

Abstract
The system of hepatocyte growth factor (HGF) and its receptor c‐Met plays a critical role in tumor invasive growth and metastasis. The mortality rate of colorectal cancer (CRC), one of the most commonly diagnosed malignancies, is increased by it gradual development into metastasis, most frequently in the liver. Overexpression of c‐Met, the protein tyrosine kinase receptor for the HCF/scatter factor, has been implicated in the progression and metastasis of human colorectal carcinoma. In this study, we aimed to investigate the role of c‐Met in CRC liver metastasis and illustrate the clinical impact of regulating HGF/c‐Met signaling in patients with CRC liver metastasis. We found that (I) higher levels of c‐Met expression (mRNA and Protein) in CRC liver metastasis than primary CRC by assessing the patient tissue samples; (II) a positive correlation of c‐Met expression with tumor stages of CRC liver metastasis, as well as c‐Met expression in CRC, live metastasis concurred with regional lymph node metastasis; (III) the clinical impact of downregulation of HGF/c‐Met signaling on the reduction of proliferation and invasion in CRC liver metastasis. Therefore, we demonstrate that the regulation of HGF/c‐Met pathways may be a promising strategy in the treatment of patients with CRC liver metastasis.
Keywords: annexin V‐FITC, c‐Met RNA interference (RNAi), colorectal cancer (CRC) cell, hepatocyte growth factor (HGF), metastasis
1. INTRODUCTION
Colorectal cancer (CRC) is recognized as the third leading cause of cancer‐related deaths worldwide.1, 2, 3, 4 CRC incidence and mortality rates are rapidly increasing over time, especially in low‐income and middle‐income countries, and the global burden of CRC is expected to surge by 60% to more than 2.2 million new cases and 1.1 million deaths by 2030.5, 6, 7, 8, 9 CRC fatality is commonly caused by metastasis. Liver metastasis has been shown as one of the leading causes of death in patients with CRC. Previous studies have shown that about 25% of patients with CRC are clinically diagnosed with liver metastasis at the initial diagnosis, and approximately 50% of patients with CRC develop symptoms of liver metastasis during the course of their disease. Around 30% to 50% of patients with CRC display recurrent liver metastasis after radical resection and more than 50% succumb to the disease.10, 11, 12, 13, 14 However, the current therapies for patients with CRC live metastasis, including surgery, radiotherapy, and chemotherapy treatment in the CRC metastasis, remain unsatisfactory due to rapid CRC metastasis deterioration and drug resistance.10, 11
Hepatocyte growth factor (HGF) and its receptor, mesenchymal‐epithelial transition (c‐Met), play an important role in several biological processes.15, 16, 17, 18, 19, 20, 21 Under normal physiological conditions, the interaction of HGF/c‐Met strongly mediates cellular organization and cell division, such as epithelial cell proliferation and migration, angiogenesis, and wound healing.18, 20 Whereas, the HGF/c‐Met pathway is also involved in different types of malignant tumors and plays critical roles in tumor proliferation, invasion, and metastasis.22, 23, 24, 25 Despite the increasing knowledge of the pathway bases of HGF/c‐Met progression, the exact relationship between HGF/c‐Met signaling and CRC and CRC liver metastasis remains a largely controversial topic. Therefore, in this study, we first determine whether the c‐Met expression is correlated with CRC liver metastasis and then determine whether regulating HGF/c‐MET signaling would be a new promising intervention to attenuate/inhibit the progression of CRC metastasis.
2. MATERIALS AND METHODS
2.1. Patients groups
With institutional review and ethical approval of the Institutional Review Board of Shaanxi Provincial People's Hospital Cancer Center and with written patient consent, pathological specimens were obtained by surgical resection with patient contents. Then the specimens were quick‐frozen in liquid nitrogen and preserved at −80°C. The cohort comprised 54 men and 42 women. The median age was 66 (range: 52‐80) years. Based on the clinical symptoms, all 48 CRC liver metastasis patients underwent surgical removal of both primary CRC tumors and resectable live metastasis; while all 48 patients with CRC only removed CRC tumor sites. All patients with surgical resection were diagnosed by postoperative pathological and immunohistochemical examination with auxiliary treatments such as chemotherapy and radiotherapy.
2.2. Validation of gene expression by real‐time polymerase chain reaction
Total RNA was extracted from the specimens by using TRIzol according to the manufacturer's protocol. The mRNA was then reversely transcribed into cDNA using a synthesis kit (M‐MLV; Promega, WI). The expression of c‐Met was detected by RT‐PCR using the upstream primer: 5′‐AGTCATAGGAAGAGGGCATT‐3′ and downstream primer: 5′‐CTTCACTTCGCAGGCAGA‐3′. The GAPDH gene was used as an endogenous control; upstream primer: 5′‐TGACTTCAACAGCGACACCCA‐3′ and downstream primer: 5′‐CACCCTGTTGCTGTAGCCAAA‐3′. The 20 µL reaction consists of SYBR Premix ExTaq: 10.0 µL, upstream primer (2.5 µM) 0.5 µL, downstream primer (2.5 µm) 0.5 µL, cDNA 1.0 µL, and RNase‐free H2O 8.0 µL. The reaction was carried out as follows: 95°C initial denaturation‐30 seconds, PCR reaction: 95°C melting‐5 seconds, 60°C annealing‐30 seconds, 55°C extending‐30 seconds, for a total of 45 cycles. The relative mRNA expression of c‐Met is calculated.
2.3. Western blot
RIPA buffer supplemented with protease and phosphatase inhibitors (Cat: 78441B; Thermo‐Fisher, MA) was used for collecting about 10 mg protein estimated by the bicinchoninic acid method from the tissue that was frozen in liquid nitrogen. Total protein was denatured at 100°C for 5 minutes and resolved on 8% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (25 µg protein/lane) and transferred to polyvinylidene fluoride membrane. The membrane was blocked for 1 hour at room temperature with 5% milk in Tris‐buffered saline supplemented with Tween‐20, followed by probing with rabbit anti‐human c‐Met antibody (1:1000; Invitrogen, CA) at 4°C overnight. Subsequently, the membrane was incubated with the horseradish peroxidase‐conjugated secondary antibody (1:2000) for 1 hour at room temperature. The immunoreactive bands were developed using the enhanced chemiluminescence detection method and analyzed by the Image J after scanning.
2.4. Cell culture
CRC cell lines including Lovo, SW480, SW620 and HT‐29, and normal cell line FHC purchased from the American Type Culture Collection (Manassas, VA) were grown in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS) supplemented with 100 µL/mL penicillin and 100 µg/mL streptomycin.26, 27 The incubator was maintained at 5% CO2, saturated humidity, and 37°C. When the cells reached 80% confluence, the cells were collected by trypsinization for the subsequent experiments.
2.5. CRC live metastasis organoid culture
All patients were newly diagnosed and had not been treated with chemotherapy before our receipt of the samples. Pathological specimens were immediately placed in DMEM/F12+GlutaMAX (CAT #31331‐028; ThermoFisher) basal medium supplemented with 1% penicillin/streptomycin. Simple disaggregates were plated in both a 96‐well plate and a 24‐well plate mixed with Matrigel Matrix (Product Number: 354234; Corning, NY). For Matrigel mixed samples, an additional 100 µL Matrigel was added into each well and mixed well. DMEM/F12+GlutaMAX containing StemPro, ROCKs inhibitor, R‐Spondin‐1, Noggin, WNT3A, epithelial growth factor, insulin‐like growth factor 1, fibroblast growth factor 10, endothelin 3, and fibroblast growth factor‐basic was added to the samples. For passaging organoids, cultures should grow enough to 7 to 10 days. The media was then removed from the well and add ice‐cold PBS to melt the matrigel. Cultures were then centrifuged and dissociated by pipetting from disrupting organoids into small pieces and signal cells. After removing the supernatant, the left crypt pellets were then resuspended in PBS and counted under a light microscope. Approximately 50 crypt‐like pellets were resuspended in Matrigel and followed by plating, as above. The passage number of cultures between 8 and 16 was used for further analysis.
2.6. 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide assay
A hundred microliters of CRC live metastasis organoids, in logarithmic phase and at a density of 4 × 103 cells/well, was inoculated in a sterile 96‐well plate. The cells were divided into the control group (0 ng/mL, HGF) and different concentrations (20, 40, and 70 ng/mL) of HGF as the experimental groups. Each group was supplemented with different concentrations of HGF for 0, 24, 48, 72, and 96 hours, followed by 10 µL of 5 mg/mL 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) into each well. The reaction was quenched with 100 µL/well dimethyl sulfoxide. The absorbance was measured at 490 nm (A‐value) against the enzyme standard using HGF duration as the abscissa. The values of the cell proliferation curve were plotted.
2.7. Transwell assay
The cells treated with HGF were harvested as a single cell suspension at a density of 2 × 105cells/mL in the presence of low‐serum culture medium. Hundred microliters of this suspension was inoculated in the upper chamber of the transwell and 30% FBS was added into the lower chamber, and the assembly was incubated at 37°C, 5% CO2 for 6 hours. The nontransferred cells were removed by a cotton swab. The cells in the lower chamber were fixed and stained using crystal violet. An average of five enumerations was utilized for the evaluation of cell invasion.
2.8. c‐Met RNA interference lentivirus design, packaging, and transduction
The short‐hairpin RNA (shRNA) design software (OriGene; OriGene Technologies Inc., MD) was used to determine the target gene sequence 5′‐TCAACTTCTTTGTAGGCAATA‐3′, as the candidate; and the synthetic sequence 5′‐TTCTCCGAACGTGTCACGT‐3′ as a control. The candidate sequence did not show homology with any known human gene sequence. The sequence of the single‐stranded DNA was synthesized including the interference sequence. It was cloned into the GV‐115 vector, and the product was transformed into DH5α cells. PCR screening and DNA sequencing confirmed the coding sequence of the recombinant plasmid lentivirus particles peroxisome proliferator‐activated receptor gamma coactivator‐lenti viral (PGC‐LV) and its auxiliary packaging vector plasmids helper 1.0, helper 2.0. These constructs were transfected into 293T cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. The virus drops were evaluated by fluorescence shRNA‐Met‐Lv was transduced into cells, and after 72 hours, the green fluorescent protein (GFP) expression was observed using a fluorescence microscope. A fluorescence rate >80% was considered positive. The cells were collected for further qPCR detection.
2.9. Flow cytometry
Transfected cells were collected after 72 hours. Configured mark fluid: The fluid could tag 1 × 105~1 × 106 cells per 100 µL, including Annexin V‐fluorescein isothiocyanate (FITC) 1 µL and propidium iodide 10 µL. Cells were incubated for 5 minutes away from light at room temperature after re‐suspension and then mixed. Four hundred microliters of buffer was added and the cell apoptosis rate detected by flow cytometry under 488 nm excitation wavelength. Red fluorescence (>600 nm) and side scatter were measured. 20 000 events were collected at least.
2.10. Statistical analysis
The statistical analyses were performed using the SPSS16.0 software. The data were presented as means + SD. A comparison between the two groups was carried out using the independent t test and between multiple groups using the single factor analysis of variance. A P value less than 0.05 was considered statistically significant for all analyses.
3. RESULTS
3.1. Expression of c‐Met in primary CRC and liver metastasis
We assess the c‐Met expression in CRC and the corresponding colon mucosa, CRC live metastasis, and normal live tissues. To do this, we first compared the c‐Met mRNA expression between groups by using real‐time quantitative reverse transcription polymerase chain reaction (qPCR). Quantitative analysis revealed that c‐Met mRNA expression significantly increased in specimens of CRC (the mean Ct = 22.736 ± 0.111) compared with corresponding colon mucosa; nevertheless, the CRC live metastasis exhibited even higher c‐Met expression than primary CRC groups (the mean Ct = 32.327 ± 0.093) (Figure 1A). To confirm this difference, we then assess the c‐Met protein expression between groups by Western blot analysis. We consistently found that c‐Met protein expression in CRC cases with live metastasis was clearly highest among all the groups (Figure 1B and 1C). These findings indicated that c‐Met was activated in CRC and continue to be activated during the course of the CRC metastasis.
Figure 1.
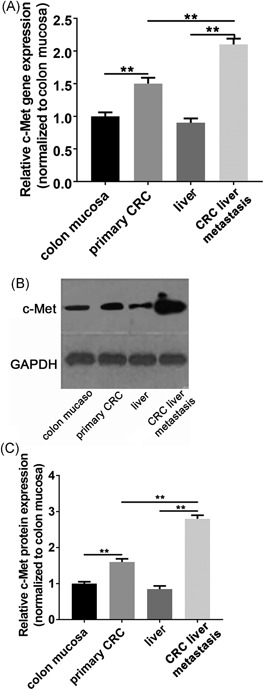
The c‐Met expression in primary CRC and CRC liver metastasis. A, The qPCR analysis revealed a significant increase in the relative c‐Met mRNA levels in CRC and CRC liver metastasis samples compared with either colon mucosa or liver controls, in which the CRC liver metastasis have the highest c‐Met expression. B, The representative image indicating c‐Met protein levels were upregulated in CRC and CRC liver metastasis samples as seen by immunoblot analysis. C, The quantification analysis of western blot. Data present as mean ± SEM. ** represents P < 0.01. CRC, colorectal cancer
3.2. Prognostic significance of c‐Met expression in different clinicopathologic parameters
To investigate the prognostic significance of c‐Met in CRC live metastasis, we studied the clinicopathological correlation of c‐Met expression at RNA level and protein level in 96 patients with CRC live metastasis. Univariate analysis revealed that the expression levels of RNA and protein for c‐Met positively correlated with the tumor stages and, at the same time, correlated with lymph node metastasis, indicating that they contributed to the development of liver metastasis. Multivariate analysis indicated that the gender, age, tissue differentiation degree are all independent prognostic factors (Table 1). These correlation results suggest that c‐Met expression strongly correlated with CRC live metastasis and that manipulating c‐Met signaling would be a useful strategy to suppress tumor invasion and metastasis.
Table 1.
The relationship of C‐Met mRNA and protein expression with the clinicopathologic features of CRC
| Clinicopathologic features | n | c‐MetmRNA | t/F | P | c‐Met protein | t/F | P |
|---|---|---|---|---|---|---|---|
| △Ct | c‐Met/GAPDH | ||||||
| Sex | |||||||
| Male | 54 | 5.309 ± 0.544 | 1.316 | 0.258 | 0.312 ± 0.272 | 0.802 | 0.481 |
| Female | 42 | 6.651 ± 0.593 | 0.070 ± 0.042 | ||||
| Age | |||||||
| >60 | 58 | 7.526 ± 0.882 | 1.803 | 0.169 | 0.323 ± 0.271 | 0.849 | 0.458 |
| ≤60 | 38 | 5.446 ± 0.591 | 0.071 ± 0.041 | ||||
| Tissue differentiation degree | |||||||
| Low | 29 | 6.818 ± 0.482 | 3.773 | 0.100 | 0.313 ± 0.274 | 0.881 | 0.470 |
| Medium | 36 | 8.226 ± 0.618 | 0.071 ± 0.039 | ||||
| High | 31 | 6.280 ± 0.780 | 0.325 ± 0.270 | ||||
| Malignant tumors stage | |||||||
| II | 18 | 5.450 ± 0.557 | 19.732 | 0.002 | 0.165 ± 0.090 | 5.333 | 0.047 |
| II | 22 | 12.437 ± 1.648 | 0.093 ± 0.006 | ||||
| IV | 56 | 17.558 ± 0.862 | 0.322 ± 0.013 | ||||
| Lymph node metastasis | |||||||
| With | 47 | 6.216 ± 0.738 | 3.076 | 0.037 | 0.283 ± 0.014 | 8.307 | 0.010 |
| Without | 49 | 15.113 ± 0.145 | 0.073 ± 0.006 | ||||
| Tumor site | |||||||
| Rectal cancer | 52 | 5.709 ± 0.546 | 2.070 | 0.107 | 0.252 ± 0.251 | 1.165 | 0.309 |
| Colon cancer | 44 | 7.051 ± 0.593 | 0.032 ± 0.013 | ||||
Abbreviation: CRC, colorectal cancer.
3.3. Expression of c‐Met colon cancer cell lines and CRC liver metastases organoids.
To investigate the role of c‐Met in CRC metastasis, we first assess the mRNA expressions of c‐Met in various human colon cancer cell lines as well as CRC liver metastases organoids by using real‐time PCR. The CRC liver metastases organoids were derived from Human pathological specimens with patient consent. Before resection, all six patients with CRC had progressed to clear live metastasis. In addition to CRC liver metastases, one patient had lung metastases and one patient had lymph node metastases.We then extracted genomic DNA from all the colon cell lines and CRC liver metastases organoids to analyze c‐Met copy number variations. Quantitative analysis revealed a significant increase in c‐Met relative expression, with a more than twofold increase, in CRC liver metastases organoids compared with CRC cell lines (Figure 2A). These results indicated that c‐Met expression was significantly upregulated in CRC live metastasis.
Figure 2.
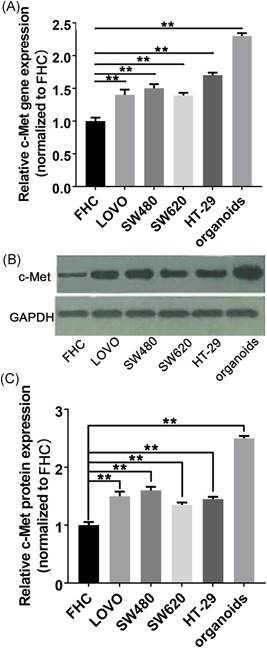
The c‐Met expression in CRC cells lines and CRC organoids. A, The qPCR analysis revealed a significant increase of the relative c‐Met mRNA levels in CRC cell lines and CRC organoids compared with FHC control cell line, in which the CRC organoids have the highest c‐Met expression. B, The representative image indicates that CRC cell lines and CRC organoids exhibited a significant upregulation of c‐Met protein expression by Western blot analysis with CRC organoids showing a more significant increase in c‐Met expression. C, The quantification analysis of Western blot. Data present as mean ± SEM. ** represents P < 0.01. CRC, colorectal cancer
We then assess c‐Met protein expression in CRC colon cell lines as well as in CRC live metastasis organoids by Western Blot analysis. The representative blot bands indicate c‐Met expressed levels in all groups. The quantitative results show that the c‐Met protein expression in CRC live metastasis organoids was significantly higher than the CRC cell lines (Figure 2B and 2C). Taken together, these data confirmed that c‐Met expression was significantly increased in CRC live metastasis.
3.4. High concentrations of HGF promotes CRC metastasis proliferation and invasion
Since the HGF/c‐Met signal pathway plays an important role in tumor proliferation, invasion, and metastasis, we then explore whether HGF is also related to the proliferation and invasion progression of cancer cells. For this, patient‐derived CRC liver metastases organoids were seeded in 96‐well plates with various concentration of HGF, and their proliferation was analyzed using the MTT assay for the next 4 days. In comparison with other lower HGF, cells treated with a concentration of 70 ng/mL HGF exhibited a significantly higher optical density (OD) value. Notably, we also found that SW480 cells reach their plateau of OD value at 48 hours at almost all four different HGF concentration according to the according to the growth curve (Figure 3A), suggesting that interfering cell proliferation of CRC liver metastases organoids by anti‐HGF should start well before this 48‐hour time window.
Figure 3.
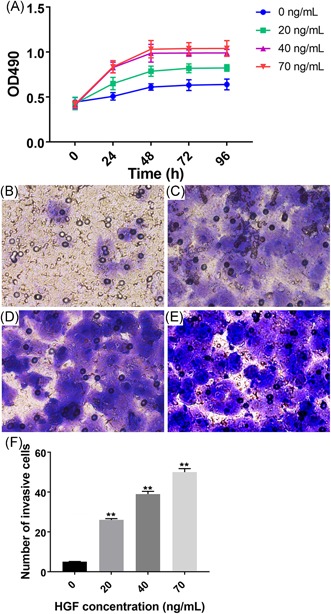
HGF administration increases the proliferation and metastasis of CRC liver metastasis organoids. A, Cell proliferation was increased in CRC liver metastasis organoids by HGF administration. B‐E, Representative images of transwell assays indicate cell migration was progressively increased with HGF concentration at 96 h after administration. F, The quantitative analysis revealed that cell migration is HGF‐dependent, with the greatest number of invasive cells in liver organoids treated with 70 ng/mL of HGF. Data present as mean ± SEM. ** represents P < 0.01. CRC, colorectal cancer; HGF, hepatocyte growth factor; OD, optical density
To examine the effects of HGF on cell migration, a transwell chamber assay was performed. The number of migrated cells was monitored after HGF treatment with four different concentration of for the next 4 days. In line with a previous observation in cell proliferation, cell treated with a concentration of 70 ng/mL HGF showed the greatest number of migrated cells compared to other lower concentrations of HGF treatment (Figure 3B‐F), indicating that HGF treatment also increases cell migration in a dose‐dependent fashion in CRC liver metastases organoids.
3.5. Downregulation of c‐Met expression in CRC liver metastases organoids inhibits cell proliferation and migration
Since we found that increased HGF‐induced c‐Met expression was able to enhance cell proliferation and migration in CRC liver metastases organoids, it can be reasonably hypothesized that downregulation of c‐Met expression would suppress tumor cell proliferation and invasion. Thus, we used RNA interference (RNAi) technology to reduce c‐Met levels in CRC liver metastases organoids. We established lentivirus constructs that express shRNA with scramble sequence (c‐Met‐siCon), and four shRNAs directed against the c‐Met mRNA (c‐Met‐siRNA). To determine the transduction efficacy of all four c‐Met‐siRNA constructs, we analyzed c‐Met mRNA expression by qPCR after transfection. As expected, the results demonstrate that the c‐Met‐siRNA‐4 significantly downregulated the c‐Met expression with an outstanding effect among other constructs (Figure 4A). Therefore, we chose c‐Met‐siRNA‐4 for the further experimental use. To further validate the interfering effect of c‐Met‐siRNA‐4, we further monitor c‐Met expression at the protein level. Similarly, cells transfected with c‐Met‐siRNA‐4 dramatically reduced c‐Met protein expression compared with blank organoids or organoids expressing c‐Met‐siCon (Figure 4B and 4C).
Figure 4.
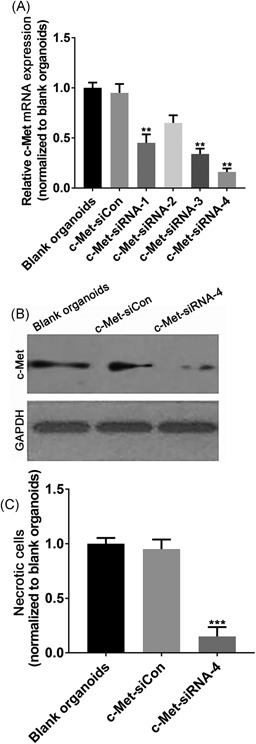
shRNA‐mediated knockdown of c‐Met expression in CRC liver metastasis organoids. A, The qPCR analysis demonstrates a reduction of c‐Met mRNA in c‐Met‐siRNA‐infected organoids compared with uninfected blank organoids or organoids infected with c‐Met‐siRNA‐Con, in which the c‐Met‐siRNA‐4 exert a significantly better inhibition effect than others. B, Immunoblot analysis showing that c‐Met‐siRNA‐4 significantly reduced c‐Met protein expression in CRC liver metastasis organoids compared with blank organoids or organoids infected with c‐Met‐siRNA‐Con. C, The quantitative analysis of Western blot. Data present as mean ± SEM. ** represents P < 0.01. CRC, colorectal cancer; shRNA, short‐hairpin RNA
We then evaluated the functional effect of downregulating c‐Met expression on the transfected CRC liver metastases organoids. To explore whether downregulation of c‐Met inhibits tumor cell proliferation, the apoptosis assay of liver metastases organoids in different RNAi conditions was carried out by flow cytometry. At 72 hours after transfection, the CRC liver metastases organoids cells transfected with c‐Met‐siRNA‐4 construct exhibited a significant increase in the apoptosis rate compared with control cells or the infecting cells with negative constructs. A quantitative analysis of the apoptosis rate revealed an approximately 20% increase in c‐Met siRNA‐4 CRC liver metastases organoids compared with c‐Met‐Con CRC liver metastases organoids or the untreated CRC liver metastases organoids (Figure 5A‐D). These results suggest that downregulating c‐Met expression inhibits tumor cell proliferation in vitro.
Figure 5.
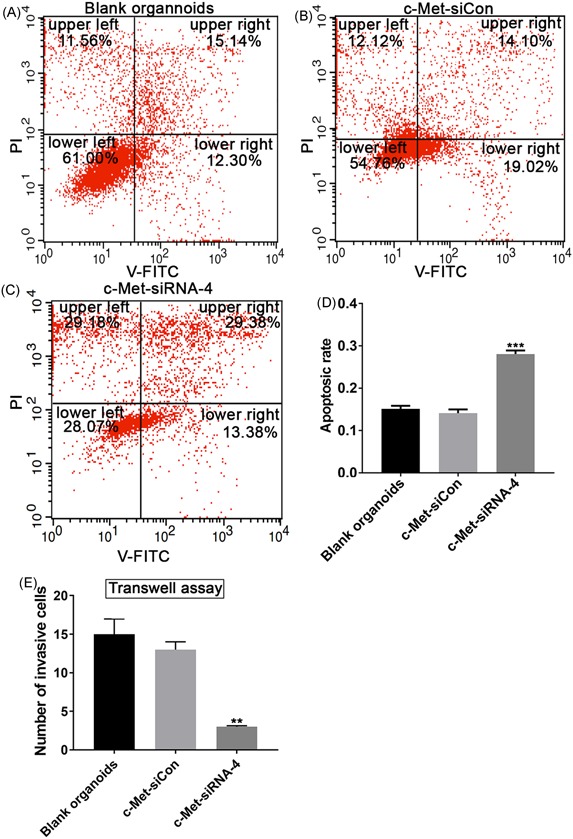
shRNA‐mediated knockdown of c‐Met expression inhibited proliferation and invasion of CRC liver metastasis. A‐C, Resuspended organoids were incubated with Annexin V‐FITC and PI and were analyzed using flow cytometry. The results showed apoptosis rate in blank CRC liver organoids (A) or organoids infected by c‐Met‐siCon (B) or c‐Met‐siRNA‐4 (C). In each panel, the lower left quadrant shows the percentage of V‐FITC negative and PI negative cells, and the upper left indicates only PI positive cells which are necrotic. The lower right shows V‐FITC only positive cells, which indicate early apoptotic cells and the upper right represents the rate of V‐FITC/PI double positive cells which are late apoptotic cells. D, The quantitative analysis of the percentage of necrotic cells. E, The quantitative analysis of invasive changes by transwell assay in blank CRC liver organoids, or organoids treated with c‐Met‐siCon or c‐Met‐siRNA‐4, in which the knockdown of c‐Met expression by c‐Met‐siRNA‐4 showed a significant reduction in the number of invasive cells. Data present as mean ± SEM. ** represents P < 0.01. *** represents P < 0.001. CRC, colorectal cancer; FITC, fluorescein isothiocyanate; PI, propidium iodide
In addition, we also take the advantage of the transwell chamber assay to assess tumor cell migration in response to RNAi, which downregulates c‐Met expression in CRC liver metastases organoids. There was a significant decrease in the number of migrated cell in c‐Met‐siRNA‐4‐infecting cells compared with CRC liver metastases organoids or CRC liver metastases organoids transfected by negative control (Figure 5E), indicating that downregulating c‐Met expression inhibits tumor cell migration in vitro.
4. DISCUSSION
In the current study, we first found a positive correlation between c‐Met expression and the clinicopathological factors of CRC tumor stages and lymph node metastasis, indicating that c‐Met expression would be a significant prognostic factor in CRC metastasis. Using CRC metastasis patient tissue samples and cell lines, we identified that high c‐Met expression (mRNA and protein level) was associated with CRC live metastasis. We finally modulated c‐Met expression in CRC live metastasis organoids in vitro and found that upregulation of c‐Met expression in CRC liver metastasis organoids through high HGF treatment enhanced tumor cell proliferation, invasion and migration, and vice versa upo downregulation of c‐Met expression in CRC liver metastases organoids by RNAi technology. Taken together, the results of our study indicate that targeting c‐Met receptors may be a promising therapeutic strategy for CRC metastasis patients to attenuate CRC metastasis.
It is well known that the lymph node metastasis incidence and its depth of tumor invasion are prognostic factors in malignant metastasis, including CRC metastasis.18, 28, 29 Recently, HGF/c‐Met signaling in various malignant tumors has been reported to be effective biologic markers for monitoring malignant potential.30, 31, 32 In the current study, we have revealed the importance of c‐Met expression in the prognosis and treatment of CRC metastasis. We demonstrated that c‐Met expression was correlated with lymph node metastasis and the depth of tumor invasion. Although no clear relationship between serum HGF levels and clinical symptoms has been identified,33, 34, 35 high serum HGF secreted in the tumor microenvironment has been thought to contribute to various aggressive disease behavior and progression. The relationship between serum HGF levels and malignant metastasis was not assessed in this study but would have an interesting link.
Studies have reported that the c‐Met overexpression is associated with both primary tumors and in nodal metastasis. However, whether c‐Met expression significantly increased in CRC live metastasis remains largely uncertain. To understand the potential role of c‐Met amplification in CRC liver metastases, we took advantage of qPCR and western blot analysis and revealed a significant increase of c‐Met expression in live metastasis patient specimens and organoids compared with primary patients with CRC and CRC cell lines, respectively. Although substantial evidence has been shown that c‐Met expression is associated with invasion and tumor budding in CRC,15, 18, 21 to the best of our knowledge, this is convincing evidence showing that c‐Met expression is significantly higher in CRC liver metastasis than human primary CRC, indicating that c‐Met expression is highly correlated with the depth of CRC tumor invasion and penetration. Our results are also in line with other recent findings that c‐Met amplification or overexpression is able to facilitate colonization of the liver.
A growing body of evidence‐based research shows that c‐Met tyrosine kinase inhibitors can block c‐Met signaling and reverse tumor growth in different cancers.15, 36, 37, 38 Recent studies have shown that MET signaling in lung cancer and gastric cancer is sensitive to inhibition using a specific tyrosine kinase inhibitor.39 These findings suggested that overexpressed c‐Met may act as an important biomarker for CRC targeted therapy. We therefore test this possibility in our study. Our data have shown that upregulation of HGF/c‐Met signaling increased CRC live metastasis organoids proliferation and invasion, whereas, downregulation of HGF/c‐Met signaling in organoids significantly inhibit the tumor growth and migration.
In conclusion, our study indicates the association with advanced stage CRC and liver metastases, suggesting that c‐Met gene amplification plays an important role in CRC progression and metastases. The intervention of HGF/c‐Met signaling may provide an invaluable target for exploring novel drug therapies in CRC metastasis. Collectively, these data demonstrate that a small reduction in c‐Met protein levels leads to profound biological effects, and potential c‐Met inhibitors may be of therapeutic value in the treatment of colon cancer.
ACKNOWLEDGMENT
The present study was funded by the grants from the National Natural Science Foundation of China (grant no. 81101874 and no. 81172362) and the Social Development Foundation of Shaanxi Province (grant no. 2017SF‐101).
CONFLICT OF INTERESTS
The authors declare that there are no conflicts of interest.
AUTHOR CONTRIBUTIONS
J‐FY contributed to the experimental design, the analysis and interpretation of data, and the drafting of the manuscript. X‐JL, L‐KY, SH, and J‐BZ contributed to the acquisition of the data and analysis. X‐LZ, P‐HZ, and LZ contributed to the patient recruitment and data collection. G‐BW provided editorial support for the manuscript. X‐JS provided the funding support, experimental design support, and editorial support for this study. All authors approved the final manuscript.
Yao J‐f, Li X‐j, Yan L‐k, et al. Role of HGF/c‐Met in the treatment of colorectal cancer with liver metastasis. J Biochem Mol Toxicol. 2019;33:e22316 10.1002/jbt.22316
References
REFERENCES
- 1. Danese E., Montagnana M., Ann. Transl. Med. 2017, 5(13), 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Berretta M., Alessandrini L., De Divitiis C., Nasti G., Lleshi A., Di Francia R., Facchini G., Cavaliere C., Buonerba C., Canzonieri V., Crit. Rev. Oncol. Hematol. 2017, 111, 103. [DOI] [PubMed] [Google Scholar]
- 3. Nakayama G., Tanaka C., Kodera Y., Gastrointest. Tumors 2013, 1(1), 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pisani P., Parkin D. M., Bray F., Ferlay J., Int. J. Cancer 1999, 83(1), 18. [DOI] [PubMed] [Google Scholar]
- 5. Ullah M. F., Fleming C. A., Mealy K., The Surgeon. 2018, 16, 350. [DOI] [PubMed] [Google Scholar]
- 6. Jin H., Liu X., Li V., Ding Y., Yun S., Liu F., Zhou S., Song Y., Ni M., Int. J. Colorectal Dis. 2009, 24(1), 41. [DOI] [PubMed] [Google Scholar]
- 7. Stagnitti A., Barchetti F., Barchetti G., Pasqualitto E., Sartori A., Glorioso M., Gigli S., Buonocore V., Monti M. L., Marini A., Mele C., Stagnitti F., Laghi A., Eur. Rev. Med. Pharmacol. Sci. 2015, 19(9), 1645. [PubMed] [Google Scholar]
- 8. Zarzavadjian Le Bian A., Denet C., Tabchouri N., Donatelli G., Wind P., Louvet C., Bennamoun M., Christidis C., Perniceni T., Fuks D., Gayet B., Langenbecks Arch. Surg. 2018, 403, 443. [DOI] [PubMed] [Google Scholar]
- 9. Arnold M., Sierra M. S., Laversanne M., Soerjomataram I., Jemal A., Bray F., Gut 2017, 66(4), 683. [DOI] [PubMed] [Google Scholar]
- 10. Chinese Journal of Cancer, Chin. J. Cancer 2017, 36(1), 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lampe J. W., J. Am. Coll. Nutr. 2011, 30(5 Suppl 1), 464S. [DOI] [PubMed] [Google Scholar]
- 12. Nguyen K. T., Gamblin T. C., Geller D. A., Ann. Surg. 2009, 250(5), 831. [DOI] [PubMed] [Google Scholar]
- 13. Sotelo M. J., García‐Paredes B., Aguado C., Sastre J., Díaz‐Rubio E., World J. Gastroenterol. 2014, 20(15), 4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De Greef K., Rolfo C., Russo A., Chapelle T., Bronte G., Passiglia F., Coelho A., Papadimitriou K., Peeters M., World J. Gastroenterol. 2016, 22(32), 7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schweiger T., Starkl V., Glueck O., Glogner C., Traxler D., Jedamzik J., Liebmann‐Reindl S., Birner P., Streubel B., Klepetko W., Hoetzenecker K., Eur. J. Cardiothorac. Surg. 2016, 49(4), 1103 discussion 1111. [DOI] [PubMed] [Google Scholar]
- 16. Woo J. K., Kang J. H., Kim B., Park B. H., Shin K. J., Song S. W., Kim J. J., Kim H. M., Lee S. J., Oh S. H., Oncotarget 2015, 6(27), 24047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li F., Zhu Y. T., Cell. Signal. 2015, 27(4), 860. [DOI] [PubMed] [Google Scholar]
- 18. Sun Y., Liu W., Ma G., Gao D., Jiang Y., Liu Q., Du J., Int. J. Med. Sci. 2013, 10(5), 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hiscox S. E., Hallett M. B., Jiang W. G., Puntis M. C. A., Nakamura T., Cancer Invest. 1997, 15(6), 513. [DOI] [PubMed] [Google Scholar]
- 20. Tahara E., J. Cancer Res. Clin. Oncol. 1993, 119(5), 265. [DOI] [PubMed] [Google Scholar]
- 21. Giordano S., di Renzo M. F., Olivero M., Mondino A., Zhen Z., Medico E., Comoglio P. M., Eur. J. Cancer Prev. 1992, 1(Suppl 3), 45. [DOI] [PubMed] [Google Scholar]
- 22. Osada S., Gotoh A., Yokoi R., Tsuchiya H., Sakuratani T., Sasaki Y., Okumura N., Hayashi H., Mukai T., Anticancer Res. 2018, 38(2), 737. [DOI] [PubMed] [Google Scholar]
- 23. Mira A., Morello V., Céspedes M. V., Perera T., Comoglio P. M., Mangues R., Michieli P., Oncotarget 2017, 8(24), 38193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Seneviratne D., Ma J., Tan X., Kwon Y. K., Muhammad E., Melhem M., DeFrances M. C., Zarnegar R., Gastroenterology 2015, 148(1), 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun Y., Li Z., Xu Z., Zhonghua Wei Chang Wai Ke Za Zhi 2014, 17(6), 598. [PubMed] [Google Scholar]
- 26. Budinska E., Wilding J., Popovici V. C., Missiaglia E., Roth A., Bosman F., Delorenzi M., Bodmer W., Tejpar S., J. Clin. Oncol. 2013, 31(15 [Google Scholar]
- 27. Morelli M. P., Meyer A. M., Tentler J. J., Pitts T. M., Eur. J. Cancer Supplements 2008, 6(12), 111. [Google Scholar]
- 28. Peyravian N., Larki P., Gharib E., Nazemalhosseini‐Mojarad E., Anaraki F., Young C., McClellan J., Ashrafian Bonab M., Asadzadeh‐Aghdaei H., Zali M., Biomedicines 2018, 6(1), 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lou X., Qi X., Zhang Y., Long H., Yang J., J. Surg. Oncol. 2013, 108(4), 230. [DOI] [PubMed] [Google Scholar]
- 30. Bradley C. A., Dunne P. D., Bingham V., McQuaid S., Khawaja H., Craig S., James J., Moore W. L., McArt D. G., Lawler M., Dasgupta S., Johnston P. G., Van Schaeybroeck S., Oncotarget 2016, 7(48), 78932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matsui S., Osada S., Tomita H., Komori S., Mori R., Sanada Y., Takahashi T., Yamaguchi K., Yoshida K., Int. J. Oncol. 2010, 37(2), 289. [DOI] [PubMed] [Google Scholar]
- 32.S. Benvenuti, M. Milan, P. M. Comoglio. (2018) Discovery and function of the HGF/MET and the MSP/RON kinase signaling pathways in cancer. Extracellular targeting of cell signaling in cancer: strategies directed at MET and RON receptor tyrosine kinase pathways. Wiley, USA.
- 33. Ma T. H., Gao C. C., Xie R., Yang X. Z., Dai W. J., Zhang J. L., Yan W., Wu S. N., Neoplasma 2017, 64(6), 880. [DOI] [PubMed] [Google Scholar]
- 34. Moore A. E., Greenhough A., Roberts H. R., Hicks D. J., Patsos H. A., Williams A. C., Paraskeva C., Carcinogenesis 2009, 30(10), 1796. [DOI] [PubMed] [Google Scholar]
- 35. Otte J. M., Schmitz F., Kiehne K., Stechele H. U., Banasiewicz T., Krokowicz P., Nakamura T., Fölsch U. R., Herzig K. H., Digestion 2000, 61(4), 237. [DOI] [PubMed] [Google Scholar]
- 36. Long Z. H., Bai Z. G., Song J. N., Zheng Z., Li J., Zhang J., Cai J., Yao H. W., Wang J., Yang Y. C., Yin J., Zhang Z. T., Anticancer Res. 2017, 37(8), 4345. [DOI] [PubMed] [Google Scholar]
- 37. de Klerk D., Honeywell R., Jansen G., Peters G., Cancers 2018, 10(12), 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roskoski R., Pharmacol. Res. 2018, 133, 35. [DOI] [PubMed] [Google Scholar]
- 39. Al‐Maghrabi J., Emam E., Gomaa W., Saggaf M., Buhmeida A., Al‐Qahtani M., Al‐Ahwal M., BMC. Cancer 2015, 15, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]