

Abstract
Avelumab, a human anti–programmed death ligand 1 immunoglobulin G1 antibody, has shown efficacy and manageable safety in multiple tumors. A two‐compartment population pharmacokinetic model for avelumab incorporating intrinsic and extrinsic covariates and time‐varying clearance (CL) was identified based on data from 1,827 patients across three clinical studies. Of 14 tumor types, a decrease in CL over time was more notable in metastatic Merkel cell carcinoma and squamous cell carcinoma of the head and neck, which had maximum decreases of 32.1% and 24.7%, respectively. The magnitude of reduction in CL was higher in responders than in nonresponders. Significant covariate effects of baseline weight, baseline albumin, and sex were identified on both CL and central distribution volume. Significant covariate effects of black/African American race, C‐reactive protein, and immunogenicity were found on CL. None of the covariate or time‐dependent effects were clinically important or warranted dose adjustment.
STUDY HIGHLIGHTS.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑This is the first published analysis of avelumab pharmacokinetics over time.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑What is the dose–exposure relationship for avelumab in cancer patients? Are there any clinically significant covariate predictors of exposure at a given dose?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑This analysis provides a population pharmacokinetics model for avelumab, elucidating its dose–exposure relationship in patients with 14 types of cancer (including the identification of time‐varying clearance in two cancer types), and identifies covariate predictors of avelumab exposure at a given dose, including body weight, baseline albumin and C‐reactive protein, and sex as well as time‐varying clearance.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑The information in this article adds to the growing body of evidence related to changes in clearance over time for monoclonal antibodies used in immuno‐oncology.
Programmed death ligand 1 (PD‐L1) has a well‐established role in the suppression of T‐cell responses and is strongly correlated with cancer prognosis.1, 2, 3 Blockade of the PD‐L1/programmed death 1 interaction therefore presents a rational strategy for cancer immunotherapy. By blocking the interaction between PD‐L1 and programmed death 1, antitumor cluster of differentiation (CD)8–positive T cells are released from the suppressive effects of PD‐L1, restoring cytotoxic T‐cell responses. Preclinical studies suggest that avelumab can also induce antitumor effects mediated by innate immune effector cells.4 Avelumab (Bavencio, EMD Serono; Rockland, Massachusetts) is a human immunoglobulin G1 (IgG1) anti–PD‐L1 monoclonal antibody with a wildtype fragment crystallizable (Fc) region, which has been approved in various countries for the treatment of patients with metastatic Merkel cell carcinoma (mMCC) and for patients with platinum‐treated advanced urothelial carcinoma. Avelumab is also in clinical development for the treatment of other cancer types, including renal cell carcinoma, non‐small cell lung cancer (NSCLC), gastric cancer, and ovarian cancer.4, 5
Avelumab was initially approved at a dose of 10 mg/kg administered intravenously every 2 weeks, and its single‐dose pharmacokinetics (PK) has been reported previously.6 Recently, a time‐dependent decrease in clearance (CL) has been reported for anti–programmed death 1 (nivolumab and pembrolizumab)7, 8 and anti–PD‐L1 (atezolizumab) antibodies,9 which was associated with the magnitude of tumor response.8 In this article, we (i) describe the population PK analysis of avelumab across 14 different cancer types, (ii) assess the impact of intrinsic and extrinsic factors that affect the PK of avelumab, and (iii) assess the impact of treatment response on the PK of avelumab.
Materials and Methods
Patients
Concentration‐time data from three clinical trials were available for analysis. JAVELIN Solid Tumor6, 10 (EMR100070‐001; NCT01772004) was a phase I, open‐label, multiple–ascending‐dose trial to investigate the safety, tolerability, PK, and biological and clinical activities of avelumab in patients with metastatic or locally advanced solid tumors, including an expansion to additional indications, and contributed the data of 1,688 individuals to this analysis. JAVELIN Solid Tumor JPN11, 12 (EMR100070‐002; NCT01943461) was a phase I, open‐label trial to investigate the tolerability, safety, PK, and biological and clinical activities of avelumab in Japanese patients with metastatic or locally advanced solid tumors, with an expansion phase in Japanese patients with gastric cancer, and it contributed data from 51 patients. Finally, JAVELIN Merkel 2005, 13, 14 (EMR100070‐003; NCT02155647) was a phase II, open‐label, multicenter trial to investigate the clinical activity and safety of avelumab in patients with Merkel cell carcinoma and contributed data from 88 patients. In total, 10,637 avelumab serum‐concentration values from 1,827 patients were used in this analysis. All three trials received ethical approval from the relevant institutional review boards, and the procedures followed were in accordance with the Declaration of Helsinki. Table 1 provides a summary of the demographics by study.
Table 1.
Demographic and disease‐related covariates in the studied population
| Covariate | JAVELIN Solid Tumor | JAVELIN Solid Tumor JPN | JAVELIN Merkel 200 | Total population in analysis |
|---|---|---|---|---|
| N | 1,688 | 51 | 88 | 1,827 |
| Nominal dose, n (%) | ||||
| 1 mg/kg | 4 (0.237) | 0 | 0 | 4 (0.219) |
| 3 mg/kg | 13 (0.77) | 5 (9.8) | 0 | 18 (0.985) |
| 10 mg/kg | 1,650 (97.7) | 40 (78.4) | 88 (100) | 1,778 (97.3) |
| 20 mg/kg | 21 (1.24) | 6 (11.8) | 0 | 27 (1.48) |
| Median treatment duration, daysa | 85.1 {NC} (0–951) [0] | 98 {67.8} (0.0429–714) [0] | 140 {NC} (0–617) [0] | 89.8 {NC} (0–951) [0] |
| Median baseline age, yearsa | 63 {60.5} (19–91) [0] | 62 {60.1} (30–77) [0] | 72.5 {68.7} (33–88) [0] | 63 {60.9} (19–91) [0] |
| Median baseline body weight, kga | 71.2 {71.5} (30.4–204) [2] | 55.5 {55.3} (35.2–89.3) [0] | 82.8 {81} (47–153) [0] | 71 {71.4} (30.4–204) [2] |
| Sex, n (%) | ||||
| Male | 854 (50.6) | 35 (68.6) | 65 (73.9) | 954 (52.2) |
| Female | 834 (49.4) | 16 (31.4) | 23 (26.1) | 873 (47.8) |
| Race, n (%) | ||||
| White | 1,311 (77.7) | 0 | 81 (92) | 1,392 (76.2) |
|
Black or African American |
87 (5.15) | 0 | 0 | 87 (4.76) |
| Asian | 152 (9) | 51 (100) | 3 (3.41) | 206 (11.3) |
|
American Indian or Alaska Native |
5 (0.296) | 0 | 0 | 5 (0.274) |
|
Native Hawaiian or other Pacific Islander |
4 (0.237) | 0 | 0 | 4 (0.219) |
| Other | 129 (7.64) | 0 | 1 (1.14) | 130 (7.12) |
| Missing | 0 | 0 | 3 (3.41) | 3 (0.164) |
| AST, U/La | 22 {23.4} (2–210) [0] | 24 {24.1} (12–122) [0] | 26 {27.7} (10–113) [0] | 22 {23.6} (2–210) [0] |
| ALT, U/La | 19 {NC} (0–185) [0] | 16 {16.4} (6–70) [0] | 18.5 {18.3} (5–62) [0] | 19 {NC} (0–185) [0] |
| Albumin, g/La | 39 {38.3} (10–52) [0] | 37 {39} (21–310) [0] | 40.4 {39.8} (24.1–53) [0] | 39 {38.4} (10–310) [0] |
| CRP, mg/La | 11.1 {NC} (0–1,770) [27] | 3.2 {3.58} (0.2–97.1) [0] | 5.9 {7.26} (0.5–275) [2] | 10.7 {NC} (0–1,770) [29] |
| Platelets, 109/La | 251 {251} (71–1,130) [0] | 227 {233} (95–907) [0] | 201 {203} (73–398) [0] | 247 {248} (71–1,130) [0] |
| Total protein, g/La | 70 {70} (45–105) [0] | 64 {64.8} (49–77) [0] | 69 {68.9} (51–89) [0] | 70 {69.8} (45–105) [0] |
| eGFR, mL/minute/1.73 m2 a | 85.9 {84.2} (21.8–398) [0] | 106 {102} (51.4–178) [0] | 75.2 {78.4} (32.6–177) [0] | 85.9 {84.3} (21.8–398) [0] |
| Renal impairment, n (%) | ||||
| None | 699 (41.4) | 18 (35.3) | 25 (28.4) | 742 (40.6) |
| Mild | 637 (37.7) | 20 (39.2) | 43 (48.9) | 700 (38.3) |
| Moderate | 346 (20.5) | 13 (25.5) | 20 (22.7) | 379 (20.7) |
| Severe | 4 (0.237) | 0 | 0 | 4 (0.219) |
| Missing | 2 (0.118) | 0 | 0 | 2 (0.109) |
| Hepatic impairment, n (%) | ||||
| None | 1,455 (86.2) | 42 (82.4) | 67 (76.1) | 1,564 (85.6) |
| Mild | 219 (13) | 9 (17.6) | 20 (22.7) | 248 (13.6) |
| Moderate | 3 (0.178) | 0 | 1 (1.14) | 4 (0.219) |
| Severe | 0 | 0 | 0 | 0 |
| Missing | 11 (0.652) | 0 | 0 | 11 (0.602) |
| PD‐L1 expression, n (%) | ||||
| Negative (<5%) | 672 (39.8) | 31 (60.8) | 54 (61.4) | 757 (41.4) |
| Positive (≥5%) | 447 (26.5) | 9 (17.6) | 20 (22.7) | 476 (26.1) |
| Missing | 569 (33.7) | 11 (21.6) | 14 (15.9) | 594 (32.5) |
| Tumor type, n (%) | ||||
| Adrenocortical carcinoma | 50 (2.96) | 0 | 0 | 50 (2.74) |
| Castration‐resistant prostate cancer | 18 (1.07) | 0 | 0 | 18 (0.985) |
| Colorectal cancer | 21 (1.24) | 0 | 0 | 21 (1.15) |
| Gastric and gastroesophageal junction cancer | 252 (14.9) | 34 (66.7) | 0 | 286 (15.7) |
| Squamous cell carcinoma of the head and neck | 153 (9.06) | 0 | 0 | 153 (8.37) |
| Melanoma | 51 (3.02) | 0 | 0 | 51 (2.79) |
| Merkel cell carcinoma | 0 | 0 | 88 (100) | 88 (4.82) |
| Mesothelioma | 53 (3.14) | 0 | 0 | 53 (2.9) |
| Metastatic breast cancer | 168 (9.95) | 0 (0) | 0 (0) | 168 (9.2) |
| Non‐small cell lung cancer | 340 (20.1) | 0 | 0 | 340 (18.6) |
| Ovarian cancer | 228 (13.5) | 0 | 0 | 228 (12.5) |
| Renal cell carcinoma | 52 (3.08) | 0 | 0 | 52 (2.85) |
| Solid tumors | 53 (3.14) | 17 (33.3) | 0 | 70 (3.83) |
| Urothelial carcinoma | 249 (14.8) | 0 | 0 | 249 (13.6) |
| Tumor size, mma | 60 {56.8} (10–750) [26] | 55.5 {58.5} (15–195) [1] | 62 {65.8} (10–404) [1] | 60 {57.2} (10–750) [28] |
| Number of nontarget lesions, n (%) | ||||
| 1 | 346 (20.5) | 20 (39.2) | 13 (14.8) | 379 (20.7) |
| 2 | 333 (19.7) | 11 (21.6) | 12 (13.6) | 356 (19.5) |
| 3 | 257 (15.2) | 10 (19.6) | 9 (10.2) | 276 (15.1) |
| 4 | 139 (8.23) | 3 (5.88) | 10 (11.4) | 152 (8.32) |
| 5 | 97 (5.75) | 2 (3.92) | 13 (14.8) | 112 (6.13) |
| >5 | 168 (9.95) | 0 | 23 (26.1) | 191 (10.5) |
| Missing | 348 (20.6) | 5 (9.8) | 8 (9.09) | 361 (19.8) |
| ECOG performance status, n (%) | ||||
| Fully active (0) | 626 (37.1) | 35 (68.6) | 49 (55.7) | 710 (38.9) |
| Restricted in physically strenuous activity (1) | 1,055 (62.5) | 16 (31.4) | 39 (44.3) | 1,110 (60.8) |
| Ambulatory, capable of self‐care but unable to work (2) | 6 (0.355) | 0 | 0 | 6 (0.328) |
| Capable of only limited self‐care (3) | 1 (0.0592) | 0 | 0 | 1 (0.0547) |
| Missing | 0 | 0 | 0 | 0 |
| Immunogenicity status for ADAs, n (%) | ||||
| Never positive (0) | 1,539 (91.2) | 48 (94.1) | 85 (96.6) | 1,672 (91.5) |
| Ever positive (1) | 70 (4.15) | 3 (5.88) | 3 (3.41) | 76 (4.16) |
| Missing | 79 (4.68) | 0 | 0 | 79 (4.32) |
| Concomitant acetaminophen/paracetamol, n (%) | ||||
| No | 53 (3.14) | 1 (1.96) | 0 | 54 (2.96) |
| Yes | 1,635 (96.9) | 50 (98) | 88 (100) | 1,773 (97) |
| Concomitant ibuprofen, n (%) | ||||
| No | 1,322 (78.3) | 51 (100) | 73 (83) | 1,446 (79.1) |
| Yes | 366 (21.7) | 0 | 15 (17) | 381 (20.9) |
| Concomitant acetylsalicylic acid, n (%) | ||||
| No | 1,407 (83.4) | 51 (100) | 68 (77.3) | 1,526 (83.5) |
| Yes | 281 (16.6) | 0 | 20 (22.7) | 301 (16.5) |
| Concomitant opioid, n (%) | ||||
| No | 540 (32) | 32 (62.7) | 32 (36.4) | 604 (33.1) |
| Yes | 1,148 (68) | 19 (37.3) | 56 (63.6) | 1,223 (66.9) |
| Concomitant systemic corticosteroid, n (%) | ||||
| No | 1,091 (64.6) | 33 (64.7) | 63 (71.6) | 1,187 (65) |
| Yes | 597 (35.4) | 18 (35.3) | 25 (28.4) | 640 (35) |
| Concomitant biologic, n (%) | ||||
| No | 1,077 (63.8) | 36 (70.6) | 84 (95.5) | 1,197 (65.5) |
| Yes | 611 (36.2) | 15 (29.4) | 4 (4.55) | 630 (34.5) |
| Previous biologic, n (%) | ||||
| No | 1,221 (72.3) | 40 (78.4) | 88 (100) | 1,349 (73.8) |
| Yes | 467 (27.7) | 11 (21.6) | 0 | 478 (26.2) |
ADAs, antidrug antibodies; ALT, alanine transaminase; AST, aspartate transaminase; CRP, C‐reactive protein; ECOG, Eastern Cooperative Oncology Group; eGFR, estimated glomerular filtration rate; NC; not calculable; PD‐L1, programmed death ligand 1.
Continuous covariates are reported as median {geometric mean} (range) [missing].
Analytical methods
Avelumab concentrations were quantified using an immunoassay sandwich method. Interrun precision was ≤16.1% coefficient of variation (CV), interrun accuracy was ≤15.5% bias (absolute value), and interrun total error was ≤19.1%. The lower limit of quantification was 0.2 μg/mL.
Data analysis
The model was built using NONMEM (version 7.3.0; ICON Development Solutions, Dublin, Ireland) with GNU Fortran (GNU Compiler Collection version 4.7.2; Free Software Foundation, Boston, MA) on an Intel‐based cluster running 64‐bit SUSE Linux Enterprise Server (version 11 SP3; SUSE, Nürnberg, Germany) and an Univa Grid Engine (version 8.2; Univa Corporation, Chicago, IL). Perl‐speaks‐NONMEM (version 4.4.8)15, 16 was used to manage NONMEM runs and perform some computational tasks. R (version 3.2.2)17 was used together with Xpose (version 4.5.3)18 for exploratory analysis, postprocessing of NONMEM output, and some data manipulation. All software was installed in a validated GxP environment.
Model selection was informed by the use of the objective function, a goodness‐of‐fit criterion equivalent to minus twice the log likelihood of the data given in the model, and by the evaluation of parameter estimates (including precision), graphical goodness of fit, and scientific and physiological plausibility. The differences in objective function between a full and a reduced model are approximately χ2 distributed. Differences of ≥3.84 in the objective function were regarded as significant, corresponding to a confidence level of P < 0.05 assuming degree of freedom (the norm for comparing nested models).
Interindividual variability (IIV) on the model parameters was assumed to be normally distributed, with a mean of zero and variance of ω2. Residual variability, arising from unspecified within‐patient variability, model misspecification, and experimental error, was estimated using additive and proportional random‐effects parameters.
Two‐compartment models incorporating empirical mechanisms for time‐varying CL were explored to explain potential change in the PK of avelumab over time.7, 19 A model published by Gibiansky et al.19 for obinutuzumab included parallel linear and time‐varying CL processes, whereas another more complex alternative, published by Liu et al.7 to describe the PK of nivolumab, modeled the decrease in CL as a sigmoid maximal inhibitory response process. The base avelumab model selected was built using the latter approach and explained the time‐dependent increase in CL observed in the data, as described in Eq. (1):
| (1) |
Here, CLi,t is CL in individual i at time t, TVCL is the typical value of CL in the population, Imax,i is the maximal possible change in CL relative to baseline for individual i, t i is the time after first dose in individual i, T 50 is the time at which 50% of Imax is reached, γ describes the shape of the relationship, and ηCL,i is IIV in CL for individual i, defined as being normally distributed with a mean of zero and variance of . Target‐mediated drug disposition (TMDD) was investigated, but its inclusion was not supported by the data.
In addition to CL, IIV was included on central volume of distribution (V 1), peripheral volume of distribution (V 2), and Imax. Covariances for CL, V 1, and V 2 were estimated.
Covariate relationships were assessed using the full model approach, in which all covariates were tested in the model simultaneously,20 on CL and V 1. Body weight, age, albumin, estimated glomerular filtration rate (eGFR), hepatic impairment, tumor size, tumor type, PD‐L1 expression (yes or no), Eastern Cooperative Oncology Group performance status, immunogenicity (antidrug antibody (ADA) if a positive result was ever obtained), C‐reactive protein, and platelet count (all at baseline), sex, and race as well as time‐varying formulation were predefined. Baseline alanine transaminase, aspartate transaminase, bilirubin, total protein, concomitant medications (acetaminophen/paracetamol, ibuprofen, acetylsalicylic acid, opioids, corticosteroids, and other biologics), and previous treatment with biologics were evaluated graphically by plotting against individual IIV parameters from the base model and included only if there was a strong indication (through a trend in the plots) that a relationship was likely.
Categorical covariates were tested using a linear function, as in Eq. (2).
| (2) |
PARi is the parameter value for individual i, PAR is the typical value of the parameter in the population, and θPAR,COV is an estimated parameter corresponding to the unique value of the categorical covariate in individual i. For the largest category, θPAR,COV is defined as 0. Covariate categories containing <80 patients were not separately tested but instead, were lumped in with the reference case (the category with highest frequencies).
Continuous covariate relationships were tested using a power function, as described in Eq. (3).
| (3) |
PARi and PAR are as previously defined. COVi is the value of the covariate in individual i. COVmed is the median value of the covariate in the population, and θPAR,COV is a parameter describing the shape of the relationship of the covariate to the parameter. Covariate ranges were capped to exclude extreme values.
Parameters controlling the time variability of CL were assessed individually by tumor type. No covariates were tested on V 2 or intercompartmental clearance (Q).
In addition, the parameters controlling the time variance of CL (Imax, T 50, and γ) were estimated separately by tumor type in the full model.
To yield a reduced model, covariate relationships were removed in a single step if both of the following criteria were met: the 95% confidence interval (CI) for the parameter estimate that included the covariate effect overlapped the null value and the 95% CI for the parameter that included the covariate effect was completely enclosed within the no‐effect range (defined as 75–125% of the point estimate of the covariate).
The 95% CIs for the covariate relationships were calculated using parameter standard errors. The covariate inclusion criteria were judged by comparing the value of the parameter (including covariate effects) with the parameter's reference value using a forest plot.
Any covariate relationships included in the reduced model after the reduction step that were no longer suitable for inclusion based on the rules noted previously were also removed in an additional final step, which resulted in the final reduced model. For tumor types in which Imax was estimated to overlap 0, no time effect on CL was assumed.
Visual predictive checks were performed at key model‐development decision points to evaluate predictive performance. The visual predictive check evaluates the model's ability to reproduce the same data used in its development. Concentration measurements were simulated 400 times using the dose and covariate data from the patients in the analysis using the same sampling times. Medians and 5% and 95% quantiles (the prediction intervals) were obtained from the distributions of simulated values in each of a range of binned time intervals and plotted against those obtained from the original observations. The 95% CIs for the prediction intervals were obtained by taking 5th, 50th, and 95th percentiles in each time‐interval bin of each of the 400 simulated data sets separately and computing the 2.5% and 97.5% percentiles of the resulting percentile distributions.
Results
Final model
The base model was two‐compartmental, with the change in CL over time parameter described according to the model of Liu et al.7 The addition of time‐varying CL produced a pronounced improvement in the model fit (reduction in the objective function value of 1,026). Log‐normally distributed IIV was included on CL, V 1, and V 2. Normally distributed (additive) IIV was included on the maximal change in CL relative to baseline (Imax) to allow it to vary nonmonotonically. The variance–covariance matrix for IIV included covariances between IIV on CL (), IIV on V 1 (), and IIV on V 2 (). The residual error was described by a combined additive and proportional error model.
Based on the literature, known effects of body weight were included a priori as a covariate on CL and V 1 (a full list of the covariates tested is provided in the Materials and Methods). The final model included the covariate effects on CL of body weight, albumin, eGFR, tumor size, C‐reactive protein, platelet count, and aspartate transaminase, all at baseline, and age, sex, race, treatment‐emergent ADAs, concomitant opioid use, and previous use of biologics. Baseline body weight and albumin, sex, and previous use of biologics were included on V 1. No covariates were included on Q or V 2. The IIV for CL reduced from 33.9% to 23.1% after the inclusion of the covariates in the final model.
The extent of time‐varying CL was substantially different for mMCC and squamous cell carcinoma of the head and neck (SCCHN) relative to the other 12 tumor types studied. For patients with mMCC, CL was reduced by a maximum of 32.1% (from 0.0308 to 0.0209 L/hour), with an associated T 50 value of 131 days and a γ value of 1.68. In the population with SCCHN, CL was reduced by a maximum of 24.7% (from 0.0308 to 0.0232 L/hour), with an associated T 50 value of 68.4 days and a γ value of 0.73. For all other tumor types, the typical value of Imax was zero, implying that CL could increase and decrease over time within individual patients through its IIV random effect but also that the typical change in CL in these groups was zero. The mean maximal reduction in CL reported for patients with mMCC in the current analysis is slightly different than that found in a previous analysis (41.7%; reported in US prescribing information for avelumab), which was based on a smaller data set (n = 1,629) and a linear population PK model. Nevertheless, the changes in CL are not clinically relevant and do not warrant dose adjustment.
Forest plots for the final model showing the relationships between parameters and covariates for CL, V 1, and Imax are shown in Figure 1. A complete list of model parameter estimates is provided in Table 2. Visual predictive checks for the 10 mg/kg dose group are shown in Figure 2, and Figure 3 shows the predicted variation in CL over time by patient and tumor type. Diagnostic plots are shown in Figure S1 and S2 (Figure Supplementary Material).
Figure 1.
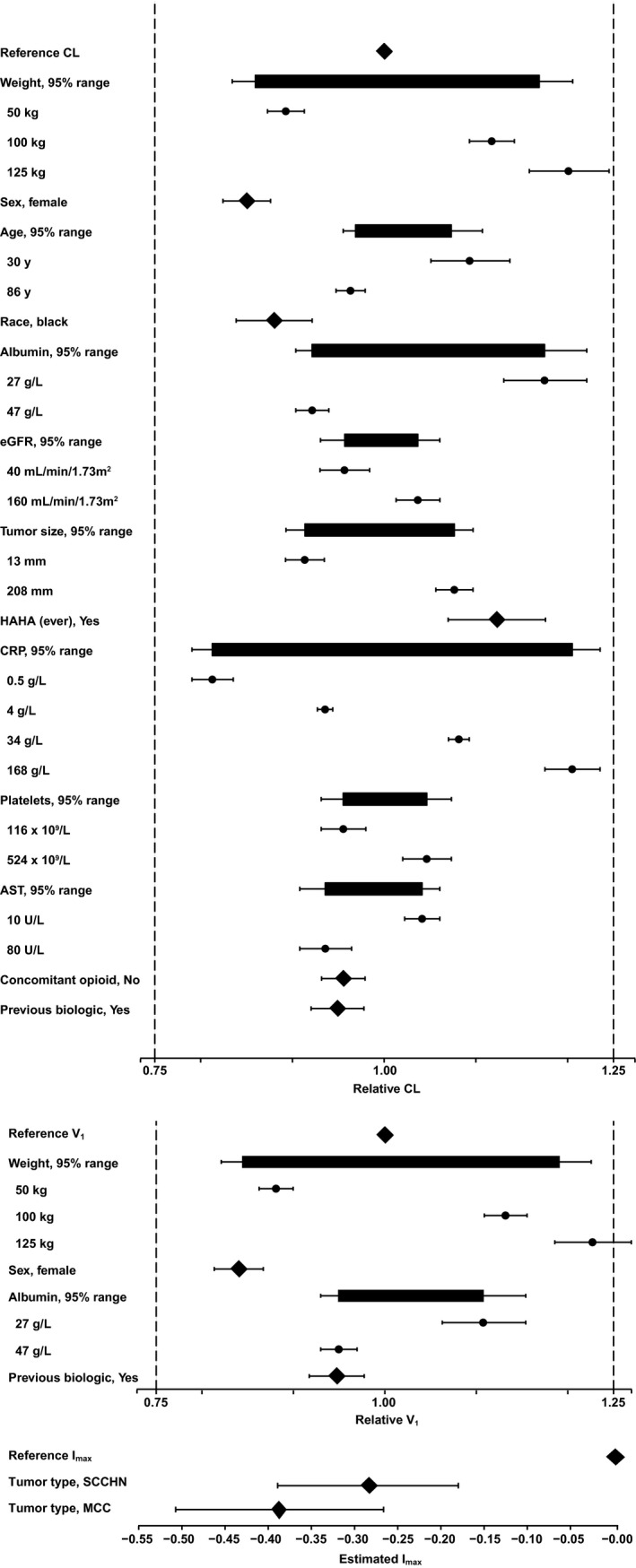
Forest plots illustrating the effects of covariates in the final reduced model. AST, aspartate transaminase; CL, clearance; CRP, C‐reactive protein; eGFR, estimated glomerular filtration rate; HAHA, human anti‐human antibody; Imax, maximal change in CL relative to baseline; MCC, Merkel cell carcinoma; SCCHN, squamous cell carcinoma of the head and neck; V1, central volume of distribution.
Table 2.
Parameter estimates for the final model
| Parameter | Estimate | %RSE | 95% CI | Shrinkage |
|---|---|---|---|---|
| CL, L/hour | 0.0308 | 1.36 | 0.03; 0.0316 | |
| Central volume, L | 3.42 | 1.20 | 3.34; 3.5 | |
| Peripheral volume, L | 0.918 | 6.75 | 0.796; 1.04 | |
| Intercompartmental CL, L/hour | 0.0313 | 5.92 | 0.0277; 0.0349 | |
| Imax SCCHN | −0.284 | 18.8 | −0.389; −0.180 | |
| Imax mMCC | −0.387 | 15.9 | −0.507; −0.266 | |
| Imax for all other tumor types | 0 | Fixed | Fixed | |
| T 50 mMCC | 131 | 12.2 | 99.6; 162 | |
| T 50, for all other tumor types | 68.4 | 2.54 | 65.0; 71.8 | |
| γ for SCCHN | 0.73 | 22.0 | 0.415; 1.05 | |
| γ for mMCC | 1.68 | 14.1 | 1.22; 2.15 | |
| γ for all other tumor types | 2.91 | 6.90 | 2.52; 3.31 | |
| CL | ||||
| Body weight on CL (θCL,Wt)1 | 0.324 | 10.1 | 0.260; 0.388 | |
| Female sex on CL (θCL,Sex)2 | −0.15 | 8.85 | −0.176; −0.124 | |
| Age on CL (θCL,Age)1 | −0.12 | 22.5 | −0.173; −0.0671 | |
| Black race on CL (θCL,Race)2 | −0.12 | 17.6 | −0.161; −0.0785 | |
| Albumin on CL (θCL,Alb)1 | −0.438 | 12.2 | −0.543; −0.333 | |
| eGFR on CL (θCL,eGFR)1 | 0.0579 | 32.6 | 0.0209; 0.0949 | |
| Tumor burden on CL (θCL,TS)1 | 0.0592 | 13.0 | 0.0441; 0.0744 | |
| Ever positive for ADA on CL (θCL,ADA)2 | 0.123 | 22.0 | 0.0698; 0.176 | |
| CRP on CL (θCL,CRP)1 | 0.0677 | 6.82 | 0.0586; 0.0767 | |
| Platelets on CL (θCL,Plat)1 | 0.0603 | 28.5 | 0.0266; 0.094 | |
| AST on CL (θCL,AST)1 | −0.0514 | 23.2 | −0.0747; −0.028 | |
| Not taking concomitant opioids on CL (θCL,COpi)2 | −0.0445 | 27.2 | −0.0683; −0.0208 | |
| Previous use of biologics on CL (θCL,PBio)2 | −0.0508 | 28.9 | −0.0796; −0.022 | |
| V 1 | ||||
| Body weight on V 1 ()1 | 0.362 | 8.50 | 0.301; 0.422 | |
| Female sex on V 1 2 | −0.160 | 8.57 | −0.187; −0.133 | |
| Albumin on V 1 1 | −0.278 | 20.6 | −0.390; −0.165 | |
| Previous use of biologics on V 1 2 | −0.0526 | 29.1 | −0.0827; −0.0226 | |
| IIV | ||||
| IIV on CL (, variance) | 0.0535 | 6.71 | 0.0464; 0.0605 | 19.7 |
| cov(CL,V 1) (, covariance) | 0.0171 | 13.8 | 0.0125; 0.0218 | |
| IIV on V 1 (, variance) | 0.0332 | 5.31 | 0.0297; 0.0366 | 36.3 |
| cov(V 1,V 2) (ωCL,V1, covariance) | 0.0633 | 25.2 | 0.0321; 0.0946 | |
| cov(CL,V 2) (ωCL,V1, covariance) | −0.014 | −105 | −0.0429; 0.0148 | |
| IIV on V 2 (, variance) | 0.858 | 11.8 | 0.659; 1.06 | 54.7 |
| IIV on Imax (, variance) | 0.0596 | 9.51 | 0.0485; 0.0707 | 38.1 |
| Residual variability | ||||
| Proportional residual error (σadd) | 0.162 | 0.521 | 0.161; 0.164 | 13.5 |
| Additive residual error (σadd), μg/mL | 2.43 | 0.904 | 2.39; 2.48 | 13.5 |
ADA, antidrug antibody; AST, aspartate transaminase; CI, confidence interval; CL, clearance; cov, covariate; CRP, C‐reactive protein; eGFR, estimated glomerular filtration rate; IIV, interindividual variability; Imax, maximal change in CL relative to baseline; mMCC, metastatic Merkel cell carcinoma; RSE, relative standard error; SCCHN, squamous cell carcinoma of the head and neck; T50, time at which 50% of change in CL has occurred; V 1, central volume of distribution.
Figure 2.
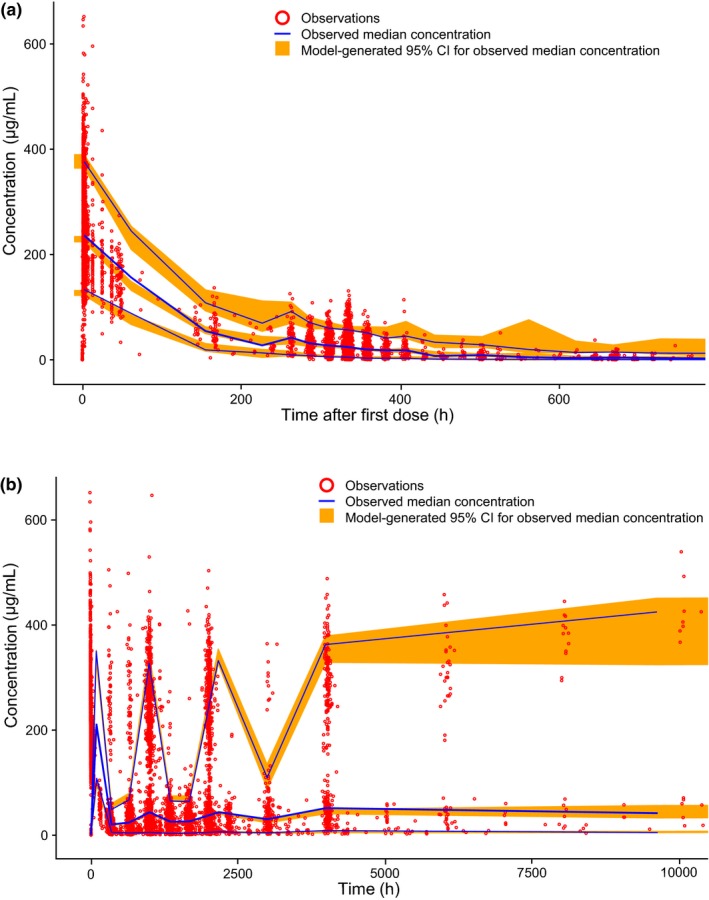
Visual predictive check for the final reduced model: (a) single cycle and (b) across the complete time course. CI, confidence interval; h, hour.
Figure 3.
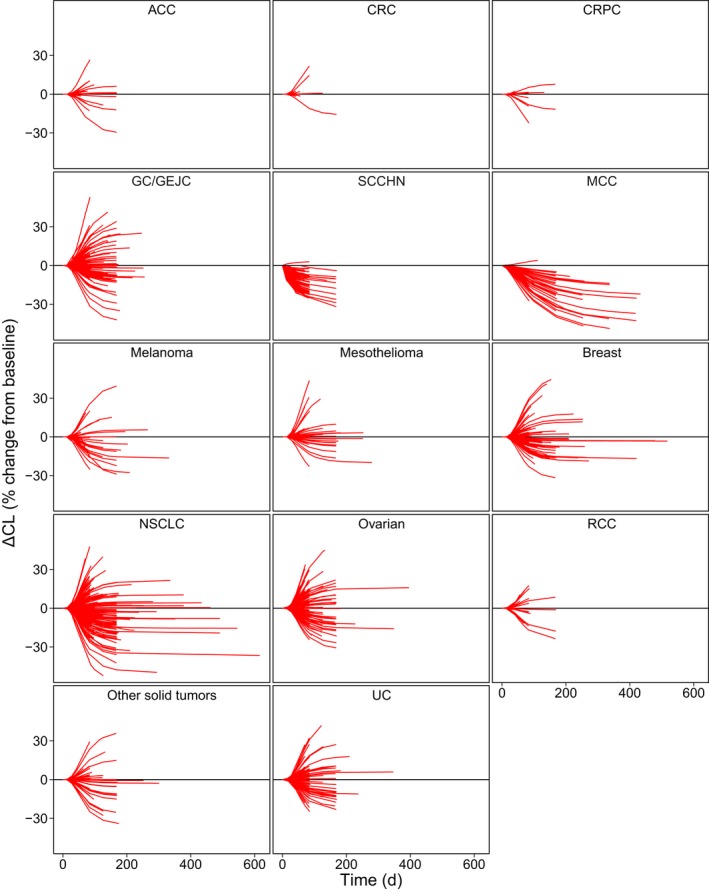
Change in clearance (CL) over time by tumor type predicted by the final reduced model. Lines are individual patients. ACC, adrenocortical carcinoma; CRC, colorectal cancer; CRPC, castration‐resistant prostate cancer; GC, gastric cancer; GEJC, gastroesophageal junction cancer; d, day; MCC, Merkel cell carcinoma; NSCLC, non‐small cell lung cancer; RCC, renal cell carcinoma; SCCHN, squamous cell carcinoma of the head and neck; UC, urothelial carcinoma.
Geometric mean half‐life (t 1/2) at baseline (i.e., after a single dose), calculated from empirical Bayes estimates of individual model parameters, was estimated to be 4.5 days (CV × 100, 36.7%) for the total population. After 26 weeks of biweekly dosing, t 1/2 was estimated to increase to 6.8 days (CV 42.7%) in patients with mMCC and to 5.7 days (CV 30.3%) for patients with SCCHN; for other tumor types, t 1/2 remained constant during this period. The parameter estimates reported in the current analysis are slightly different than those reported in the US prescribing information and US Food and Drug Administration submission package (t 1/2 of 6.1 days)21 because of the smaller data set (n = 1,629) and different PK model used for the previous analysis, as noted previously.
Effect of response on PK
For patients with mMCC, the mean maximum change in CL relative to baseline was 32.1%. This change in CL over time may have been the result of improvement in disease status following effective treatment, as theorized in previous work on nivolumab and pembrolizumab.7, 9 The magnitude of reduction in CL was observed to be higher in responders than in nonresponders (Figure 4), and the magnitude of reduction in CL was higher for responding and nonresponding patients in the mMCC population than for responding and nonresponding patients in the urothelial carcinoma population (Figure 5).
Figure 4.
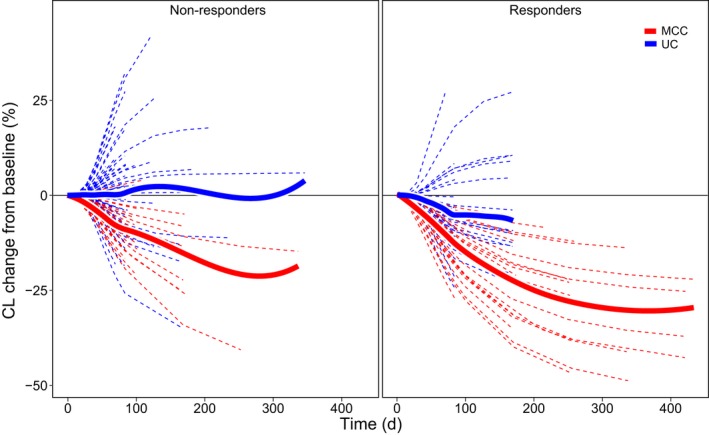
Estimated clearance (CL) over time relative to baseline stratified by response and tumor. d, day; MCC, Merkel cell carcinoma; UC, urothelial carcinoma.
Figure 5.
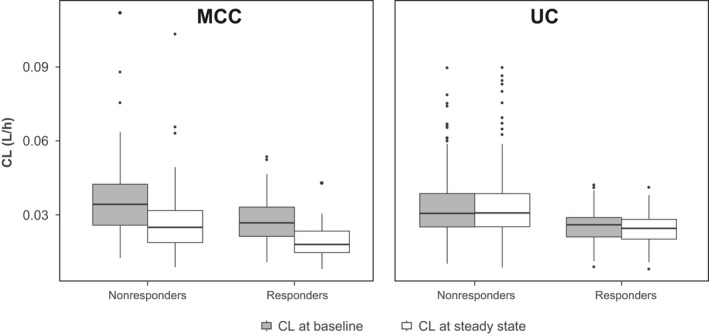
Estimated clearance (CL) at baseline and after 26 weeks of biweekly treatment, comparing mMCC and UC tumor types. h, hours; MCC, Merkel cell carcinoma; UC, urothelial carcinoma.
Discussion
The PK of avelumab is best described by a two‐compartment model with time‐varying CL using the approach described for nivolumab by Liu et al.7 TMDD was investigated during model development, but its inclusion in the final model was not supported by the data. The parameters were generally well estimated except for covariance between CL and V 2, which was quite imprecise. Shrinkages for all variability parameters were relatively high, implying that caution should be exercised in interpreting goodness‐of‐fit plots.22
The phenomenon of CL changing over time has recently been identified in similar monoclonal antibodies, such as nivolumab,7 pembrolizumab,8 and atezolizumab,9 and has been theorized to be a consequence of disease response to treatment corresponding with a decrease in cancer‐related cachexia.7, 8, 9, 23, 24 The mechanistic explanation for time‐varying CL for antineoplastic monoclonal antibodies is thought to be related to a reduction in inflammation status and subsequent changes to protein catabolism, including a decrease in the catabolism of therapeutic antibodies caused by the therapeutic effects of avelumab and other checkpoint inhibitors.23 This implies that patients with the most pronounced response to treatment should also show the most pronounced decreases in CL over time (or lower baseline CL), and this is indeed borne out by our results (see Figure 4 and Figure 5). Another contributing factor may be decreased antigen burden associated with tumor shrinkage, which would also gradually decrease CL with effective treatment. These findings may have strong implications for any future attempts to implement therapeutic drug monitoring with avelumab or any other agents that exhibit this type of change in CL over time.
A significant decrease in CL could be identified in only 2 of the 14 cancer types included in the analysis, namely mMCC, which showed a maximum decrease in CL of 32.1% relative to baseline, and SCCHN, which showed a maximum decrease of 24.7%. Patients with other tumor types also showed changes in CL over time (including both increases and decreases), but the mean CL in these groups remained similar to baseline. This phenomenon has been observed with other similar monoclonal antibodies, including nivolumab, pembrolizumab, and atezolizumab, for which mean maximum decreases of 24.5% (nivolumab in metastatic NSCLC, advanced renal cell carcinoma, and SCCHN),7 20% (pembrolizumab in melanoma and NSCLC),8 and 17.1% (atezolizumab in NSCLC) have been reported.9 Consistent with other similar monoclonal antibodies, no dose adjustment is warranted for avelumab because clinical studies have shown that the benefit/risk balance is maintained with long‐term treatment.14
In patients with Merkel cell carcinoma, the time‐dependent effect on CL may reflect that these patients were followed longer than patients with other tumor types. The mMCC group had a median treatment duration of 105 days vs. 74 days in the total population. In evaluations of PK observations only, the difference is even larger: the mMCC group had a median final PK observation after 84 days, whereas the median final PK observation in the total population was after 56 days. Considering that the estimated time at which 50% of the change in CL had occurred (T 50) varied between 68 days and 131 days (the latter for patients with mMCC), this longer follow‐up may explain why no time‐varying CL was seen in most other groups: they were not followed long enough for any time‐dependent effect on CL to be clearly detectable. Alternatively, the larger decrease may reflect greater susceptibility of this tumor type to immunotherapy.
For SCCHN, time‐varying CL was identified with a relatively short T 50 (68 days compared with 131 days). The reason for this effect is not known, but it may be related to potentially faster or more pronounced resolution of inflammation in response to treatment in this tumor type. SCCHN is often associated with human papillomavirus infection and chronic inflammation.25 It is possible that inhibition of the PD‐L1 pathway in this tumor type may be associated with both tumor shrinkage and resolution of chronic human papillomavirus–mediated inflammation at the site of the tumor, both of which may amplify the impact of reduced inflammation on antibody catabolism and time‐varying CL.26
The estimate of typical elimination t 1/2 of avelumab obtained from this analysis is 6.1 days (146 hours, CV 91.5%) for patients who received 10 mg/kg once every 2 weeks. This value is shorter than what has been observed for endogenous IgG1 (30 days)27 and some other monoclonal antibodies, such as ipilimumab (13 days), adalimumab (14 days), belimumab (19 days), and infliximab (8.0–9.5 days).28 The observed shorter t 1/2 relative to endogenous IgG1 and other human IgG1 monoclonal antibodies could be because of the higher isoelectric point of avelumab (8.5–9.3) when compared with other monoclonal antibodies. Antibodies with higher isoelectric points have been observed to have faster and increased tissue distribution, including into tumor tissue,29 leading to an observed shorter t 1/2 in the periphery. Despite the shorter t 1/2, the mean predose target occupancy on circulating CD3‐positive T cells immediately prior to the second dose on day 15 in the phase Ia study of avelumab was observed to be 76% in the 1 mg/kg cohort (n = 3), 90% in the 3 mg/kg cohort (n = 7), 93% in the 10 mg/kg cohort (n = 4), and 87% in the 20 mg/kg cohort (n = 7), suggesting that peak target occupancy had been achieved at the recommended doses of 10 mg/kg every 2 weeks.6
A total of 17 covariate relationships were identified, but none of these effects were considered clinically relevant, and no dose adjustments were warranted. Body weight was of primary interest given its impact on the dosing regimen; power exponents describing the effects of baseline weight for CL and V 1 were 0.324 and 0.362, respectively. Both weight effect exponents were well estimated and were robust to changes in other aspects of the model, and body weight–related changes in CL and V 1 did not exceed 25%, even at the extremes in the range. The magnitude of the weight exponent on CL30 suggested that a fixed, “flat” dose would be as effective and less variable as a weight‐based dose, and subsequent simulation work has shown this to be true (personal communication Ana M. Novakovic [A.M.N.], Justin J. Wilkins [J.J.W.], Haiqing Dai [H.D.], Janet R. Wade [J.R.W.], Berend Neuteboom [B.N.], Satjit Brar [S.B.], Carlo L. Bello [C.L.B.], Pascal Girard [P.G.], Akash Khandelwal [A.K.]). As a result, the currently approved dose of avelumab in the United States is 800 mg every 2 weeks.
Both CL and V 1 were lower in female patients independent of weight (by 15% and 16%, respectively). CL was also 12% lower in black or African American patients relative to the rest of the population.
CL and V 1 decreased significantly with increasing albumin. The neonatal Fc receptor facilitates IgG recycling and albumin homeostasis, protecting IgG (as well as antibody drugs) from catabolism. Hypoalbuminemia could be a marker for elevated neonatal Fc receptor–mediated protein turnover, resulting in higher CL and lower exposure.31, 32, 33 In addition, both albumin and C‐reactive protein are markers of baseline inflammation status, and their associations with CL at baseline (negative and positive, respectively) are therefore not surprising in the context of the proposed reasons for time‐varying CL, as discussed previously. These effects do not warrant dose adjustment because the changes are not considered to be clinically relevant.
CL in the 76 patients (4.16%) who were positive for ADAs at any timepoint was 12.3% higher than in the patients in whom ADAs were never positive or were missing. ADA seroconversion is an outcome of drug treatment rather than a baseline patient characteristic. For avelumab, the higher CL in patients with ADA at any timepoint was evident from the first dose, prior to ADA seroconversion. Therefore, a causal relationship between ADA seroconversion and CL is unlikely. In any event, this effect was not considered clinically relevant given the relatively low magnitude of IIV on CL of 23%.
The population PK analysis examined the potential influence of renal impairment by evaluating the influence of eGFR as a continuous covariate on the parameters of the population PK model across a wide range of renal function impairment, including none (n = 742), mild (n = 700), moderate (n = 379), and severe (n = 4). There was no influence of eGFR on avelumab CL in the final population PK model. Although a limited number of patients with severe renal impairment were studied, renal impairment is not expected to affect the avelumab PK because the molecular weight of avelumab is much higher than the glomerular filtration cut‐off. Although fewer patients with sever hepatic impairment were enrolled, severe hepatic impairment is unlikely to affect avelumab PK given the elimination pathway of avelumab. Thus, it is reasonable to conclude that severe hepatic or renal impairment will not influence avelumab elimination.
A higher tumor burden might increase nonlinear elimination via TMDD and therefore increase the total CL and produce lower exposures, although data did not support the inclusion of a TMDD component in the model, most likely because the target was expected to be saturated at doses of 10 mg/kg. Although a history of biologics use was associated with small reductions in both CL and V 1, these differences were not considered to be clinically relevant. The effects of age, tumor size, baseline platelet count, baseline aspartate transaminase, and concomitant opioid use were noted for CL, but these were small even at extreme values and were considered to be of low clinical interest.
In conclusion, this analysis describes avelumab population PK over time in 1,827 patients with various types of cancer. None of the covariates included in the final reduced model were found to warrant dosing modifications. Similar to other monoclonal antibodies used in the treatment of cancer, time‐varying CL was identified in some tumor types and was associated with posttreatment effects but was not considered to be clinically important.
Funding
This work was supported by Merck Healthcare KGaA, Darmstadt, Germany, and its subsidiary, EMD Serono, Rockland, Massachusetts, USA as well as Pfizer Inc., New York, New York, USA.
Conflict of Interest/Disclosure
J.J.W. and J.R.W. were employed as consultants by Merck Healthcare KGaA at the time the analysis was performed. B.B. and A.K. are employees of Merck Healthcare KGaA. H.D., Y.V., J.T.W., and B.N. are employees of EMD Serono, business of Merck Healthcare KGaA. P.G. is an employee of Merck Serono S.A., Lausanne, Switzerland, an affiliate of Merck Healthcare KGaA. S.B. and C.L.B. are employees of Pfizer Inc.
Author Contributions
J.J.W. wrote the manuscript. J.J.W., B.B., and A.K. performed the research. J.J.W., B.B., H.D., Y.V., J.T.W., S.B., C.L.B., B.N., J.R.W., P.G., and A.K. analyzed the data.
Supporting information
Figure S1. Basic diagnostic plots for the final reduced model.
Figure S2. Distributions of random effects in the final reduced model.
Data S1. Model code.
Acknowledgments
The authors thank the patients, their families, the investigators, coinvestigators, and study teams who participated in the clinical trials that provided the data.
References
- 1. Dong, H. , et al Tumor‐associated B7‐H1 promotes T‐cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 8, 793–800 (2002). [DOI] [PubMed] [Google Scholar]
- 2. Iwai, Y. , et al Involvement of PD‐L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD‐L1 blockade. Proc. Natl. Acad. Sci. USA 99, 12293–12297 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hirano, F. , et al Blockade of B7‐H1 and PD‐1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 65, 1089–1096 (2005). [PubMed] [Google Scholar]
- 4. Hamilton, G. & Rath, B. Avelumab: combining immune checkpoint inhibition and antibody‐dependent cytotoxicity. Expert Opin. Biol. Ther. 17, 515–523 (2017). [DOI] [PubMed] [Google Scholar]
- 5. Kaufman, H.L. , et al Avelumab in patients with chemotherapy‐refractory metastatic Merkel cell carcinoma: a multicentre, single‐group, open‐label, phase 2 trial. Lancet Oncol. 17, 1374–1385 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Heery, C.R. , et al Avelumab for metastatic or locally advanced previously treated solid tumours (JAVELIN Solid Tumor): a phase 1a, multicohort, dose‐escalation trial. Lancet Oncol. 18, 587–598 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu, C. , et al Association of time‐varying clearance of nivolumab with disease dynamics and its implications on exposure response analysis. Clin. Pharmacol. Ther. 101, 657–666 (2017). [DOI] [PubMed] [Google Scholar]
- 8. Li, H. , et al Time dependent pharmacokinetics of pembrolizumab in patients with solid tumor and its correlation with best overall response. J. Pharmacokinet. Pharmacodyn. 44, 403–414 (2017). [DOI] [PubMed] [Google Scholar]
- 9. Center for Drug Evaluation and Research . BLA 761041 clinical pharmacology review: atezolizumab. <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/761041Orig1s000ClinPharmR.pdf> (2016). Accessed November 20, 2017.
- 10. Gulley, J.L. , et al Avelumab for patients with previously treated metastatic or recurrent non‐small‐cell lung cancer (JAVELIN Solid Tumor): dose‐expansion cohort of a multicentre, open‐label, phase 1b trial. Lancet Oncol. 18, 599–610 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shitara, K. et al Phase I, open‐label, multi‐ascending dose trial of avelumab (MSB0010718C), an anti‐PD‐L1 monoclonal antibody, in Japanese patients with advanced solid tumors. J. Clin. Oncol. 33 (15 suppl.), abstract 3023 (2016). [Google Scholar]
- 12. Doi, T. et al Avelumab (anti–PD‐L1) in Japanese patients with advanced gastric or gastroesophageal junction cancer (GC/GEJC): updated results from the phase Ib JAVELIN solid tumour JPN trial. Ann. Oncol. 29 (suppl.), abstract 659P (2018). [Google Scholar]
- 13. Kaufman, H.L. , et al Updated efficacy of avelumab in patients with previously treated metastatic Merkel cell carcinoma after ≥1 year of follow‐up: JAVELIN Merkel 200, a phase 2 clinical trial. J. Immunother. Cancer. 6, 4–10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ngheim, P. et al Two‐year efficacy and safety update from JAVELIN Merkel 200 part A: a registrational study of avelumab in metastatic Merkel cell carcinoma progressed on chemotherapy. J. Clin. Oncol. 36 (suppl.), abstract 9507 (2018). [Google Scholar]
- 15. Lindbom, L. , et al Perl‐speaks‐NONMEM (PsN) – a Perl module for NONMEM related programming. Comput. Methods Programs Biomed. 75, 85–94 (2004). [DOI] [PubMed] [Google Scholar]
- 16. Lindbom, L. , et al PsN‐Toolkit—a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput. Methods Programs Biomed. 79, 241–257 (2005). [DOI] [PubMed] [Google Scholar]
- 17. R Core Team . R: A Language and Environment for Statistical Computing. <https://www.R-project.org/> (2016). Accessed August 31, 2018. [Google Scholar]
- 18. Jonsson, E.N. & Karlsson, M.O. Xpose—an S‐PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput. Methods Programs Biomed. 58, 51–64 (1999). [DOI] [PubMed] [Google Scholar]
- 19. Gibiansky, E. , et al Population pharmacokinetics of obinutuzumab (GA101) in chronic lymphocytic leukemia (CLL) and non‐Hodgkin's lymphoma and exposure‐response in CLL. CPT Pharmacometrics Syst. Pharmacol. 3, e144 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gastonguay, M. Full covariate models as an alternative to methods relying on statistical significance for inferences about covariate effects: a review of methodology and 42 case studies <www.page-meeting.org/?abstract=2229> (2011).
- 21. Bavencio (avelumab) [package insert] , Darmstadt, Germany: Merck Healthcare KGaA; (2017). [Google Scholar]
- 22. Karlsson, M.O. & Savic, R.M. Diagnosing model diagnostics. Clin. Pharmacol. Ther. 82, 17–20 (2007). [DOI] [PubMed] [Google Scholar]
- 23. Turner, D.C. et al Pembrolizumab exposure‐response assessments challenged by association of cancer cachexia and catabolic clearance. Clin. Cancer Res. 24, 5841–5849 (2018). [DOI] [PubMed] [Google Scholar]
- 24. Bajaj, G. , et al Model‐based population pharmacokinetic analysis of nivolumab in patients with solid tumors. CPT Pharmacometrics Syst. Pharmacol. 6, 58–66 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tezal, M. , et al Local inflammation and human papillomavirus status of head and neck cancers. Arch. Otolaryngol. Head Neck Surg. 138, 669–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ryman, J.T. , et al Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst. Pharmacol. 6, 576–588 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mankarious, S. , et al The half‐lives of IgG subclasses and specific antibodies in patients with primary immunodeficiency who are receiving intravenously administered immunoglobulin. J. Lab. Clin. Med. 112, 634–640 (1988). [PubMed] [Google Scholar]
- 28. Dostalek, M. , et al Pharmacokinetics, pharmacodynamics and physiologically‐based pharmacokinetic modelling of monoclonal antibodies. Clin. Pharmacokinet. 52, 83–124 (2013). [DOI] [PubMed] [Google Scholar]
- 29. Bumbaca, D. , et al Physiochemical and biochemical factors influencing the pharmacokinetics of antibody therapeutics. AAPS J. 14, 554–558 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang, D.D. , et al Fixed dosing versus body size‐based dosing of monoclonal antibodies in adult clinical trials. J. Clin. Pharmacol. 49, 1012–1024 (2009). [DOI] [PubMed] [Google Scholar]
- 31. Kim, J. , et al Kinetics of FcRn‐mediated recycling of IgG and albumin in human: pathophysiology and therapeutic implications using a simplified mechanism‐based model. Clin. Immunol. 122, 146–155 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fasanmade, A.A. , et al Serum albumin concentration: a predictive factor of infliximab pharmacokinetics and clinical response in patients with ulcerative colitis. Int. J. Clin. Pharmacol. Ther. 48, 297–308 (2010). [DOI] [PubMed] [Google Scholar]
- 33. Liu, L. Pharmacokinetics of monoclonal antibodies and Fc‐fusion proteins. Protein Cell. 9, 15–32 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Basic diagnostic plots for the final reduced model.
Figure S2. Distributions of random effects in the final reduced model.
Data S1. Model code.