

Abstract
Precision medicine aims to use patient genomic, epigenomic, specific drug dose, and other data to define disease patterns that may potentially lead to an improved treatment outcome. Personalized dosing regimens based on tumor drug penetration can play a critical role in this approach. State‐of‐the‐art techniques to measure tumor drug penetration focus on systemic exposure, tissue penetration, cellular or molecular engagement, and expression of pharmacological activity. Using in silico methods, this information can be integrated to bridge the gap between the therapeutic regimen and the pharmacological link with clinical outcome. These methodologies are described, and challenges ahead are discussed. Supported by many examples, this review shows how the combination of these techniques provides enhanced patient‐specific information on drug accessibility at the tumor tissue level, target binding, and downstream pharmacology. Our vision of how to apply tumor drug penetration measurements offers a roadmap for the clinical implementation of precision dosing.
Precision medicine in oncology entails tailored drug treatment for individual patients. Personalized dosing regimens based on tumor drug penetration play a critical role in this approach. Technologies necessary for this endeavor, such as in vivo molecular, functional imaging, and ex vivo mass spectrometry, have matured. Tools providing enhanced patient‐specific information on drug accessibility at tumor tissue level, target binding, and downstream pharmacology are crucial to understand exposure‐response relationships and guide precision dosing to improve treatment outcome.
Precision dosing as a missing piece of the personalized cancer treatment puzzle
Precision medicine aims to use patient's genomic, epigenomic, specific drug dose, and other data to define disease patterns that may potentially lead to an improved treatment outcome.1 Advances in precision medicine have been especially apparent in the field of oncology. An increasing number of targeted and “classic” cytotoxic agents can now be tailored to patient's cancer characteristics, such as targeting the BCR‐ABL fusion gene translocation in chronic myelogenous leukemia or dose reduction in polymorphism in enzymes involved in drug metabolism to reduce the risk of toxicity (e.g., UGT1A1*28 in irinotecan‐based chemotherapy). In addition, some progress has been made to identify biomarkers that predict response to these agents. The human epidermal growth factor receptor 2 (HER2; also known as ERBB2/neu) has been one of the most well‐documented successes in targeted cancer treatment. The HER2 gene is amplified in 15–20% of patients with breast cancer. The approval of HER2 targeted agents, led by trastuzumab in 1998, has improved outcomes in curative and noncurative breast cancers.2 Despite the success of targeted agents, such as those for HER2, improved long‐term outcomes from precision medicine are still comparatively rare, with over 600,000 Americans expected to die of cancer in 2018,3 and a predicted global burden of 13 million cancer deaths by 2030.4
A potential missing piece of the puzzle for precision medicine in cancer treatment may be an integrated “precision dosing” approach that tailors to patients’ tumor characteristics, as well as the extent of drug penetration in tumor tissue. In current dose escalation study designs, a correlation is sought between treatment dosage or systemic (plasma) exposure and treatment response.5 Most often, drugs are assumed to distribute relatively homogeneously in the tumor tissue. However, distribution of drugs into tumor tissue is in fact highly variable and may not correlate with dose or plasma concentrations. Such variability of drug penetration into tumor tissues may result in suboptimal treatment responses and yet its significance is often neglected.
Thus, an important parameter in precision dosing is drug tumor penetration, which can be assessed by measuring accessibility of the target and drug penetration in tumors at macroscopic and/or microscopic levels. At the macroscopic level, drug penetration is often heterogeneous within one single tumor lesion and even more so across different metastatic sites in the same patient. At the microscopic level, sanctuary sites may result in heterogeneity in drug concentrations leading to a proportion of neoplastic cells not receiving the required therapeutic dose. Anticancer drug distribution will be investigated at four levels, in the context of drug accessibility and downstream pharmacologic effects (Figure 1), bridging the gap between drug dose and the pharmacological link with a solid tumor's clinical outcome.
Figure 1.
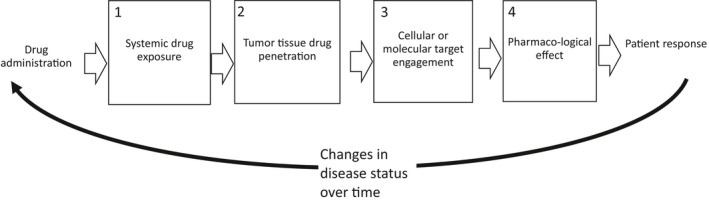
The pathway of drug administration to the tumor response is affected by tumor drug penetration at four levels: (1) the systemic level (the concentration of the drug in the blood pool, which determines how much of the drug is available for tumor penetration), (2) the tissue level (e.g., is the drug able to distribute throughout the tumor tissue, as influenced by the tumor microenvironment), (3) the cellular or molecular engagement level (where the drug is able to engage and interact with its target at the cellular/molecular level, a proximal or direct measure of drug mechanism of action), and (4) the expression of pharmacological activity following target engagement (a distal or indirect measure of drug pharmacodynamics). All these levels will be affected by responses to treatment (bottom).
In this review, we will focus on the state‐of‐the‐art tools, including imaging techniques that provide patient‐specific information on drug accessibility at the tumor tissue level, target binding, and downstream pharmacology in the context of precision dosing. Although these four drug distribution levels resemble the “three pillars of survival” framework, described by others,6 that includes drug exposure at the target site, target binding, and expression of pharmacological activity, additional consideration of spatial drug distribution in tumor tissue is added here. These four levels and their key considerations are further described below.
Systemic drug exposure
Assessment of systemic exposure ensures that the drug achieves a blood concentration during treatment that, in principle, permits optimal penetration and target binding into the tumor tissue, allowing selection of a dose with the best probability to reach the maximal receptor occupancy in the tumor.7 Ideally, this would be the dose that results in the number of bound target receptors/proteins in the tumor close to the maximum attainable, which is not always the maximum tolerated dose. Below, we discuss separately assessment of systemic exposure from measurements at the site of action (tumor tissue).
Tumor tissue drug penetration
At the tissue level, state‐of‐the‐art techniques may be used to visualize whether the drug is able to homogeneously distribute throughout the tumor. A range of factors (e.g., vascularity, hypoxia, or drug efflux transporters) may influence drug penetration, depending on characteristics of the drug. For immunotherapies, where immune cells are the effectors, the presence of specific immune cells and/or ligands in relation/proximity to tumor cells may affect the immune function and subsequent outcomes.8, 9 These studies suggest that before onset of an immune response, the presence and colocalization of immune and tumor cells, should be assessed,8, 9 in addition to drug tissue penetration. Changes in the tumor microenvironment in response to drug treatment can be investigated. Last, for small molecules, the free fraction of the drug (i.e., the pharmacologically active fraction) may be different in tissues vs. the circulation, and, therefore, whenever possible, total and free drug concentrations should be measured.6
Cellular/molecular target engagement
At the cellular/molecular level, it is possible to assess the presence and accessibility of the target in the right conformation to allow drug binding (i.e., target engagement).10 As such, drug binding and target availability should be assessed within and across tumor lesions. Furthermore, for some drugs, it is important to assess temporal changes in drug‐target engagement. For example, when drug binding results in target internalization and the pharmacologic effect causes target downregulation or upregulation.
Expression of pharmacological activity
Biomarkers that reflect downstream disease cascading effects or treatment effects can also be measured through target binding at the site of action. These distal measurements of pharmacodynamics (PDs) can indirectly demonstrate that sufficient levels of target modulation are being achieved at the site of action, in addition to providing a bridge toward quantification of drug efficacy and/or resistance.6
A multitude of state‐of‐the‐art technologies can be applied to assess the four levels of drug penetration, as shown in Table 1. Each of these techniques differs in the information provided and its potential role to inform clinical decision making and to guide optimal treatment strategies. We will mostly discuss implications for large molecules, although many of these concepts hold for, and can be extended to, small molecules as well. Techniques for drug characteristics to improve drug penetration at each level (e.g., association to albumin to prolong systemic half‐life, convection‐enhanced delivery to improve tumor tissue penetration or changing affinity to the target to enhance target engagement) are beyond the scope of this article.11, 12, 13
Table 1.
State‐of‐the‐art technologies that can be applied to assess specific aspects of drug penetration related to the systemic level, the tumor tissue level, and the cellular or molecular level
| Levels | Aim | Tools | Potential clinical relevance | Examples | |
|---|---|---|---|---|---|
| Macroscopic level | Microscopic level | ||||
| 1. Systemic exposure | Ensure optimal bioavailability in blood to reach the maximal binding capacity in tumor tissues | * PK measurements in blood:
|
Optimize dose (to overcome the tissue sink) |
* 89Zr‐Trastuzumab PET imaging and plasma PK to understand the tissue sink effect18
* Plasma PK of RG7356, an anti‐CD44 humanized antibody to define optimal dose for phase II study instead of MTD19 * Linear plasma PK of nivolumab and durvalumab may reflect severity of the disease, and may not be useful to guide dose adjustments21, 22 |
|
| 2. Tissue penetration | Assess tumor vascularization, immune infiltration and other factors in the tumor microenvironment |
* (Labeled drug)‐ molecular imaging:
* Microdialysis * Optical imaging |
✓ IHC/immunofluorescence ✓ MALDI‐MSI ✓ Multiplexed ion beam imaging |
Optimize treatment selection and understand mechanism of action |
* Microdialysis of methotrexate40
* Immunofluorescence imaging T‐DM141 * Fluorescent labeled bevacizumab/cetuximab‐guided surgery44, 45 * Immunotherapies: radiolabeled PD‐L146 or granzyme B PET imaging48 |
| 3. Cellular/molecular concentrations | Ensure the presence/accessibility of the target |
* Labeled drug‐molecular imaging: ✓ PET/SPECT Imaging barriers of target engagement ✓ Genomics |
Biopsy‐based assay to detect the presence of the target and the presence of factors that limit target: ✓ IHC/immunofluorescence |
Optimize treatment selection |
Macroscopic imaging: * 89Zr trastuzumab and T‐DM149 * Dose escalation guided by89Zr cetuximab50 * Somatostatin receptor imaging (e.g., 68Ga for imaging and 177Lu‐Dotatate treatment)54 Interference factors: * ICD/ECD HER2 expression58 MUC4 and trastuzumab60 * TAM uptake of lipidic nanoparticles63 * ABCB1 polymorphism anthracycline and cytarabine67 |
| 4. Expression of pharmacology | Ensure that sufficient target modulation has been achieved, assess drug efficacy and predict drug resistance |
Molecular imaging: * PET/SPECT |
* Imaging of PD markers ✓ For example, platinum adduct by immunofluorescence |
Change treatments, and optimize dosing |
* 18F‐fluorodihydrotestosterone androgen receptor imaging post apalutamide70 and enzalutamide69
* 18FES imaging post‐RAD1901/fulvestrant/Z‐endoxifen73, 74, 75, 76 * 89Zr trastuzumab HER2 response imaging post‐HSP90 inhibitor78 * Platinum adducts after carboplatin administration79, 81, 82 |
18FES, FES16α‐[18F]‐fluoro‐17β‐estradiol; CT, computed tomography; DCE‐MRI, dynamic contrast‐enhanced magnetic resonance imaging; HP‐LC, high‐performance liquid chromatography; HSP90, heat shock protein 90; ICD/ECD HER2, intracellular or extracellular domains of the human epidermal growth factor receptor; IHC, immunohistochemistry; LC‐MS, liquid chromatography‐mass spectrometry; MALDI‐MSI, matrix‐assisted laser desorption ionization mass spectrometry imaging; MTD, maximum tolerated dose; PD, pharmacodynamic; PD‐L1, programmed cell death‐ligand 1; PET, positron emission tomography; PK, pharmacokinetics; SPECT, single photon emission computed tomography; TAM, tumor‐associated macrophage; T‐DM1, ado‐trastuzumab emtansine.
As each of these four levels of biological organization is linked, information acquired from each component cannot be viewed separately. In addition, dosing recommendations require coupling quantification of heterogeneity in drug penetration and target engagement with a drug's PD link and long‐term outcome. In silico methods can help bridge the gap from all these data into a more comprehensive understanding. Modeling and simulation can be applied to integrate available information on accessibility, target engagement, PD effects, and outcomes at multiple scales.14 In oncology, the previously proposed concept of model‐informed precision dosing (MIPD)15 can be extended to include data from drug penetration studies. When properly validated, these models can be used in principle to predict and individualize doses.
Studying systemic exposure: Illustration and tools
As mentioned previously, drugs need to achieve systemic concentrations in the blood that permit adequate penetration and target binding into the tumor tissues.7 Assessment of drug concentration in blood or plasma alone (plasma pharmacokinetics (PKs)) may provide valuable information to guide drug dosing. PKs can be influenced by factors related to the patient, such as age, body weight, activity of drug transporters and metabolizing enzymes, and renal or liver function. Additionally, for monoclonal antibodies or other large molecules, target binding, immunogenicity, affinity for the neonatal Fc receptor, nonspecific uptake followed by proteolytic degradation and catabolism, and deconjugation determine the plasma PK‐time profile.16 Furthermore, these plasma PK profiles can be profoundly different depending on the drug dose administered to the patient. At a low dose, nontarget specific (e.g., Fc receptor in liver) or nontumor but specific (e.g., circulating target in blood) binding may decrease the drug's systemic exposure, leading to less drug target binding at the tumor site (the so‐called antigen sink). Higher doses may saturate the nonspecific binding sites, and lead to relatively high, nondose‐proportionate, increases in systemic exposure compared to low doses, an effect called target‐mediated drug disposition (TMDD).17 Therefore, evidence of nonlinear clearance by assessment of plasma PK profiles of monoclonal antibodies and other high affinity drugs can sometimes be used as a tool to predict the maximum binding capacity of the accessible drug target.7
State‐of‐the‐art tools can be used to assess systemic exposure. During drug development, plasma PK profiles are routinely assessed using techniques, such as liquid chromatography mass spectrometry (LC‐MS), for small molecules or immunoassays for monoclonal antibodies. Thus, validated methods for measuring drug concentrations in blood should be available during drug development. Less commonly, positron emission tomography (PET) imaging with radiolabeled drugs has been applied to quantify systemic exposure and TMDD.
By way of example, an 89Zr‐trastuzumab PET imaging study18 demonstrates the interplay between systemic and tumor exposure in the presence of nonspecific binding resulting in large tissue sinks. This study showed that reaching a minimal systemic exposure is necessary to improve drug penetration into the tumor and engage the target.18 At 10 mg (1 mCi) tracer dose, rapid 89Zr‐trastuzumab clearance was observed in trastuzumab naïve patients, and only after administration of 50 mg of 89Zr‐trastuzumab (replenished with nonradioactive trastuzumab), tumor penetrance was observed using PET imaging, indicating the ability to overcome the normal tissue sink. In patients undergoing treatment with trastuzumab (up to 6 mg/kg) at the time of tracer injection, a dose of 10 mg tracer was sufficient to indicate tumor distribution, suggesting that during treatment, some saturation of (nonspecific) drug's elimination pathways (e.g., via catabolism or plasma levels of extracellular domains shed by HER2) occurs. This also indicates that there are remaining free receptors in the tumor (we discuss saturation of the tumor sink in the next section).
Among many reported studies, one example of using TMDD to optimize dosing in drug development is shown in the analysis of the early phase study of RG7356, an anti‐CD44 humanized antibody, in patients with acute myeloid leukemia.19 At low doses, nonlinear PK was observed, which plateaued at 1,200 mg, at which point the maximum binding capacity of the accessible target was reached. This dose, as opposed to the maximum tolerated dose, was used to define the optimal dose for further phase II studies.19
This methodology is directly applicable to drug development: in 2017, the European Medicines Agency suggested in EMEA/CHMP/SWP/28367/07, revision 1, assessment of target saturation in early phase clinical trials and proposed that, instead of maximum tolerated dose, “maximum exposure” is considered, that provides complete inhibition or activation of the target and no further therapeutic effect is to be expected by increasing the dose.
In contrast to the TMDD characterization in the 89Zr‐trastuzumab and RG7356 examples, the PK profile of monoclonal antibodies in blood at clinical doses (at maximum binding capacity) may be prognostic of disease rather than predictive of the binding capacity. Post hoc analyses of trastuzumab in a phase III study in gastric cancer (ToGa) showed that patients in the lowest quartile of trastuzumab serum trough concentration had shorter overall survival than patients in the higher serum trough concentration quartiles.20 More recently, clearance of the immunotherapeutic monoclonal antibody, nivolumab (a programmed cell death 1 (PD‐1) blocking immunotherapeutic antibody) was shown to decrease when patients responded to therapy,21 and durvalumab (a PD‐L1 blocking antibody) change in clearance over time correlated with the change in tumor size during therapy and with the decrease in nonspecific protein catabolic rate in patients who benefited from therapy.22 This correlation suggests that the steep drug exposure‐tumor response relationship observed in these studies may be confounded by prognosis and treatment outcome.21, 22 Reasons for the correlation between PK and performance status of the patient is an active area of research and needs to be studied further. It may be related to cachexia, antibody cleavage by the tumor or systemic inflammation.21 These monoclonal antibodies when used in clinical doses generally show linear and dose‐proportional pharmacokinetics.21, 22, 23 Therefore, it is less likely that TMDD contributed to the disease‐specific PK. As a result, increasing the drug dose based on exposure at linear plasma PK in patients with poorer prognostic factors may not improve their outcomes. The HELOISE study – a randomized study of high‐dose trastuzumab (8 mg/kg + 10 mg/kg) vs. standard of care trastuzumab (8 mg/kg + 6 mg/kg) – indeed showed that higher doses of trastuzumab in patients predicted to have low exposure (based on an Eastern Cooperative Oncology Group score of 2) did not improve outcomes.24
Studying tumor tissue penetration: Illustration and tools
Tumor tissue penetration is largely determined by a variety of factors in the tumor microenvironment.25 This includes tumor vascular architecture, the composition and structure of the extracellular matrix, and the stroma. Aggressive proliferation of tumor cells and associated overexpression of pro‐angiogenic factors often leads to the development of poorly organized vascular and lymphatic architecture in tumors. As a result, the irregular blood flow and increased interstitial fluid pressure can severely affect drug distribution within the tumor and limit delivery of anticancer drugs to cells distal from the vasculature, depending on characteristics of the drugs. Stroma proteins in the extracellular matrix may give rise to a dense network of tumor matrix components that forms a physical barrier to tumor drug penetration.26 Small, hydrophilic compounds with low protein binding are expected to diffuse rapidly from the cellular rim into less vascularized or necrotic tissues (as seen in caseous tuberculosis),27 whereas drugs with high lipophilicity, poor solubility, and high number of aromatic rings are more likely to bind and not diffuse.27 For macromolecules, extravasation across the relatively permeable tumor vasculature is a limiting step for tumor penetration.7, 25
State‐of‐the‐art tools can be used to assess tumor tissue penetration. At a macroscopic level, information on vascularity and vascular‐related drug uptake can be obtained using dynamic contrast‐enhanced (DCE) magnetic resonance imaging (MRI), PET, or single photon emission computed tomography (SPECT) perfusion studies. In these noninvasive methods, molecules that pass through the blood vessel walls of the tumor and enter the extracellular extravascular space without penetrating the cellular membrane can provide information on vascular‐related drug uptake (e.g., blood volume, blood flow, and/or vascular permeability).28 In DCE MRI, gadolinium chelates are contrast agents that alter the relaxation time of water protons in tissues to create a contrast in imaging. In PET or SPECT, radiolabeled tracers, such as 15O‐water, 13N‐ammonia for PET,29 and 99mTc‐sestamibi for SPECT,30 are used. In addition, microdialysis, a semi‐invasive method can be used. During microdialysis, a catheter is placed in the vicinity of the tumor to allow measurement of the extracellular, nonprotein bound drug in accessible solid tumors at multiple timepoints postdose.31
At a microscopic level, large molecules or nonionizable agents can be analyzed in a section of the biopsy by immunofluorescence (IF) or multiplexed ion beam imaging, if secondary fluorescent antibodies or those contain isotopically pure elemental metal are available.32, 33 To assess the spatial distribution of unlabeled small molecules (and their metabolites) in clinical tissue samples, matrix‐assisted laser desorption ionization‐mass spectrometry imaging (MALDI‐MSI) can be applied34, 35 to a section of a fresh frozen biopsies.35, 36 By multiplexing (multiple stains per section), other histological assays can be applied to detect other factors in the tumor microenvironment influencing drug penetration (e.g., CD31 immunohistochemistry (IHC) staining for blood vessels).
Recently, it has become feasible to study microscopic drug penetration in in vitro cultured tumor cells in a 3D setting, resembling the in vivo architecture and tumor microenvironment of the tumor.37 Such tumor‐on‐a‐chip models are a subset of organ‐on‐a‐chip models. Therefore, tumor‐on‐a‐chip models can easily be combined with imaging (e.g., confocal microscopy) to study how microscopic drug penetration is influenced (e.g., interstitial flow), by the leakiness of the endothelial barrier or the density of the collagen matrix.37 As an example, a tumor‐on‐a‐chip was used for screening optimal nanoparticle designs prior to in vivo studies.38 In this study, the effect of nanoparticle size and interstitial flow rate on tissue accumulation was confirmed in murine tumor models.38
In the clinical setting, optical imaging can be used to assess tissue penetration. In fluorescence‐guided surgery, fluorescently labeled drugs are administered prior to surgery to delineate tumor margin and to visualize drug penetration in solid tumors.39 The optical technique can also be used to visualize the penetration of the fluorescently labeled drug in the resected tumor. An advantage of using surgical material to understand drug penetration over a 2D biopsy section‐based assay is that the latter may be representative only of the immediate tissue surroundings. Multiple sections obtained during fluorescence‐guided surgery may provide macroscopic and microscopic 3D information. Although the information on microscopic drug penetration may not be applied directly to optimize dosing in the same patient, data from these studies may help dose optimization in patients with similar disease characteristics and contribute to the understanding of mechanisms of drug penetration.
An example of macroscopic quantification by microdialysis is provided by a study measuring methotrexate in the extracellular fluid of brain tumors of four patients with recurrent high‐grade gliomas after high‐dose methotrexate (12 g/m2).40 Methotrexate levels were considerably higher in two patients who showed contrast enhancing regions of the brain by DCE MRI compared to two patients with nonenhancing brain regions.40 The results of this small study suggest that methotrexate penetration into brain tumors is variable and that combining drug measurements with DCE MRI can be applied to predict drug penetration as a function of tumor perfusion.40 That being said, clinical studies using microdialysis are currently too small to provide information on whether drug penetration relates with clinical outcomes.
Another example of assessing spatial drug gradients at a microscopic level with potential clinical implications has been provided for ado‐trastuzumab emtansine (T‐DM1), an antibody‐drug conjugate (ADC) that binds to receptors of cells in close proximity to the vasculature.41, 42 The T‐DM1 drug penetration images in an HER2‐positive xenograft mouse model (Figure 2 a–c) show that, at clinically relevant doses, the binding of T‐DM1 to the HER2 target occurs at a faster rate than diffusion across tissues, with the drug becoming immobilized immediately outside of blood vessels, a phenomenon commonly referred to as the “binding site barrier.” Lowering the drug payload to antibody ratio, by co‐administering unconjugated antibody (trastuzumab) with T‐DM1 at the same payload dose level, caused a larger fraction of antibody to compete and occupy receptors. This in turn allowed binding and internalization of the toxic payload to receptors across a larger number of tumor cells highly expressing the HER2 target (Figure 2 d–f). This method of lowering the drug payload to antibody ratio to improve homogeneity in payload penetration among tumor cells has been shown to increase response in the animals.41, 43 Although this study visualizes limited penetration of an ADC with a highly potent payload when target expression is high, the best strategy to homogenize the penetration of ADCs in patient tumors should be studied further in patients and furthermore correlated with patient outcome.
Figure 2.
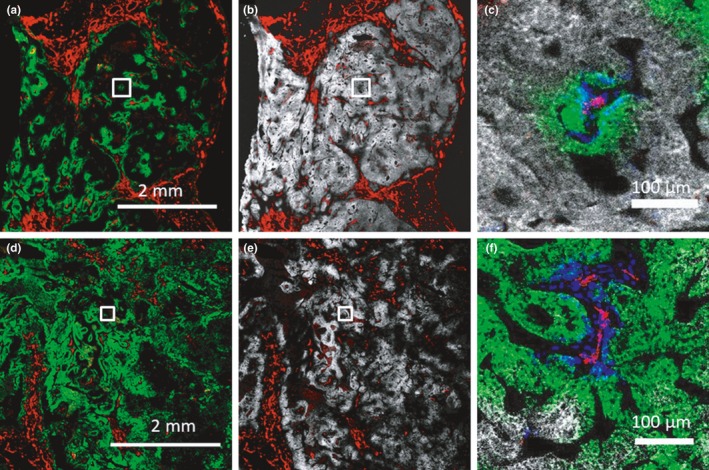
At clinically relevant doses, the binding of ado‐trastuzumab emtansine (T‐DM1) to human epidermal growth factor receptor 2 (HER2) expressing tumor cells is limited to the cells near functional blood vessels, and much higher doses are needed to provide a more homogeneous penetration, as shown at the microscopic level in an HER2 expressing xenograft tumor model (NCI‐N87 xenograft). (a) An immunofluorescence image of a tumor 24 hours following administration of 3.6 mg/kg of Alexa Fluor 680 tagged T‐DM1 ‐ a dose comparable to the dose used in patients ‐ to nude mice bearing NCI‐N87 flank tumors (green). Immunofluorescence staining with CD31‐AF555 (red) shows tumor vasculature, and intravenous administration and visualization of Hoechst 33342 shows functional vessels (blue) using multiplexed imaging. (b) HER2 expression (ex vivo staining with trastuzumab) in the same tumor section (white) and enlarged (c), indicating the uptake in the tumor was only sufficient to target a few cell layers. Images d, e, f show the same visualizations 24 hours following administration of 3.6 mg/kg of Alexa Fluor 680 tagged T‐DM1 and 10.8 mg/kg unlabeled trastuzumab (14.4 mg/kg total in a 1:3 ratio), indicating a more homogenous tumor penetration of T‐DM1. This dose reached many cells but did not occupy all accessible receptors in the tumor. Much higher doses up to 32 mg/kg of a combination of T‐DM1 and trastuzumab, in a 1:8 ratio (the latter to avoid antibody‐drug conjugate toxicity and improve penetration) were required in this animal model (with high HER2 expression, ~1 million receptors/cell) to reach all cells (data not shown). Red = CD31 + staining; green = 3.6 mg/kg T‐DM1‐AlexaFluor 680 (a–c) or 3.6 mg/kg T‐DM1‐AlexaFluor 680 + 10.8 mg/kg untagged trastuzumab (d–f); white = HER2 (trastuzumab labeling of histology slide); blue = functional vessels (intravenous Hoechst 33342). [Colour figure can be viewed at wileyonlinelibrary.com]
Two recent studies sought to visualize microscopic drug penetration in resected tissues after fluorescence‐guided surgery. The accumulation of fluorescently labeled bevacizumab, an antivascular endothelial growth factor antibody, was found to correlate with the pathological Bloom–Richardson–Elston tumor grade (a score that indicates cancer aggressiveness)44 in resected breast tissues from 19 patients with breast cancer. In another study, the level of fluorescently labeled cetuximab, an anti‐epidermal growth factor receptor (EGFR), accumulated within the resected tumor tissues of nine patients with head and neck squamous cell carcinoma, was correlated with cytokeratin (a measure of tumor density), EGFR expression, and factor VIII (vascular density).45 The latter biomarkers show that biological characteristics of the tumor may influence antibody (peri‐) tumoral distribution and target binding.
Immunotherapy poses new challenges for assessing drug efficacy. The key event for successful immunotherapies is the ability to attract both the drug and the immune cells into the tumor microenvironment; for example, immune cells, as opposed to drugs, thus become the mediator of antitumor effect. Tumor expression of PD‐L1 might be required for response to anti–PD‐1/programmed cell death‐ligand 1 (PD‐L1)‐targeted therapies, but also the accessibility of tumor‐antigen specific (PD‐1 suppressed) immune cells to the tumor space is another important driver of immune checkpoint inhibitor efficacy. In patients with melanoma, the spatial distribution and colocalization of immune cells immediately adjacent to PD‐1/PD‐L1 expressing tumor cells correlated with outcome of anti‐PD1 therapy.8, 9 The distribution was assessed using histological sections of tumor biopsies collected from patients before anti‐PD‐1 therapy with outcome of anti‐PD‐1 therapy.8, 9 Molecular imaging of immunotherapies may show the presence and accessibility of the target, but its use is still in its infancy.46 PET imaging with 89Zr‐atezolizumab in an imaging trial NCT02453984 or with 89Zr‐pembrolizumab in another trial NCT02760225 prior to the start of immunotherapy may be able to demonstrate the value of immune‐PET imaging for patient selection. Furthermore, the presence of other immune‐suppressive cells in the tumor microenvironment may predict resistance to immunotherapy.47 Multiple other cell types may contribute to tumor‐mediated immune suppression, including regulatory T cells, type 2 natural killer T cells, tumor‐associated macrophages (TAMs), tumor‐associated neutrophils, and myeloid‐derived suppressor cells and, therefore, may influence the efficacy of PD‐1‐based therapies.47 Importantly, an imaging biomarker of cytotoxic T‐cell activity may be more valuable for predicting response to cancer immunotherapy than biomarkers characterizing the entire immune infiltrate. Accordingly, PET imaging of radiolabeled granzyme B, a protease released from CD8+ T cells inducing apoptotic death of target cells, is currently under development.48 In conclusion, a combination of target engagement/activation imaging and assessment of spatial heterogeneity in PD‐1/PD‐L1 expressing immune cells and tumor cells in the tumor microenvironment at the microscopic level may advance the prediction of response to immunotherapies.
Studying target binding at a cellular/molecular level: Illustration and tools
When systemic exposure (in blood) has been optimized and the drug has been shown to penetrate tumor tissues, the subsequent step is to demonstrate target engagement.
Depending on the mechanism of action, target engagement can occur either intracellularly or extracellularly. For targeted drugs requiring internalization to be effective, intracellular accumulation should also be assessed.
State‐of‐the‐art tools can be used to assess target engagement. At a macroscopic level, in vivo whole‐body imaging (e.g., molecular imaging) may help to determine the presence of the target in the tumor lesion as well as the heterogeneity of the target expression across all lesions. Noninvasive in vivo imaging can be combined with standard pathology methods to provide absolute expression level (receptors/cell).
Molecular imaging has been applied to study target engagement through the visualization of the target's presence and accessibility (including the right conformation). It has largely been based on radionuclide imaging in the form of SPECT and/or PET. Other techniques, such as optical, spectroscopy, or photo‐acoustic imaging, are also in clinical development.
At a microscopic level, standard pathology procedures on a tumor sample (ex vivo), such as immunohistochemistry (IHC) or immunofluorescence (IF), gene, and RNA or protein expression measurements, are used to determine the presence of targets for therapy. However, these measurements should be appropriately validated allowing some quantification of these markers, as described in the Challenges/Prospects section.
Target engagement imaging could allow optimal drug selection and drug dosing.
One example of optimal drug selection is the ZEPHIR trial (NCT01565200). It is the first prospective clinical study that sought to explore the clinical utility of HER2 imaging as a predictive biomarker to optimize treatment selection in advanced HER2‐positive breast cancer. The study examined if low/absent radiolabeled trastuzumab tumor engagement, due to lack of target accessibility and/or drug penetration, could predict poor treatment response to HER2 targeted therapy, in this case, T‐DM1.49
In the trial, 89Zr‐trastuzumab PET/computed tomography (CT; HER2 PET/CT) and 18F‐fluorodeoxyglucose (FDG) PET/CT imaging were performed at baseline prior to T‐DM1 initiation. In an IHC/fluorescence in situ hybridization (FISH) confirmed HER2‐positive population, one‐third of patients were found to be “HER2‐negative” based on HER2 PET/CT. Accordingly, the median time to treatment failure of the latter group was three times shorter than the ones with a more homogenously positive HER2 PET/CT. Figure 3 shows a patient in which 89Zr‐trastuzumab drug does not reach its anticipated target. This patient's lung metastasis was tested HER2‐positive by IHC, but HER2 PET/CT showed a lack of penetration into the biopsied tumor. Response imaging on18F‐FDG PET/CT after three courses of T‐DM1 showed progressive disease.
Figure 3.
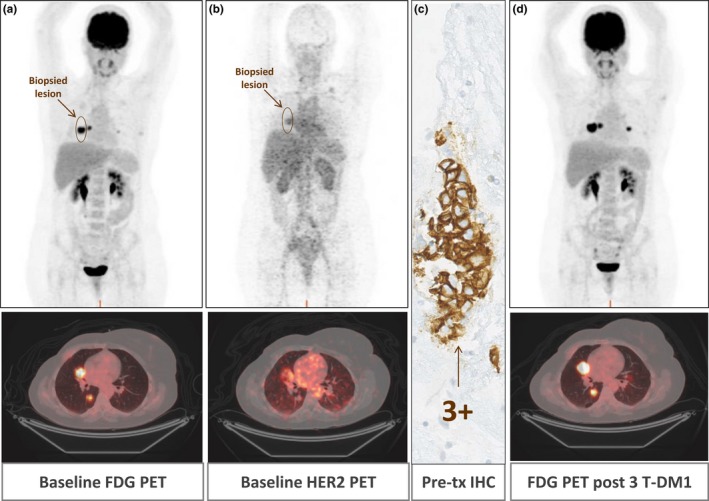
Lack of correlation between human epidermal growth factor receptor 2 (HER2) assessed by immunohistochemical (IHC) and 89Zr‐trastuzumab uptake in the same lesion of a patient in the ZEPHIR trial (NCT01565200). An HER2‐positive tumor of a patient with metastatic breast cancer with lung metastasis was visualized using (a) 18Fluorodeoxyglucose (FDG) positron emission tomography (PET)/computed tomography (CT), (a marker of tumor metabolism) but not with (b) HER2 PET/CT (non‐significant tracer uptake). Pre‐treatment (tx) biopsy of a right metastasis in the middle lobe (c) shows IHC 3 + staining (antibody recognizing the intracellular domain of the receptor). Response assessment (d) with FDG‐PET/CT shows progressive disease after three courses of ado‐trastuzumab emtansine (T‐DM1). [Colour figure can be viewed at wileyonlinelibrary.com]
Two ongoing clinical trials (study NCT02117466 and NCT01691391) show that dosing based on imaging of target engagement is feasible. In these studies, the uptake of 89Zr‐cetuximab assessed at day 6 after treatment onset is tested as a potential predictive biomarkers for early benefit of cetuximab as an EGFR targeted drug in treatment with colorectal cancer. The first results show that interpatient variability in PK and tumor uptake of 89Zr‐cetuximab only allowed dose escalation of cetuximab in 6 of 44 patients with metastatic colorectal cancer.50
Another example is the use of target engagement for drug selection in neuroendocrine tumors (NETs). Somatostatin receptor (SSTR)‐based molecular imaging has been used to detect NETs overexpressing SSTRs (initially using SPECT and more recently also using PET) and for the selection of candidates for therapies directed against these receptors. Moreover, SSTR imaging may also be used to optimize drug dosing (i.e., dosimetry) when radiolabeled somatostatin analogue based treatment (i.e., peptide‐receptor radionuclide therapy (PRRT)) is considered in advanced, well‐differentiated somatostatin expressing NETs.51, 52, 53 In addition, PRRT can be used as theranostic,10 using the same peptide labeled with diagnostic nuclide, such as 68 Ga‐, 111In for imaging, and 177Lu or 90Y for radiotherapy. However, the dosimetry approach is still under debate due to conflicting results in dose‐effect relationships.54, 55 Most PRRTs are still given according to a fixed activity administration scheme,56 or use pretreatment scans to adjust dosing based on organ uptake to avoid toxicity.57 In addition, randomized studies comparing fixed vs. tumor image‐driven dosing are lacking.
Measurement of expression of the target may not always translate into a correct prediction of target engagement due to many interfering factors. An example of a factor that precludes optimal target engagement prediction based on standardized target expression measurements comes from a comparison of HER2 epitopes. A recent study58 using a quantitative IF technique demonstrated the importance of the binding epitope on the target, in HER2‐positive breast cancer. A comparison was made between quantification of HER2 based on its intracellular domain (ICD; one used for IHC in clinical setting) epitope vs. its extracellular domain (ECD; binding epitope of trastuzumab) and their relation to treatment outcome of adjuvant chemotherapy and trastuzumab. This comparison showed that ECD was the most important predictor for a favorable treatment outcome, rather than ICD.58 This study shows that caution should be applied when opting to characterize binding to a target using molecular imaging tools binding an epitope different than the therapeutic. When available, optimizing the pathology platform for assessing target expression based on multiple epitopes or pathways can be used to optimize treatment selection, like for HER2 directed drugs against multiple epitopes or downstream pathways.
The presence of high molecular weight mucins may mask the binding epitope on the target and, thus, impede target engagement. Transmembrane mucin MUC4 has been reported to hinder the accessibility and, hence, the binding of trastuzumab to HER2 ECD, thereby impairing sufficient binding of trastuzumab to tumor cells.59 Therefore, reducing MUC4‐masking with mucolytic drugs improved HER2 accessibility, resulting in a higher antitumor effect of trastuzumab in HER2/MUC4‐positive xenograft models.60 Similarly, altered glycosylation in cancer cells increases sialic acids and carbohydrate structures called “tumor‐associated carbohydrate antigens” within the cell surface's sugar coating, or glycocalyx, which may prevent immune cells to trigger or evade immunological recognition. Targeting the glycocalyx by sialidase conjugation to trastuzumab has been shown to preclinically enhance the cell‐mediated cytotoxicity preclinically,61 and vaccines against tumor‐associated carbohydrate antigens are being developed.62 Clinical studies are needed to show whether reducing mucin masking or targeting the glycocalyx is applicable in patients.
Uptake of drugs by TAMs could be another barrier to target engagement. Pegylated liposomes were taken up primarily by macrophages in the tumor, whereas the same liposomes containing anti‐HER2 antibody on the surface distributed over HER2 overexpressing tumor cells with similar overall tumor tissue accumulation.63 Therefore, the interest for TAMs in oncology is not limited to their role in suppressing antitumor immune therapy response, but extends to the fact that they may limit drug target binding through macrophage‐directed drug clearance64 especially of lipophilic drugs,27 or by removal of immunotherapeutic antibodies from immune cells.65
Drug transporters add another layer of complexity in intracellular target engagement.66 P‐glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) have established roles in conferring multidrug resistance by limiting intracellular drug accumulation in tumor cells. For example, polymorphisms in these efflux transporters and an increase in messenger RNA expression correlated with relapse and survival in 263 Chinese patients with intermediate‐risk acute myeloid leukemia treated with anthracycline and cytarabine.67 However, up until now, it is unclear whether transporter‐mediated drug efflux by P‐glycoprotein and breast cancer resistance protein leading to reduced intracellular drug accumulation actually occurs in tumor cells in patients.68 Imaging agents need to be developed to quantify drug accumulation in tumor cells in patients. Unfortunately, the development of inhibitors of specific drug transporters has failed to provide benefit in the clinic to date.68 A more targeted approach to stratify patients based on multidrug transporter expression and/or function should be considered. Further research is needed to understand how transporter expression can be used to provide information on dose selection.
Studying expression of pharmacological activity following target binding: Illustration and tools
We distinguish between proximal target binding and subsequent distal expression of pharmacological activity (e.g., antibody target binding vs. downstream signaling response). This distinction may clarify how the drug mechanism of action modulates the biological effects following successful binding and provide insight into the drug's downstream effects. This may inform the existence and extent of a pharmacological link with outcome. Pharmacological activity at the protein or RNA expression level or downstream pathway activation can be assessed using the tools as described in previous sections.
In recent first‐in‐human studies of drugs directed at the androgen receptor (AR), such as enzalutamide69 and apalutamide70 – two nonsteroidal anti‐androgens – molecular imaging was used to determine the optimal biological dose. The uptake of 18F‐fluoro‐5α‐dihydrotestosterone (FDHT; an endogenous dihydrotestosterone analogue) reflects AR expression and binding capacity. Therefore, the PD biomarker FDHT in PET/CT imaging gauges PD response to these treatments.69, 70 Uptake of FDHT reached a plateau at a dose of 120 mg apalutamide70 and 150 mg for enzolutamide,69 suggesting maximal AR binding capacity and, thus, achievement of the optimal drug concentration. The recommended—and later US Food and Drug Administration‐approved—dose of apalutamide based on this study was much lower than the maximum tolerated dose.70
The 16α‐[18F]‐fluoro‐17β‐estradiol (18FES) imaging has been used to predict responders to endocrine therapies targeting the estradiol receptor (ER): the absence of FES uptake at baseline may predict endocrine treatment failure in patients with ER‐positive breast cancer.71, 72 In a feasibility study assessing ER availability before and during fulvestrant treatment, incomplete reduction of the ER target was observed after fulvestrant administration in 6 of 16 patients with metastatic breast cancer (38%).73 In addition, FES was used as a biomarker to assess efficacy of novel ER treatments, such as Z‐endoxifen (the most potent of the metabolites of tamoxifen), RAD1901 (a novel, oral, ER ligand), and GDC‐0810 (a selective ER degrader).74, 75, 76 For the latter, a phase II study was designed with an optimal dose of GDC‐0810 selected using the ER target engagement measurements.76
HER2 imaging with 89Zr‐trastuzumab might be a surrogate of the efficacy of novel agents like the heat shock protein 90 (HSP90) inhibitor luminespib (NVP‐AUY922). HER2 is a sensitive client protein of HSP90, and was shown to be depleted by HSP90 inhibition with luminespib in preclinical experiments.77 The 89Zr‐trastuzumab PET was used to determine the in vivo degradation of HER2 caused by the drug. The change between tumor uptake on baseline and early 89Zr‐trastuzumab PET after 3 weeks of treatment with this HSP90 inhibitor had a moderate positive correlation with change in tumor size on CT after 8 weeks of treatment in 29 lesions of 5 patients, showing that HER2 imaging can be used to assess target degradation and response to novel agents, such as luminespib.78
Another example of imaging PD markers is the measurement of platinum‐adduct formation (the covalent binding of carboplatin/cisplatin or oxaliplatin to nuclear DNA) by IF,79 by LC‐MS80 or inductively coupled plasma‐mass spectrometry.81 Platinum adducts in tumors are highly variable between patients, and small studies show that these may be more predictive of treatment response than platinum exposures in plasma.79, 81, 82
The role of modeling and simulation to integrate multiscale information and provide dosing guidance
To understand the effect of drug penetration at all levels on treatment response, information obtained from multiple levels and at temporal scales needs to be simultaneously considered. Multiscale models integrating information of drug distribution in spatial and temporal scales will be needed to understand macroscopic and microscopic distribution of drugs and to optimally guide personalized dosing14 (Figure 4).
Figure 4.
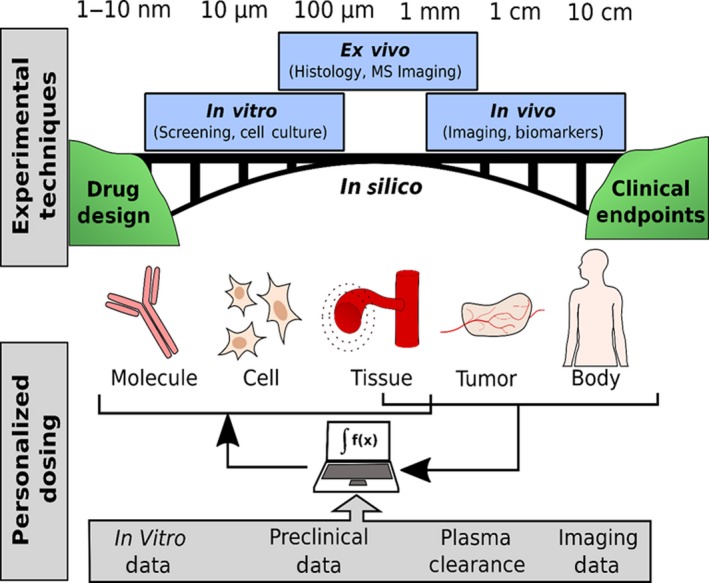
Drug development typically proceeds by optimizing molecular properties of target engagement and access (e.g., biophysical binding and cell culture methods) followed by preclinical studies (ex vivo and in vivo measurements) and eventual human trials to determine clinical endpoints. Here, we present a vision of how we can use in silico methods to help bridge the gap between these methods to a more comprehensive understanding (top). These same approaches can be used to integrate personalized data (imaging, plasma clearance, and biopsies) with computational models containing preclinical and in vitro data to develop personalized dosing schemes (bottom). MS, mass spectrometry. [Colour figure can be viewed at wileyonlinelibrary.com]
Preclinical information derived from either in vitro or in vivo experiments, such as receptor internalization rate, binding affinity, and the affinity for drug transporters (measured in cell culture), can be paired with clinical plasma PK,17 along with preclinical43 and clinical imaging data,10, 83 to simulate events at molecular, cellular, and tissue levels based on data from imaging, blood samples, and biopsies (Figure 4 , top). The diversity in the tumor environment should then be linked to spatial heterogeneity in the cellular states across the tumor.84 In addition, mechanistic information from preclinical pharmacology models can be used to further understand drug dose‐drug penetration‐drug activity relationships.85 When experimental data containing spatial information are obtained, image processing can reconstruct the relative order, geometry, topology, patterns, and dynamics of the 2D tissue sections. A 3D tumor is created, which can then be used to simulate temporal growth and evolution (Figure 4 , middle).14 By integrating all drug penetration information into spatiotemporal models, (Figure 4 , bottom) one can predict the dose needed for the optimal response (maximal binding of target receptors) using prior drug information and data from both the individual and a similar population of patients. In such spatiotemporal models, the microscopic and macroscopic spatial scales as well as the temporal scales should be considered.14 These temporal scales may encompass milliseconds for molecular interactions, hours for PK changes, days for tumor growth, and weeks to years for disease evolution.14 In such a model, the effect of drug transporters and specific factors that hinder target engagement, or change target expression during treatment, can be tested at all spatiotemporal levels.
The PK/PD modeling approaches provide a powerful tool to integrate time‐dependent exposure and response data to predict treatment outcomes.15 However, in most current PK/PD models, drugs are assumed to distribute homogeneously in the tumor tissue. To describe the spatial distribution of drugs in tumors, common mathematical models involving ordinary or partial differential equations or agent‐based modeling can be applied.14 The agent‐based modeling is an increasingly popular modeling approach in which individual discrete “agents” are simulated that can interact according to some prespecified rules; agents can simulate spatial heterogeneity by moving along a 3D lattice, thus accounting for spatial and temporal information.
An example of a multiscale model is the Oncosimulator.86 In this model, clinical, imaging, molecular, and treatment schedules are combined to predict response to anticancer treatments and radiotherapy.86 MRI images (T1 with contrast enhancement, T2, and T2 flair) of patients with nephroblastoma before treatment onset and after 4 weeks of chemotherapy were used to validate the model‐predicted tumor size changes. Model‐predicted tumor sizes were compared with an automated segmentation of the MRI scans of the tumors and with clinical experts’ annotation.86 Inclusion of models assessing the heterogeneity in tumor drug penetration in the Oncosimulator models may further improve the predictive value of the models. Other examples show how the spatial measurements at three levels (systemic, tissue, and cellular level) can be combined. First, Ribba et al.87 used longitudinal data from multiple state‐of‐the‐art techniques to describe the tumor drug penetration of an immune‐stimulatory drug Cergutuzumab amunaleukin. The nonlinear plasma concentration‐time profiles and interleukin‐2R‐positive cells in peripheral blood of 50 patients were described using a TMDD model. A Krogh cylinder model (a model describing the spatial drug gradients from the tumor blood vessels) was used to describe the extravasation and diffusion of the drug, thereby predicting the expansion of the target cells in the tumor by immune activation. The predicted tumor drug penetration was validated using measurements of drug uptake in tumor lesions following administration of 89Zr‐labeled Cergutuzumab amunaleukin at 3 sequential timepoints in 14 patients. The final model was used to identify a dosing regimen with an optimal antibody tumor uptake in patients. Quantification of radiolabeled drugs per tumor site was accomplished here using an uptake scaling factor at the level of the extravasation processes, the rate limiting process of drug uptake. In future studies, such a quantification can conceivably be expanded by using tumor vascularization and expression data to determine the temporal microscopic distribution and response in each lesion.41 Such models could be extended by using information about the molecular aspects of the drug of interest. For example Checkley et al.88 used a cell cycle model and incorporated mechanisms of DNA damage and repair based on in vitro and in vivo tumor growth experiments to describe the effects of an ataxia telangiectasia and rad3‐related (ATR) kinase inhibitor (AZD6738) and ionizing radiation. When information about mechanism of action and spatial drug distribution at all four levels are combined, models, such as those we presented, can bridge the gap between preclinical experiments and clinical observations. When these models are correlated with clinical outcomes, the model structures may have re‐usability across drugs with the same mechanistical properties.89
Challenges/prospects
As all four levels of biological organization we described are linked, understanding each aspect will inevitably lead to a cascade of interactions. To make precision dosing a clinical reality, optimization of all these processes is needed simultaneously.
Assessment of systemic exposure can become an integral part of precision dosing when adequate PK assays and data analyses are used to estimate the individual PK‐profiles in blood. Nonlinear mixed effects models combine structural models with estimates of nested variability in clinical observations, enabling estimation of means and variances of the statistical distributions of model parameters. Such a population approach may be used to calculate individual PK‐parameters in blood, limiting the need to design and plan for very specific sampling strategies. Moreover, this approach utilizes both individual PK parameters and population estimates to quantify nonlinearity in drug clearance and TMDD.
Assessment of drug tissue penetration can become an integral part of precision dosing when carefully timed tumor biopsies during treatment or alternative noninvasive techniques are available. A challenge when assessing drug exposures is that the biopsy should be performed at specific times after dosing, chosen to be relevant to the temporal evolution of the drug's action. In addition, in a significant proportion of patients, biopsies cannot be performed, owing to difficult tumor (metastasis) locations and low percentage of cancer cells in some samples,90 specifically when these biopsies are taken during an effective treatment. Although the I‐SPY 2 TRIAL shows that it may be feasible to take a biopsy during treatment,91 noninvasive techniques for macroscopic visualization of drug penetration or combination with other techniques, such as fluorescence‐guided surgery, may help collection of the optimal study samples. A limitation of techniques, such as image intensities by MALDI‐MSI, IHC, and immunofluorescence, is that the resulting images do not allow comparison between patients. These measurements rely heavily on the specific settings used and show large variability among different assessments.92 Efforts toward quantitative MALDI‐MSI measurements of drug have been demonstrated in preclinical samples93; however, drug detection still requires the MALDI‐MSI methods be developed and optimized for each drug of interest. An example that standardization of MALDI‐MSI is possible is provided in the application of MALDI to detect the presence of bacteria in infectious diseases.94 If appropriately validated, estimated absolute levels (e.g., drug or proteins/cell) would provide a dramatic improvement in uniformity across laboratories and comparisons between targets.
Target engagement molecular imaging can be used to perform precision dosing when both feasibility and benefit are confirmed in prospective clinical trials. One of the limitations of target engagement molecular imaging using PET/SPECT is the fact that radiolabeled molecules will irradiate the patient for a certain time conforming to the decay of the chosen radionuclide. Using long‐lived radioisotopes, which are required for large molecules with long circulation time, will, therefore, result in a higher radiation dose to the patient. One approach to avoid this is to use smaller protein scaffolds (affibodies, diabodies, and nanobodies). An example of the latter is given by 68 Gallium HER2 nanobodies,95 single domain antigen‐binding fragments that exhibit rapid targeting and fast blood clearance, high solubility, high stability, easy cloning, and modular nature compared to radiolabeled HER2 antibodies. These tactics may result in different distribution relative to the therapeutic drug.96, 97 Binding of a targeted drug is localized onto a specific domain (i.e., epitope). Therefore, the target presence does not guarantee target engagement. However, opting for these approaches may in fact be more suitable for determining the expression of pharmacological activity (discussed in the Studying Expression of Pharmacological Activity Following Target Binding: Illustration and Tools) rather than target engagement. Current tools to assess factors that hinder target engagement and the downstream pharmacologic effect provide mechanistic insight when predicting target engagement. However, with the current knowledge gap and lack of pharmacological tools to eliminate these factors, we can only speculate how assessing drug transporters (e.g., MUC4), or other factors may help with dose selection in the future.
Feasibility challenges of executing target engagement imaging include the cost of implementing molecular imaging in the clinic (imaging equipment, radiolabeled probes, and personnel costs for the required expertise).10, 98 A close collaboration among the nuclear medicine department, clinical pharmacologists, and medical oncologists is needed to implement target engagement imaging. In addition, when multicenter studies are performed, evidence that the final radiolabeled drug products and manufacturing processes are comparable between preparing institutions should be provided.98 A multicenter trial like SAKK 56/07, where validated PET or MRI scanning technique are applied in multicenter trial for response evaluation show that molecular imaging can be applied in larger populations.99 The widespread use and reimbursement of 18FDG‐PET in solid tumor diagnoses and assessment of treatment response shows that standardized, radiolabeled techniques are able to influence how we diagnose and treat patients.100 Given the advances in target engagement imaging, one day target imaging may replace some of the current pathology techniques for treatment guidance. A clinical trial investigating this hypothesis is the IMPACT‐MBC (NCT01957332). This study compares the impact of FES‐PET and 89Zr‐trastuzumab–PET with the gold standard (tumor biopsy) on treatment decision and outcomes in newly diagnosed metastatic breast cancer. The results of this trial will show whether target engagement imaging improves treatment outcomes compared to standard pathology.
The last level is expression of pharmacological activity following target binding, which allows the classification of a drug's PDs between proximal (direct measures of target engagement) and distal (indirect measures of effect) measurements. A disconnect between positive observed target engagement and negative expression of pharmacology may suggest a partial understanding of the interconnected pathways the drug is expected to modulate, and in turn an incomplete understanding of the underlying biology. Currently, a variety of detection platforms and assays are used to determine pharmacological activity, making validation among platforms and between laboratories crucial. Degradation or instability of proteins and (micro)RNA may limit the interpretation of the data.101 As an example, in the Analysis for Therapy Choice (NCI‐MATCH), a national signal‐finding precision medicine study that relies on genomic assays to screen and enroll patients with relapsed or refractory cancer after standard treatments, a next‐generation sequencing RNA and DNA assay was validated in multiple laboratories prior to study onset.102 In contrast, the presence of PD‐L1 by IHC, which is used to select or stratify patients for PD‐1/PD‐L1‐related studies, is not yet standardized and different cutoff values and scoring systems are used. These factors may explain some differences in the correlation between PD‐L1 expression and outcome seen among studies.103 This suggests a need for standardization and more sensitive and specific diagnostic tests.104
To bring all sources of data together, modeling and simulation may be used to perform precision dosing after prospective validation and clinical implementation. Darwich et al.15 describe ample evidence to support the use of MIPD tools to derive therapeutic recommendations for individual patients, but also stress that there is little evidence of its use and impact within clinical care. Although this review did not specifically address the issue of studying heterogeneity in drug penetration, many of the suggestions to improve clinical implementation apply. Examples of improvement are the need for extensive model validation; prospective clinical evaluation; the perspective of developing MIPD as a companion tool together with other diagnostic tools, such as imaging probes and other biomarkers in the early stage of drug development.15 Preclinical information used in these mechanistic models to inform personalized cancer treatment may be biased due to lack of translatability between preclinical experiments and patients. Therefore, each assumption needs to be validated (e.g., by performing sensitivity analyses or prospective validation). Furthermore, spatiotemporal models have not been extensively validated in the clinic, and many steps are needed before these models can provide individualized dosing information.
A vision to design prospective clinical trials including drug penetration measurements
Multiple tools have been identified to help inform optimal treatment strategies, (pre)clinical studies provide evidence that drug measurements are the key to successful personalized dosing, and the key challenges for clinical implementation have been defined, so the last step is to discuss optimal implementation of measurements at a systemic, tissue, and cellular/molecular level into clinical oncology practice to create the premise for precision dosing.
We envision that, before treatment, noninvasive imaging‐based measurements using the radiolabeled drug can assess the presence of the target in the target lesion, and the heterogeneity in abundance of the target in all lesions (Figure 5 , left). In addition, measurements related to the tumor microenvironment (e.g., vascularity, hypoxia, tumor stiffness, and immune cells) may provide information to predict drug behavior and allow optimal drug selection. During treatment, performing plasma PK sampling may guide optimal systemic exposure and help assessing the maximum binding capacity (Figure 5 , right). When a biopsy is available during treatment or surgical resection is performed, this tissue material can be used to visualize drug penetration at a microscopic level. Additional imaging during treatment may inform on drug‐response and/or drug resistance, either through target binding or downstream expression of pharmacology, or (preferably) both. The information gathered during treatment can be used to decide whether drug dosing should be de‐escalated or escalated. When this information is integrated with preclinical and prior knowledge using multiscale models, this may further support adaptation of the treatment decision (Figure 5 , bottom).
Figure 5.
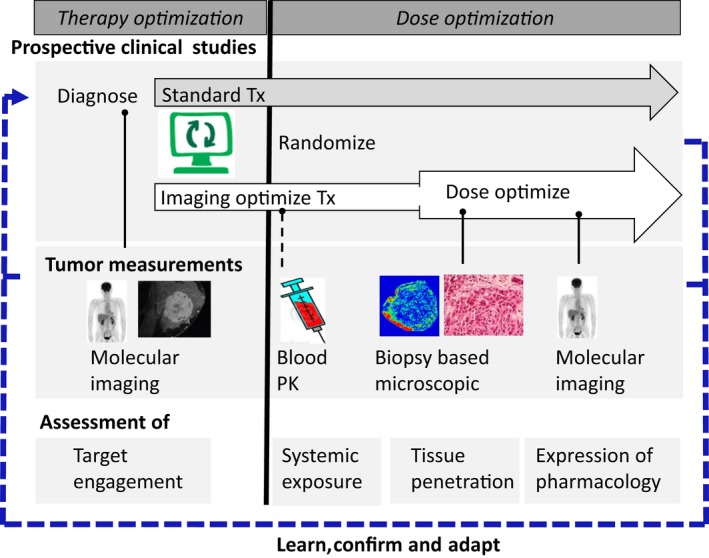
A vision for incorporating tumor drug penetration imaging to guide precision dosing. Noninvasive and invasive measurements can be applied to optimize treatment selection (prior to treatment initiation, left side) and to monitor and optimize drug dosing (during treatment right side. See text for further details). PK, pharmacokinetic; Tx, treatment. [Colour figure can be viewed at wileyonlinelibrary.com]
The use of image‐based treatment selection and dose optimization, as proposed in Figure 5, needs to be supported by prospective studies to: (i) assess whether image‐based or standard assessment guided treatment provide better outcomes and (ii) assess whether drug doses can be modified according to intratumor drug measurements. Freidlin and Korn105 provide the methodologies to efficiently incorporate such biomarker‐driven enrichment strategies, with the most efficient example provided by enrichment designs, in which only patients who show high target engagement, high tissue penetration, and high systemic exposures in imaging studies are to be randomized over a new treatment vs. standard of care. Furthermore, a Bayesian approach106 can be used to integrate clinical and preclinical data (prior information) to optimally inform dosing and speed up decision making. Then, an adaptive design can be used to efficiently test optimized dosing strategies in patients.
Conclusions
Recent advances in technologies at a macroscopic and microscopic level improve the visualization and assessment of drug penetration in solid tumors at the systemic, tumor tissue, and cellular or molecular level and the expression of pharmacological activity following target engagement. Individual (pre)clinical studies of tumor drug penetration measurements to date, although small and generally retrospective in nature, suggest that “precision dosing” (i.e., personalized dosing based on drug penetration in a solid tumor), may improve outcomes in patients. Unambiguous assessment of the benefits of precision dosing require clinical investigation of anticancer drug distribution in randomized trials at the four biological levels that we outlined, with the results being analyzed using integrative and mechanistic models, including spatial and temporal understanding of drug penetration. This review shows that in today's era of potent targeted drugs, precision dosing remains the missing piece of the current oncology precision medicine puzzle.
Funding
No funding was received for this work.
Conflict of Interest
Y.Z. and P.V. are employees of MedImmune and own stock and/or stock interests in AstraZeneca. I.H.B. was an employee of MedImmune and owned stock and/or stock interests in AstraZeneca at the time of the research. Other authors have no competing interests or other interests that might be perceived to influence the results and/or discussion reported in this article.
Acknowledgments
The authors thank Prof. Dr. E.G.E de Vries for her contribution and advice to the manuscript.
References
- 1. Precision Medicine Initiative. <https://www.fda.gov/ScienceResearch/SpecialTopics/PrecisionMedicine/default.htm>.
- 2. Slamon, D.J. et al Human breast cancer: correlation of relapse and survival with amplification of the HER‐2/neu oncogene. Science 235, 177–182 (1987). [DOI] [PubMed] [Google Scholar]
- 3. American Cancer Society Cancer Facts & Figures 2017. 1–182. <https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2017/cancer-facts-and-figures-2017.pdf> (2017).
- 4. Bray, F. , Jemal, A. , Grey, N. , Ferlay, J. & Forman, D. Global cancer transitions according to the Human Development Index (2008–2030): a population‐based study. Lancet Oncol. 13, 790–801 (2012). [DOI] [PubMed] [Google Scholar]
- 5. Adjei, A.A. What is the right dose? The elusive optimal biologic dose in phase I clinical trials 1. J. Clin. Oncol. 24, 4054–4055 (2006). [DOI] [PubMed] [Google Scholar]
- 6. Morgan, P. et al Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving phase II survival. Drug Discov. Today 17, 419–424 (2012). [DOI] [PubMed] [Google Scholar]
- 7. Glassman, P.M. & Balthasar, J.P. Mechanistic considerations for the use of monoclonal antibodies for cancer therapy. Cancer Biol. Med. 11, 20–33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tumeh, P.C. et al PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Taube, J.M. et al Colocalization of inflammatory response with B7‐h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 4, 127ra37 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moek, K.L. et al Theranostics using antibodies and antibody‐related therapeutics. J. Nucl. Med. 58, 83S–90S (2017). [DOI] [PubMed] [Google Scholar]
- 11. Sleep, D. Albumin and its application in drug delivery. Expert Opin. Drug Deliv. 12, 793–812 (2015). [DOI] [PubMed] [Google Scholar]
- 12. Goins, B. , Phillips, W.T. & Bao, A. Strategies for improving the intratumoral distribution of liposomal drugs in cancer therapy. Expert Opin. Drug Deliv. 13, 1–17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Khawar, I.A. , Kim, J.H. & Kuh, H.‐J. Improving drug delivery to solid tumors: priming the tumor microenvironment. J. Control. Release 201, 78–89 (2015). [DOI] [PubMed] [Google Scholar]
- 14. Yankeelov, T.E. et al Multi‐scale modeling in clinical oncology: opportunities and barriers to success. Ann. Biomed. Eng. 44, 2626–2641 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Darwich, A.S. et al Why has model‐informed precision dosing not yet become common clinical reality? Lessons from the past and a roadmap for the future. Clin. Pharmacol. Ther. 101, 646–656 (2017). [DOI] [PubMed] [Google Scholar]
- 16. Keizer, R.J. , Huitema, A.D.R. , Schellens, J.H.M. & Beijnen, J.H. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin. Pharmacokinet. 49, 493–507 (2010). [DOI] [PubMed] [Google Scholar]
- 17. Mager, D.E. Target‐mediated drug disposition and dynamics. Biochem. Pharmacol. 72, 1–10 (2006). [DOI] [PubMed] [Google Scholar]
- 18. Dijkers, E.C. et al Biodistribution of 89Zr‐trastuzumab and PET imaging of HER2‐positive lesions in patients with metastatic breast cancer. Clin. Pharmacol. Ther. 87, 586–592 (2010). [DOI] [PubMed] [Google Scholar]
- 19. Vey, N. et al Phase I clinical study of RG7356, an anti‐CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget 7, 32532–32542 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cosson, V.F. , Ng, V.W. , Lehle, M. & Lum, B.L. Population pharmacokinetics and exposure–response analyses of trastuzumab in patients with advanced gastric or gastroesophageal junction cancer. Cancer Chemother. Pharmacol. 73, 737–747 (2014). [DOI] [PubMed] [Google Scholar]
- 21. Liu, C. et al Association of time‐varying clearance of nivolumab with disease dynamics and its implications on exposure response analysis. Clin. Pharmacol. Ther. 101, 657–666 (2017). [DOI] [PubMed] [Google Scholar]
- 22. Baverel, P.G. et al Population pharmacokinetics of durvalumab in cancer patients and association with longitudinal biomarkers of disease status. Clin. Pharmacol. Ther. 103, 631–642 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bernadou, G. et al Influence of tumour burden on trastuzumab pharmacokinetics in HER2 positive non‐metastatic breast cancer. Br. J. Clin. Pharmacol. 81, 941–948 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shah, M.A. et al HELOISE: phase IIIb randomized multicenter study comparing standard‐of‐care and higher‐dose trastuzumab regimens combined with chemotherapy as first‐line therapy in patients with human epidermal growth factor receptor 2‐positive metastatic gastric or gast. J. Clin. Oncol. 35, 2558–2567 (2017). [DOI] [PubMed] [Google Scholar]
- 25. Thurber, G.M. & Weissleder, R. A systems approach for tumor pharmacokinetics. PLoS One 6, e24696 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu, P. , Weaver, V.M. & Werb, Z. The extracellular matrix: a dynamic niche in cancer progression. J. Cell Biol. 196, 395–406 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sarathy, J.P. et al Prediction of drug penetration in tuberculosis lesions. ACS Infect. Dis. 2, 552–563 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tofts, P.S. et al Estimating kinetic parameters from dynamic contrast‐enhanced T(1)‐weighted MRI of a diffusable tracer: standardized quantities and symbols. J. Magn. Reson. Imaging 10, 223–232 (1999). [DOI] [PubMed] [Google Scholar]
- 29. Mullani, N.A. et al Tumor blood flow measured by PET dynamic imaging of first‐pass 18F‐FDG uptake: a comparison with 15O‐labeled water‐measured blood flow. J. Nucl. Med. 49, 517–523 (2008). [DOI] [PubMed] [Google Scholar]
- 30. Cai, J. & Li, F. Single‐photon emission computed tomography tracers for predicting and monitoring cancer therapy. Curr. Pharm. Biotechnol. 14, 693–707 (2013). [DOI] [PubMed] [Google Scholar]
- 31. Zhou, Q. & Gallo, J.M. In vivo microdialysis for PK and PD studies of anticancer drugs. AAPS J. 7, E659–E667 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Angelo, M. et al Multiplexed ion beam imaging of human breast tumors. Nat. Med. 20, 436–442 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Giesen, C. et al Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422 (2014). [DOI] [PubMed] [Google Scholar]
- 34. Sugihara, Y. et al A new look at drugs targeting malignant melanoma–an application for mass spectrometry imaging. Proteomics 14, 1963–1970 (2014). [DOI] [PubMed] [Google Scholar]
- 35. Bartelink, I.H. et al Heterogeneous drug penetrance of veliparib and carboplatin measured in triple negative breast tumors. Breast Cancer Res. 19, 107 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marko‐Varga, G. et al Drug localization in different lung cancer phenotypes by MALDI mass spectrometry imaging. J. Proteomics 74, 982–992 (2011). [DOI] [PubMed] [Google Scholar]
- 37. van den Brand, D. , Massuger, L.F. , Brock, R. & Verdurmen, W.P.R. Mimicking tumors: toward more predictive in vitro models for peptide‐ and protein‐conjugated drugs. Bioconjug. Chem. 28, 846–856 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Albanese, A. , Lam, A.K. , Sykes, E.A. , Rocheleau, J.V. & Chan, W.C.W. Tumour‐on‐a‐chip provides an optical window into nanoparticle tissue transport. Nat. Commun. 4, 2718 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rosenthal, E.L. et al Safety and tumor specificity of cetuximab‐IRDye800 for surgical navigation in head and neck cancer. Clin. Cancer Res. 21, 3658–3666 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Blakeley, J.O. et al Effect of blood brain barrier permeability in recurrent high grade gliomas on the intratumoral pharmacokinetics of methotrexate: a microdialysis study. J. Neurooncol. 91, 51–58 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cilliers, C. , Guo, H. , Liao, J. , Christodolu, N. & Thurber, G.M. Multiscale modeling of antibody‐drug conjugates: connecting tissue and cellular distribution to whole animal pharmacokinetics and potential implications for efficacy. AAPS J. 18, 1117–1130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Juweid, M. et al Micropharmacology of monoclonal antibodies in solid tumors: direct experimental evidence for a binding site barrier. Cancer Res. 52, 5144–5153 (1992). [PubMed] [Google Scholar]
- 43. Cilliers, C. , Menezes, B. , Nessler, I. , Linderman, J. & Thurber, G.M. Improved tumor penetration and single‐cell targeting of antibody–drug conjugates increases anticancer efficacy and host survival. Cancer Res. 78, 758–768 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Koch, M. et al Threshold analysis and biodistribution of fluorescently labeled bevacizumab in human breast cancer. Cancer Res. 77, 623–631 (2017). [DOI] [PubMed] [Google Scholar]
- 45. de Boer, E. et al In vivo fluorescence immunohistochemistry: localization of fluorescently labeled cetuximab in squamous cell carcinomas. Sci. Rep. 5, 10169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shields, A.F. et al Immune modulation therapy and imaging: workshop report. J. Nucl. Med. 59, 410–417 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jenkins, R.W. , Barbie, D.A. & Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 118, 9–16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Larimer, B.M. et al Granzyme B PET imaging as a predictive biomarker of immunotherapy response. Cancer Res. 77, 2318–2327 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gebhart, G. et al Molecular imaging as a tool to investigate heterogeneity of advanced HER2‐positive breast cancer and to predict patient outcome under trastuzumab emtansine (T‐DM1): the ZEPHIR trial. Ann. Oncol. 27, 619–624 (2016). [DOI] [PubMed] [Google Scholar]
- 50. Van Helden, E.J. et al Pharmacokinetics of cetuximab and tumor uptake of 89Zr‐cetuximab as potential predictive biomarkers for benefit of cetuximab in patients with advanced colorectal cancer. J. Clin. Oncol. 35, (suppl.), abstract e15117 (2017). [Google Scholar]
- 51. Strosberg, J. et al Phase 3 Trial of 177 Lu‐dotatate for midgut neuroendocrine tumors. N. Engl. J. Med. 376, 125–135 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Krenning, E.P. et al Somatostatin receptor scintigraphy with 111In‐DTPA‐d‐Phe1‐ and 123I‐Tyr3‐octreotide: the Rotterdam experience with more than 1000 patients. Eur. J. Nucl. Med. 20, 716–731 (1993). [DOI] [PubMed] [Google Scholar]
- 53. Kwekkeboom, D.J. & Krenning, E.P. Peptide receptor radionuclide therapy in the treatment of neuroendocrine tumors. Hematol. Oncol. Clin. North Am. 30, 179–191 (2016). [DOI] [PubMed] [Google Scholar]
- 54. Ilan, E. et al Dose response of pancreatic neuroendocrine tumors treated with peptide receptor radionuclide therapy using 177Lu‐dotatate. J. Nucl. Med. 56, 177–182 (2015). [DOI] [PubMed] [Google Scholar]
- 55. Kulkarni, H.R. & Baum, R.P. Patient selection for personalized peptide receptor radionuclide therapy using Ga‐68 somatostatin receptor PET/CT. PET Clin. 9, 83–90 (2014). [DOI] [PubMed] [Google Scholar]
- 56. Bison, S.M. et al Peptide receptor radionuclide therapy using radiolabeled somatostatin analogs: focus on future developments. Clin. Transl. Imaging 2, 55–66 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chalkia, M.T. et al Patient‐specific dosimetry in peptide receptor radionuclide therapy: a clinical review. Australas. Phys. Eng. Sci. Med. 38, 7–22 (2015). [DOI] [PubMed] [Google Scholar]
- 58. Carvajal‐Hausdorf, D.E. et al Measurement of domain‐specific HER2 (ERBB2) expression may classify benefit from trastuzumab in breast cancer. J. Natl. Cancer Inst. 107, djv136 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Price‐Schiavi, S.A. et al Rat Muc4 (sialomucin complex) reduces binding of anti‐ErbB2 antibodies to tumor cell surfaces, a potential mechanism for herceptin resistance. Int. J. Cancer 99, 783–791 (2002). [DOI] [PubMed] [Google Scholar]
- 60. Wimana, Z. et al N‐Acetylcysteine breaks resistance to trastuzumab caused by MUC4 overexpression in human HER2 positive BC‐bearing nude mice monitored by 89Zr‐trastuzumab and 18F‐FDG PET imaging. Oncotarget 8, 56185–56198 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xiao, H. , Woods, E.C. , Vukojicic, P. & Bertozzi, C.R. Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc. Natl. Acad. Sci. USA 113, 10304–10309 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hutchins, L.F. et al Targeting tumor‐associated carbohydrate antigens: a phase I study of a carbohydrate mimetic‐peptide vaccine in stage IV breast cancer subjects. Oncotarget 8, 99161–99178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kirpotin, D.B. et al Antibody targeting of long‐circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 66, 6732–6740 (2006). [DOI] [PubMed] [Google Scholar]
- 64. Pei, Y. & Yeo, Y. Drug delivery to macrophages: challenges and opportunities. J. Control. Release 240, 202–211 (2016). [DOI] [PubMed] [Google Scholar]
- 65. Arlauckas, S.P. et al In vivo imaging reveals a tumor‐associated macrophage‐mediated resistance pathway in anti‐PD‐1 therapy. Sci. Transl. Med. 9, eaal3604 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rizk, M.L. , Zou, L. , Savic, R.M. & Dooley, K.E. Importance of drug pharmacokinetics at the site of action. Clin. Transl. Sci. 10, 133–142 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. He, H. et al Association of ABCB1 polymorphisms with prognostic outcomes of anthracycline and cytarabine in Chinese patients with acute myeloid leukemia. Eur. J. Clin. Pharmacol. 71, 293–302 (2015). [DOI] [PubMed] [Google Scholar]
- 68. Shaffer, B.C. et al Drug resistance: still a daunting challenge to the successful treatment of AML. Drug Resist. Updat. 15, 62–69 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Scher, H.I. et al Antitumour activity of MDV3100 in castration‐resistant prostate cancer: a phase 1‐2 study. Lancet (London, England) 375, 1437–1446 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rathkopf, D.E. et al Phase I study of ARN‐509, a novel antiandrogen, in the treatment of castration‐resistant prostate cancer. J. Clin. Oncol. 31, 3525–3530 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Peterson, L.M. et al A phase 2 study of 16α‐[18F]‐fluoro‐17β‐estradiol positron emission tomography (FES‐PET) as a marker of hormone sensitivity in metastatic breast cancer (MBC). Mol. Imaging Biol. 16, 431–440 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dehdashti, F. et al PET‐based estradiol challenge as a predictive biomarker of response to endocrine therapy in women with estrogen‐receptor‐positive breast cancer. Breast Cancer Res. Treat. 113, 509–517 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. van Kruchten, M. et al Measuring residual estrogen receptor availability during fulvestrant therapy in patients with metastatic breast cancer. Cancer Discov. 5, 72–81 (2015). [DOI] [PubMed] [Google Scholar]
- 74. Vries, E.de. et al A Phase 1 study of RAD1901, an oral selective estrogen receptor degrader, in ER positive, HER2 negative, advanced breast cancer patients. J. Clin. Oncol. 34, TPS627 (2016). [Google Scholar]
- 75. Lin, F.I. et al Utility of 18F‐fluoroestradiol (18F‐FES) PET/CT imaging as a pharmacodynamic marker in patients with refractory estrogen receptor‐positive solid tumors receiving Z‐endoxifen therapy. Eur. J. Nucl. Med. Mol. Imaging 44, 500–508 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang, Y. et al (18)F‐Fluoroestradiol PET/CT measurement of estrogen receptor suppression during a phase I trial of the novel estrogen receptor‐targeted therapeutic GDC‐0810: using an imaging biomarker to guide drug dosage in subsequent trials. Clin. Cancer Res. 15, 3053–3060 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tillotson, B. et al Hsp90 (heat shock protein 90) inhibitor occupancy is a direct determinant of client protein degradation and tumor growth arrest in vivo. J. Biol. Chem. 285, 39835–39843 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gaykema, S.B.M. et al 89Zr‐trastuzumab and 89Zr‐bevacizumab PET to evaluate the effect of the HSP90 inhibitor NVP‐AUY922 in metastatic breast cancer patients. Clin. Cancer Res. 20, 3945–3954 (2014). [DOI] [PubMed] [Google Scholar]
- 79. Nel, I. et al Formation and repair kinetics of Pt‐(GpG) DNA adducts in extracted circulating tumour cells and response to platinum treatment. Br. J. Cancer 109, 1223–1229 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Brouwers, E.E.M. et al Inductively coupled plasma mass spectrometric analysis of the total amount of platinum in DNA extracts from peripheral blood mononuclear cells and tissue from patients treated with cisplatin. Anal. Bioanal. Chem. 391, 577–585 (2008). [DOI] [PubMed] [Google Scholar]
- 81. Bonetti, A. et al Inductively coupled plasma mass spectroscopy quantitation of platinum‐DNA adducts in peripheral blood leukocytes of patients receiving cisplatin‐ or carboplatin‐based chemotherapy. Clin. Cancer Res. 2, 1829–1835 (1996). [PubMed] [Google Scholar]
- 82. Kim, E.S. et al Tissue platinum concentration and tumor response in non‐small‐cell lung cancer. J. Clin. Oncol. 30, 3345–3352 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wang, Y.‐X. & Deng, M. Medical imaging in new drug clinical development. J. Thorac. Dis. 2, 245–252 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gay, L. , Baker, A.‐M. & Graham, T.A. Tumour cell heterogeneity. F1000Res 5, 238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ogilvie, L.A. , Kovachev, A. , Wierling, C. , Lange, B.M.H. & Lehrach, H. Models of models: a translational route for cancer treatment and drug development. Front. Oncol. 7, 219 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Stamatakos, G. et al The technologically integrated Oncosimulator: combining multiscale cancer modeling with information technology in the in silico oncology context. IEEE J. Biomed. Heal. Inform. 18, 840–854 (2014). [DOI] [PubMed] [Google Scholar]
- 87. Ribba, B. et al Prediction of the optimal dosing regimen using a mathematical model of tumor uptake for immunocytokine‐based cancer immunotherapy. Clin. Cancer Res. 24, 3325–3333 (2018). [DOI] [PubMed] [Google Scholar]
- 88. Checkley, S. et al Bridging the gap between in vitro and in vivo: dose and schedule predictions for the ATR inhibitor AZD6738. Sci. Rep. 5, 13545 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ruiz‐Cerdá, L. , Asín‐Prieto, E. , Parra‐Guillen, Z.P. & Trocóniz, I.F. The long neglected player: modeling tumor uptake to guide optimal dosing. Clin. Cancer Res. 24, 3236–3238 (2018). [DOI] [PubMed] [Google Scholar]
- 90. Arnedos, M. et al Precision medicine for metastatic breast cancer—limitations and solutions. Nat. Rev. Clin. Oncol. 12, 693–704 (2015). [DOI] [PubMed] [Google Scholar]
- 91. Barker, A.D. et al I‐SPY 2: an adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clin. Pharmacol. Ther. 86, 97–100 (2009). [DOI] [PubMed] [Google Scholar]
- 92. Leeuwenburgh, M.M.N. et al Accuracy and interobserver agreement between MR‐non‐expert radiologists and MR‐experts in reading MRI for suspected appendicitis. Eur. J. Radiol. 83, 103–110 (2014). [DOI] [PubMed] [Google Scholar]
- 93. Groseclose, M.R. & Castellino, S. A mimetic tissue model for the quantification of drug distributions by MALDI imaging mass spectrometry 1. Anal. Chem. 85, 10099–10106 (2013). [DOI] [PubMed] [Google Scholar]
- 94. Prideaux, B. et al High‐sensitivity MALDI‐MRM‐MS imaging of moxifloxacin distribution in tuberculosis‐infected rabbit lungs and granulomatous lesions. Anal. Chem. 83, 2112–2118 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Keyaerts, M. et al Phase I study of 68Ga‐HER2‐nanobody for PET/CT assessment of HER2 expression in breast carcinoma. J. Nucl. Med. 57, 27–33 (2016). [DOI] [PubMed] [Google Scholar]
- 96. Stern, L.A. , Case, B.A. & Hackel, B.J. Alternative non‐antibody protein scaffolds for molecular imaging of cancer. Curr. Opin. Chem. Eng. 2, 425–432 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zhang, L. , Bhatnagar, S. , Deschenes, E. & Thurber, G.M. Mechanistic and quantitative insight into cell surface targeted molecular imaging agent design. Sci. Rep. 6, 25424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mankoff, D.A. , Farwell, M.D. , Clark, A.S. & Pryma, D.A. Making molecular imaging a clinical tool for precision oncology: a review. JAMA Oncol. 3, 695–701 (2017). [DOI] [PubMed] [Google Scholar]
- 99. Montemurro, M. et al Long‐term outcome of dasatinib first‐line treatment in gastrointestinal stromal tumors: a multicenter two stage phase II trial SAKK 56/07. Ann. Oncol. 25, iv496–iv497 (2014). [Google Scholar]
- 100. Zhu, A. , Lee, D. & Shim, H. Metabolic positron emission tomography imaging in cancer detection and therapy response. Semin. Oncol. 38, 55–69 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Jung, M. et al Robust microRNA stability in degraded RNA preparations from human tissue and cell samples. Clin. Chem. 56, 998–1006 (2010). [DOI] [PubMed] [Google Scholar]
- 102. Lih, C.‐J. et al Analytical validation of the next‐generation sequencing assay for a nationwide signal‐finding clinical trial. J. Mol. Diagn. 19, 313–327 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Garon, E.B. Cancer immunotherapy trials not immune from imprecise selection of patients. N. Engl. J. Med. 376, 2483–2485 (2017). [DOI] [PubMed] [Google Scholar]
- 104. Topalian, S.L. , Taube, J.M. , Anders, R.A. & Pardoll, D.M. Mechanism‐driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 16, 275–287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Freidlin, B. & Korn, E.L. Biomarker enrichment strategies: matching trial design to biomarker credentials. Nat. Rev. Clin. Oncol. 11, 81–90 (2013). [DOI] [PubMed] [Google Scholar]
- 106. Gupta, S.K. Use of Bayesian statistics in drug development: advantages and challenges. Int. J. Appl. Basic Med. Res. 2, 3–6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]