

Abstract
Standard therapy for advanced Prostate Cancer (PCa) consists of antiandrogens, which provide respite from disease progression, but ultimately fail resulting in the incurable phase of the disease: mCRPC. Targeting PCa cells before their progression to mCRPC would greatly improve the outcome. Combination therapy targeting the DNA Damage Response (DDR) has been limited by general toxicity, and a goal of clinical trials is how to target the DDR more specifically. We now show that androgen deprivation therapy (ADT) of LNCaP cells results in increased expression of TLK1B, a key kinase upstream of NEK1 and ATR and mediating the DDR that typically results in a temporary cell cycle arrest of androgen responsive PCa cells. Following DNA damage, addition of the TLK specific inhibitor, thioridazine (THD), impairs ATR and Chk1 activation, establishing the existence of a ADT > TLK1 > NEK1 > ATR > Chk1, DDR pathway, while its abrogation leads to apoptosis. Treatment with THD suppressed the outgrowth of androgen‐independent (AI) colonies of LNCaP and TRAMP‐C2 cells cultured with bicalutamide. Moreover, THD significantly inhibited the growth of several PCa cells in vitro (including AI lines). Administration of THD or bicalutamide was not effective at inhibiting long‐term tumor growth of LNCaP xenografts. In contrast, combination therapy remarkably inhibited tumor growth via bypass of the DDR. Moreover, xenografts of LNCaP cells overexpressing a NEK1‐T141A mutant were durably suppressed with bicalutamide. Collectively, these results suggest that targeting the TLK1/NEK1 axis might be a novel therapy for PCa in combination with standard of care (ADT).
Short abstract
What's new?
Standard therapy for advanced Prostate Cancer (PCa) consists of anti‐androgens, which only provide temporary respite from disease progression to metastatic castrate‐resistant prostate cancer (mCRPC). Here, the authors show in the LNCaP cell model that the increased expression with ADT of TLK1B, a prosurvival checkpoint pathway that is enacted before conversion to androgen‐independent growth, offers a unique target for attacking more specifically PCa cells before their conversion to CRPC. Moreover, they suggest to re‐purpose thioridazine or other phenothiazine antipsychotic drugs as inhibitors of the TLK1 > Nek1 > ATR > Chk1 DNA Damage Response (DDR) axis for the early treatment of advanced PCa still responsive to ADT.
Introduction
Prostate cancer (PCa) is diagnosed in over 200,000 men in the US each year and results in approximately 25,000 deaths. The mainstay therapy for patients with locally advanced prostate cancer, metastatic prostate cancer and biochemically recurrent disease after failure of localized treatments, is androgen‐deprivation therapy (ADT) with gonadropin‐releasing hormone analogs and antiandrogens. ADT is known to provide remission of the disease, best evidenced by a decline of prostate‐specific antigen (PSA) in about 90% of patients. After a mean time of 2–3 years, however, the disease progresses despite continuous hormonal manipulation. This type of cancer is known as metastatic castrate‐resistant prostate cancer (mCRPC). mCRPC is associated with a poor prognosis and mean survival time of only 16–18 months.1 Thus, the best window of opportunity is before development of mCRPC, and this requires a clear understanding of the process of PCa cells’ mechanisms of adaptation to ADT. The best characterized model so far for studying this is the LNCaP. Androgen deprivation of LNCaP cells results in loss of AR function with a compensatory prosurvival activation of mTOR2 and concomitant implementation of a cell division arrest by activation of the DNA Damage Response (DDR) mediated by ATR‐Chk13 or ATM‐Chk2.4 However, it is not well understood what signals the activation of the DDR and ATR (rev. in Ref. [5]). Then, after a quiescent period of ADT adaptation of 2–3 weeks, androgen independent (AI) colonies begin to form.6 An attractive strategy to prevent this process would be to bypass the cell cycle arrest via inhibition of ATM or ATR, causing the cells to undertake replication with damaged DNA that would cause mitotic catastrophe, a strategy that was in fact implemented in LNCaP treated concomitantly with bicalutamide and ATM inhibition.4 But a limitation of this approach is how to make the inhibition of ATM or ATR specific to PCa cells.
The Tousled Like kinases (TLK1 and TLK2) function in several processes including chromatin assembly, replication, transcription, DNA repair, and chromosome segregation (rev. in7). We recently uncovered the proteome target of TLK1 and identified the NIMA‐related kinase, NEK1, as a major substrate,8 a key kinase which in turn activates ATR and the DDR.9, 10 Inhibition of the TLK1/NEK1 axis with thioridazine (THD), a rather specific inhibitor of TLKs,11 suppresses the DDR. THD was previously shown to improve therapeutic response in combination with DNA damaging agents.11, 12, 13, 14, 15 Furthermore, THD was shown to cause growth inhibition of ovarian cancer cells via suppression of AKT activity16 by an unidentified mechanism downstream of PI3K and mTOR, but which we now propose could be the result of inhibition of TLK1 and another of its targets, AKTIP, which is a direct activator of AKT. What is noteworthy is that the concomitant inhibition of TLK1 along with ADT (the current standard of care for advanced PCa) can result in a specific killing of Androgen Responsive PCa cells, instead of the more generalized, and thus more prone to side effects, inhibition of ATR or ATM. A similar strategy targeting a specific AR/CDC6‐ATR‐Chk1 signaling pathway has been used in a recent preclinical model,17 but in that case, androgen deprivation resulted in reduction of CDC6 expression (a AR target gene), whereas in our work we show that ADT results in increased expression of TLK1B, a positive prosurvival mechanism, leading to activation of NEK1 and the DDR, that can be optimally targeted in PCa. Indeed, treatment with THD bypasses cell cycle arrest elicited by ADT and causes unencumbered replication‐induced DNA damage, leading to apoptosis in vitro and in mouse xenografts models.
Materials and Methods
Cell lines
All PCa cell lines were obtained from the ATCC, recently authenticated, and cultured according to their guidelines. LNCaP cells stably overexpressing Nek1 (wt and mut) were generated as previously described.18
Animal studies
All animals used in this study received humane care based on guidelines set by the American Veterinary, and approved by the Institutional Animal Care and Use Committee of LSU Health Sciences Center at Shreveport. Male NOD‐SCID (Taconic) mice (5 per group) were used. For xenografts, 1 × 106 LNCaP cells were resuspended in 100 μL of matrigel solution (Corning Matrigel Basement Membrane Matrix High Concentration, Catalog Number 354248) and injected in 1‐month‐old mice subcutaneously into the both flanks. After appearance of palpable tumors (∼2 weeks) the mice were divided into the 4 study groups making sure that the tumors match in size at the start of the study. Tumor size and body weight were measured twice weekly and tumor volume were calculated by the formula V = (length × Widtĥ2)/2. Tumor volumes and weights were statistically analyzed with ANOVA. Mice were treated with THD (dissolved in sterile water—2.5 mg/mL) at dosages similar to those reported previously in noncancer mouse studies every other day by ip injection or oral gavage—0.1 mL). Bicalutamide was dissolved in DMSO and diluted in corn oil and administered at a dose of 100 mg kg−1 by intraperitoneal injection twice a week.
Immunohistochemistry (IHC) of tumors
Sectioning and processing of the tissues were carried out in the FWCC Histology Service, using automated processes and equipment to provide uniform and standardized results. Indirect labeling was with ABC Elite: RTU Vectastain Elite Reagent, Vector #PK‐7100; DAB: ImmPact DAB, Vector #SK‐4105. Light counterstaining was with hematoxylin. Representative IHC images were taken at 40× magnification with the help of Olympus BX51 microscope. Positive staining were quantified by NIH ImageJ software with an average of three fields. Two‐way ANOVA analysis was carried to compare the intensity of staining,
Results
Androgen deprivation results in translational increase in TLK1B expression
Our lab identified a splice variant of TLK1 that encodes a 65 kDa protein (named TLK1B) that becomes translationally upregulated in response to genotoxic stress via the mTOR‐4EBP1 pathway.19 TLK1B is similar to TLK1 but lacks the first 169 amino acids. TLK1 and TLK1B share the same catalytic domain and we sometime refer them to as TLK1/1B. TLK1B overexpression protects cells from the genotoxic effects of IR.20 We now show that androgen deprivation in LNCaP cells results in a rapid mTOR‐dependent, rapamycin suppressible, strong increase in translation of TLK1B, whereas its mRNA does not change (Figs. 1 a and 1b and Supporting Information Fig. S1A). Consistent with this mechanism, the phosphorylation of 4EBP1 increases in cells incubated in charcoal‐stripped‐serum (CSS) and this is suppressible with rapamycin (Supporting Information Fig. S1 b). Notably, the TLK1 gene was identified by co‐expression analysis using WGCNA as a key driver of PCa, highly enriched among candidate genes collected from expression Quantitative Trait Loci (eQTL), somatic copy number alterations (SCNA), and prognostic analyses.21 But since expression of TLK1B is largely controlled at the post‐transcriptional level it may have been largely underestimated in transcriptomics analyses of PCa. Recently we have identified the proteome complement of TLK1/1B, and have identified the protein kinase Nek1 as one of its principal targets.18 TLK1/1B activates Nek1, by phosphorylating T141, and we established that this mechanism is an early DDR mediator upstream of ATR and Chk1 upon oxidative damage or replication arrest,18 extending earlier studies on Nek1.10 It seemed possible that a similar pathway becomes activated upon AR inhibition by ADT, leading to a prosurvival cell cycle arrest.
Figure 1.
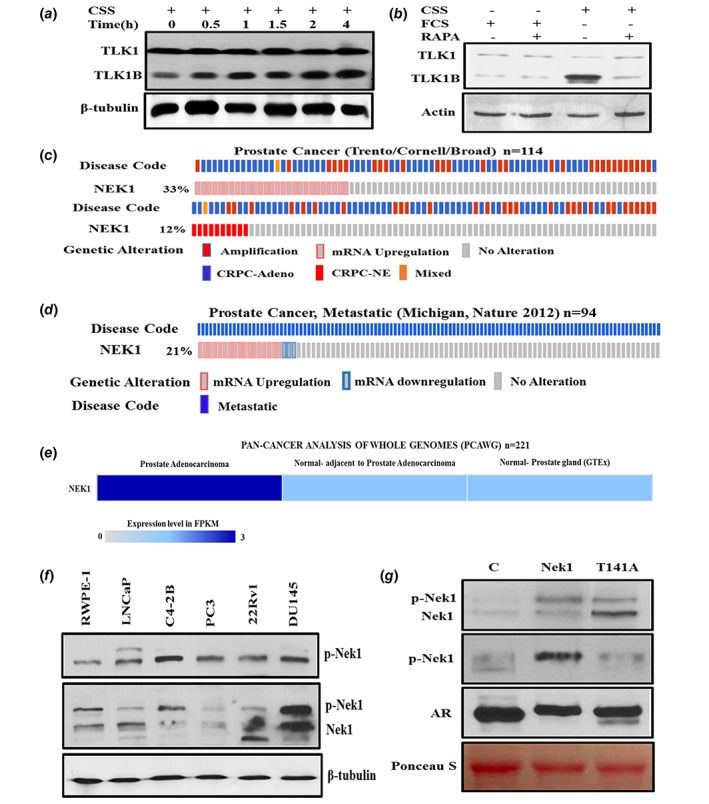
(a) Androgen Deprivation results in time‐dependent increase in TLK1B expression correlated with 4EBP1 phosphorylation. LNCaP Cells were grown in medium containing FCS (t = 0) then transferred to CSS during a 4 h time‐course. (b) The increased expression of TLK1B after ADT is mTOR dependent and suppressible with rapamycin. LNCaP cells were shifted to CSS for 17 h with and without rapamycin (20 nM) to inhibit mTOR, and cell extracts were prepared and probed with TLK1 antiserum. (c)–(e) Nek1 expression and Gene Amplification in PCa samples analyzed as described in Ref. 38. (c) Nek1 mRNA is upregulated and amplified in patients in CRPC‐Neuroendocrine Prostate Cancer dataset. (d) Nek1 mRNA is upregulated in patients with Metastatic Prostate Cancer. (e) Nek1 is upregulated in PCa patients in PAN‐CANCER ANALYSIS OF WHOLE GENOME Dataset. (f)–(g) Expression and phosphorylation of Nek1 in a panel of PCa lines and in LNCaP cells overexpressing Nek1 (wt) and T141A mutant. (f) Top panel probed with anti‐pNek1 (T141) antiserum. Middle panel was probed with pan‐Nek1 antiserum. (g) WB of Nek1. Top panel probed with pan‐Nek1 antiserum. Second panel was probed with anti‐pNek1 (T141) antiserum. Third panel was probed for the Androgen receptor (AR). [Color figure can be viewed at wileyonlinelibrary.com]
Nek1 expression in PCa and its TLK1‐dependent phosphorylation/activation
The involvement of Nek1 in PCa development or progression is unknown, but interestingly C‐Bioportal analysis of Nek1 revealed upregulation in 33% of patients and gene amplification in 12% of patients with CRPC‐Neuroendocrine Prostate Cancer (NEPC, a type of AR negative CRPC). Moreover, Nek1 mRNA is upregulated in 21% of patients with metastatic PCa. Finally, Nek1 is upregulated in PCa patients in PAN‐CANCER analysis of whole genome Dataset (Figs. 1 c–1e). It is tempting to speculate that Nek1 may be involved is some mechanism(s) leading to survival of PCa cells particularly at the stage of mCRPC and importantly in AR‐negative NEPC. We thus analyzed the Nek1 protein expression and its phosphorylation status (pT141‐ the site that is phosphorylated by TLK1) with a custom‐made antibody (Pierce) in a panel of human PCa lines (Fig. 1 f). This analysis revealed that Nek1 is ubiquitously expressed and is phosphorylated at T141 in all cells, including PC3 that do not express TLK122 although PC3 express TLK2, therefore suggesting that TLK2 can replace TLK1 in phosphorylating Nek1‐T141. The specificity of the phospho‐antibody (pAb) is shown in Figure 1 g, in which we have analyzed two LNCaP derivatives overexpressing wt‐Nek1 or a T141A mutant. While overexpression of wt‐Nek1 resulted in a strong signal with the pAb, overexpression of the T141A gave a weak signal, likely attributable to the endogenous Nek1. Parallel probing for the AR and Ponseau S staining of the blots confirmed similar protein loading. Our main hypothesis is that ADT of LNCaP cells (with CSS or bicalutamide) should result in increased expression of TLK1B, and in turn in increased level of phosphorylated Nek1. Moreover, the specific phosphorylation of T141 by TLK1B should be suppressible with THD, a specific inhibitor of TLKs. These expectation were confirmed in Figure 2 a. Moreover, treatment with THD of LNCaP overexpressing wt‐Nek1 resulted in greatly reduced signal of pNek1 compared to bicalutamide‐treated cells (Fig. 2 b), whereas in cells overexpressing the T141A mutant, only the overexpressed unphosphorylated form of Nek1 was detected (note that this blot was probed with crude antiserum that reacts also with unphosphorylated Nek1, or the T141A mutant).
Figure 2.
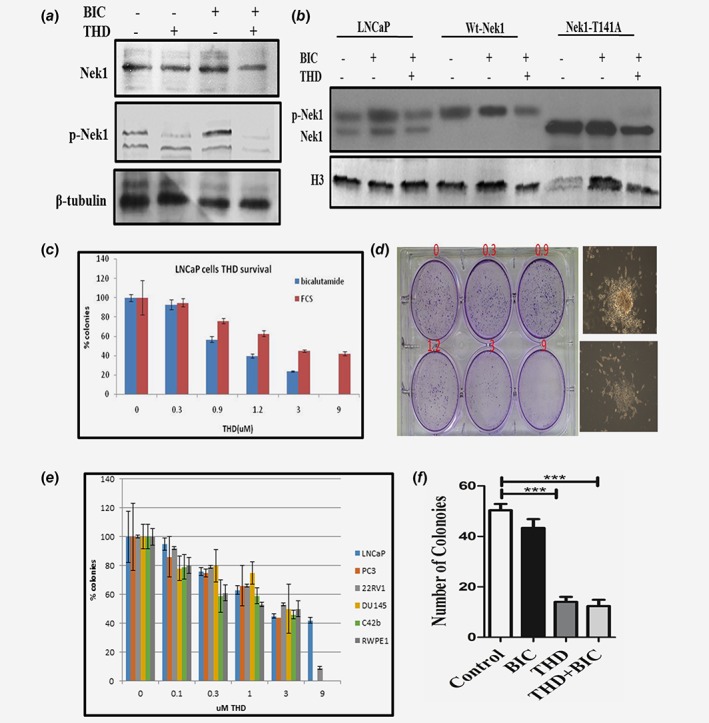
(a)–(b) Phosphorylation of Nek1 in parental LNCaP cells and overexpressing Nek1 (wt) and T141A mutant after incubation with bicalutamide +/− THD. The WB in panel b was probed with the pNek1 antiserum before affinity purification, so that it also reacts with the unphosphorylated Nek1, although more weakly. (c)–(d) AI colonies do not form with THD (concentration dependent). LNCap cells were plated in duplicates in 6‐well plates at 4000 cells with bicalutamide (10 nM) to score developing AI colonies, or 400 cells in control medium (FCS) to monitor for general clonogenic inhibition by THD. (d) An example of crystal violet‐stained plate from which the quantitation in panel C was derived. Here the AI colonies incubated with increasing concentrations of THD were scored after 3 weeks. After 1 month, a typical AI colony is shown in d (right top) without THD, or with addition of 3 μM THD (right bottom). (e) Percent of colonies treated with different conc. of THD in different PCa cell lines with increasing concentrations of THD. F. TRAMP‐C2 cells do not form AI colonies when cultured with THD. Clonogenic assays of TRAMP‐C2 cells untreated (control) or treated with bicalutamide (10 μM), THD (5 μM), or combination are displayed. [Color figure can be viewed at wileyonlinelibrary.com]
Treatment of LNCaP cells with ADT + THD prevents formation of AI colonies
Our hypothesis is that the activation of the DDR and cell cycle checkpoint via the TLK1/Nek1 axis,18 in this case in response to ADT in the Androgen‐sensitive (AS) line LNCaP, is a protective effect resulting in a quiescent state, before these cells convert to a Androgen‐Independent (AI) growth. We postulate that this DDR axis is critical for survival of the cancer cells. We have tested this by monitoring the inhibition of emergence of AI colonies following treatment of LNCaP cells with bicalutamide (antiandrogen) and increasing concentrations of THD. AI colonies were scored after 3 weeks. At a concentration of 3–9 μM THD, which inhibits growth by only 50% in androgen containing medium (FCS), few or no AI colonies emerged (Figs. 2 c and 2d). In addition, after 4 weeks with bicalutamide+3 μM THD the remaining cells became apoptotic and lifted from the plate—see example in 2D right panel where we compare a colony without (top) and with THD treatment (bottom). On the other hand, THD has been reported to have anticancer activity, although the precise mechanisms for this have not been elucidated. We thus also tested a panel of AI cell lines, in addition to LNCaP, in androgen containing medium—FCS. This resulted in a dose‐dependent inhibition of colonies formation with maximal efficacy around 10 μM (Fig. 2 e). Note however, that the ‘normal’ prostate cell line RWPE1 was largely insensitive to THD, except at the highest concentration. This suggests that THD may indeed be a more targeted anticancer agent, or it is possible that the TLK1/Nek1 axis remains critical even for CRPC cells. However, we previously reported that THD delays, but does not eventually prevent, the growth of PC3 subcutaneous tumors.11 Note also that LNCaP cells grown in FCS‐medium were not much affected by THD, again suggesting that a large part of inhibition of the AI growth of these cells in CSS could be mediated via the TLK1/Nek1 DDR axis. We have also tested the AI growth of TRAMP‐C2 cells (see below) in Figure 2 f.
Treatment of LNCaP cells with ADT + THD results in apoptosis
We propose that bypass of the DDR induced by ADT by inhibition of TLK1/Nek1 with THD results in abrogation of the checkpoint and in accumulation of replication‐dependent DNA damage and induction of apoptosis. To establish if this is the case, we analyzed the cell cycle distribution of LNCaP cells treated with bicalutamide, THD, or combination for 24 h to determine if there is cell cycle arrest or induction of apoptotic—sub‐G1 cells (Fig. 3 a). As expected, treatment with bicalutamide alone resulted in accumulation of cells in G1, 77% vs 61 (untreated). In contrast, THD (10 μM) did not induce accumulation of cells in G1, but when combined with bicalutamide it resulted in a drastic ∼30% fraction of apoptotic cells. This result can well explain the fact that AI cells are unlikely to form in these conditions (see Figs. 2 c and d).
Figure 3.
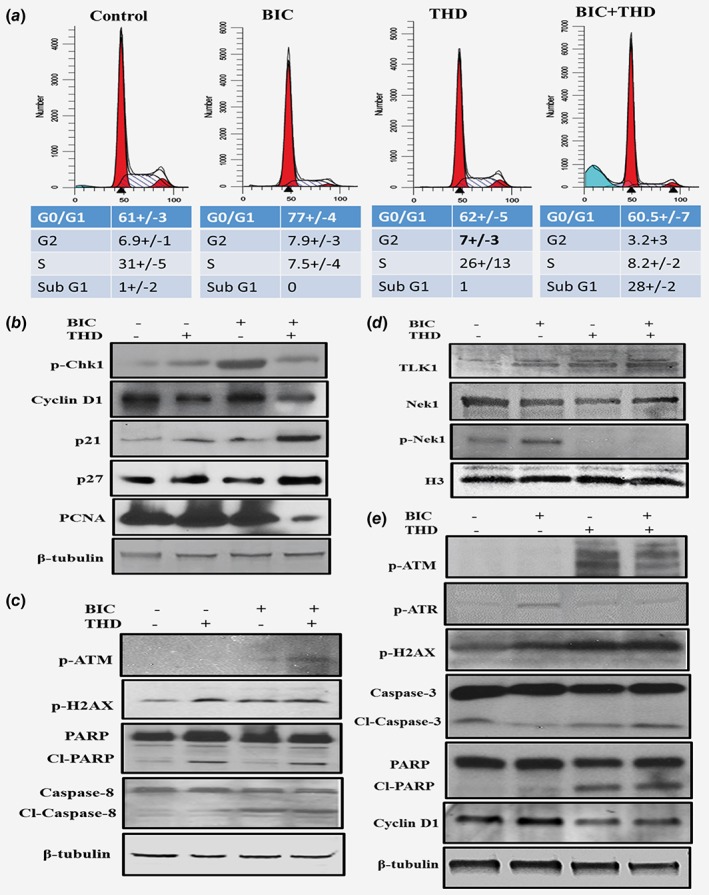
(a) Treatment of LNCaP cells with ADT + THD results in apoptosis. LNCaP cells were incubated bicalutamide (10 μM), THD (10 μM), or bicalutamide + THD for 24 h for cell cycle analysis of PI‐stained cells. (b)–(c) LNCaP‐Western Blots of cell cycle and apoptotic indicators. Cells from the four treatment groups as in A were analyzed by WB for several indicators of DNA damage/apoptosis and cell cycle arrest/proliferation. (d)–(e) TRAMP‐C2‐Western blots of several proteins involved in cell cycle and apoptosis. [Color figure can be viewed at wileyonlinelibrary.com]
To solidify these results, we collected the cells from the four treatment groups and determined by WB several indicators of DNA damage/apoptosis and cell cycle arrest/proliferation (Figs. 3 b and 3c). Treatment with bicalutamide resulted in the anticipated increase in pChk1 (Fig. 3 b), an indicator of its activation, and consistent with the G1‐arrest shown above. In contrast, the simultaneous addition of THD suppressed Chk1 phosphorylation by abrogating the TLK1 > NEK1 > ATR > Chk1 pathway as we previously showed.18 Bicalutamide also caused an increase in pH2AX (Fig. 3 c), an indicator of DNA damage, all consistent with previous reports of activation of the DDR and ATR or ATM upon ADT of LNCaP cells.3 In addition, combining THD with bicalutamide lead to activation of ATM (pS1981, Fig. 3 c), suggesting that bypass of the Nek1 > ATR > Chk1 checkpoint may result in replication forks collapse and leading to ATM activation. THD also caused an increase in the appearance of a fraction of cleaved PARP, which is consistent with the fact that it induced an increase in apoptotic cells. However, the combination treatment did not result in further increase in cleaved PARP, although there was a significant increase in cleaved Caspase 8 in comparison to THD alone. PCNA, a marker of proliferating cells, was not strongly reduced in cells treated with bicalutamide, unlike what we had anticipated judging from the increase in G1‐arrested cells. However, it was drastically reduced in the combination treatment group. Cyclin D1 was not changed with bicalutamide treatment (Fig. 3 b), but was reduced in the THD and in the combination group. Finally, it seemed possible that bypass of the DDR elicited by bicalutamide via THD‐mediated inhibition of the TLK1/Nek1 axis18 would cause the emergency activation of the P53 > p21 pathway, as shown for treatment of LNCaP‐C42B cells with a Chk1 inhibitor.17 Indeed, there was a strong increase in p21 expression in the combination treatment group (Fig. 3 b). P27 was also slightly increased in the THD treated groups.
The ADT > TLK1 > NEK1 > ATR, DDR axis is conserved in mouse TRAMP‐C2 cells and is likely fundamental in PCa progression
It was our desire to be able to extend our observations to additional PCa models. Unfortunately, other than LNCaP there are not other human cell lines that can recapitulate the AS to AI conversion in culture in a short time (most are AI for growth as they were derived from metastatic lesions of ADT‐patients). However, the TRAMP‐C2 cells convert in culture from AS to AI growth, displaying a NE phenotype, and they are wt PTEN unlike LNCaP—we were concerned that the bicalutamide mediated activation of the AKT > mTOR>TLK1 axis would be a peculiar response to loss of PTEN. We thus tested if the DDR axis we uncovered in LNCaP cells following ADT was conserved in TRAMP‐C2 cells. Treatment of TRAMP‐C2 cells with bicalutamide for 16 h resulted in a strong increase in TLK1 expression (Fig. 3 d—note that the equivalent of the TLK1B splice variant is not reported in mouse EST data). This result indicates that the compensatory prosurvival pathway of mTOR activation following ADT is conserved in the TRAMP mouse‐derived model. Next, we tested if the remainder of the DDR pathway is also conserved. First we checked pNek1, whose kinase domain, including the T141 residue is completely conserved between mouse and human (94% identity). As for LNCaP, treatment of TRAMP‐C2 cells with bicalutamide resulted in increased phosphorylation of Nek1‐T141 without change in total Nek1 (Fig. 3 d). In contrast, addition of THD completely abolished pNek1, even when combined with bicalutamide. ATR followed the same pattern of activation (Fig. 3 e—pATR) consistent with its dependence on Nek1. However, THD treatment in TRAMP‐C2 cells resulted in significant ATM activation (Fig. 3 e—ATM‐pS1981) even in the absence of bicalutamide, suggesting that loss of the TLK1 > Nek1 > ATR axis in these cells may result in replication forks collapse and induction of some DSBs, as also demonstrated by the presence of pH2AX. In fact, THD and more so in combination with bicalutamide resulted Caspase3 and PARP cleavage (Fig. 3 e), suggesting that the inhibition of this DDR axis eventually results in apoptosis. Cyclin D1 was reduced in the THD and combination group, just as in LNCaP cells, an indication that proliferation was also impaired. In fact, clonogenic assays of C2 cells treated with bicalatumide, THD, or combination showed that whereas AI colonies (bicalutamide resistant) formed readily within 2 weeks, THD and THD + bicalutamide strongly impaired their growth or was toxic (Fig. 2 f). It is important to point out that TRAMP‐C2 cells, unlike LNCaP, carry a truncation mutant of TP53,23 and therefore our observations seem to apply to at least two cell models regardless of TP53 status.
Combination bicalutamide and THD suppresses growth of LNCaP xenografts via suppression of the pNek1‐pATR‐pChk1 DDR pathway
We wanted to establish if the results obtained with LNCaP in culture could be translated in vivo as a method to suppress emergence of CRPC in a model of LNCaP cells xenograft. Following formation of sizeable tumors (∼200 mm3), castration or antiandrogens halt the progression of LNCaP xenografts for some time (2–3 weeks), while typically the subsequent re‐growth is refractory to ADT (AI). Therefore, we injected LNCaP cells in matrigel subcutaneously to both of the flank in NOD‐SCID mice, and then randomly assigned them to four treatment groups, as shown in Figures 4 a–4c. As expected, bicalutamide arrested the growth of tumors for 10 days, but later the tumors began growing again, although slowly. Importantly, treatment with bicalutamide resulted in an increase in TLK1B expression in the tumors of 3/3 mice tested compared to controls (Supporting Information Fig. S2D), consistent with the results previously shown in vitro. Interestingly, treatment with THD alone resulted is a similar temporary suppression of tumor growth (***p < 0.005), showing that THD can have antitumor activity by itself. Importantly, the combination treatment with THD and bicalutamide resulted in complete suppression of tumor growth (***p < 0.005) and actually upon removal of the tumors, both volume and weight showed some regression compared to the beginning of the treatment Figures 4 b and 4c.
Figure 4.
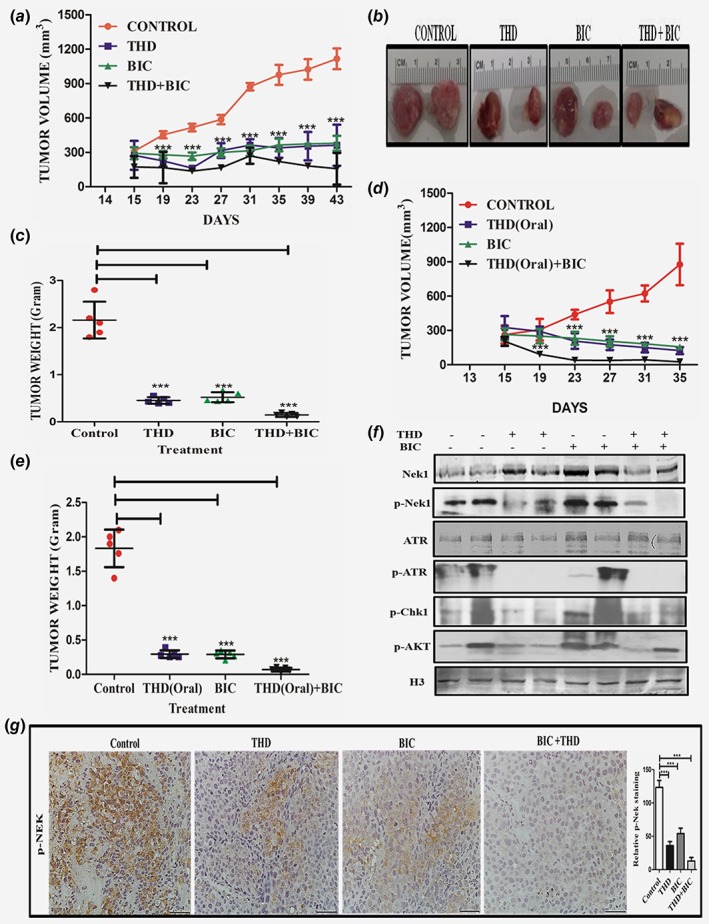
(a)–(e) Combination bicalutamide and THD suppresses growth of LNCaP xenografts. A. Time course of tumor growth of LNCaP cells xenografts in 4 treatment groups. Treatment started after 2 weeks of implantation when the tumors measure ~200 mm3. Bicalutamide was injected IP twice/week and THD IP an alternate days. (b) Examples of tumor size at the end of the experiment. (c) Tumor weights at the end of the experiment. (d) Same as in (a) but the mice were treated with THD given orally an alternate days. Same as in C but the mice were treated with THD given orally. (e) Tumor weights. (f) Proteins extracted from the tumors of two representative mice in each group were analyzed by western blot for the indicated key proteins of cell cycle checkpoint and also for pAKT and histone H3. (g) Representative sections from tumors resected from mice in the 4 treatment groups analyzed by IHC for pNek1. Note the absence of pNek1 stain in the bicalutamide+THD tumor, consistent with the results shown in F. [Color figure can be viewed at wileyonlinelibrary.com]
As THD is normally given orally for schizophrenic individuals, we also tested the efficacy of THD through oral administration in the LNCaP xenografts model. We found that oral route of THD administration alone or in combination with bicalutamide (IP) was also very effective (***p < 0.005) in reducing the tumor burden (volume and weight) as shown in Figures 4 d and 4e. Oral administration would be a much better method of treatment for PCa patients in conjunction with ADT. Further, we checked the status of the pNek1, pATR and pChk1 axis in protein extracted from residual tumors after 3 weeks of treatment, and found a significant decrease in this DDR pathway in THD alone or in combination with bicalutamide. In particular, ATR was highly phosphorylated in tumors from control mice, possibly the result of replication stress induced DDR. THD resulted in complete loss of pATR and reduced level of pNek1 and pChk1 in vivo (Fig. 4 f), suggesting that abrogation of the DDR in the growing tumors may have forced their demise by inducing apoptosis, as we have shown in tissue culture. In support of this, immunohistochemistry analysis of tissue sections from LNCaP xenografts revealed that THD treatment alone or in combination with bicalutamide inhibits pNek1 (Fig. 4 g) and decreases Ki67 (Fig. 5 a) and Proliferating Cell Nuclear Antigen (PCNA) (Supporting Information Fig. 2C), which are markers of proliferation. The combination treatment also induces apoptosis (as shown by cleaved PARP [Fig. 5 b] and cleaved Caspase 3 [Fig. 5 c]) and results in increased DNA damage, as indicated by γH2AX staining (Fig. 5 d), which can be attributed to a failure to properly activate the DDR. Representative tumor sections H&E stained are shown in Supporting Information Fig. S2B. In addition, and consistent with what was reported for treatment of ovarian cancer cells with THD,16 tumors isolated from mice treated with THD and in combination showed a decrease in pAKT (Fig. 4 f), an event that was linked to decreased viability of ovarian cancer cells.16 It is noteworthy that AKT‐interacting‐protein (AKTIP) is a target of TLK1/1B18 and we are investigating if THD may inhibit AKT phosphorylation via a TLK1 > AKTIP pathway. We also determined that bicalutamide and THD treatments did not cause apparent behavioral changes or feeding habits nor lead to significant changes in body weights (Supporting Information Fig. S2A), although we have not checked for specific organ toxicity since both bicalutamide and THD have been already extensively tested in mice and humans.
Figure 5.
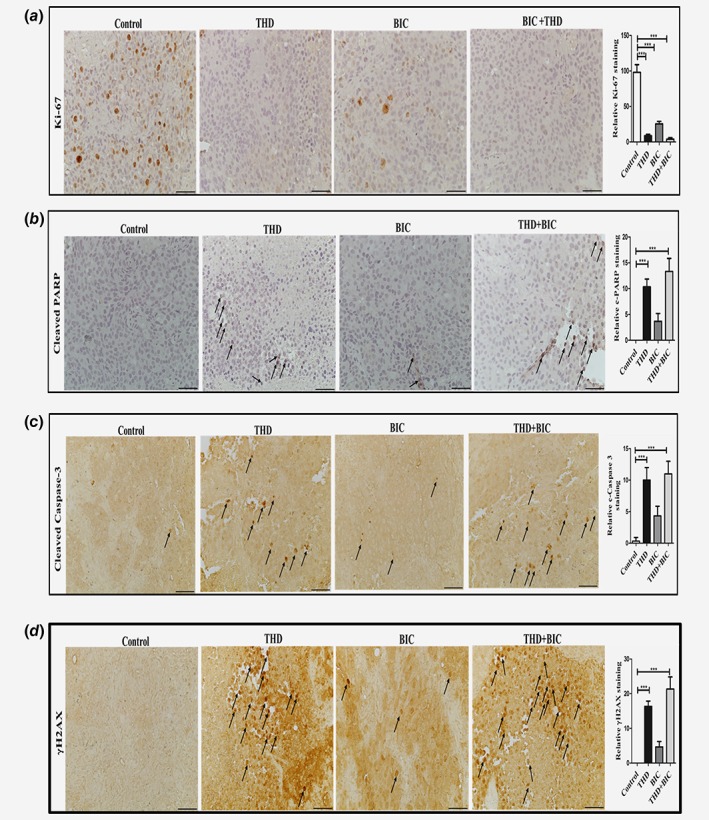
(a)–(d) IHC analysis of markers of proliferation (Ki67—(a)), apoptosis (Cleaved PARP and Caspase 3—(b), (c)), and presence of γH2AX (an indicator of DSBs—da)). [Color figure can be viewed at wileyonlinelibrary.com]
Overexpression of wt‐Nek1, but not the T141A mutant, counters the growth arrest from bicalutamide and the apoptosis induced by combination with THD.
Treatment of LNCaP cells with bicalutamide results in a temporary growth arrest and morphological changes already noticeable the next day (Fig. 6, LNCaP parental). Eventually, the cells become AI independent for growth after about 2 weeks and form colonies (Fig. 2), although in combination with THD the cells instead undergo apoptosis. This can be seen in Fig. 6 (BIC + THD), which shows few residual cells with evidence of blebbing and nuclear condensation. In stark contrast, LNCaP cells overexpressing (OE) Nek1 do not appear to growth arrest when incubated with bicalutamide or even in combination with THD, suggesting that Nek1 overexpression (or increased activity) could be a key factor in the conversion to AI growth of at least LNCaP cells. Consistent with the fact that it is not just the overexpression of Nek1, but that also the kinase activity that is critical for the AI growth, we show that overexpression of the T141A mutant, previously shown to be significantly less catalytically active,18 was unable to confer AI growth in presence of bicalutamide and resistance to apoptosis when combined with THD.
Figure 6.
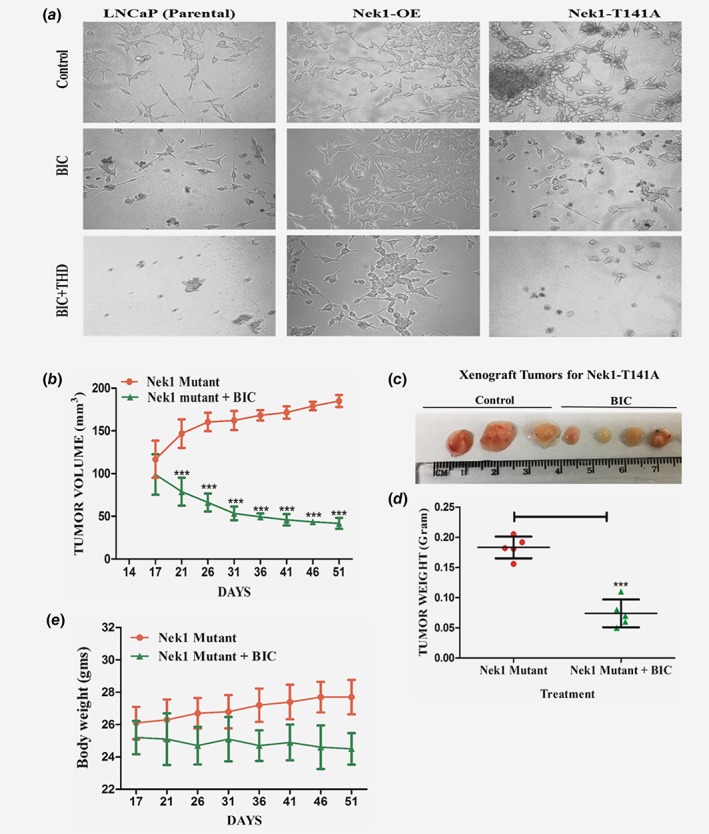
(a) Overexpression of wt‐Nek1, but not the T141A mutant, counters the growth arrest from bicalutamide and the apoptosis induced by combination with THD. LNCaP parental cells and overexpressing Nek1 (wt or T141A mutant) were plated at low density and incubated for 1 week without or with bicalutamide and +/− THD. (b) LNCaP overexpressing Nek1‐T141A form slow‐growing tumor xenografts which remain sensitive to ADT. Time course of tumor growth LNCaP cells xenografts in mice treated or not with bicalutamide. Treatment started after 2 weeks of implantation when the tumors measure ~100 mm3 (note that at this time, tumors from parental LNCaP had already reached 200 mm3—Fig. 4). (c) Examples of tumor size at the end of the experiment. (d) Tumor weights at the end of the experiment. (e) Body weights of mice treated or not with bicalutamide. [Color figure can be viewed at wileyonlinelibrary.com]
LNCaP overexpressing Nek1‐T141A mutant form slow‐growing tumor xenografts which remain sensitive to ADT
Overexpression of Nek1‐T141A in LNCaP cells did not alter their doubling time (∼32 h) or their cell cycle profile (not shown, but also see)18 in FCS‐containing medium (i.e., with androgen). In contrast, these cells had very different properties when grown as tumor xenografts (Fig. 6). First of all, in untreated animals the tumors grew much more slowly than those from parental LNCaP cells (Fig. 6 b). In fact, at the end of the experiment (51 days from time of implantation) the tumors had just reached the size of the start point for LNCaP tumors (200 mm3—compare with Fig. 4, day 16). Moreover, in mice treated bicalutamide starting at day 17, the tumors actually regressed. This was clearly visible at the end of the experiments, where all the LNCaP‐T141A tumors isolated from mice treated with bicalutamide were clearly smaller than those in the untreated group (Figs. 6 b–6d). Note that the average body weights were a little less in the mice treated with bicalutamide, but all still in the normal range for their age (Fig. 6 e).
Discussion
Use of phenothiazines TLK inhibitors as adjuvant to standard PCa treatment
TLK1/1B and Nek1 are functionally linked kinases18 involved in DNA repair and are frequently overexpressed in PCa cell lines and patients’ samples. The DDR is activated during ADT and is a mechanism to ensure survival from replication forks arrest induced damage. During a high throughput screen to identify inhibitors of TLK1 we have found several in the class of phenothiazine (PTH) antipsychotics, most notably THD,11 which we propose to repurpose for the treatment of PCa to improve response to ADT. PTH antipsychotics have been used extensively for the treatment of severe psychiatric disorders for over 30 years, for prolonged periods with relatively low side effects. These include a small increased risk of cardiac arrhythmia with long term use,24 and extrapyramidal toxicity that can be potentially permanent. Notably, several PTH antipsychotics were reported to have significant antitumoral effects with several cancer cell lines25 and their repurposing has been proposed26 even though their cellular targets have not been identified, and they were generally assumed to work largely through inhibition of dopamine receptors.27 Interestingly, an analysis from five independent studies of PCa incidence in individuals with schizophrenia revealed a significant decrease in Standardized Incidence Ratio ranging from 0.49 to 0.76 (95% CI).28 Proposed explanations for this were: specific genetic factors; antipsychotic drug effects, either by being cancer protective or decreasing testosterone; and lifestyle differences. The most compelling study specifically pointed to the use of PTH as the possible cause.29 We propose that this is in fact the result of inhibition of TLK1/1B by PTH antipsychotics. PCa cells are subject to elevated DNA damage from replication stress, which we propose require the activity of the TLK1/NEK1 axis to repair the damage and mediate the checkpoint/DDR. Hence, inhibition of the axis will result in lethal levels of damage, causing suppression of PCa emergence in schizophrenic patients. Given these considerations, we believe that the relatively small increased risk for side effects from the use of PTH inhibitors of the TLK1/NEK1 axis can be outweighed by a more complete response to ADT, perhaps preventing progression to mCRPC altogether.
Combination ADT and THD suppresses growth of androgen responsive PCa cells via suppression of the pNek1‐pATR‐pChk1 DDR pathway
We previously showed that treatments that result in activation of the DDR, like oxidative stress and replication arrest, are mediated via the TLK1 > Nek1 > ATR > Chk1 pathway,18 and that its suppression with THD results in checkpoint bypass and then induction of apoptosis. Likewise, treatment of androgen‐responsive LNCaP and TRAMP‐C2 cells with ADT results in activation of the DDR,3 as shown here. We have shown that combination treatment of ADT and THD results in increased apoptosis and in complete arrest of progression to AI growth in vitro (formation of colonies). Moreover, we have shown that in xenografts, treatment of LNCaP tumors with THD resulted in bypass of ATR and Chk1 activation, resulting in increased apoptosis and suppression of tumor growth. In addition there was an increase in markers of apoptosis, like cleaved Caspase 3 and PARP, and presence of numerous γH2AX‐expressing cells, a marker of DNA damage and especially of presence of DSBs. This is in support of our model that bypass of the DDR with THD can result in progression into mitosis with damaged DNA, resulting in accumulation of unrepaired DSBs and then cell death (see model in Supporting Information Fig. S3). However, we should stress that the mechanism(s) we are proposing is not quite proven, and that THD could have other effects. Nonetheless, this proposed mechanism appears to be evolutionarily conserved, as both LNCaP and TRAMP‐C2 gave similar results. Moreover, we have shown in LNCaP cells that overexpression of wt‐Nek1 counteracts the growth inhibition of bicalutamide and loss of viability in presence of THD, whereas the T141A mutant (a single amino acid change in Nek1) does not. Since this is a complete bypass of the TLK1/1B activity on Nek1 phosphorylation/activation, it seems like a direct effect, although we cannot rule out some other activity of Nek1 in addition to its regulation of the DDR.
NEK1 and TLK1/1B in DNA repair and DDR
The founding member of the NIMA family of protein kinases was originally identified in Aspergillus nidulans as a protein kinase essential for mitosis.30 NIMA related kinases (NEKs) have adapted to a variety of cellular functions in addition to mitosis.31 In human cells, 11 NEKs were identified that are involved in several functions. For example Nek2 is critical for centrosome duplication,31 whereas Nek6, 7, 9 are regulators of the mitotic spindle and cytokinesis.32 Nek19, 33, 34 and Nek435 have been linked to the DDR and DNA repair pathways. Nek1 promotes Chk1 activation via its association with ATR‐ATRIP and primes ATR for the DDR.10 Nek1‐deficient cells fail to activate the checkpoint kinases Chk1 and Chk2, and fail to arrest properly at G1/S or G2/M‐phase checkpoints after DNA damage. Nek1‐deficient cells suffer major errors in mitotic chromosome segregation and cytokinesis.36 However, it was not known what regulates Nek1 activity and its nuclear and cytoplasmic functions. We have recently shown that TLK1 is a key regulator of Nek1 activity, particularly in the DDR.18 While specific inhibitors of Nek1 have not been identified so far,37 we have now shown that reduction of Nek1 activity via TLK1/1B inhibition (or the T141A mutant) can sensitize PCa cells and tumors to ADT. While similar strategies have been proposed recently for treatment with ADT in combination with ATM or Chk1 inhibitors,3, 17 one problem that was apparent with those studies is how to limit the effects of general inhibitors of the DDR pathway to make them more PCa cells‐specific. We are now showing that the increased expression of TLKB with ADT in LNCaP cells, a prosurvival checkpoint pathway that is enacted before conversion to AI growth, offers a unique target for attacking more specifically PCa cells before their conversion to CRPC. Moreover, we suggest the use of THD or similar PTH derivatives, which have already a long history of relatively safe use with patients suffering from severe psychotic conditions, for the early treatment of advanced PCa still responsive to ADT.
Authors’ Contributions
V.S., P.J., I.G., H.K., X.Y. and A.D.B.: Conception and design
V.S., P.J., I.G., H.K., X.Y. and A.D.B.: Development of methodology
V.S., P.J., I.G. and A.D.B.: Acquisition of data
V.S. and P.J.: Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis)
V.S., P.J., I.G., H.K., X.Y. and A.D.B.: Writing, review, and/or revision of the manuscript
V.S., P.J., I.G., H.K., X.Y. and A.D.B.: Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases)
H.K., X.Y. and A.D.B.: Study supervision
Supporting information
Supplemental Methods
Fig. S1 S1A. Normalized expression of TLK1B splice variant. LNCaP cells were shifted for 24 h to charcoal‐stripped‐serum (CSS) or maintained in FCS. After preparation of cDNA the abundance of TLK1B transcript was determined relative to GAPDH mRNA. The Cq mean was 26.4 for TLK1B and 23.8 for GAPDH.
S1B. The phosphorylation of 4EBP1 was monitored in parallel with the experiment in Figure 1B via its change in mobility.
Fig. S2 S2A. Body weights (LNCaP xenografts) do not differ statistically in mice treated with bicalutamide and/or THD.
S2B. Representative H&E‐stained section of tumors from mice from the four groups. Note that in the THD + bicalutamide tumors there appear to be gaps in the tumor tissue and many cells look shrunk.
S2C. Representative tumor sections stained for PCNA.
S2D. Tumors from mice treated with bicalutamide display an increase in TLK1B proteins.
Fig. S3 Model for the combination of ADT and inhibition of the TLK1‐Nek1, DDR axis with bicalutamide and THD, culminating in abrogation of the checkpoint and then cell death.
Acknowledgements
We thank Paula Polk and the Research Core Facility‐Genomics and the Histopathology Core at LSU Health‐Shreveport, which is supported in part by the LSU Health Shreveport Foundation, the Biomedical Research Foundation, the Center for Molecular and Tumor Virology, the Center for Cardiovascular Diseases and Sciences, and the Feist‐Weiller Cancer Center (FWCC). This work was supported primarily by a FWCC bridge award and DoD‐PCRP grant W81XWH‐17‐1‐0417 to A. De Benedetti.
V.S. and P.K.J. contributed equally to this work.
Conflict of interest: No potential conflicts of interest were disclosed.
References
- 1. Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013;32:5501–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carver BS, Chapinski C, Wongvipat J, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN‐deficient prostate cancer. Cancer Cell 2011;19:575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chiu YT, Liu J, Tang K, et al. Inactivation of ATM/ATR DNA damage checkpoint promotes androgen induced chromosomal instability in prostate epithelial cells. PLoS One 2012;7:e51108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reddy V, Wu M, Ciavattone N, et al. ATM inhibition potentiates death of androgen receptor‐inactivated prostate cancer cells with telomere dysfunction. J Biol Chem 2015;290:25522–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Karanika S, Karantanos T, Li L, et al. DNA damage response and prostate cancer: defects, regulation and therapeutic implications. Oncogene 2015;34:2815–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yu P, Duan X, Cheng Y, et al. Androgen‐independent LNCaP cells are a subline of LNCaP cells with a more aggressive phenotype and androgen suppresses their growth by inducing cell cycle arrest at the G1 phase. Int J Mol Med 2017;40:1426–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Benedetti A. The tousled‐like‐kinases as guardians of genome integrity. ISRN Mol Biol 2012;2012:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feige E, Shalom O, Tsuriel S, et al. Nek1 shares structural and functional similarities with NIMA kinase. Biochim Biophys Acta 2006;1763:272–81. [DOI] [PubMed] [Google Scholar]
- 9. Chen Y, Chen CF, Riley DJ, et al. Nek1 kinase functions in DNA damage response and checkpoint control through a pathway independent of ATM and ATR. Cell Cycle 2011;10:655–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu S, Ho CK, Ouyang J, et al. Nek1 kinase associates with ATR‐ATRIP and primes ATR for efficient DNA damage signaling. Proc Natl Acad Sci U S A 2013;110:2175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ronald S, Awate S, Rath A, et al. Phenothiazine inhibitors of TLKs affect double‐Strand break repair and DNA damage response recovery and potentiate tumor killing with radiomimetic therapy. Genes Cancer 2013;4:39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shen J, Ma B, Zhang X, et al. Thioridazine has potent antitumor effects on lung cancer stem‐like cells. Oncol Lett 2017;13:1563–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Spengler G, Csonka A, Molnar J, et al. The anticancer activity of the old neuroleptic phenothiazine‐type drug Thioridazine. Anticancer Res 2016;36:5701–6. 10.21873/anticanres.11153. [DOI] [PubMed] [Google Scholar]
- 14. Seo SU, Cho HK, Min KJ, et al. Thioridazine enhances sensitivity to carboplatin in human head and neck cancer cells through downregulation of c‐FLIP and Mcl‐1 expression. Cell Death Dis 2017;8:e2599 10.1038/cddis.2017.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jin X, Zou B, Luo L, Zhong C, Zhang P, Cheng H, Guo Y, Gou M: Codelivery of thioridazine and doxorubicin using nanoparticles for effective breast cancer therapy. Int J Nanomedicine 2016, 11:4545–4552.doi: 10.2147/IJN.S104635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rho SB, Kim BR, Kang S. A gene signature‐based approach identifies thioridazine as an inhibitor of phosphatidylinositol‐3′‐kinase (PI3K)/AKT pathway in ovarian cancer cells. Gynecol Oncol 2011;120:121–7. [DOI] [PubMed] [Google Scholar]
- 17. Karanika S, Karantanos T, Li L, et al. Targeting DNA damage response in prostate cancer by inhibiting androgen receptor‐CDC6‐ATR‐Chk1 signaling. Cell Rep 2017;18:1970–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Singh V, Connelly ZM, Shen X, et al. Identification of the proteome complement of humanTLK1 reveals it binds and phosphorylates NEK1 regulating its activity. Cell Cycle 2017;16:915–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sunavala‐Dossabhoy G, Fowler M, De Benedetti A. Translation of the radioresistance kinase TLK1B is induced by gamma‐irradiation through activation of mTOR and phosphorylation of 4E‐BP1. BMC Mol Biol 2004;5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sunavala‐Dossabhoy G, Balakrishnan S, Sen S, et al. The radioresistance kinase TLK1B protects the cells by promoting repair of double strand breaks. BMC Mol Biol 2005;6:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang J, Jia P, Zhao Z, et al. Key regulators in prostate cancer identified by co‐expression module analysis. BMC Genomics 2014;15:1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ronald S, Sunavala‐Dossabhoy G, Adams L, et al. The expression of tousled kinases in CaP cell lines and its relation to radiation response and DSB repair. Prostate 2011;71:1367–73. [DOI] [PubMed] [Google Scholar]
- 23. Bhadury J, Nilsson L, López M, et al. Trp53 mutational status correlates with tumorigenicity in TRAMP‐C mouse prostate cancer cell lines. Sciencematters 2016. 10.19185/matters.201607000007. [DOI] [Google Scholar]
- 24. Wu CS, Tsai YT, Tsai HJ. Antipsychotic drugs and the risk of ventricular arrhythmia and/or sudden cardiac death: a nation‐wide case‐crossover study. J Am Heart Assoc 2015;4:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jaszczyszyn A, Gasiorowski K, Swiatek P, et al. Chemical structure of phenothiazines and their biological activity. Pharmacol Rep 2012;64:16–23. [DOI] [PubMed] [Google Scholar]
- 26. Lee JK, Nam DH, Lee J. Repurposing antipsychotics as glioblastoma therapeutics: potentials and challenges. Oncol Lett 2016;11:1281–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yin TAO, He S, Shen G, et al. Dopamine receptor antagonist thioridazine inhibits tumor growth in a murine breast cancer model. Mol Med Rep 2015;12:4103–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Torrey EF. Prostate cancer and schizophrenia. Urology 2006;68:1280–3. [DOI] [PubMed] [Google Scholar]
- 29. Mortensen PB. The incidence of cancer in schizophrenic patients. J Epidemiol Community Health 1989;43:43–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Osmani SA, Pu RT, Morris NR. Mitotic induction and maintenance by overexpression of a G2‐specific gene that encodes a potential protein kinase. Cell 1988;53:237–44. [DOI] [PubMed] [Google Scholar]
- 31. Meirelles GV, Perez AM, de Souza EE, et al. "stop ne(c)king around": how interactomics contributes to functionally characterize Nek family kinases. World J Biol Chem 2014;5:141–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moniz L, Dutt P, Haider N, et al. Nek family of kinases in cell cycle, checkpoint control and cancer. Cell Div 2011;6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen Y, Chen PL, Chen CF, et al. Never‐in‐mitosis related kinase 1 functions in DNA damage response and checkpoint control. Cell Cycle 2008;7:3194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pelegrini AL, Moura DJ, Brenner BL, et al. Nek1 silencing slows down DNA repair and blocks DNA damage‐induced cell cycle arrest. Mutagenesis 2010;25:447–54. [DOI] [PubMed] [Google Scholar]
- 35. Basei FL, Meirelles GV, Righetto GL, et al. New interaction partners for Nek4.1 and Nek4.2 isoforms: from the DNA damage response to RNA splicing. Proteome Sci 2015;13:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen Y, Chen CF, Chiang HC, et al. Mutation of NIMA‐related kinase 1 (NEK1) leads to chromosome instability. Mol Cancer 2011;10:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moraes EC, Meirelles GV, Honorato RV, et al. Kinase inhibitor profile for human nek1, nek6, and nek7 and analysis of the structural basis for inhibitor specificity. Molecules 2015;20:1176–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jaiswal PK, Koul S, Shanmugam PST, et al. Eukaryotic translation initiation factor 4 gamma 1 (eIF4G1) is upregulated during prostate cancer progression and modulates cell growth and metastasis. Sci Rep 2018;8:7459 10.1038/s41598-41018-25798-41597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Methods
Fig. S1 S1A. Normalized expression of TLK1B splice variant. LNCaP cells were shifted for 24 h to charcoal‐stripped‐serum (CSS) or maintained in FCS. After preparation of cDNA the abundance of TLK1B transcript was determined relative to GAPDH mRNA. The Cq mean was 26.4 for TLK1B and 23.8 for GAPDH.
S1B. The phosphorylation of 4EBP1 was monitored in parallel with the experiment in Figure 1B via its change in mobility.
Fig. S2 S2A. Body weights (LNCaP xenografts) do not differ statistically in mice treated with bicalutamide and/or THD.
S2B. Representative H&E‐stained section of tumors from mice from the four groups. Note that in the THD + bicalutamide tumors there appear to be gaps in the tumor tissue and many cells look shrunk.
S2C. Representative tumor sections stained for PCNA.
S2D. Tumors from mice treated with bicalutamide display an increase in TLK1B proteins.
Fig. S3 Model for the combination of ADT and inhibition of the TLK1‐Nek1, DDR axis with bicalutamide and THD, culminating in abrogation of the checkpoint and then cell death.