ratio (Supporting Information, Figures S1–S3).
The spectroscopic characterization of the Prot‐NCs with different metal composition showed the expected signatures from the metal NCs.12, 15, 17, 21a, 30 The UV/Vis spectra of the Prot‐NCs showed the presence of a peak at 370 nm corresponding to the absorption of the NCs, along with the characteristic protein absorption at 280 nm (Figure 2 B). The fluorescence emission spectra of WHW‐CuNCs, WHW‐AuNCs, and WHW‐AgNCs showed single peaks with maxima at 435, 445, and 455 nm, respectively, when excited at their excitation maximum wavelengths (Figure 2 C). Thorough control experiments confirmed that the fluorescence was emitted by the NCs, stabilized by the WHW protein upon sodium ascorbate reduction, since the fluorescence emission was observed only when protein, metal, and reducing agent were present (Supporting Information, Figure S4). The quantum yields (QYs) of WHW‐CuNCs, WHW‐AuNCs, and WHW‐AgNCs were 9.5, 8.3, and 6.3 %, respectively, using anthracene as a reference. The QYs of this set of Prot‐NCs are reasonably high compared to those of other protein‐capped photoluminescent metal NCs (CuNCs 0.2–4.0 %, AuNCs 3.5–6.0 %, AgNCs 1.2–1.7 %).12, 15, 17, 21a, 30 To have a better understanding of the excited‐state dynamics of the Prot‐NCs, time‐resolved PL measurements were performed. Multi‐exponential fits of the PL decay curves (Supporting Information, Figure S5), provide 1.5, 1.5, and 1.6 ns amplitude‐weighted average lifetimes for WHW‐CuNCs, WHW‐AuNCs, and WHW‐AgNCs, respectively. It is noted that lifetimes in this range (from picoseconds to a few nanoseconds) are characteristic of transitions between singlet states in metal NCs.2, 3c, 4, 5, 6, 10d, 14, 31
Figure 2.
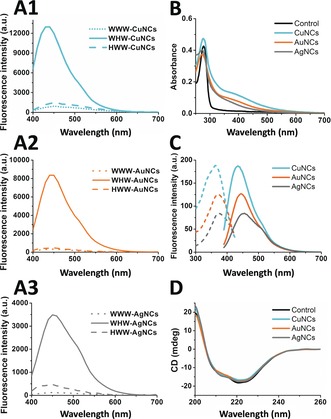
A) Fluorescence spectra of Prot‐NCs: CuNCs (A1, cyan), AuNCs (A2, orange), and AgNCs (A3, gray) synthesized with WWW control protein (••••), WHW (—), and HWW (‐ ‐ ‐ ‐) under the same experimental conditions. All spectra were acquired using a λ exc: 370 nm. B) UV/Vis spectra of WHW‐NCs: WHW control (black line), WHW‐CuNCs (cyan line), WHW‐AuNCs (orange line), and WHW‐AgNCs (gray line). C) Fluorescence excitation (‐ ‐ ‐ ‐) and emission (—) spectra of WHW‐CuNCs (cyan), WHW‐AuNCs (orange), and WHW‐AgNCs (gray) acquired using λexc=363, 371, and 376 nm for Cu, Au, and Ag NCs, respectively. D) CD spectra of WHW‐NCs: WHW protein (black line), WHW‐CuNCs (cyan line), WHW‐AuNCs (orange line), and WHW‐AgNCs (gray line).
The structural integrity of the protein template upon NCs stabilization, critical for future applications, was monitored by circular dichroism (CD). CD spectra of the Prot‐NCs showed that the protein retained α‐helical structure, indicating that the synthesis of metal NCs did not affect the structure of the scaffolds (Figure 2 D).
The valence states of these Prot‐NCs were investigated by XPS, showing that the metal cores of CuNCs, AuNCs, and AgNCs were mainly composed of Cu0, Au0/AuI, and Ag0, respectively (Figure 3 A). The XPS spectrum of CuNCs showed two prominent peaks at 932.6 and 952.5 eV, assigned to Cu 2p3/2 and Cu 2p1/2, which are characteristic Cu0 peaks. The absence of peak owing to CuII at 942 eV indicated that CuNCs did not contain CuII (Figure 3 A).32 As the binding energy of Cu0 is only 0.2 eV lower than that of CuI,2d it is not possible to fully exclude the presence of small amounts of CuI in the CuNCs, as has been previously observed.32, 33 The XPS spectrum of AuNCs showed a peak at the binding energy of 84.6 eV (Au 4f7/2), which is between the typical binding energies of Au foil (84.0 eV) and AuI‐L3 complex (86.0 eV). This suggests that both Au0 and AuI coexist in the AuNCs, as has been previously observed (Figure 3 A).34 The XPS spectrum of AgNCs showed two binding energy values at 368.2 eV for Ag 3d5/2 and 374.3 eV for Ag 3d3/2, confirming the presence of elemental Ag0 in the AgNCs, consistent with the standard spectrum for AgNCs (Figure 3 A).35 The Ag 3d5/2 peak centered at 368.2 eV lies in between the value for Ag0 (368.0 eV)36 and that of AgI salt (368.4 eV),36 which suggests the presence of AgI in AgNCs, as has been previously observed.36, 37
Figure 3.
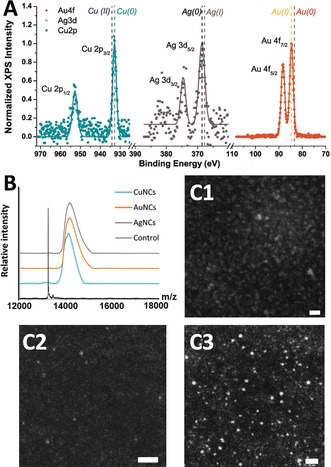
Characterization of Protein‐NCs complexes. A) XPS spectra of WHW‐CuNCs (left), WHW‐AgNCs (center), and WHW‐AuNCs (right). B. MALDI‐TOF mass spectra of WHW control protein (black) WHW‐CuNCs (cyan), WHW‐AuNCs (orange), and WHW‐AgNCs (gray). C) HAADF STEM images of the WHW‐CuNCs (C1), WHW‐AuNCs (C2), and WHW‐AgNCs (C3). Scale bar: 10 nm.
MALDI‐TOF, LC‐ESI‐TOF, and ICP‐MS mass spectrometry were used to determine the number of metal atoms in the Prot‐NCs. The MALDI‐TOF and LC‐ESI‐TOF mass spectra of the Prot‐NCs showed broad peaks with a clear shift with respect to the protein, which confirmed the coordination of metals by the protein and indicated that the metal NCs present certain polydispersity in size (Figure 3 B; Supporting Information, Table S1, Figure S6). An average of 14±7 copper atoms, 5±2 gold atoms, and 9±4 silver atoms per protein were calculated for the Prot‐NCs from MALDI spectra, in agreement with the size range obtained by LC‐ESI‐TOF (Supporting Information, Table S1). Accordingly, ICP‐MS analysis of Cu, Au, and Ag from the digested Prot‐NCs showed 15±1 Cu atoms per protein in the WHW‐CuNCs, 5±1 Au atoms per protein in the WHW‐AuNCs and 9±1 Ag atoms per protein in the WHW‐AgNCs (Supporting Information, Table S2). These results are consistent with the size range of previously reported blue‐PL Prot‐NCs.17, 31, 32, 38 Additionally, SDS‐PAGE gel electrophoresis of the Prot‐NCs showed the PL signal of metal NCs at the molecular weight that of the protein monomer. The fact that the NCs retained the PL even after gel electrophoresis, demonstrates the remarkable stability of the NC coordination (Supporting Information, Figure S7). The size of the Prot‐NCs were evaluated by scanning transmission electron microscopy (STEM), taking advantage of the chemical contrast of Cu, Au, and Ag obtained in high‐angle annular dark field (HAADF). Indeed, metal NCs with diameters of 1.2±0.4, 1.0±0.3, and 1.1±0.5 nm were detected for WHW‐CuNCs, WHW‐AuNCs, and WHW‐AgNCs, respectively (Figure 3 C; Supporting Information, Figure S8). These size ranges are consistent with the size range of previously reported blue‐PL metal NCs.17, 31, 33b, 39 However, it should be noted that TEM images could show larger metal NCs than those obtained from the MS analysis, as the larger metal NCs more clearly observed by TEM are more difficult to ionize and cannot be detected by the MS,33a, 40 and the NCs can aggregate on carbon grids.32, 41
For bioimaging and biolabeling applications, the Prot‐NCs need to have good storage and work stabilities, good photostability and biocompatibility. The Prot‐NCs are stable over a month under storage conditions (PBS at 4 °C; Supporting Information, Figure S9), and over seven days under physiological conditions (PBS and human plasma‐HP at 37 °C; Supporting Information, Figure S10). In HP the fluorescence intensity showed a slight increase, which has been previously reported in blood serum and related to the reducing the nature of the environment in the serum.32 Furthermore, the Prot‐NCs are stable under a temperature range from 20 °C to 70 °C, a broad pH range of pH 5–12, an ionic strength range of 0.15–1.0 m of NaCl, to biothiol concentration up to 1.0 mm of Cys, and in the presence of different metal ions (50–150 μm; Supporting Information, Figures S11–S15). Additionally, the PL of the Prot‐NCs remained nearly constant (85–90 %) after 10 min under continuous irradiation (Supporting Information, Figure S16). In comparison, the PL of an organic fluorophore (DAPI) and a fluorescent protein (GFP) decreased to a greater extent (60 % and 35 %, respectively). This excellent resistance to photobleaching of Prot‐NCs, makes them suitable for their use in imaging applications.
The performance of the Prot‐NCs as PL probes in cell cultures was evaluated on account of their biocompatibility and notable fluorescence properties. MDA‐MB‐231 breast cancer cells were incubated with different concentrations of Prot‐NCs (1–20 μm) under standard cell culture conditions. Following 5 days of incubation, none of the Prot‐NCs presented any cytotoxic effect even at the highest concentration tested (Supporting Information, Figure S17). To test the suitability of these Prot‐NCs for cell labeling and bioimaging, MDA‐MB‐231 cells were incubated with Prot‐NCs for 16 h in DMEM at 37 °C and then incubated with nuclear marker (NucRed Live 647) for another 30 min. Finally, the cells were imaged by live confocal fluorescence microscopy (Supporting Information, Figure S18 A–D). All of the cells treated with Prot‐NCs exhibited blue fluorescence (Supporting Information, Figures S18 B–D), compared to non‐treated cells which did not show any fluorescence (Supporting Information, Figure S18 A). To better understand the internalization of the Prot‐NCs, live confocal fluorescence microscopy studies incubating MDA‐MB‐231 cells with fluorescein‐labeled WHW‐CuNCs, and fluorescein‐labeled free WHW protein were performed. Figure 4 shows the internalization of the Prot‐NCs and the co‐localization of fluorescence signal of both protein and NCs. Z‐stack images of the complete cell volume further confirmed the Prot‐NC internalization into the cells, and the observed dotted fluorescence signal may indicate endosomal uptake mechanism (Supporting Information, Figures S19, S20). These results clearly demonstrate the efficacy of Prot‐NCs to label live cells. Taking into account that the plasma membrane is generally impervious to proteins42 and, specifically, that the fluorescein‐labeled protein alone cannot be internalized by cells (Figure 4 A), as previously shown for a similar CTPR variant,43 these results demonstrate that the internalization of CTPR‐metal NCs is promoted by the presence of the metal NCs coordinated to the CTPR structure. This work reveals the dual activity of the NCs as transfection and labeling agent.
Figure 4.
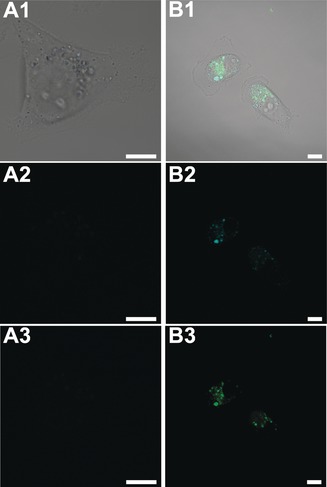
Live confocal fluorescence microscopy images of MDA‐MB‐231 breast cancer cells incubated with A) fluorescein‐labeled WHW protein and B) fluorescein‐labeled WHW‐CuNCs. 1. Merged image of DIC, blue and green channel; 2. Blue channel (λexc: 405 nm); 3. Green channel (λexc: 488 nm). Scale bars: 10 μm.
In summary, a simple versatile approach to design proteins for the sustainable synthesis and stabilization of highly fluorescent metal NCs has been developed by the incorporation of specific metal binding sites. A strategically positioned pair of His at i, i+4 positions on an α‐helix of a repeat protein scaffold was able to stabilize Cu, Ag, and Au NCs, showing the versatility of the approach. The local environment of the metal coordination site affected the ability of cluster formation and an optimal cluster‐stabilizing unit has been defined. The resulting Prot‐NCs are produced through simple reduction of inorganic metal salts and have excellent fluorescent properties, photostability, storage‐stability, and biocompatibility. Furthermore, the Prot‐NCs are able to enter into living cells without any permeabilization treatment, damage to the cells, or effect on their fluorescent properties, making them useful tools for live cell imaging and labeling. This versatile method can be used as a general approach to incorporate specific metal binding sites into proteins to obtain functional Prot‐NCs complexes.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors gratefully acknowledge support from the European Research Council (Grant ProNANO ERC‐CoG‐648071, A.L.C.), from the Ministerio de Economía y Competitividad (BIO2016‐77367‐C2‐1‐R, A.L.C.), (MAT2014‐57652‐C2‐1‐R LAPSEN and PCIN‐2015‐169‐C02‐01 MOFSENS, J.C.‐G.), and the Basque Government (Elkartek KK‐2017/00008, A.L.C). We thank Dr. L. Yate, Dr. J. Calvo, and Dr. D. Otaegui at CIC biomaGUNE for support with XPS and mass spectrometry measurements. We thank Dr. M. Liutkus for careful reading and insightful comments on the manuscript. This work was performed under the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency—Grant No. MDM‐2017‐0720. IMDEA Nanociencia acknowledges support from the “Severo Ochoa” Programme for Centres of Excellence in R&D (MINECO, Grant SEV‐2016‐0686).
A. Aires, I. Llarena, M. Moller, J. Castro-Smirnov, J. Cabanillas-Gonzalez, A. L. Cortajarena, Angew. Chem. Int. Ed. 2019, 58, 6214.
References
- 1.
- 1a. Sau T. K., Rogach A. L., Adv. Mater. 2010, 22, 1781–1804; [DOI] [PubMed] [Google Scholar]
- 1b. Cobley C. M., Chen J., Cho E. C., Wang L. V., Xia Y., Chem. Soc. Rev. 2011, 40, 44–56; [DOI] [PubMed] [Google Scholar]
- 1c. Guo S., Wang E., Nano Today 2011, 6, 240–264. [Google Scholar]
- 2.
- 2a. Zheng J., Zhang C., Dickson R. M., Phys. Rev. Lett. 2004, 93, 077402; [DOI] [PubMed] [Google Scholar]
- 2b. Huang C. C., Yang Z., Lee K. H., Chang H. T., Angew. Chem. Int. Ed. 2007, 46, 6824–6828; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 6948–6952; [Google Scholar]
- 2c. Zheng J., Dickson R. M., J. Am. Chem. Soc. 2002, 124, 13982–13983; [DOI] [PubMed] [Google Scholar]
- 2d. Wei W., Lu Y., Chen W., Chen S., J. Am. Chem. Soc. 2011, 133, 2060–2063; [DOI] [PubMed] [Google Scholar]
- 2e. Tanaka S., Miyazaki J., Tiwari D. K., Jin T., Inouye Y., Angew. Chem. Int. Ed. 2011, 50, 431–435; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 451–455. [Google Scholar]
- 3.
- 3a. Chen S., Ingram R. S., Hostetler M. J., Pietron J. J., Murray R. W., Schaaff T. G., Khoury J. T., Alvarez M. M., Whetten R. L., Science 1998, 280, 2098–2101; [DOI] [PubMed] [Google Scholar]
- 3b. Peyser L. A., Vinson A. E., Bartko A. P., Dickson R. M., Science 2001, 291, 103–106; [DOI] [PubMed] [Google Scholar]
- 3c. Zheng J., Nicovich P. R., Dickson R. M., Annu. Rev. Phys. Chem. 2007, 58, 409–431; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Jin R., Zhu Y., Qian H., Chem. Eur. J. 2011, 17, 6584–6593; [DOI] [PubMed] [Google Scholar]
- 3e. Shang L., Dong S., Nienhaus G. U., Nano Today 2011, 6, 401–418; [Google Scholar]
- 3f. Lu Y., Chen W., Chem. Soc. Rev. 2012, 41, 3594–3623; [DOI] [PubMed] [Google Scholar]
- 3g. Diez I., Ras R. H., Nanoscale 2011, 3, 1963–1970; [DOI] [PubMed] [Google Scholar]
- 3h. Díez I., Ras R., Advanced Fluorescence Reporters in Chemistry and Biology II, Springer, Berlin, 2010. [Google Scholar]
- 4.
- 4a. Herzing A. A., Kiely C. J., Carley A. F., Landon P., Hutchings G. J., Science 2008, 321, 1331–1335; [DOI] [PubMed] [Google Scholar]
- 4b. Turner M., Golovko V. B., Vaughan O. P., Abdulkin P., Berenguer-Murcia A., Tikhov M. S., Johnson B. F., Lambert R. M., Nature 2008, 454, 981–983; [DOI] [PubMed] [Google Scholar]
- 4c. Muhammed M. A., Verma P. K., Pal S. K., Kumar R. C., Paul S., Omkumar R. V., Pradeep T., Chem. Eur. J. 2009, 15, 10110–10120; [DOI] [PubMed] [Google Scholar]
- 4d. Xie J., Zheng Y., Ying J. Y., Chem. Commun. 2010, 46, 961–963; [DOI] [PubMed] [Google Scholar]
- 4e. Li J., Zhong X., Cheng F., Zhang J. R., Jiang L. P., Zhu J. J., Anal. Chem. 2012, 84, 4140–4146; [DOI] [PubMed] [Google Scholar]
- 4f. Hussain A. M., Sarangi S. N., Kesarwani J. A., Sahu S. N., Biosens. Bioelectron. 2011, 29, 60–65. [DOI] [PubMed] [Google Scholar]
- 5. Varnavski O., Ispasoiu R. G., Balogh L., Tomalia D., Goodson T., J. Chem. Phys. 2001, 114, 1962–1965. [Google Scholar]
- 6. Bao Y., Yeh H.-C., Zhong C., Ivanov S. A., Sharma J. K., Neidig M. L., Vu D. M., Shreve A. P., Dyer R. B., Werner J. H., Martinez J. S., J. Phys. Chem. C 2010, 114, 15879–15882. [Google Scholar]
- 7. Liu G., Shao Y., Wu F., Xu S., Peng J., Liu L., Nanotechnology 2013, 24, 015503. [DOI] [PubMed] [Google Scholar]
- 8. Slocik J. M., Moore J. T., Wright D. W., Nano Lett. 2002, 2, 169–173. [Google Scholar]
- 9. Chevrier D. M., Chatt A., Zhang P., J. Nanophotonics 2012, 6, 064504. [Google Scholar]
- 10.
- 10a. Cui Y., Wang Y., Liu R., Sun Z., Wei Y., Zhao Y., Gao X., ACS Nano 2011, 5, 8684–8689; [DOI] [PubMed] [Google Scholar]
- 10b. Gregersen S., Vosch T., Jensen K. J., Chem. Eur. J. 2016, 22, 18492–18500; [DOI] [PubMed] [Google Scholar]
- 10c. Zaytsev D. V., Morozov V. A., Fan J., Zhu X., Mukherjee M., Ni S., Kennedy M. A., Ogawa M. Y., J. Inorg. Biochem. 2013, 119, 1–9; [DOI] [PubMed] [Google Scholar]
- 10d. Morozov V. A., Ogawa M. Y., Inorg. Chem. 2013, 52, 9166–9168. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Clem Gruen L., Biochim. Biophys. Acta Protein Struct. 1975, 386, 270–274; [DOI] [PubMed] [Google Scholar]
- 11b. Mandal S., Phadtare S., Sastry M., Curr. Appl. Phys. 2005, 5, 118–127. [Google Scholar]
- 12. Xie J., Zheng Y., Ying J. Y., J. Am. Chem. Soc. 2009, 131, 888–889. [DOI] [PubMed] [Google Scholar]
- 13. Chen Y., Wang Y., Wang C., Li W., Zhou H., Jiao H., Lin Q., Yu C., J. Colloid Interface Sci. 2013, 396, 63–68. [DOI] [PubMed] [Google Scholar]
- 14. Le Guével X., Daum N., Schneider M., Nanotechnology 2011, 22, 275103. [DOI] [PubMed] [Google Scholar]
- 15. Wei H., Wang Z., Yang L., Tian S., Hou C., Lu Y., Analyst 2010, 135, 1406–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu J. M., Chen J. T., Yan X. P., Anal. Chem. 2013, 85, 3238–3245. [DOI] [PubMed] [Google Scholar]
- 17. Kawasaki H., Hamaguchi K., Osaka I., Arakawa R., Adv. Funct. Mater. 2011, 21, 3508–3515. [Google Scholar]
- 18. Liu C. L., Wu H. T., Hsiao Y. H., Lai C. W., Shih C. W., Peng Y. K., Tang K. C., Chang H. W., Chien Y. C., Hsiao J. K., Cheng J. T., Chou P. T., Angew. Chem. Int. Ed. 2011, 50, 7056–7060; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7194–7198. [Google Scholar]
- 19. Wen F., Dong Y., Feng L., Wang S., Zhang S., Zhang X., Anal. Chem. 2011, 83, 1193–1196. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Liu L., Jiang H., Wang X., Nano Res. 2018, 11, 311–322; [Google Scholar]
- 20b. Xu Y., Sherwood J., Qin Y., Crowley D., Bonizzoni M., Bao Y., Nanoscale 2014, 6, 1515–1524; [DOI] [PubMed] [Google Scholar]
- 20c. Maity B., Abe S., Ueno T., Nat. Commun. 2017, 8, 14820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Couleaud P., Adan-Bermudez S., Aires A., Mejías S. H., Sot B., Somoza A., Cortajarena A. L., Biomacromolecules 2015, 16, 3836–3844; [DOI] [PubMed] [Google Scholar]
- 21b. Chall S., Mati S. S., Das I., Kundu A., De G., Chattopadhyay K., Langmuir 2017, 33, 12120–12129. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Grove T. Z., Regan L., Cortajarena A. L., J. R. Soc. Interface 2013, 10, 20130051; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b. Mejías S. H., Sot B., Guantes R., Cortajarena A. L., Nanoscale 2014, 6, 10982–10988; [DOI] [PubMed] [Google Scholar]
- 22c. Kajava A. V., J. Struct. Biol. 2001, 134, 132–144. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Jackrel M. E., Cortajarena A. L., Liu T. Y., Regan L., ACS Chem. Biol. 2010, 5, 553–562; [DOI] [PubMed] [Google Scholar]
- 23b. Cortajarena A. L., Liu T. Y., Hochstrasser M., Regan L., ACS Chem. Biol. 2010, 5, 545–552; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23c. Cortajarena A. L., Kajander T., Pan W., Cocco M. J., Regan L., Protein Eng. Des. Sel. 2004, 17, 399–409. [DOI] [PubMed] [Google Scholar]
- 24. D'Andrea L. D., Regan L., Trends Biochem. Sci. 2003, 28, 655–662. [DOI] [PubMed] [Google Scholar]
- 25. Geng X., Grove T. Z., RSC Adv. 2015, 5, 2062–2069. [Google Scholar]
- 26.
- 26a. Kajander T., Cortajarena A. L., Mochrie S., Regan L., Acta Crystallogr. Sect. D 2007, 67, 800–811; [DOI] [PubMed] [Google Scholar]
- 26b. Main E. R., Xiong Y., Cocco M. J., D'Andrea L., Regan L., Structure 2003, 11, 497–508. [DOI] [PubMed] [Google Scholar]
- 27. Arnold F. H., Haymore B. L., Science 1991, 252, 1796–1797. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Kumarevel T., Nucleic Acids Res. 2005, 33, 5494–5502; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. Chakrabarti P., Protein Eng. 1990, 4, 57–63; [DOI] [PubMed] [Google Scholar]
- 28c. Carrera F., Marcos E. S., Merkling P. J., Chaboy J., Munoz-Paez A., Inorg. Chem. 2004, 43, 6674–6683. [DOI] [PubMed] [Google Scholar]
- 29. Lin Y.-F., Cheng C.-W., Shih C.-S., Hwang J.-K., Yu C.-S., Lu C.-H., J. Chem. Inf. Model. 2016, 56, 2287–2291: [DOI] [PubMed] [Google Scholar]; http://bioinfo.cmu.edu.tw/MIB/.
- 30.
- 30a. Wang C., Wang C., Xu L., Cheng H., Lin Q., Zhang C., Nanoscale 2014, 6, 1775–1781; [DOI] [PubMed] [Google Scholar]
- 30b. Goswami N., Giri A., Bootharaju M. S., Xavier P. L., Pradeep T., Pal S. K., Anal. Chem. 2011, 83, 9676–9680. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Sahu D. K., Sahu K., J. Photochem. Photobiol. A 2017, 347, 17–25; [Google Scholar]
- 31b. Anand U., Ghosh S., Mukherjee S., J. Phys. Chem. Lett. 2012, 3, 3605–3609. [DOI] [PubMed] [Google Scholar]
- 32. Ghosh R., Sahoo A. K., Ghosh S. S., Paul A., Chattopadhyay A., ACS Appl. Mater. Interfaces 2014, 6, 3822–3828. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a. Wang C., Ling L., Yao Y., Song Q., Nano Res. 2015, 8, 1975–1986; [Google Scholar]
- 33b. Feng J., Chen Y., Han Y., Liu J., Ma S., Zhang H., Chen X., ACS Omega 2017, 2, 9109–9117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang L., Chen J., Huang T., Huang L., Sun Z., Jiang Y., Yao T., Wei S., J. Mater. Chem. C 2017, 5, 4448–4454. [Google Scholar]
- 35. Yang X., Gan L., Han L., Wang E., Wang J., Angew. Chem. Int. Ed. 2013, 52, 2022–2026; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2076–2080. [Google Scholar]
- 36. Volkov I. L., Smirnova A., Makarova A. A., Reveguk Z. V., Ramazanov R. R., Usachov D. Y., Adamchuk V. K., Kononov A. I., J. Phys. Chem. B 2017, 121, 2400–2406. [DOI] [PubMed] [Google Scholar]
- 37. Petty J. T., Sergev O. O., Ganguly M., Rankine I. J., Chevrier D. M., Zhang P., J. Am. Chem. Soc. 2016, 138, 3469–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.
- 38a. Yuan X., Setyawati M. I., Tan A. S., Ong C. N., Leong D. T., Xie J., NPG Asia Mater. 2013, 5, e39; [Google Scholar]
- 38b. Yu Y., Luo Z., Teo C. S., Tan Y. N., Xie J., Chem. Commun. 2013, 49, 9740. [DOI] [PubMed] [Google Scholar]
- 39.
- 39a. Ding H., Li H., Wang X., Zhou Y., Li Z., Hiltunen J. K., Shen J., Chen Z., Chem. Mater. 2017, 29, 8440–8448; [Google Scholar]
- 39b. Le Guével X., Spies C., Daum N., Jung G., Schneider M., Nano Res. 2012, 5, 379–387. [Google Scholar]
- 40. Jia X., Li J., Wang E., Small 2013, 9, 3873–3879. [DOI] [PubMed] [Google Scholar]
- 41. Lin Y.-J., Chen P.-C., Yuan Z., Ma J.-Y., Chang H.-T., Chem. Commun. 2015, 51, 11983–11986. [DOI] [PubMed] [Google Scholar]
- 42. Leifert J. A., Whitton J. L., Mol. Ther. 2003, 8, 13–20. [DOI] [PubMed] [Google Scholar]
- 43. Cortajarena A. L., Yi F., Regan L., ACS Chem. Biol. 2008, 3, 161–166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary