

Abstract
As a relatively new discipline, quantitative systems pharmacology has seen a significant increase in the application and utility of drug development. One area that could greatly benefit from such an approach is in the proarrhythmia assessment of new drugs. The Comprehensive In Vitro Proarrhythmia Assay (CiPA) Initiative is a global public–private partnership project that has developed an integrated approach using mechanistic in silico models for proarrhythmia risk prediction. Progress to date has led to the formation of the International Council on Harmonisation Implementation Working Group to revise regulatory guidelines via the Questions‐and‐Answers process to address the best practices for proarrhythmia models and how they can impact clinical drug development. This article reviews the CiPA in silico model‐development process, focusing on its unique development and validation strategy, and summarizes the lessons learned as consideration points for the ongoing implementation of CiPA‐like in silico models in drug development.
In 2011, the National Institutes of Health sponsored two workshops to review the state of the art in systems biology and (quantitative) pharmacology, which jointly published a white paper proposing a merger of these two approaches via the emerging discipline of quantitative (and) systems pharmacology (QSP).1 Since then, there has been a dramatic increase of interest from academia and the pharmaceutical industry to apply this system‐level modeling approach to drug discovery and development.2 This approach was supported by global regulatory agencies. Both the European Medicines Agency and the US Food and Drug Administration (FDA) published guidance documents about physiologically‐based pharmacokinetic (PBPK) modeling,3, 4 a subdiscipline of QSP. Recently, an International Council on Harmonisation (ICH) guideline addendum (E11 (R1)) was published that introduces modeling and simulation, including QSP approaches, to clinical investigation processes for pediatric drugs across major regulatory agencies around the world.4
The mechanistic nature of QSP models requires some degree of quantitative understanding of the underlying system (molecules, cells, organs, and disease states) to build a mechanistic “platform” model to represent the physiology/pathophysiology and their interplay with the drugs.5 One of the biological systems that has received extensive quantitative investigation through both experimental and computational approaches is the cellular apparatus responsible for cardiac electrophysiology,6 which can be disturbed by drugs resulting in arrhythmias (proarrhythmia). Because of the long history and relatively mature status of cardiac electrophysiology modeling, there has been increasing interest in applying in silico approaches with QSP‐type models to cardiac safety prediction, especially for proarrhythmia risk assessment.6 In line with this trend, in 2013 a think tank jointly sponsored by the Cardiac Safety Research Consortium, Health and Environmental Sciences Institute, and FDA proposed a new cardiac safety paradigm—the Comprehensive In Vitro Proarrhythmia Assay (CiPA)—in which mechanistic in silico models are proposed to be the primary tool to predict the risk of drug‐induced arrhythmias.7 Subsequently, the In Silico Working Group (ISWG), together with other CiPA working groups, was convened by the public–private partnership CiPA initiative that includes partners from global regulatory agencies, industry, and academia.8 The recent progress9 resulted in the formation of an ICH Implementation Working Group to develop Questions and Answers to the ICH S7B (nonclinical) and E14 (clinical) guidelines on how novel approaches, such an mechanistic in silico models, can aid in determining the proarrhythmic risk of a drug and inform clinical development.10
This document will give a brief overview of the history of the assessment of drug‐induced heart rate–corrected QT (QTc) prolongation and torsade de pointes (TdP), the mechanistic understanding of its biology and pharmacology, the development of a prototype model under CiPA, and the implementation considerations for using CiPA‐type models in the new cardiac safety regulatory paradigm. The focus will be on the distinct features of the CiPA in silico approach and lessons learned regarding using QSP models to inform decision making, as these are easily transportable to other areas of QSPs. The technical details of cardiac electrophysiology modeling will not be discussed extensively here; however, interested readers could refer to excellent reviews, such as Davies et al.6
TdP and Current Regulatory Paradigm
Since the 1920s, certain cardiac drugs (such as quinidine) were found to cause a rare but severe adverse event of sudden loss of consciousness and cardiac death.11 It was not until the 1960s when this phenomenon was associated with a distinctive form of polymorphic ventricular tachycardia, which was termed torsade de pointes to characterize the typical twisting appearance of QRS complexes on the electrocardiogram (ECG).12 Since then, it was discovered that a wide range of drugs with various indications have the potential to cause TdP, and one of the most common features shared by these torsadogenic drugs is blocking the potassium channel encoded by the human ether‐à‐go‐go related gene (hERG), which subsequently leads to delayed ventricular repolarization and a prolonged QT interval on ECG.11 It was estimated that 12% of ambulatory sudden death may be caused by drug‐induced TdP.13 The wide recognition of this fatal adverse event led to a series of drug withdrawals between the 1990s and early 2000s and subsequent establishment of two ICH regulatory guidelines in 2005 for cardiac proarrhythmia assessment: the nonclinical guideline S7B and clinical guideline E14.14
ICH S7B describes a nonclinical strategy to assess drug‐induced delay in ventricular repolarization.15 The core battery of test assays under S7B include the following two types of studies: electrophysiology studies to test a pharmaceutical agent's potency of blocking the hERG channel, the cardiac ion channel responsible for the potassium current IKr (rapidly activating delayed rectifier potassium current), and in vivo studies to investigate the test substance's impact on ECG changes, especially QT prolongation. ICH E14 provides guidelines about conducting clinical trials to evaluate a drug's potential to delay cardiac repolarization, focusing on thorough QT studies to assess drug effects on the QT or QTc interval.16
The current cardiac safety paradigm described in ICH S7B and E14, which focuses on hERG block potency and delayed cardiac repolarization as measured by QTc interval prolongation, has been highly successful in preventing withdrawals of newly approved drugs because of an unacceptable risk of TdP when these drugs are widely used in the patient population.7 However, it is recognized that this paradigm is sensitive but not specific for predicting whether a drug will cause TdP.7, 17, 18 For instance, some drugs (such as verapamil and ranolazine) that significantly block hERG and/or prolong QTc and would have been red flagged by the current S7B and/or E14 guidelines probably have little TdP risk.7 Mechanistic markers that directly measure the risk of TdP rather than that of delayed ventricular repolarization (QT prolongation) are needed.
Comprehensive Understanding of TdP and the Use of In Silico Models in Proarrhythmia Risk Assessment
Tremendous progress has been made in our understanding of the cellular machinery that controls ionic flows and membrane voltages of cardiac cells under normal and pathophysiological conditions. Now it is known that, in addition to the hERG potassium channel, many other ion channels can critically contribute to shaping the trajectory of membrane voltage during a heartbeat (action potential). Some ion channels allow the flow of positive charge into the cell, such as Nav1.5 (peak/late sodium currents, or INa/INaL) and Cav1.2 (L‐type calcium current, or ICaL). Others (including hERG) allow the flow of positive charge out of the cell, such as KvLQT1/mink (slow‐delayed rectifier potassium current, or IKs), Kir2.1 (inward rectifier potassium current, or IK1), and Kv4.3 (transient outward potassium current, or Ito). Although classifying cross‐membrane ionic flows into inward and outward currents may be an oversimplification of the complex system underlying cellular electrophysiology, as there are cardiac currents that are composed of multiple types of charged ions that flow in both directions (such as the sodium‐calcium exchanger current19), it provides a conceptual framework to understand the mechanisms of repolarization and arrhythmia through the balance of two opposing forces. If this balance is tilted toward inward current during repolarization, either through a block of outward current(s) or increase of inward current(s), repolarization may be significantly slowed (delayed repolarization) or even reversed into premature depolarization (early afterdepolarization).11, 20 Early afterdepolarizations from subregions of the heart can propagate to other regions, triggering shifting focal activations and amplifying action potential heterogeneity to create vulnerable regions of reentry,11, 12, 20 which were implicated as the primary mechanisms underlying various ventricular arrhythmias including TdP and ventricular fibrillation.20
The deep understanding of the proarrhythmia mechanisms and the long history of cardiac electrophysiology modeling have prompted the use of in silico models to predict drug‐induced arrhythmias in recent years. Typically, a drug's potency of blocking selected inward and outward currents was measured through in vitro ion channel experiments to derive half‐inhibition concentrations (IC50s), which were then input into a computational model to output a quantitative marker (metric) indicating the drug's propensity of inducing arrhythmias.6 The complexity of the models used by different studies varies dramatically, ranging from empirical models using statistical or machine‐learning algorithms to directly link pharmacology measurements to risk classification,21, 22, 23 to more QSP‐like action‐potential models simulating the physiological interaction between various ion channels within individual cardiomyocytes,17, 24, 25, 26, 27, 28 and to more complex systems where whole‐heart‐level simulation (>20 million simulated cells) was used to recapitulate drug‐induced TdP directly in silico.29
The Cipa In Silico Approach
Encouraged by the promising results from earlier modeling studies, the CiPA initiative launched the in silico workstream in 2013 to develop mechanistic models for TdP risk assessment.7 To keep the balance between biological reality and model complexity, a QSP‐type approach was selected in which the base model or “platform” of physiology is based on a widely used model of a mathematically detailed representation of adult human ventricular cardiomyocyte (O'Hara Rudy model30). Because of the intended use of CiPA models in the regulatory setting, the CiPA in silico approach adopts a development and validation pipeline that is different from most other in silico studies. Typically, risk‐prediction models are validated by cross‐validation, where the data set used for model development is randomly divided into training and validation subsets in an iterative fashion. As usually done in modeling studies for TdP risk prediction, a series of potential metrics (or regression models with different formats) will be evaluated by leave‐one‐out cross‐validation, and the metric with the best performance will be selected.17, 21, 24 Because during repeated data set divisions a given drug will be assigned to a training subset in some but validation subset in other iterations, such a strategy will blur the boundary between training and validation and effectively use information from the whole data set for metric selection. An alternative strategy is to use a prospective design in which the validation data set is kept “hidden” and not available for model development and metric selection during the training phase. The use of such an independent or “holdout” validation data set is recommended by principles or guidelines from related fields, such as complex model reliability assessment31 and biomarker qualification.32
The CiPA in silico workstream adopted the latter validation strategy. A dedicated Cardiac Safety Research Consortium Compound Selection Team for CiPA selected 28 CiPA compounds of known clinical TdP liabilities and divided them into three TdP risk categories (high, intermediate, and low) according to publicly available data and consensus expert opinion within the group.8 Of note, the 28 CiPA drugs were culled from a longer list of candidates based on which drugs could be associated with a consensus TdP risk category among the Compound Selection Team. Although a relatively small number of drugs was used, this primarily data‐driven approach for the CiPA paradigm is distinct from the expert opinion–based ICH S7B/E14 paradigm. This list of drugs was further separated into a training set of 12 and a validation set of 16 drugs. Of note, the number of training drugs11 was predetermined,7 and the partition of drugs into the two sets ensured that both sets included drugs from the three TdP risk categories and contained desired electrophysiological properties. The data for 16 validation drugs were not used (remained “hidden”) until model training was completed. A stepwise approach (Figure 1) was taken to ensure the strict separation and formal documentation of each step during model training and validation. The selected metric qNet, which represents the electric charge carried by (the area under the curve of) the net current Inet (the difference between the four selected outward currents IKr, IKs, IK1, and Ito and the two selected inward currents INaL and ICaL), is not only mechanistically indicative of the distance from early afterdepolarization but also was selected based entirely on 12 training drugs by comparing with alternative metrics.33 Also, predetermined by the training drugs were the model structure, parameters, and any calculation/simulation methods.33, 34, 35 Together, these prespecified features effectively “froze” the model before validation and prevented it from being affected (informed) by the validation data.
Figure 1.

The Comprehensive In Vitro Proarrhythmia Assay (CiPA) in silico model validation strategy. The CiPA in silico model validation strategy is shown as a flowchart. The model training process includes model optimization and metric development using published human cardiomyocyte experimental data originally used for O'Hara Rudy model development and newly acquired in vitro drug block data against various cardiac currents for the 12 training compounds. A “freezing” step makes sure the model structure and parameters, the metric, the simulation procedure, classification thresholds, and acceptable performance measures were all prespecified prior to the model validation process, where the torsade de pointes risk profiles of the 16 validation drugs were predicted. After successful validation, the classification thresholds for risk prediction can be updated using all 28 drugs. This procedure was approved by the CiPA Steering Committee and time stamped in the validation strategy document (Supplementary Text 1 of ref. 36) before the actual validation took place. The figure was adapted from ref. 36 and is licensed under CC BY 4.0. ©2018 The authors.
Before the model was “frozen,” it went through a fine‐tuning process in which different aspects of the model were gradually determined based on training data. The first determined was the structure, the physiological (gating) parameters, and parameterization procedure for the pharmacological (drug‐binding) parameters, of the hERG dynamic model.35 Following this, the other physiological parameters (ion channel conductance) of the cardiomyocyte model were adjusted and the metric qNet was selected.33 Building on these calibration steps, a formal uncertainty quantification process was established in which the calculation methods for pharmacodynamic parameters (drug binding and potency) and the qNet metric were updated to derive probability distributions of predicted risk.34 This uncertainty quantification procedure also made the observation that, to achieve a low prediction error with acceptable uncertainty, one could use pharmacological data from four of the seven currents originally selected by CiPA8 and focus on plasma concentrations that are around the maximum free therapeutic concentration (Cmax).34 Based on these observations, the scoring system (torsade metric score, defined as the qNet value averaged across 1×, 2×, 3×, and 4× Cmax), the number of tested ion currents as drug‐specific model input (IKr, INa, INaL, and ICaL), along with all other aspects of the model determined by previous publications, were “frozen” by the validation strategy document (Supplementary Text 1 in ref. 36) immediately prior to validation.
Another distinct feature of the CiPA in silico approach is a more mechanistically detailed pharmacodynamic component. Traditionally, drug actions on ion channels are simplified by a pore‐block model using the IC50s and Hill coefficients to scale down conductance of each ionic current. Although easy to interpret, this “static” view of drug block is not realistic, as drug block potency is dependent on the experimental conditions (such as voltage protocol) used to elucidate it.37 A more realistic representation is to use a mechanistically detailed model (such as a Markov model) to describe the drug binding/unbinding kinetics on different states (conformations) of the channel, which provides intrinsic information not dependent on the experimental protocol.38, 39 The ISWG replaced the hERG/IKr component of the base O'Hara Rudy model with such a Markov submodel (Figure 2 a), which can distinguish drugs with different binding kinetics captured by a dynamic voltage protocol35 (Figure 2 b). Together with other model adjustments based on training data,33 this resulted in a prototype model CiPAORdv1.034 to be evaluated by the validation data set later.36
Figure 2.
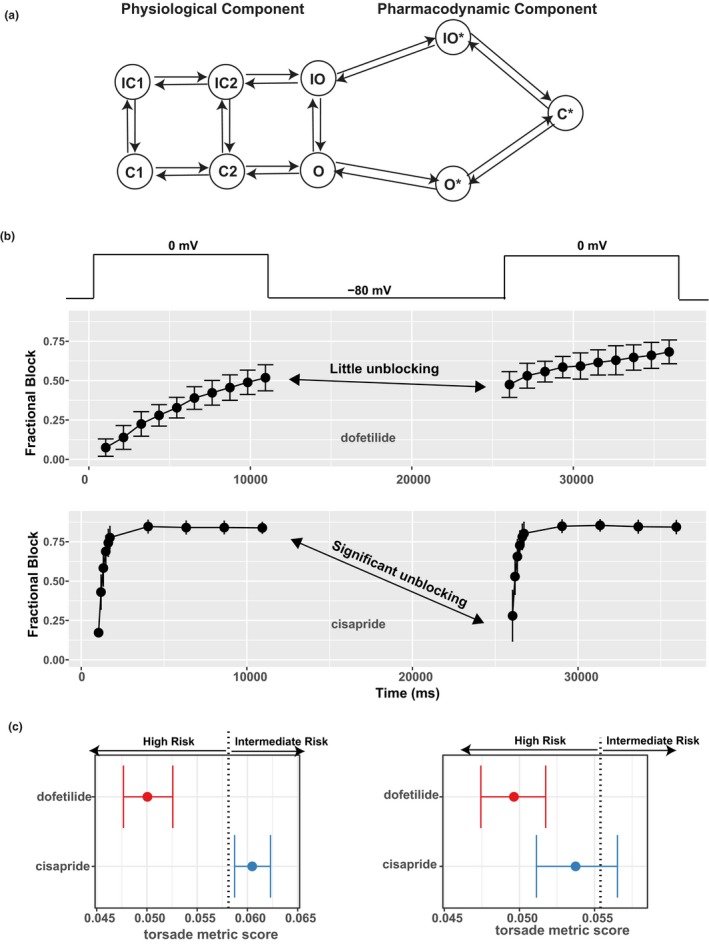
The pharmacodynamic submodel of drug–human ether‐à‐go‐go related gene (hERG) dynamic interaction and its impact on risk prediction. (a) A diagram of the hERG submodel describing the channel‐gating (physiological component) and drug‐binding (pharmacodynamic component) processes of the hERG–drug interaction. Each node represents a distinct state (conformation) of the channel. States with an asterisk indicate drug‐bound states, whereas those without are drug‐free channel states. O, open state; C, closed state; IO, inactivated open state; IC, inactivated closed state; O*, open‐bound state; IO*, inactivated open‐bound state; C*, closed‐bound state. Note that there are two inactivated closed (IC) and closed (C) states, and they are distinguished by a numeric suffix (such as IC1, IC2 for the two IC states). (b) Using a dynamic voltage protocol to manifest the different binding kinetics of two selective hERG blockers, dofetilide (at 30 nM) and cisapride (at 300 nM). The voltage protocol is briefly shown in the top panel as voltage steps alternating between −80 and 0 mV. For the main panels, x‐axis is the time in milliseconds (ms) after the protocol was applied to the cell. y‐axis is fractional block 1 − I drug/I control, where I drug is the measured hERG current after drug application, and I control is the hERG current without drug. During the 0 mV steps, the channel is accumulated in the open state, and the binding/blocking process (O → O* → C*) in a takes place and can be measured as the development of fractional block. During the −80 mV step, the unbinding/unblocking process (C* → O* → O) takes place, and some blocking effect can be relieved. Dofetilide tends to be “trapped” in the C* state, resulting in little unblocking during the −80 mV step (fractional block changed little as shown by the double‐headed arrow). Cisapride has less tendency to be trapped, resulting in significant unblocking during the −80 mV step (fractional block decreased a lot as shown by the double‐headed arrow). Note that only two 0 mV steps are shown, but the actual protocol contains 10 such steps. (c) The use of the hERG‐binding kinetic information, but not half‐inhibition concentrations (IC50s), can distinguish different torsade de pointes risk liabilities associated with dofetilide and cisapride. Left: The hERG‐binding submodel in a was parameterized by the full data obtained from the dynamic protocol in b across multiple concentrations and then used within the O'Hara Rudy model to calculate the torsade metric scores (qNet values averaged across 1×, 2×, 3×, and 4× maximum free therapeutic plasma concentration (Cmax)).34 The torsade metric scores for the 12 training drugs were used by ordinal logistic regression to determine the classification thresholds. One of the thresholds (value 0.058 as published in ref. 36) that separates high risk from intermediate risk is shown as a vertical dotted line. Dofetilide and cisapride are correctly classified on the left and right of this threshold, respectively. Right: The same practice as on the left but IC50s, instead of hERG‐binding kinetics, were used within the O'Hara Rudy model to calculate the torsade metric scores. The hERG IC50s were calculated using the fractional block at the last timepoint of the dynamic protocol across multiple concentrations in b. The vertical dotted line is the classification threshold determined the same way as on the left panel, the only difference being the use of IC50s instead of binding kinetics within the model. Under this situation without considering the binding kinetics, dofetilide and cisapride both have most of their score distributions in the high‐risk category. Note that the prediction in left, but not right, is consistent with the Comprehensive In Vitro Proarrhythmia Assay (CiPA) risk categories.
A third feature of the CiPA in silico modeling approach is the development of statistical methods to characterize and quantify in vitro data variability and translate that into uncertainty in risk prediction. Experimentally measured drug actions on ion channels unavoidably show variability even when collected by the same protocol within the same laboratory.40 Such variability can be quantified as probability distributions of drug‐specific model input (uncertainty characterization) and propagated through the model to derive the probability distributions of the model output (metric), indicating uncertainty in the predicted risk (uncertainty propagation).41 The ISWG developed such a method to estimate the joint distributions of pharmacodynamic parameters (binding kinetics, IC50s, etc.) based on training data34 and then applied it to validation.36 In contrast, most of the other published models for TdP risk prediction did not consider variability and uncertainty.17, 21, 22, 23, 24, 27, 28, 29 A few studies considered intersubject variability in physiological parameters,25, 26 similar to the “virtual population” approach commonly used by other areas of QSP applications such as PBPK.42 However, how to estimate the joint distribution and interdependency of such a large number of parameters is still an open question.43 Although future versions of proarrhythmia models under CiPA may incorporate such population variabilities, the uncertainty in pharmacology measurements directly affects drug‐specific model inputs and reflects the baseline confidence in the predicted risk. An example of using confidence intervals of qNet from CiPAORdv1.0 to assess TdP risk is shown in Figure 2 c.
The strict design of the validation strategy of the CiPA in silico approach resulted in a clear “document trail” of model development. Although most studies developing TdP risk prediction models publish model calibration (training) and performance evaluation (validation) together within a single document (usually publications), the CiPA in silico model training process alone was documented by a series of publications33, 34, 35, 44 as well as FDA Advisory Committee Meeting minutes.45 After the training was completed and before the validation began, the model, metric, validation data set and methodology, plus performance measures and acceptable performance levels were all “frozen” in a time‐stamped validation strategy document (Supplementary Text 1 in ref. 36). Subsequently, the model was taken to predict validation drugs, and the validation report document (Supplementary Text 2 in ref. 36) indicated that the preregistered model CiPAORdv1.0 and metric qNet were able to meet all prespecified performance measures and outperform other alternative models/metrics tested. Such a rigorous design provides high confidence that the prototype model developed by ISWG has acceptable prediction accuracy for TdP risk assessment under the new CiPA paradigm.
Lessons Learned and Considerations for Implementation
The successful completion of the validation stage using the prototype model CiPAORdv1.0, along with progress made by other workstreams of CiPA and similar research worldwide, led to ICH forming an Implementation Working Group to develop Questions and Answers for the S7B and E14 guidelines.10 At this early stage of CiPA implementation, it is worthwhile to revisit some lessons learned from the recent efforts in developing TdP risk‐prediction models. This could provide some points for consideration not only for implementing CiPA‐like approaches in the forthcoming new cardiac safety paradigm but also for other areas where it is desirable to use QSP models to inform clinical decision making (such as risk assessment).
Model development should follow a prespecified validation plan and be monitored by a multidisciplinary team of experts
Within the CiPA initiative, even though the ISWG is the main working group responsible for developing and validating the in silico model according to a prespecified plan, multiple groups with different expertise contributed to this process. The Ion Channel Working Group selected cardiac ion currents and designed the in vitro voltage protocols to generate experimental data as drug‐specific input to the model. The Cardiac Safety Research Consortium Compound Selection Team selected reference drugs, designated training and validation data sets, and performed risk categorization. The steering committee designed the validation strategy, coordinated the effort from different groups, routinely monitored the model development progress, and preapproved the validation strategy before the model validation was initiated. Outside of CiPA, the progress made by various CiPA working groups has been periodically presented to external experts across academia, industry, and regulatory agencies through dedicated conferences and workshops for evaluation and discussion.9 Such a multidisciplinary effort brought together experts from different areas such as computational modeling, statistics, electrophysiology, safety pharmacology, regulatory science, and clinical cardiology, which ensured both the stringency of the validation process and the applicability of the developed model. After the development of the prototype model CiPAORdv1.0, a community‐wide, multidisciplinary approach is again being used to establish general principles for not only updating the prototype model but also validating new models for proarrhythmia risk prediction.
Such a multidisciplinary approach with a prospective validation strategy could be used by other areas of QSP. Although the term validation is sometimes used in different contexts with different meanings in the QSP literature,42, 46 the statistical meaning of “validation” or “model assessment” is to evaluate a model's prediction error on new data.48 Consistent with the statistical considerations for the evidentiary framework for biomarker qualification,32 the QSP model development process could use a prospective design similar to the CiPA strategy that prespecifies training and validation processes, as it provides the strongest level of evidence to support the predictivity of a model.32 CiPA's multidisciplinary evaluation process, conceptually similar to the qualification process for drug development tools,49 could also be used by other QSP projects to aid in the evaluation of the regulatory acceptance of developed models.
Model input data should be obtained from experimental assays that follow standardized protocols and use robust quality control criteria
The CiPA TdP risk assessment model is dependent on the quality of the experimental data (e.g., IC50 values or binding kinetics for cardiac ion channels) used for drug‐specific model input. Therefore, it is essential that experimental conditions are robust to give a valid measurement of these pharmacodynamic parameters and the training and validation drugs use the same experimental conditions to generate the input data. Currently, most of the published TdP risk‐prediction models combined previously reported pharmacology data collected with various experimental protocols for model building and validation.17, 24, 25, 28 As measured drug block potency is known to be dependent on the experimental protocols,37 the use of literature data for model input may affect the internal consistency of the model (i.e., a drug may appear to have different block potency on the same channel because of differences in protocols). In addition, our internal investigation suggests quality control also plays a large role in creating data consistency.36 To address this issue, the CIPA ion channel and cardiac myocyte workstreams are collaborating with subject matter experts to develop scientific white paper documents that describe the standardized experimental protocols and quality control criteria.
Although for some QSP model applications such as PBPK, an individual model is developed for a specific drug, other QSP models are intended to be used for many different drugs, similar to the CiPA TdP risk‐prediction model. For these models, experimental standardization may also be needed to ensure that different drugs are assessed in a consistent manner.
The uncertainty in pharmacodynamic measurements (i.e., drug‐specific model input values) should be quantified and considered when predicting clinical outcomes
As shown by the CiPA in silico approach,34, 36 the uncertainty in nonclinical data can be quantified and translated into uncertainty in the predicted risk (metric score). Typically, a drug's metric score will be compared with predetermined threshold(s) for risk classification. The uncertainty in the metric score, represented as a confidence interval in Figure 2 c, provides a means to assess the uncertainty in risk prediction. For example, the distribution of the metric scores for a certain drug could be compared with the threshold by reporting the probability (fraction of the distribution) in each category, respectively, as was done in the CiPA in silico validation study.36 Alternatively, an arbitrarily defined boundary of the confidence interval could be compared with the classification threshold. For instance, in Figure 2 c (left or right panel) the lower confidence limit of the torsade metric score (for the metric qNet, the lower the more dangerous) may be compared with the thresholds to assign each drug into a distinct risk category. The exact method (distribution, boundary, etc.) to express the uncertainty in risk prediction could be optimized to fit the purpose of each model.
QSP models usually have large numbers of physiological parameters to simulate the underlying healthy or diseased conditions. Although such parameter uncertainty (e.g., intersubject variability) may contribute significantly to the final prediction uncertainty, appropriate statistical methodology to estimate the joint distributions of such large numbers of parameters is still an open research field.46 The CiPA's strategy of focusing on the probability distributions of pharmacodynamic parameters, which are directly input to the model, may be a good starting point for many other QSP models.
The uncertainty in pharmacokinetics needs to be considered to explore a range of clinical exposures and their impact on risk assessment
Another factor that has uncertainty is clinically relevant concentrations to be explored in the model for risk assessment. Similar to many other adverse events, the propensity of a drug to cause TdP is dependent on the drug concentration.11 Typically, proarrhythmia models in the model development stage would classify a drug into a distinct risk category based on fixed concentrations for the ease of performance evaluation (such as the average of 1× to 4× maximum free therapeutic concentration used by CiPAORdv1.036). However, when evaluating the TdP risk for a new drug, a model should explore a range of clinically relevant drug concentrations. Importantly, the range of exposure for risk profiling will need to be determined for each drug based on its pharmacokinetic properties. For some drugs, exposures can substantially increase because of drug–drug interactions (e.g., terfenadine18), cardiac tissue accumulation, a decrease in protein binding, or drug overdoses (e.g., loperamide49).
For other QSP models where concentration‐dependent risk/efficacy profiles are to be established, this principle to determine the appropriate clinical exposure for model assessment based on each drug's pharmacokinetic profile also applies.
The amount of experimental data to be collected by the nonclinical assays may be tailored for individual drugs
Although CiPA initially aimed to test a drug's ability to inhibit a comprehensive list of cardiac ion channels with in vitro assays to generate a complete profile of drug action to inform a risk‐prediction model, it is likely that not all ion channels play an equally important role for TdP risk assessment. Based on the pharmacology of the 28 CiPA drugs, it was determined that four cardiac currents (IKr/hERG, INa, INaL, ICaL) were essential currents for TdP risk prediction and warranted a standard pharmacological investigation.34, 36 Drugs that potentially block the other ion channels as off‐targets were not likely to be selected into the reference drug list. This suggests that the selection of four “essential currents” may not be generalizable to all drugs, as TdP can be caused by inhibition of other cardiac ion channels or by other mechanisms such as the inhibition of hERG channel trafficking to the cell membrane. This calls for a triaged approach, where the amount of additional experimental data (e.g., number of additional ion channels to be assayed) are determined for each drug based on the concordance between nonclinical assays of the four essential currents and in vivo ECG findings.
This individualized approach is consistent with the principles suggested by the current S7B guideline15 and also applicable to other QSP models that rely on a series of nonclinical assays to capture the pharmacodynamic effects and assess clinical outcomes such as risk.
The consistency between nonclinical and clinical data needs to be assessed
CiPA‐like mechanistic models use pharmacodynamic parameters from nonclinical experimental data to predict clinical risk profiles. There are many factors, such as the selection of experimental protocol, the data quality, drug metabolism, formation of active metabolites, and distribution of the drug into cardiac tissue, that may result in a discrepancy between nonclinical and clinical ECG and safety data. Under those circumstances, the possible reasons for such inconsistencies as well as the impact of accepting a model prediction in the face of data discrepancy will need to be evaluated. The use of clinical ECG parameters and new ECG biomarkers to assess potential discrepancies in the model TdP risk prediction are essential components of CiPA's integrated, multicomponent approach.8
Similar checkpoints could be applied to many other QSP models that use nonclinical data as model input, which can be compared against clinical observations, when available, to evaluate whether the pharmacodynamic effects captured by nonclinical assays can be translated to clinical settings.
Promising Roles and Open Issues of Qsp Models in a New Cardiac Safety Paradigm
With the ongoing discussion of ICH S7B/E14 Questions and Answers, one could envision that a new international cardiac safety paradigm is approaching, where mechanistic and quantitative interpretation of nonclinical data through QSP‐type models may be used to help clinical assessment of TdP risk and de‐risk drugs that prolong QTc but actually have low TdP liability.50 Other than the proposed role of minimizing the use and informing the design of clinical trials like thorough QT studies, there are other ways CiPA‐type QSP models could be used in drug development in the context of cardiac safety. Models could be developed to predict the risk of other types of arrhythmias beyond TdP or cover proarrhythmic mechanisms other than direct ion channel block. The mechanistic nature of QSP models makes it possible to shed light on specific pathways perturbed by a drug, even if these pathways are secondary to the direct drug targets. Similarly, pharmacodynamic drug–drug interaction could be explored in a QSP model by integrating diverse proarrhythmic mechanisms in a systematic manner. In addition, QSP models could help with dose adjustments by defining at what clinical exposure a toxicity might occur. Doing this requires exploring a wide range of clinical exposures beyond those already studied and have data available, which is straightforward with QSP models, but difficult, sometimes impossible, for certain empirical models (e.g., the MICE (Multiple Ion Channel Effects) model for TdP prediction21). When the uncertainty quantification is expanded to include intersubject variability, QSP models could be used to simulate virtual populations and identify susceptible subpopulations or individuals who are most at risk to develop drug‐induced arrhythmia events.
To fill these promising roles, different types of QSP models may need to be developed with different contexts of use, and even for existing models that have passed an initial validation stage (such as CiPAORdv1.0), they may need to go through an iterative process of identifying knowledge gaps, collecting/refining data, and model updating, as commonly done with many QSP models.46 This raises a series of questions, such as how to evaluate the regulatory acceptability of different models in a consistent manner and how to maintain a “validated” status during the updating of an existing model. These open questions probably need a community‐wide discussion. As a first step, some general principles for validating proarrhythmia risk‐prediction models were discussed at a CiPA in silico breakout session51 and a white paper is being developed to start the discussion of such a topic.
Concluding Remarks
The severe adverse event of arrhythmias, especially TdP, offers a unique opportunity for QSP‐type modeling approaches to realize their promise of helping drug development by reducing the attrition rate along the development pipeline and informing clinical development.2 The rarity of TdP makes it difficult to be directly captured during clinical trials, and for many drugs this arrhythmia is not seen during clinical development but appears several years postapproval.52 The CiPA in silico approach represents a model‐informed drug‐development strategy that offers a predictive solution at both early and late drug‐development stages to help the selection of the right candidates to move forward and prevent the premature termination of drugs with perceived cardiac safety concern based on less specific surrogate markers under the current paradigm.7 The successful validation of the CiPAORdv1.0 model by a highly stringent, clinical trial–like validation strategy, and the lessons learned from the development of not only CiPAORdv1.0 but also related TdP risk‐prediction models in the field are helping to pave the way for the development and implementation of the next generation of QSP‐based approaches53 for cardiac safety assessment.
Funding
The Comprehensive In Vitro Proarrhythmia Assay (CiPA) in silico work was supported by the Research Participation Program at the Center for Drug Evaluation and Research, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the US Department of Energy and the US Food and Drug Administration.
Conflict of Interest
The authors declared no competing interests for this work.
Disclaimer
This report is not an official US Food and Drug Administration guidance or policy statement. No official support or endorsement by the US Food and Drug Administration is intended or should be inferred.
References
- 1. Quantitative and Systems Pharmacology Workshop Group. NIH white paper <https://www.nigms.nih.gov/Training/Documents/SystemsPharmaWPSorger2011.pdf> (2011).
- 2. Knight‐Schrijver, V.R. , Chelliah, V. , Cucurull‐Sanchez, L. & Le Novere, N. The promises of quantitative systems pharmacology modelling for drug development. Comput. Struct. Biotechnol. J. 14, 363–370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. European Medicines Agency. Qualification and reporting of physiologically based pharmacokinetic (PBPK) modeling and simulation (draft guideline) <https://www.ema.europa.eu/en/qualification-reporting-physiologically-based-pharmacokinetic-pbpk-modelling-simulation;> (2016).
- 4. US Food and Drug Administration. Physiologically based pharmacokinetic analysis – format and content (guidance for industry) <https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM531207.pdf> (2018).
- 4. US Food and Drug Administration. E11 (R1) addendum: clinical investigation of medicinal products in the pediatric population: ICH <https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM530012.pdf> (2018).
- 5. Musante, C.J. , Ramanujan, S. , Schmidt, B.J. , Ghobrial, O.G. , Lu, J. & Heatherington, A.C. Quantitative systems pharmacology: a case for disease models. Clin. Pharmacol. Ther. 101, 24–27 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davies, M.R. et al Recent developments in using mechanistic cardiac modelling for drug safety evaluation. Drug Discov. Today 21, 924–938 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sager, P.T. , Gintant, G. , Turner, J.R. , Pettit, S. & Stockbridge, N. Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium. Am. Heart J. 167, 292–300 (2014). [DOI] [PubMed] [Google Scholar]
- 8. Colatsky, T. et al The Comprehensive in Vitro Proarrhythmia Assay (CiPA) initiative—update on progress. J. Pharmacol. Toxicol. Methods 81, 15–20 (2016). [DOI] [PubMed] [Google Scholar]
- 9. Strauss, D.G. et al Comprehensive In Vitro Proarrhythmia Assay (CiPA) update from a Cardiac Safety Research Consortium/Health and Environmental Sciences Institute/FDA Meeting. Ther. Innov. Regul. Sci. 10.1177/2168479018795117.[e‐pub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 10. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. Questions & answers: clinical and non‐clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential (concept paper) <https://www.ich.org/products/guidelines/efficacy/article/efficacy-guidelines.html#13-3> (2018).
- 11. Roden, D.M. Cellular basis of drug‐induced Torsades de Pointes. Br. J. Pharmacol. 154, 1502–1507 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murakawa, Y. Focal and reentrant mechanisms of Torsades de Pointes: EAD, reentry, or chimera? J. Arrhythm. 27, 28–37 (2011). [Google Scholar]
- 13. Bayes de Luna, A. & Coumel, P. , Leclercq, J.F. Ambulatory sudden cardiac death: mechanisms of production of fatal arrhythmia on the basis of data from 157 cases. Am. Heart J. 117, 151–159 (1989). [DOI] [PubMed] [Google Scholar]
- 14. Vicente, J. et al Mechanistic model‐informed proarrhythmic risk assessment of drugs: review of the “CiPA” initiative and design of a prospective clinical validation study. Clin. Pharmacol. Ther. 103, 54–66 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. The nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals S7B. (2005). [PubMed]
- 16. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. The clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs E14. (2005).
- 17. Lancaster, M.C. & Sobie, E.A. Improved prediction of drug‐induced Torsades de Pointes through simulations of dynamics and machine learning algorithms. Clin. Pharmacol. Ther. 100, 371–379 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Redfern, W.S. et al Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc. Res. 58, 32–45 (2003). [DOI] [PubMed] [Google Scholar]
- 19. Viswanathan, P.C. & Rudy, Y. Pause induced early afterdepolarizations in the long QT syndrome: a simulation study. Cardiovasc. Res. 42, 530–542 (1999). [DOI] [PubMed] [Google Scholar]
- 20. Weiss, J.N. , Garfinkel, A. , Karagueuzian, H.S. , Chen, P.S. & Qu, Z. Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm 7, 1891–1899 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kramer, J. et al MICE models: superior to the HERG model in predicting Torsade de Pointes. Sci. Rep. 3, 2100 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mistry, H.B. Complex versus simple models: ion‐channel cardiac toxicity prediction. PeerJ. 6, e4352 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Parikh, J. , Gurev, V. & Rice, J.J. Novel two‐step classifier for Torsades de Pointes risk stratification from direct features. Front. Pharmacol. 8, 816 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mirams, G.R. et al Simulation of multiple ion channel block provides improved early prediction of compounds' clinical torsadogenic risk. Cardiovasc. Res. 91, 53–61 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Passini, E. et al Human in silico drug trials demonstrate higher accuracy than animal models in predicting clinical pro‐arrhythmic cardiotoxicity. Front. Physiol. 8, 668 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abbasi, M. , Small, B.G. , Patel, N. , Jamei, M. & Polak, S. Early assessment of proarrhythmic risk of drugs using the in vitro data and single‐cell‐based in silico models: proof of concept. Toxicol. Mech. Methods 27, 88–99 (2017). [DOI] [PubMed] [Google Scholar]
- 27. McMillan, B. , Gavaghan, D.J. & Mirams, G.R. Early afterdepolarisation tendency as a simulated pro‐arrhythmic risk indicator. Toxicol. Res. (Camb). 6, 912–921 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krogh‐Madsen, T. , Jacobson, A.F. , Ortega, F.A. & Christini, D.J. Global optimization of ventricular myocyte model to multi‐variable objective improves predictions of drug‐induced Torsades de Pointes. Front. Physiol. 8, 1059 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Okada, J. et al Screening system for drug‐induced arrhythmogenic risk combining a patch clamp and heart simulator. Sci. Adv. 1, e1400142 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. O'Hara, T. , Virag, L. , Varro, A. & Rudy, Y. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput. Biol. 7, e1002061 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Council, N.R. Assessing the Reliability of Complex Models: Mathematical and Statistical Foundations of Verification, Validation, and Uncertainty Quantification (The National Academies Press, Washington, DC, 2012). [Google Scholar]
- 32. US Food and Drug Administration. Biomarker qualification: evidentiary framework <https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM628118.pdf> (2018).
- 33. Dutta, S. et al Optimization of an in silico cardiac cell model for proarrhythmia risk assessment. Front. Physiol. 8, 616 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chang, K.C. et al Uncertainty quantification reveals the importance of data variability and experimental design considerations for in silico proarrhythmia risk assessment. Front. Physiol. 8, 917 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li, Z. et al Improving the in silico assessment of proarrhythmia risk by combining hERG (human ether‐a‐go‐go‐related gene) channel‐drug binding kinetics and multichannel pharmacology. Circ. Arrhythm. Electrophysiol. 10, e004628 (2017). [DOI] [PubMed] [Google Scholar]
- 36. Li, Z. et al Assessment of an in silico mechanistic model for proarrhythmia risk prediction under the CiPA Initiative. Clin. Pharmacol. Ther. 105, 466–475 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kirsch, G.E. et al Variability in the measurement of hERG potassium channel inhibition: effects of temperature and stimulus pattern. J. Pharmacol. Toxicol. Methods 50, 93–101 (2004). [DOI] [PubMed] [Google Scholar]
- 38. Di Veroli, G.Y. , Davies, M.R. , Zhang, H. , Abi‐Gerges, N. & Boyett, M.R. High‐throughput screening of drug‐binding dynamics to HERG improves early drug safety assessment. Am. J. Physiol. Heart Circ. Physiol. 304, H104–H117 (2013). [DOI] [PubMed] [Google Scholar]
- 39. Di Veroli, G.Y. , Davies, M.R. , Zhang, H. , Abi‐Gerges, N. & Boyett, M.R. hERG inhibitors with similar potency but different binding kinetics do not pose the same proarrhythmic risk: implications for drug safety assessment. J. Cardiovasc. Electrophysiol. 25, 197–207 (2014). [DOI] [PubMed] [Google Scholar]
- 40. Elkins, R.C. et al Variability in high‐throughput ion‐channel screening data and consequences for cardiac safety assessment. J. Pharmacol. Toxicol. Methods 68, 112–122 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pathmanathan, P. , Shotwell, M.S. , Gavaghan, D.J. , Cordeiro, J.M. & Gray, R.A. Uncertainty quantification of fast sodium current steady‐state inactivation for multi‐scale models of cardiac electrophysiology. Prog. Biophys. Mol. Biol. 117, 4–18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kirouac, D.C. How do we “validate” a QSP model? CPT Pharmacometrics Syst. Pharmacol. 7, 547–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mirams, G.R. , Pathmanathan, P. , Gray, R.A. , Challenor, P. & Clayton, R.H. Uncertainty and variability in computational and mathematical models of cardiac physiology. J. Physiol. 594, 6833–6847 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li, Z. , Dutta, S. , Sheng, J. , Tran, P.N. , Wu, W. & Colatsky, T. A temperature‐dependent in silicomodel of the human ether‐a‐go‐go‐related (hERG) gene channel. J. Pharmacol. Toxicol. Methods (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. US Food and Drug Administration . Meeting of the Pharmaceutical Science and Clinical Pharmacology Advisory Committee 2017 <https://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/ucm535520.htm> (accessed March 15, 2017).
- 46. Ribba, B. et al Methodologies for Quantitative Systems Pharmacology (QSP) models: design and estimation. CPT Pharmacometrics Syst. Pharmacol. 6, 496–498 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sheng, J. et al Characterization of loperamide‐mediated block of hERG channels at physiological temperature and its proarrhythmia propensity. J. Pharmacol. Toxicol. Methods 88(Pt 2), 109–122 (2017). [DOI] [PubMed] [Google Scholar]
- 50. Strauss, D.G. The potential role of CiPA on drug discovery, development, and regulatory pathways <https://cardiac-safety.org/wp-content/uploads/2018/05/03-David-Strauss-Talk-1-CiPA-Potential-Role-CSRC-5-20-2018-v2.pdf> (2018).
- 51. CiPA in silico breakout session—discussion points <https://cardiac-safety.org/wp-content/uploads/2018/05/In-Silico-Breakout-Session.pdf> (2018).
- 52. Darp ö, B. Spectrum of drugs prolonging QT interval and the incidence of Torsades de Pointes. Eur. Heat J. Suppl.. 2001;3(suppl. K):K70–K80. [Google Scholar]
- 53. Allerheiligen, S.R. Next‐generation model‐based drug discovery and development: quantitative and systems pharmacology. Clin. Pharmacol. Ther. 88, 135–137 (2010). [DOI] [PubMed] [Google Scholar]