

Abstract
Cyclic oligochalcogenides (COCs) are emerging as promising systems to penetrate cells. Clearly better than and different to the reported diselenolanes and epidithiodiketopiperazines, we introduce the benzopolysulfanes (BPS), which show efficient delivery, insensitivity to inhibitors of endocytosis, and compatibility with substrates as large as proteins. This high activity coincides with high reactivity, selectively toward thiols, exceeding exchange rates of disulfides under tension. The result is a dynamic‐covalent network of extreme sulfur species, including cyclic oligomers, from dimers to heptamers, with up to nineteen sulfurs in the ring. Selection from this unfolding adaptive network then yields the reactivities and selectivities needed to access new uptake pathways. Contrary to other COCs, BPS show high retention on thiol affinity columns. The identification of new modes of cell penetration is important because they promise new solutions to challenges in delivery and beyond.
Keywords: adaptive networks, cellular uptake, cyclic oligochalcogenides, dynamic-covalent chemistry, polysulfanes
Benzopolysulfanes (BPS) are cyclic oligochalcogenides (COCs) characterized by large rings of sulfur atoms fused to a benzene ring.1, 2, 3 Dominant are pentasulfides as in 1–3, i.e., BPS5, also referred to as pentathiepins (Figure 1).1, 2, 3 They occur as natural products—with the dopamine‐derived varacin from tunicates probably best known—, have attracted attention as a challenge in total synthesis,4 and appeared as top hits from the screening of large libraries for targets in the brain of living animals.5 Benzopolysulfane rings are not very strained (XSSX dihedral angles ≥72°),6 but excel with unique, complex chemistry.1, 2, 3, 4, 5, 6, 7 This extreme sulfur chemistry attracted our attention in our search for new ways to enter cells. From cell‐penetrating peptides (CPPs)8 to cell‐penetrating poly(disulfide)s (CPDs)9 and COCs,10, 11, 12, 13 the cellular uptake via dynamic covalent dichalcogenide exchange chemistry has come to prominence, often referred to as thiol‐mediated uptake, with exofacial thiols as initial targets and continuing on the way into the cell.10, 11, 12, 13, 14, 15 Promising results with strained cyclic disulfides10, 11 and diselenides12, 13 called for a shift of attention to the extreme sulfur chemistry of higher oligochalcogenides.1 We here report that BPS5, for example, 1, outperforms all known COCs, and, most importantly, that BPS5 penetrate cells in a new way, i.e., by in situ selection from adaptive dynamic‐covalent networks16, 17 of extreme sulfur species with high reactivity, high selectivity, and strong retention by thiols.
Figure 1.
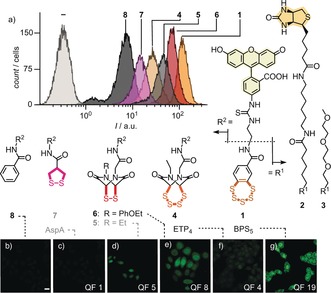
Cellular uptake of BPS5 1 into HeLa Kyoto cells compared to 4–8. a) Selected original flow cytometry data (20 μm, 45 min, Leibovitz's medium). The position of the compound numbers reflects the approximate fluorescence intensities after correction using quenching factors (QFs) of reduced COCs (note the log scale). b–g) Uncorrected CLSM images (10 μm, scale bar: 10 μm; laser power (LP): 15 %, with QFs of reduced COCs for correction if desired). Previously reported COCs are in gray.10, 11
Target molecules 1–8 were accessible by substantial multistep synthesis (Figure 1, Scheme 1, and Supporting Information, Schemes S1–S3 and S5–S8). BPS5 1 was prepared from catechol 9 following the Greer procedure.3 Dithiastannole 10, obtained in six steps from 9, was treated with S2Cl2 to give 11,3 which in turn was condensed with an amine equipped with fluorescein (FL–NH2) to yield BPS5 1. Biotinylated and TEGylated BPS5 2 and 3 were prepared correspondingly. ETP4 4 was prepared from ethylamine 12, which was converted into heterocycle 13 as described.11 Methanolysis of thioesters followed by treatment with S2Cl2,18 removal of the tert‐butyl ester in 14, and reaction with FL–NH2 yielded ETP4 4. A close congener of ETP2 5,11 ETP2 6 was newly prepared from amine 15, also to explore the advantages of a phenyl group during synthesis.
Scheme 1.
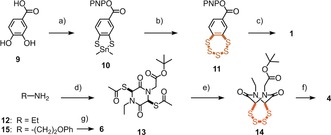
a) 6 steps, Scheme S1;3 b) S2Cl2, 0 °C to rt, 24 h, 20 %;3 c) FL–NH2, DMF, rt, 12 h, 14 %; d) 6 steps, Scheme S5;11 e) 1. NH3, MeOH, rt, 30 min, 2. S2Cl2, CH2Cl2, 0 °C to rt, 2 h, 58 %; f) 3 steps; g) 10 steps, Scheme S6. PNP=p‐nitrophenyl.
Cellular uptake of COCs into HeLa Kyoto cells was monitored by both flow cytometry and confocal laser scanning microscopy (CLSM). The flow cytometry data (Figure 1 a) were evaluated considering the different degree of fluorescence quenching by intact or reduced COCs (Supporting Information, Figures S1, S2, S6, and Table S1).12 Independent of any corrections applied, the uptake of BPS5 1 exceeded all other COCs. With correction, BPS5 1 was approximately 10‐times more active than the previous best in the sulfur series, ETP2 5,11 and approximately 140‐times more active than the best explored asparagusic acid (AspA) derivative 7.10 Tetrasulfide ETP4 4 showed less uptake than disulfide ETP2 5. This result demonstrated that simple oligomer effects in ring‐expanded COCs fail to explain the power of BPS5 1. The approximately 2.4‐times higher activity of new phenoxyethyl ETP2 6 compared to ethyl ETP2 5 suggested that the known contributions of aromatic rings to cellular uptake19 might also apply to COC‐mediated uptake. However, the inactivity of control 8 confirmed that contributions from such secondary ion–π interactions at the membrane–water interface19 to the activity of BPS5 1 are almost negligible.
CLSM images confirmed that BPS5 1 is more active than all other COCs (Figure 1 b–g). Concentration and time dependence analysis revealed binding to the plasma membrane with efficient delivery to the cytosol and particularly nucleus within one hour (Supporting Information, Figures S4 and S5). Many interpretations are possible for reduced activity at 4 °C, including hindered endocytosis, decelerated oligochalcogenide exchange kinetics, or membrane stiffening (Supporting Information, Figure S6). Insensitivity toward several inhibitors indicated the absence of uptake by clathrin‐mediated endocytosis (chlorpromazine), caveolae‐mediated endocytosis (methyl‐β‐cyclodextrin), and macropinocytosis (wortmannin, cytochalasin B; Supporting Information, Figure S7).9, 10, 11, 12, 13, 14, 15, 20 A drop in activity to 70 % upon preincubation with 2 mm Ellman's reagent (DTNB) supported contributions from thiol‐mediated dynamic covalent oligochalcogenide exchange9, 10, 11, 12, 13, 14 to the uptake of BPS5 1 (Supporting Information, Figure S8). According to the MTT assay, none of the tested COCs were cytotoxic under experimental conditions (Leibowitz, 24 h, concentrations up to 50 μm; Supporting Information, Figure S9).
Compared to other COCs, benzopolysulfanes offer different reactivity, culminating in ring contraction and expansion from trisulfides to nonasulfides, i.e., 1–3, 16–21,3 reminiscent of elemental sulfur Sn, with pentasulfides 1–3 being clearly preferred, followed by trisulfides 16 (Figure 2 a,e and Supporting Information, Figure S38).1, 2, 3 The mechanism of these reversible interconversions remains under debate, with transient ring opening by traces of nucleophilic impurities the most likely explanation.1, 2, 3, 6, 21 BPS chemistry further includes sulfur replacement, nucleophilic displacement and oxidation, radicals, metal coordination, and photochemistry,1, 2, 3 presumably much influenced, if not determined by the strings of electrophilic σ holes next to nucleophilic lone pairs on the lined‐up sulfur atoms.22 The low pK a values of thiophenols and persulfides facilitate ring opening to give interconvertible reactive intermediates, like RI‐1 and RI‐2, with preserved reactivity even in slightly acidic water. With ETP2 5 and diselenolanes, such less basic thiolates and selenolates were thought to account for mobility, i.e., their hypothetical mode of action as molecular walkers, walking along transmembrane disulfide tracks in membrane proteins.13 In a neutral, deuterated phosphate buffer, the 1H NMR spectrum of BPS5 3 remained unchanged at least for two weeks (Figure 2 b; BPSn have nearly identical spectra). In the HPLC, equilibration with BPS3 16 was detectable within hours (Figure 2 e and Supporting Information, Figure S22 and S38). In the presence of thiols (dithiothreitol, DTT, and glutathione 22, GSH), BPS5 3 transformed rapidly into multicomponent mixtures, with 1H NMR signatures changing with the substrate, time, and pH (Figure 2 c–d and Supporting Information, Figures S10–S17).
Figure 2.
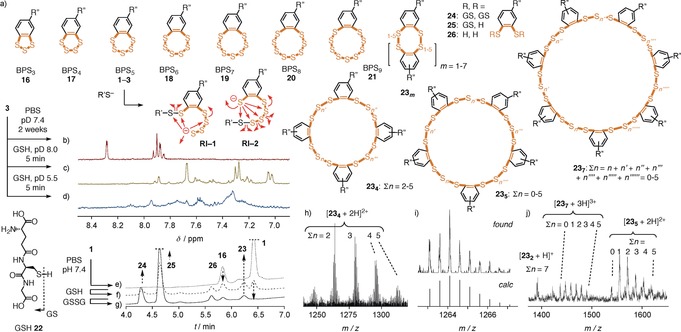
a) Possible ring expansion and contraction of BPS5, interconvertible ring‐opened intermediates RI, and selected products from intermolecular exchange. b–d) Diagnostic region of 1H NMR spectra of 3 in deuterated PBS buffer, 2 weeks (b), and with GSH, 5 min, pD 8.0 (c), and pD 5.5 (d). e–g) HPLC traces of BPS5 1 in PBS buffer, pH 7.4, 30 min (e), and with 2 equiv GSH (f), followed by 100 equiv GSSG (g). h) Signal cluster of 23 4 in UHPLC‐TOF HRMS of 1 with 22 (2 equiv), with i) a zoom and a simulated spectrum for 23 4 (Σn=2), and j) signal clusters of 23 2, 23 5, and 23 7 in LC‐MS of 1 with 22 (2 equiv). R′′: See 1, 3, Figure 1; R′: See 22.
According to HPLC analysis combined with low‐ and high‐resolution mass spectrometry (MS), the reaction of BPS5 1 with GSH affords a dynamic‐covalent network that includes unprecedented cyclic oligomers 23, from dimers 232 to heptamers 237, with up to nineteen sulfur atoms in a macrocycle of thirty‐three atoms, besides the expected di‐ and mono‐GSH‐BPS2 conjugates 24 and 25, and reduced BPS2 26 (Figure 2 and Supporting Information, Figures S24 and S39–S46). The identification of these large cyclic BPS oligomers was interesting because oligomer effects have been shown to account for thiol‐mediated uptake with ordinary disulfides.14, 23 In contrast, AspA 7 remained intact even with large excess of GSH (Supporting Information, Figure S36). This very important difference in reactivity was consistent with the weaker uptake activity of AspA, thus supporting that dynamic‐covalent networks matter for the mode of action of BPS. In agreement with the dynamic nature, the composition of the product library from BPS5 1 and GSH was altered by the subsequent addition of disulfides (GSSG, Figure 2 g and Supporting Information, Figure S27; lipoic acid, Supporting Information, Figures S29 and S48). Although less efficiently, BPS networks also formed with only disulfides (GSSG, DTNB, and lipoic acid, Supporting Information, Figures S18–S21, S25–S26, and S28), probably catalyzed by trace amounts of thiol impurities. The selectivity of the adaptive dynamic‐covalent BPS network was exemplified by the inability of amines to influence the situation (Supporting Information, Figures S30–S35).
The dynamic‐covalent networks16, 17 of extreme sulfur species obtained from BPS5 caused strong retention on thiol exchange affinity columns (Figure 3 c vs. 3a,b). Release after addition of DTT to the mobile phase exceeded initial elution by far and was unusually slow, continuing far beyond three hours (Figure 3 c, solid). Also, the application of reducing conditions in the presence of DTT led to slow elution over more than three hours (Figure 3 c, dashed). These complex chromatograms were in sharp contrast to the signatures of ETP4 4 and AspA 7. Some permanent retention of dithiolane 7 until clean release with DTT was in agreement with its dominant endosomal capture (Figures 3 a and 1 c).10 Negligible retention of ring expanded ETP4 4 as well as ETP2 6 and ETP2 5 12 was in agreement with dynamic‐covalent walking13 along thiol and disulfide tracks, through affinity columns and into the cytosol and nucleus (Figures 3 b and 1 d–f). The complementary behavior of tetrasulfide 4 and pentasulfide 1 on columns and in cells supported that the high activity of the latter is not a general property of oligosulfides but specific for the dynamic‐covalent networks produced by BPS5 (Figures 3 c,b and 1 f,g). Further supporting the importance of the dynamic‐covalent BPS network for function, preliminary observations suggest that increasing concentration of thiols in the media increase, rather than decrease, BPS‐mediated uptake.
Figure 3.
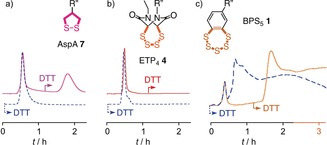
Thiol‐exchange affinity column chromatograms of a) 7,12 b) 4, and c) 1 in 10 mm Tris, 0.1 m NaCl, 1 mm EDTA, pH 7.5 with a 0–50 mm DTT gradient at t=60–70 min (solid) and constant 50 mm DTT from t=0 (dashed), R′′: See 1, 4, and 7, Figure 1.
BPS‐mediated delivery of proteins was probed first by the bioorthogonal uncaging of rhodamine 27 by artificial metalloenzyme 28 within HeLa Kyoto cells (Figure 4 and Supporting Information, Figures S49 and S50).13, 24 In this reaction, protein‐activated organometallic ruthenium complexes as in 29 cleave the allylcarbonyl protecting groups in the non‐fluorescent substrate 27 and liberate the fluorescent amine 30.25 The cell‐penetrating deallocase 28 was prepared by adding ruthenium complex 29 and biotinylated BPS5 2 to a streptavidin tetramer. Incubation of HeLa Kyoto cells first with cell‐penetrating metallodeallocase 28 and then, after washing, with the more hydrophobic, freely diffusing substrate 27 resulted in the intracellular emission from the intracellularly uncaged fluorophores 30. Emission intensities increased with increasing concentration of 28, while the BPS‐free control enzyme 31 did not cause fluorescence inside of the cells because it failed to penetrate (Figure 4 a and Supporting Information, Figure S50). These results confirmed the central importance of delivery for bioorthogonal catalysis within cells.13
Figure 4.
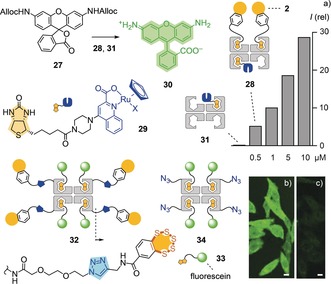
a) Fluorescence intensities in HeLa Kyoto cells treated with 31 (2.5 μm) or 28 (0.5–10 μm), then, after washing, with 27 (10 μm). CLSM images of HeLa Kyoto cells incubated with b) 32 and c) 34 (10 μm, 8 h; scale bars: 10 μm; LP: 30 %).
As a second approach to deliver proteins, we designed a cell‐penetrating streptavidin (CPS) 32 with all four biotin binding sites available for different substrates. Such constructs are desirable to fully exploit the streptavidin–biotin technology. CPS 32 was prepared by covalently linking BPS5 to a streptavidin via copper‐catalyzed azide–alkyne cycloaddition reactions followed by loading the model substrate, a biotinylated fluorescein 33. CLSM images of HeLa Kyoto cells incubated with CPS 32 revealed intense diffuse fluorescence in most parts of the cells except in the nucleoli, together with some endosome‐like punctate dots (Figure 4 b). In comparison, the uptake of control protein 34, which lacks BPS5, was negligible (Figure 4 c). Ongoing studies on the biological applications of covalent BPS–streptavidin conjugates 32 include the targeted delivery of biotinylated anti‐GFP nanobodies and biotinylated chloroalkanes together with biotinylated TAMRA fluorophores to mitochondria labelled with GFP and HaloTags, respectively.
In summary, we report that benzopolysulfanes mediate uptake into cells, better than all COCs explored so far. This activity is shown to originate from their transformation into adaptive dynamic‐covalent networks of extreme sulfur species, including cyclic oligomers with up to nineteen sulfurs in the macrocycles, for selection and possibly amplification of the best. These dynamic‐covalent BPS networks show high reactivity, high selectivity, and strong retention by thiols. While dynamic‐covalent chemistry has been explored for cellular uptake, including examples also with imines, hydrazones, and boronic esters,16, 26 high‐affinity adaptive networks evolving in situ represent a new concept for thiol‐mediated uptake of COCs and beyond. This is of interest because conceptually new ways to enter into cells have the intrinsic potential to, by acting differently, provide solutions for uptake problems that are otherwise intractable. The unusual nature of the identified dynamic‐covalent BPS network in particular could be worth considering also with regard to templated amplification for functions beyond cellular uptake that involve distance‐sensitive multivalency.27
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the NMR, the MS, and the Bioimaging platforms for services, and the University of Geneva, the NCCR Molecular Systems Engineering, the NCCR Chemical Biology, and the Swiss NSF for financial support. J.L.A. acknowledges a Curie fellowship (740288) and Y.O. acknowledges a JSPS Overseas research fellowship.
Y. Cheng, L. Zong, J. López-Andarias, E. Bartolami, Y. Okamoto, T. R. Ward, N. Sakai, S. Matile, Angew. Chem. Int. Ed. 2019, 58, 9522.
Contributor Information
Dr. Yangyang Cheng, http://www.unige.ch/sciences/chiorg/matile/
Prof. Stefan Matile, Email: stefan.matile@unige.ch.
References
- 1. Steudel R., Chem. Rev. 2002, 102, 3905–3946. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Konstantinova L. S., Rakitin O. A., Rees C. W., Chem. Rev. 2004, 104, 2617–2630; [DOI] [PubMed] [Google Scholar]
- 2b. Münchberg U., Anwar A., Mecklenburg S., Jacob C., Org. Biomol. Chem. 2007, 5, 1505–1518; [DOI] [PubMed] [Google Scholar]
- 2c. Jiang C.-S., Müller W. E. G., Schröder H. C., Guo Y.-W., Chem. Rev. 2012, 112, 2179–2207; [DOI] [PubMed] [Google Scholar]
- 2d. Filipovic M. R., Zivanovic J., Alvarez B., Banerjee R., Chem. Rev. 2018, 118, 1253–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mahendran A., Vuong A., Aebisher D., Gong Y., Bittman R., Arthur G., Kawamura A., Greer A., J. Org. Chem. 2010, 75, 5549–5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Behar V., Danishefsky S. J., J. Am. Chem. Soc. 1993, 115, 7017–7018; [Google Scholar]
- 4b. Toste F. D., Still I. W. J., J. Am. Chem. Soc. 1995, 117, 7261–7262. [Google Scholar]
- 5. Xu J., Chatterjee M., Baguley T. D., Brouillette J., Kurup P., Ghosh D., Kanyo J., Zhang Y., Seyb K., Ononenyi C., Foscue E., Anderson G. M., Gresack J., Cuny G. D., Glicksman M. A., Greengard P., Lam T. L., Tautz L., Nairn A. C., Ellman J. A., Lombroso P. J., PLoS Biol. 2014, 12, e1001923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Greer A., J. Am. Chem. Soc. 2001, 123, 10379–10386. [DOI] [PubMed] [Google Scholar]
- 7. Chatterji T., Gates K. S., Bioorg. Med. Chem. Lett. 2003, 13, 1349–1352. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Azuma Y., Imai H., Kawaguchi Y., Nakase I., Kimura H., Futaki S., Angew. Chem. Int. Ed. 2018, 57, 12771–12774; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12953–12956; [Google Scholar]
- 8b. Herce H. D., Garcia A. E., Cardoso M. C., J. Am. Chem. Soc. 2014, 136, 17459–17467; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Li M., Schlesiger S., Knauer S. K., Schmuck C., Angew. Chem. Int. Ed. 2015, 54, 2941–2944; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 2984–2987; [Google Scholar]
- 8d. Lostalé-Seijo I., Louzao I., Juanes M., Montenegro J., Chem. Sci. 2017, 8, 7923–7931; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8e. McKinlay C. J., Waymouth R. M., Wender P. A., J. Am. Chem. Soc. 2016, 138, 3510–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Morelli P., Bartolami E., Sakai N., Matile S., Helv. Chim. Acta 2018, 101, e1700266; [Google Scholar]
- 9b. Du S., Liew S. S., Li L., Yao S. Q., J. Am. Chem. Soc. 2018, 140, 15986–15996; [DOI] [PubMed] [Google Scholar]
- 9c. Zhou J., Sun L., Wang L., Liu Y., Li J., Li J., Li J., Yang H., Angew. Chem. Int. Ed. 2019, 58, 5236–5240; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5290–5294. [Google Scholar]
- 10.
- 10a. Gasparini G., Sargsyan G., Bang E.-K., Sakai N., Matile S., Angew. Chem. Int. Ed. 2015, 54, 7328–7331; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7436–7439; [Google Scholar]
- 10b. Abegg D., Gasparini G., Hoch D. G., Shuster A., Bartolami E., Matile S., Adibekian A., J. Am. Chem. Soc. 2017, 139, 231–238; [DOI] [PubMed] [Google Scholar]
- 10c. Tirla A., Rivera-Fuentes P., Biochemistry 2019, 58, 1184–1187; [DOI] [PubMed] [Google Scholar]
- 10d. Tirla A., Hansen M., Rivera-Fuentes P., Synlett 2018, 29, 1289–1292; [Google Scholar]
- 10e. Li X., Hou Y., Meng X., Ge C., Ma H., Li J., Fang J., Angew. Chem. Int. Ed. 2018, 57, 6141–6145; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6249–6253. [Google Scholar]
- 11. Zong L., Bartolami E., Abegg D., Adibekian A., Sakai N., Matile S., ACS Cent. Sci. 2017, 3, 449–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chuard N., Poblador-Bahamonde A. I., Zong L., Bartolami E., Hildebrandt J., Weigand W., Sakai N., Matile S., Chem. Sci. 2018, 9, 1860–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Bartolami E., Basagiannis D., Zong L., Martinent R., Okamoto Y., Laurent Q., Ward T. R., Gonzalez-Gaitan M., Sakai N., Matile S., Chem. Eur. J. 2019, 25, 4047–4051; [DOI] [PubMed] [Google Scholar]
- 13b. Laurent Q., Sakai N., Matile S., Helv. Chim. Acta 2019, 102, e1800209. [Google Scholar]
- 14. Shu Z., Tanaka I., Ota A., Fushihara D., Abe N., Kawaguchi S., Nakamoto K., Tomoike F., Tada S., Ito Y., Kimura Y., Abe H., Angew. Chem. Int. Ed. 2019, 58, 6611–6615; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 6683–6687. [Google Scholar]
- 15.
- 15a. Feener E. P., Shen W. C., Ryser H. J., J. Biol. Chem. 1990, 265, 18780–18785; [PubMed] [Google Scholar]
- 15b. Balakirev M., Schoehn G., Chroboczek J., Chem. Biol. 2000, 7, 813–819; [DOI] [PubMed] [Google Scholar]
- 15c. Aubry S., Burlina F., Dupont E., Delaroche D., Joliot A., Lavielle S., Chassaing G., Sagan S., FASEB J. 2009, 23, 2956–2967; [DOI] [PubMed] [Google Scholar]
- 15d. Torres A. G., Gait M. J., Trends Biotechnol. 2012, 30, 185–190; [DOI] [PubMed] [Google Scholar]
- 15e. Brülisauer L., Kathriner N., Prenrecaj M., Gauthier M. A., Leroux J.-C., Angew. Chem. Int. Ed. 2012, 51, 12454–12458; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12622–12626; [Google Scholar]
- 15f. Drake C. R., Aissaoui A., Argyros O., Thanou M., Steinke J. H. G., Miller A. D., J. Controlled Release 2013, 171, 81–90; [DOI] [PubMed] [Google Scholar]
- 15g. Oupický D., Li J., Macromol. Biosci. 2014, 14, 908–922; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15h. Bode S. A., Wallbrecher R., Brock R., van Hest J. C. M., Löwik D. W. P. M., Chem. Commun. 2014, 50, 415–417; [DOI] [PubMed] [Google Scholar]
- 15i. Hashim P. K., Okuro K., Sasaki S., Hoashi Y., Aida T., J. Am. Chem. Soc. 2015, 137, 15608–15611; [DOI] [PubMed] [Google Scholar]
- 15j. Meng X., Li T., Zhao Y., Wu C., ACS Chem. Biol. 2018, 13, 3078–3086; [DOI] [PubMed] [Google Scholar]
- 15k. Li T., Gao W., Liang J., Zha M., Chen Y., Zhao Y., Wu C., Anal. Chem. 2017, 89, 8501–8508; [DOI] [PubMed] [Google Scholar]
- 15l. Tjin C. C., Otley K. D., Baguley T. D., Kurup P., Xu J., Nairn A. C., Lombroso P. J., Ellman J. A., ACS Cent. Sci. 2017, 3, 1322–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ulrich S., Acc. Chem. Res. 2019, 52, 510–519. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Liu Y., Lehn J.-M., Hirsch A. K. H., Acc. Chem. Res. 2017, 50, 376–386; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Cougnon F. B. L., Sanders J. K. M., Acc. Chem. Res. 2012, 45, 2211–2221; [DOI] [PubMed] [Google Scholar]
- 17c. Moulin E., Cormos G., Giuseppone N., Chem. Soc. Rev. 2012, 41, 1031–1049; [DOI] [PubMed] [Google Scholar]
- 17d. Reuther J. F., Dahlhauser S. D., Anslyn E. V., Angew. Chem. Int. Ed. 2019, 58, 74–85; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 76–88; [Google Scholar]
- 17e. Li J., Nowak P., Otto S., J. Am. Chem. Soc. 2013, 135, 9222–9239; [DOI] [PubMed] [Google Scholar]
- 17f. Jin Y., Yu C., Denman R. J., Zhang W., Chem. Soc. Rev. 2013, 42, 6634–6654; [DOI] [PubMed] [Google Scholar]
- 17g. Zarra S., Wood D. M., Roberts D. A., Nitschke J. R., Chem. Soc. Rev. 2015, 44, 419–432; [DOI] [PubMed] [Google Scholar]
- 17h. Herrmann A., Chem. Soc. Rev. 2014, 43, 1899–1933; [DOI] [PubMed] [Google Scholar]
- 17i. Icli B., Solari E., Kilbas B., Scopelliti R., Severin K., Chem. Eur. J. 2012, 18, 14867–14874; [DOI] [PubMed] [Google Scholar]
- 17j. Shyshov O., Brachvogel R.-C., Bachmann T., Srikantharajah R., Segets D., Hampel F., Puchta R., Von Delius M., Angew. Chem. Int. Ed. 2017, 56, 776–781; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 794–799. [Google Scholar]
- 18. Hino T., Sato T., Chem. Pharm. Bull. 1974, 22, 2866–2874. [Google Scholar]
- 19. Yau W.-M., Wimley W. C., Gawrisch K., White S. H., Biochemistry 1998, 37, 14713–14718. [DOI] [PubMed] [Google Scholar]
- 20. Khalil I. A., Kogure K., Akita H., Harashima H., Pharmacol. Rev. 2006, 58, 32–45. [DOI] [PubMed] [Google Scholar]
- 21. Steudel R., Steudel Y., Miaskiewicz K., Chem. Eur. J. 2001, 7, 3281–3290. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Bauzá A., Frontera A., Molecules 2018, 23, 699; [Google Scholar]
- 22b. Gleiter R., Haberhauer G., Werz D. B., Rominger F., Bleiholder C., Chem. Rev. 2018, 118, 2010–2041. [DOI] [PubMed] [Google Scholar]
- 23. Chuard N., Gasparini G., Roux A., Sakai N., Matile S., Org. Biomol. Chem. 2015, 13, 64–67. [DOI] [PubMed] [Google Scholar]
- 24. Okamoto Y., Kojima R., Schwizer F., Bartolami E., Heinisch T., Matile S., Fussenegger M., Ward T. R., Nat. Commun. 2018, 9, 1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Völker T., Dempwolff F., Graumann P. L., Meggers E., Angew. Chem. Int. Ed. 2014, 53, 10536–10540; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10705–10710. [Google Scholar]
- 26.
- 26a. Priegue J. M., Crisan D. N., Martínez-Costas J., Granja J. R., Fernandez-Trillo F., Montenegro J., Angew. Chem. Int. Ed. 2016, 55, 7492–7495; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7618–7621; [Google Scholar]
- 26b. Andersen K. A., Smith T. P., Lomax J. E., Raines R. T., ACS Chem. Biol. 2016, 11, 319–323; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26c. Eising S., van der Linden N. G. A., Kleinpenning F., Bonger K. M., Bioconjugate Chem. 2018, 29, 982–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.
- 27a. Mammen M., Choi S.-K., Whitesides G. M., Angew. Chem. Int. Ed. 1998, 37, 2754–2794; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 2908–2953; [Google Scholar]
- 27b. Fasting C., Schalley C. A., Weber M., Seitz O., Hecht S., Koksch B., Dernedde J., Graf C., Knapp E.-W., Haag R., Angew. Chem. Int. Ed. 2012, 51, 10472–10498; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10622–10650. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary