

Abstract
Congenital muscular dystrophy (CMD) is a class of severe early-onset muscular dystrophies affecting skeletal/cardiac muscles as well as the central nervous system (CNS). Laminin-α2 chain-deficient congenital muscular dystrophy (LAMA2 MD), also known as merosin-deficient congenital muscular dystrophy type 1A (MDC1A), is an autosomal recessive CMD characterized by severe muscle weakness and degeneration apparent at birth or in the first 6 months of life. LAMA2 MD is the most common congenital muscular dystrophy, affecting approximately 4 in 500,000 children. The most common cause of death in early-onset LAMA2 MD is respiratory tract infection, with 30% of them dying within the first decade of life. LAMA2 MD is caused by loss-of-function mutations in the LAMA2 gene encoding for the laminin-α2 chain, one of the subunits of laminin-211. Laminin-211 is an extracellular matrix protein that functions to stabilize the basement membrane and muscle fibers during contraction. Since laminin-α2 is expressed in many tissue types including skeletal muscle, cardiac muscle, Schwann cells, and trophoblasts, patients with LAMA2 MD experience a multi-systemic clinical presentation depending on the extent of laminin-α2 chain deficiency. Cardiac manifestations are typically associated with a complete absence of laminin-α2; however, recent case reports highlight cardiac involvement in partial laminin-α2 chain deficiency. Laminin-211 is also expressed in the brain, and many patients have abnormalities on brain imaging; however, mental retardation and/or seizures are rarely seen. Currently, there is no cure for LAMA2 MD, but various therapies are being investigated in an effort to lessen the severity of LAMA2 MD. For example, antisense oligonucleotide-mediated exon skipping and CRISPR-Cas9 genome editing have efficiently restored the laminin-α2 chain in mouse models in vivo. This review consolidates information on the clinical presentation, genetic basis, pathology, and current treatment approaches for LAMA2 MD.
Keywords: LAMA2, exon skipping, genome editing, non-homologous end joining, phosphorodiamidate morpholino oligomer, CRISPR/Cas9
Introduction
Laminin-α2 chain-deficient muscular dystrophy (LAMA2 MD), or merosin-deficient congenital muscular dystrophy type 1A (MDC1A), is an autosomal recessive disorder caused by LAMA2 gene mutations that lead to loss of laminin-α2.1 The extent of laminin-α2 deficiency dictates disease severity in most cases. Complete laminin-α2 loss results in an early-onset, congenital form of LAMA2 MD characterized by severe hypotonia, muscle weakness, skeletal deformity, non-ambulation, and respiratory insufficiency.2,3 On the other hand, partial loss of laminin-α2 manifests as a late-onset, limb girdle-type muscular dystrophy form of LAMA2 MD. This presents with similar symptoms as early-onset LAMA2 MD albeit considerably milder and with wider phenotypic variability; most patients develop the ability to walk.1,2 Cardiac disease is associated with either case. There is currently no cure for LAMA2 MD.
The global prevalence of LAMA2 MD is poorly known and varies across sources. Based on available estimates, it affects about 1–9/1,000,000 individuals.2 Early-onset LAMA2 MD is the most common form of congenital muscular dystrophy (CMD) globally, affecting about 30% of the CMD patients in Europe and 6% of the patients in Japan.4–6 One study in Denmark revealed that late-onset LAMA2 MD accounts for 2.3% of the limb girdle-type muscular dystrophy cases.7
In 1994, Tomé et al first described LAMA2 MD as a form of CMD characterized by loss of laminin-211, then called merosin.8 The following year, Helbling-Leclerc et al determined this was caused by LAMA2 gene mutations.9 Laminin-α2, interacting with laminin-β1 and -γ1, forms the cruciform-like laminin-211 structure.2 There are numerous laminin isoforms, formed from various combinations of α, β, and γ chains, but laminin-211 is the major one in the neuromuscular system.10 Laminins link cells to the basement membrane via binding to cell surface receptors and also stabilize the basement membrane through interactions with each other or with extracellular matrix (ECM) proteins.2 Laminin-α2 deficiency results in a corresponding loss of laminin-211 and the disruption/absence of the basement membrane surrounding muscle fibers. While the specific molecular mechanisms are an area of active research, this ultimately leads to the observed pathology in LAMA2 MD.
Since no curative treatments are available for LAMA2 MD, current strategies in the clinic are focused on management. This usually takes the form of, among others, feeding supplementation for difficulties eating and swallowing, non-invasive ventilation support for respiratory insufficiency, and physical therapy for joint contractures, spinal defects, and other issues.1 As these only provide temporary relief, it is encouraging that many groups are currently developing therapies for LAMA2 MD. Different approaches have been devised with varying rates of success, ranging from laminin-α2 replacement to the modulation of cellular events downstream of laminin-α2 loss such as apoptosis and fibrosis. Strategies to correct the defective LAMA2 gene by genome editing or pre-mRNA using antisense oligonucleotides have also been tested in mouse models and seem promising.
In this review, we provide a comprehensive overview of LAMA2 MD, its clinical presentation, pathophysiology, as well as the approaches that have been developed to treat it.
Clinical presentation
Skeletal muscle-related features
The clinical manifestations of LAMA2 MD vary depending on the degree of laminin-α2 deficiency. Complete absence of laminin-α2 presents as severe early-onset CMD, while partial laminin-α2 deficiency often leads to mild late-onset, limb girdle-type muscular dystrophy.1 Children with severe LAMA2 MD present with a weak cry, generalized muscle weakness and profound muscle hypotonia at birth.11,12 Most of these children have delayed motor developmental milestones and very few acquire independent ambulation.3 With assistance, a small percentage of LAMA2 MD patients may be able to walk, but they invariably lose the ability later on in life. As the disease progresses, affected individuals can develop facial muscle weakness and macroglossia, which result in typical myopathic facies with protruded tongue.1
Early-onset LAMA2 MD is also characterized by respiratory involvement. Weakness of intercostal and accessory muscles results in progressive restriction of the chest wall, decreased lung volume, reduced alveolar gas exchange, and eventually restrictive respiratory insufficiency. Affected individuals also experience skeletal changes such as proximal joint contractures and scoliosis.13 During the early years, contractures tend to occur in the shoulder, elbow, hip, and knee, and progress distally. Within the first decade of life, scoliosis may result in lumbar and thoracic lordosis, which interferes with breathing.14 As a consequence, most children with severe LAMA2 MD require ventilatory support at various points in their life.3 Recurrent chest infections due to reduced secretion clearance are another common presenting feature. Respiratory tract infection is the most common cause of death in early-onset LAMA2 MD children, with 30% of them dying within the first decade of life.15
Complete absence of laminin-α2 often manifests as failure to thrive in children.16 Feeding difficulties, swallowing abnormalities, and difficulty in chewing all contribute to poor weight gain in affected children.16 On top of that, recurrent infections further exacerbate the problem. Most children with early-onset LAMA2 MD fall below the third percentile for weight, and some require enteral feeding to meet their nutritional requirement.3
Since laminin-α2 is distributed widely in the body, including the brain, central nervous system involvement in LAMA2 MD is inevitable.17 White matter abnormalities, often presenting as white matter hyperintensities on cerebral MRI, can be observed in patients at 6 months of age. This manifestation is most helpful for diagnostic purposes since it is not associated with any functional impairment.1 Structural brain changes such as bilateral occipital pachygyria or dysplastic cortical changes were reported in a small percentage of affected children and were associated with intellectual disability or epilepsy.18,19 Progressive sensorimotor neuropathy due to myelination defects in the peripheral nervous system was also reported; however, these findings are usually mild and not clinically significant.20
Individuals with partial laminin-α2 deficiency have milder disease manifestations, later onset of symptoms and are typically classified as having limb girdle-type muscular dystrophy.1,2 Affected people usually stay asymptomatic during the first few years of life, although early muscle degeneration may manifest as a delay in walking or as proximal muscle weakness. Patients may also present with elevated creatine kinase (CK) levels, typical dystrophic muscle features, respiratory insufficiency, and abnormal brain MRI.
Cardiac features
Laminin-α2 chain expression is particularly high in the heart.21 However, cardiac involvement has historically not been the focus of LAMA2 MD clinical presentation.1,2 There were only a few studies reporting cardiac manifestations in patients with LAMA2 MD and none of these were comprehensive. Lately there has been more evidence of cardiac involvement in LAMA2 MD, which raises the question of whether cardiac involvement is truly not a major complication of the disorder or is simply underreported in the literature.22,23 Similar to other muscular dystrophies, with improved ventilatory support and respiratory management, cardiac manifestations may become more important and require more attention in the treatment and management of LAMA2 MD patients.
Cardiac abnormalities are predominantly reported in patients with complete laminin-α2 deficiency. To the best of our knowledge, only two cases of partial laminin-α2 deficiency patients presenting with cardiac involvement have been reported.22,23 One study specifically investigated cardiac involvement in 16 children with CMD using two-dimensional echocardiography.24 Two of 6 children with LAMA2 MD had significant left ventricular dysfunction with ejection fractions (EFs) of less than 40%. Both of these children had complete laminin-α2 deficiency. The average EF of children with complete laminin-α2 deficiency was 43%, which was significantly lower than that of the partial deficiency group at 53%.
Another bibliographical review looked at 248 published patient cases with abnormal immunohistochemical staining of laminin-α2.11 Cardiac features were described in 20 cases, of which 7 had clinically relevant cardiac involvement. Cardiac abnormalities manifested as either a right bundle branch block, dilated cardiomyopathy or borderline changes in cardiac function. In another LAMA2 MD study, cardiac phenotypes were evaluated in 15 out of 51 patients.3 Five patients with a complete absence of laminin-α2 had cardiac abnormalities that include mitral valve regurgitation, pulmonary hypertension, palpitations, and wall motion hypokinesia as seen on the echocardiogram. Normal echocardiograms were observed in 7 cases with complete laminin-α2 deficiency and 3 cases with residual laminin-α2 expression. Documentation on the cardiac status of the remaining patients was not available.
The first case of a partial laminin-α2 defect presenting with cardiac involvement was documented in a patient with two different mutations in the LAMA2 gene.23 At 30 years of age, the first symptoms reported for this patient were palpitations and precordial pain. He also had a single episode of syncope. However, clinical evaluation did not show any signs of cardiomyopathy at the time. Electrocardiography (ECG) showed sinus rhythm, but sporadic ventricular ectopic beats were detected by 24-hr Holter ECG monitoring. Mild left ventricular dilation and reduced EF were observed on the echocardiogram. EF was confirmed to be about 39% by angiocardioscintigraphy. His cardiac status remained unchanged until age 40 when he experienced an episode of syncopal ventricular tachycardia. Long-term evaluation led to a diagnosis of dilated cardiomyopathy with a progressive decrease in ventricular function (EF of 33%), which required implantation of an intracardiac defibrillator.
The second case was also characterized as having partial laminin-α2 deficiency with severe cardiac involvement.22 Echocardiography showed impaired left ventricle contractility and mitral valve prolapse. Cardiac function progressively declined with left ventricle dysfunction and dilatation (fractional shortening of 18%). The patient was diagnosed with dilated cardiomyopathy and, eventually, congestive heart failure NYHA class II-III.
Cardiac abnormalities were previously thought to only manifest in severe early-onset LAMA2 MD with complete absence of laminin-α2.1,2 However, from the above-mentioned studies, we want to emphasize the need for routine cardiac assessment in patients with LAMA2 MD due to the potential for severe presentation even in those with residual expression of the laminin-α2 chain.
Diagnosis
In order to establish or confirm a diagnosis of LAMA2 MD, various strategies can be used including history taking, physical examination, laboratory testing, diagnostic imaging, and molecular genetic testing. For early-onset LAMA2 MD, clinical features indicative of muscle weakness and degeneration such as motor delay, muscle weakness, and profound hypotonia can be the first signs that clinicians notice upon physical examination.1,25 Growth measurement and monitoring in children are essential in the diagnosis and intervention of LAMA2 MD, since children with complete absence of laminin-α2 typically present with failure to thrive.16 As mentioned earlier, serum CK levels are usually elevated in patients with LAMA2 MD. Although serum CK is not a specific marker, it is still useful in the diagnosis and confirmation of LAMA2 MD. Expression levels of laminin-α2 can be evaluated using immunohistochemistry of skin or muscle biopsies. Antibodies against various regions of the laminin-α2 chain can be used to detect the presence of the protein as well as its level of expression. Histology studies using hematoxylin and eosin staining of muscle may show characteristic findings of muscle degeneration such as increased numbers of centrally nucleated fibers, muscle fibrosis, and fat infiltration.1 Structural brain alterations and white matter abnormalities in LAMA2 MD patients can also be revealed by brain MRI.1,2,18 Typical cardiovascular diagnostic tools such as ECG and echocardiography are useful in the evaluation of cardiac abnormalities, especially in patients with a complete absence of laminin-α2.1,21 Since LAMA2 is the only gene directly affected in LAMA2 MD, molecular genetic testing is the most definitive approach to confirm patient diagnosis.
It is essential to distinguish LAMA2 MD from other neuromuscular disorders since the clinical features and laboratory findings for LAMA2 MD can be non-specific. Early-onset LAMA2 MD needs to be differentiated from other forms of CMD, congenital myopathies and spinal muscular atrophy (SMA). Other neuromuscular disorders are not typically associated with lack of laminin-α2 staining in immunohistochemistry or white matter changes on the brain MRI.1 Histological studies prove to be most useful in distinguishing LAMA2 MD from congenital myopathies, since the latter often show pathognomonic structural abnormalities that are indicative of each condition.1 With regard to SMA as a differential diagnosis, SMA typically presents with rapid motor impairment and tongue fasciculations. A denervation-reinnervation profile from muscle biopsy findings or electromyography is suggestive of a diagnosis of SMA. Late-onset LAMA2 MD needs to be differentiated from other forms of limb girdle-type muscular dystrophy.1 Despite the overlap in clinical presentation among these diseases, protein and genetic studies can help provide a more definitive diagnosis of LAMA2 MD.
Pathophysiology
Laminin-α2 chain biology
LAMA2 MD is caused by a complete or partial deficiency of laminin-α2 chain protein. Laminin-α2 chain is encoded by the LAMA2 gene, which is transcribed and translated into a 390-kDa protein.26,27 After translation, the laminin-α2 chain is cleaved at the 2580th amino acid into a 300-kDa N-terminal fragment and an 80-kDa C terminal fragment that are non-covalently associated with each other.28,29 Laminin-α2 is a component of a heterotrimeric, cross-shaped molecule known as laminin-211 (or merosin).26,30,31 There are many laminin isomers with different compositions and arrangements of laminin subunits. Laminin-211 is the major isoform expressed in the basement membrane of cardiac and skeletal muscle.10 Laminin-211 is also found in Schwann cells and trophoblasts.10 Laminin-211 is an essential part of the dystrophin-glycoprotein complex (DGC), which provides mechanical support and stabilizes muscle cell membranes during contraction and relaxation cycles (Figure 1a).32,33 Besides laminin-α2, laminin-211 is composed of 2 other subunits: laminin-β1 and laminin-γ1. Once laminin-α2 is translated, it joins laminin-β1 and laminin-γ1 to form laminin-211.34 How laminin-211 is delivered to the muscle cell or how this process of delivery is regulated is not well understood. It has been demonstrated that the N-terminal domain of laminin-α2 is essential for laminin-211 self-assembly at the muscle cell surface.2,35 Laminin-211 networks are associated with cell surface receptors, collagen IV network, and heparan sulfate proteoglycans.2,36 The C-terminus of laminin-α2, which contains 5 laminin G (LG)-like domains (LG1-5), is also important for linking laminin-211 to the cytoskeleton in skeletal muscle cells via dystroglycan and integrin α7β1.37,38 Dystroglycan has 2 subunits: an α-dystroglycan subunit which binds laminin-211 at the LG4-5 and LG1-3 domains, and a β-dystroglycan subunit which binds dystrophin, a major protein linking the actin cytoskeleton of muscle cells to the DGC and, thus, the ECM.33,39–41 Integrin-laminin-211 association requires the LG1-3 domains of the laminin-β1, laminin-γ1, and laminin-α2 chains.2,42–45
Figure 1.
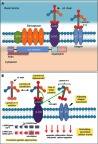
(A) Laminin-α2 and the dystrophin glycoprotein complex. Laminin-α2 interacts with the laminin β and γ chains to form laminin-211, which binds both α-dystroglycan (α-DG) and the α7β1 integrin. Other members of the dystrophin glycoprotein complex are also depicted, with the dystrophin domains shown. β-DG, β-dystroglycan; SPN: sarcospan. (B) Therapeutic strategies developed for LAMA2 MD. An overview of the various LAMA2 MD treatments (in yellow boxes) is shown. For the LAMA2 gene and pre-mRNA diagrams depicted, a red “X” represents the location of the indicated mouse model mutation.
Abbreviations: CMD, congenital muscular dystrophy; CNS, central nervous system; LAMA2 MD, Laminin-α2 chain-deficient muscular dystrophy; MDC1A, merosin-deficient congenital muscular dystrophy type 1A; NHEJ, nonhomologous end-joining; PMO, phosphorodiamidate morpholino oligomer; ECM, extracellular matrix; CK, creatine kinase; ECG, electrocardiography; EF, ejection fraction; SMA, spinal muscular atrophy; DGC, dystrophin-glycoprotein complex; LG, laminin G; CRISPR, Clustered Regularly Interspaced Short Palindromic Repeats; TA, tibialis anterior; α2LN, laminin-α2 N-terminal domain; MCK, muscle creatine kinase; CNF centrally nucleated fiber; EHS, Engelbreth-Holm-Swarm; IGF-1, insulin-like growth factor 1; EDL, extensor digitorum longus; SOL, soleus; MLC, myosin light chain; TGF-β1, transforming growth factor β1; AO, antisense oligonucleotide; DMD, Duchenne muscular dystrophy.
Genetics of LAMA2 MD
Laminin-α2 is encoded by the LAMA2 gene, which maps to chromosome 6q22.33 and is composed of 65 exons.26,27,46 Pathogenic variants in the LAMA2 gene give rise to a group of muscular diseases collectively referred to as LAMA2 MD. LAMA2 MD is inherited in an autosomal recessive fashion. As mentioned in the introduction, the prevalence of LAMA2 MD varies significantly depending on the source. LAMA2 MD has a wide mutational spectrum, ranging from changes in the coding sequence that create premature stop codons, to splice site mutations that result in the translation of pathogenic protein isoforms. In fact, the most common reported variants are those that create premature stop codons, leading to truncation of the protein.3 Loss-of-function mutations in both copies of the LAMA2 gene give rise to the more severe early-onset LAMA2 MD. These mutations were found scattered throughout the LAMA2 coding region, with 55% clustering in exons 14, 25, 26, and 27.1,3 On the other hand, missense variants, in-frame deletions and splice site mutations are often associated with late-onset LAMA2 MD where residual laminin-α2 chain expression can still be detected. It was estimated that 18.4% of the disease-causing variants in LAMA2 MD is due to large deletions and duplications.47,48
For the most part, mutations that result in complete laminin-α2 deficiency lead to a more severe phenotype. In the study of 51 patients with confirmed LAMA2 MD that we have described earlier, those with a complete deficiency of laminin-α2 showed earlier symptom onset (at or within 7 days of birth), and were more likely to never achieve independent ambulation and to require ventilatory and enteral feeding support.3 In another study of 26 LAMA2 MD patients, only 3 were able to achieve independent walking, 2 of whom harbored a missense or a single in-frame deletion mutation in one allele of the LAMA2 gene in heterozygosity with a frame-shifting mutation.49 All patients with frame-shifting mutations in both copies of the gene were unable to acquire independent ambulation. However, there are exceptions to this rule. Geranmayeh et al3 reported two individuals with complete laminin-α2 deficiency, both of whom had generally milder phenotypes, gained independent ambulation, and did not require feeding or ventilatory support.3 In-frame deletions affecting the C-terminal region of the laminin-α2 chain, which is essential for linking laminin-211 to the cytoskeleton in muscle cells, result in severe phenotypes despite the detection of residual laminin-α2.25 LAMA2 MD also shows intrafamilial clinical variability. Affected siblings with the same genotype may have different clinical manifestations.1 As previously mentioned, LAMA2 MD patients may present with brain abnormalities that are associated with intellectual disability and seizures. However, no association was found between patient genotypes and the manifestation of nervous system disease phenotypes.1,47
Pathogenesis
The primary mechanism for LAMA2 MD pathogenesis is a complete or partial deficiency of laminin-α2 in muscle. When the laminin-α2 chain is defective or absent, muscle fibers experience mechanical stress and become susceptible to tearing and fragmentation, resulting in tissue injury and degeneration. Following injury, infiltrating inflammatory cells and muscle stem cells (called satellite cells) coordinate their activities to restore tissue homeostasis.50 However, in situations with chronic tissue damage such as in LAMA2 MD, inflammatory cell infiltration and fibroblast activation persist while satellite cells are being constantly depleted due to the muscle experiencing continuous cycles of degeneration and regeneration. Eventually, the muscle tissue is deposited with excessive amounts of ECM components and is replaced by permanent scars or fibrotic tissue.51–53 Transcriptomic and proteomic studies have indicated that the most upregulated genes in LAMA2 MD encode for ECM proteins and specific isoforms of proteins that are transiently expressed during normal muscle development and regeneration.54–56
In LAMA2 MD, dystroglycan and integrin α7β1 expression levels are also altered. α-dystroglycan is reduced, while both glycosylated α-dystroglycan and β-dystroglycan levels are slightly increased.45,57–59 Although integrin α7β1 expression levels are increased, its assembly process at the muscle cell membrane is compromised.45,57,59,60 Integrin α7β1 is essential for satellite cell activation, which functions in muscle repair and regeneration.61 Reduction in integrin α7β1 activity and, subsequently, satellite cell function invariably result in impaired muscle regeneration. Additionally, integrin α7β1 plays an important role in the survival of muscle fibers.45 Integrin α7β1 dysregulation along with pathological alterations in other signaling pathways contribute to the abnormal skeletal muscle cell apoptosis observed in LAMA2 MD.2
Besides impaired regeneration, the imbalance between protein synthesis and protein breakdown is another factor leading to loss of muscle mass and muscle atrophy in LAMA2 MD. The ubiquitin-proteasome system and the autophagy-lysosome pathway, both of which function in protein degradation, are upregulated in LAMA2 MD.2 There is also evidence of integrin α7 subunit involvement in the negative regulation of these pathways.62 Decreases in integrin α7 expression, therefore, can lead to over-activation of proteasome and autophagy activity in muscle cells.63
Treatment strategies for LAMA2 MD
Therapeutic strategies for LAMA2 MD can be broadly classified into three types. The first aims to restore the structure and function of the basement membrane, as well as its interactions with adjacent cells. The second aims to modulate cellular events caused by laminin-α2 loss. Finally, the last group targets the genetic defect in LAMA2 MD, either through affecting mRNA processing or correcting the causative mutation using the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 system. We provide a summary of these approaches in Figure 1b and Table 1.
Table 1.
Summary of LAMA2 MD therapeutic strategies discussed in the review
| Purpose | Strategy | Method/drug | Comments; challenges | Main ref/s |
|---|---|---|---|---|
| Basement membrane treatment | Laminin-α2 replacement and substitution | Laminin-α2 replacement | Cell therapy a potential approach, transgenic mice show disease amelioration; laminin-α2 immunogenicity could be an issue | 64,69 |
| Laminin-α1 provision | Functionally compensates for laminin-α2; large cDNA size poses delivery issues, laminin-α1 is not exactly laminin-α2 | 74,76 | ||
| Laminin-111 therapy | Reduces dystrophy in LAMA2 MD mice, laminin-111 can be derived from EHS tumors; pharmacokinetic study recommended | 82,83 | ||
| Use of linker proteins | Mini-agrin | Enhances laminin association to α-dystroglycan; very focused, may need to be used with other linker proteins or therapies | 86 | |
| αLNND | Enhances laminin polymerization and binding to collagen IV; same challenge as for mini-agrin | 93,95 | ||
| Adjusting integrin expression | α7 integrin overexpression | Transgenic LAMA2 MD mice had moderately improved lifespan, muscle function; other integrin targets can be explored | 96 | |
| β1 integrin inhibition | RGD inhibition of β1 integrin activity improved ECM composition, myofiber stability; more research into the role of α7β1 in disease | 97 | ||
| Modulating cellular events caused by laminin-α2 loss | Enhancing cell growth | IGF-1, clenbuterol | Improved myofiber size, generally partial improvements on health and survival; treatment dose, administration, regimen needs work | 100,103 |
| Reducing apoptosis | Bax inhibition, Bcl2 expression | Disease amelioration was successfully observed in transgenic mice; need to find a way to alter expression pharmacologically | 104 | |
| Omigapil, doxycycline | Recently completed phase I clinical trial for omigapil in LAMA2 MD therapy; efficacy and other outcomes to be released | 106,108,109 | ||
| Inhibiting the immune response and fibrosis | Losartan, TXA127 | TXA127 granted Orphan Drug status for treating LAMA2 MD; exact information on safety and efficacy remains to be seen | 113,118 | |
| Prednisolone, halofuginone, GTA, FTS, C3, galectin-3, osteopontin | Various effects on LAMA2 MD disease, from helpful to harmful; each needs in-depth study, side effects have to be considered | 122–126 | ||
| Targeting other intracellular systems of regulation | Proteasome inhibition (MG-132, bortezomib) | Partially useful to having no effect at all in ameliorating LAMA2 MD; rethinking of the approach or search for more targeted inhibitors recommended | 127–129 | |
| Autophagy inhibition (3-MA) | Partially useful in ameliorating LAMA2 MD; same challenge as for proteasome inhibition | 130 | ||
| Reducing calcium levels and its effects (caldecrin, cyclophilin D inactivation) | Promising results for LAMA2 MD treatment; however, studies are very preliminary and more research into other outcomes of treatment is required | 131,132 | ||
| Targeting metabolism (metformin) | Gender-specific therapeutic effect observed; mechanism of action largely unknown | 133 | ||
| Targeting glycosylation (CT GalNAc transferase overexpression) | Ameliorates LAMA2 MD in dystrophic mice; mechanism of action also unclear, but may be linked to enhancing agrin expression | 134 | ||
| Genetic correction | Exon skipping | Lama2 exon 4 skipping PMO | Laminin-α2 protein production was restored in the dy3K model, and dystrophic symptoms were treated; PMO delivery can be improved, patient applicability currently limited | 138 |
| CRISPR/Cas9 | Exon 2 inclusion | Lama2 mutation corrected in the dy2J model, laminin-α2 production rescued, treated animals significantly improved; off-target effects are a concern, need strategies for other mutations | 140 | |
| Lama1 overexpression | Lama1 expression was induced in vitro and in vivo, can potentially be used for other attractive targets, eg agrin; assessment of functional effects lacking | 141 |
Abbreviations: IGF-1, insulin-like growth factor 1; GTA, glatiramer acetate; FTS, farnesylthiosalicylic acid; C3, complement 3; 3-MA, 3-methyladenine; CT, cytotoxic T cell; PMO, phosphorodiamidate morpholino oligomer; EHS, Engelbreth-Holm-Swarm; ECM, extracellular matrix.
Treating the basement membrane
Laminin-α2 replacement and substitution
The most straightforward way to treat LAMA2 MD is to replenish what is lost. Using the dy/dy LAMA2 MD mouse model, Vilquin et al first demonstrated partial laminin-α2 replacement by primary muscle cell culture transplantation in 1996.64 dy/dy mice carry a spontaneous mutation that results in very low to absent laminin-α2 expression in striated muscle basement membranes.65,66 Although Lama2 has been mapped to the same region as the dy locus, the exact nature and location of dy remain unknown.65 dy/dy mice exhibit progressive ataxia and muscle wasting. Histology reveals extensive fibrosis and generally smaller, fewer muscle fibers. These mice have decreased survival, with most dying by 6 months of age.67 Allogeneic transplantation of primary myoblasts from healthy mice to the tibialis anterior (TA) muscles of dy/dy mice resulted in up to 15.9% laminin-α2-positive fibers on average, with younger recipients showing more laminin-α2 rescue.64 Use of notexin and ɣ-irradiation increased the number to 27.8% on average. Syngeneic transplantation resulted in a mean 41.2% of laminin-α2-positive fibers, while transplantation of immortalized myoblasts or a fibroblast cell line yielded little to none. In a separate study, the group showed that transplantation of pure myoblasts was also successful in producing laminin-α2-positive fibers.68 Since no other assessments were done, the functional benefit of the approach cannot be determined.
Evaluation of the benefits of transgenic LAMA2 overexpression for LAMA2 MD treatment was reported by Kuang et al (1998).69 Instead of dy/dy, they used the milder dy2J/dy2J and the more severe dyW/dyW mouse models. The dy2J/dy2J, dyW/dyW, and dy3K/dy3K (to be described later) mouse models have entirely different mutations from dy/dy mice; however, the dy nomenclature was retained for ease of classification. The dy2J/dy2J model has a spontaneous G-to-A donor splice site mutation in intron 2 of the Lama2 gene.70 This excludes exon 2 from the pre-mRNA and creates an in-frame deletion of the laminin-α2 N-terminal domain (α2LN) responsible for polymerization. Laminin-α2 mRNA and protein expression are only slightly reduced in dy2J/dy2J mice, contributing to the decreased severity of phenotypes observed in this model. In contrast, the mutation in dyW/dyW mice was generated by targeted disruption of the Lama2 gene, which led to severely reduced laminin-α2 expression.69 dyW/dyW mice, therefore, have a worsened dystrophic phenotype, with most dying 2–4 weeks after birth or, in more recent reports, a median survival of ~8–14 weeks.71
Homozygous dy2J and dyW mice that were heterozygous for the human LAMA2 transgene were examined.69 The transgene was controlled by the muscle creatine kinase (MCK) promoter, which specifically expresses it in striated muscles. Transgenic LAMA2 improved the overall phenotype of dyW/dyW mice, significantly increasing body weight and prolonging survival to at least 8 months. Less improvement was observed for transgenic dy2J/dy2J mice, due to their already mild phenotype. Histologically, both transgenic dyW/dyW and dy2J/dy2J mice had skeletal muscles that appeared nearly wild-type, with evidence of only a mild myopathy due to the appearance of a few centrally nucleated fibers (CNFs). This corresponded to a significant reduction in serum CK activity in these mice, to at least 50% of the non-transgenic levels.
A potential issue associated with laminin-α2 therapy in LAMA2 MD patients is the induction of an immune response against laminin-α2 itself.59 Most patients have no laminin-α2 since birth, and so any introduced laminin-α2 will be seen as foreign and may elicit an immune response. To overcome this, groups have looked into treating LAMA2 MD through laminin-α1 (LAMA1) overexpression. Out of all the α laminins, laminin-α1 is the most structurally similar to laminin-α2.72,73 However, laminin-α1 is not expressed in the adult neuromuscular system, being found mostly during early embryogenesis and having decreased expression in most adult tissues except the epithelia, kidneys, testes, and liver.21,73 Exogenous provision of laminin-α1 or activation of silenced LAMA1 promoters in muscles and nerves is therefore necessary.
Gawlik et al (2004) produced transgenic dy3K/dy3K mice with the mouse Lama1 cDNA driven by a cytomegalovirus (CMV) enhancer and a chicken β-actin promoter.74 The dy3K/dy3K model was also created by targeted Lama1 gene disruption, resulting in a complete deficiency of laminin-α1.75 Severe dystrophic phenotypes and increased cell death were observed, accompanied by significant growth retardation and a shortened lifespan of typically less than 5 weeks. A transgenic dy3K/dy3K line that expressed Lama1 the highest in skeletal muscle had significantly higher body weight, was as active as wild-type, and are fertile. Muscle basement membranes were restored, and dystrophic histopathology was ameliorated. Lifespan was increased to beyond 10 weeks, and a more longitudinal study76 showed ~63% can survive up to 1.5 to 2 years while maintaining Lama1 expression and restored skeletal/cardiac muscle morphology. A different transgenic dy3K/dy3K line that expressed Lama1 in skeletal muscles and peripheral nerves was also studied and showed amelioration of neurological phenotypes (eg, demyelination).77
Transgenic overexpression of Lama1 likewise improved dystrophic phenotypes in dy2J/dy2J mice;78 it may also positively affect fertility, as shown in dy3K/dy3K mice.79 While promising, the laminin-α1 substitution approach is hindered by the large size of the LAMA1 cDNA (~9.6 kbp). This size is difficult to package into viral vectors. LAMA2 cDNA shares a similar size and faces the same issue. High-capacity adenoviral vectors overcome this size limit,80 but their application remains to be tested for LAMA2 MD. Another option is electroporation, which has been done previously for LAMA2-containing plasmids,81 if its efficiency can be improved.
One way around this issue is by using protein therapy. Laminin-α1, with laminin-β1 and -γ1, forms laminin-111 in basement membranes and functions similarly as laminin-211.82 Laminin-111 provision may be a feasible treatment for LAMA2 MD. Intraperitoneal injections of laminin-111 (10 mg/kg/week) derived from Engelbreth-Holm-Swarm (EHS) mouse tumors were previously done in dyW/dyW mice.82 Treatment increased lifespan 3.5-fold, with a median survival at ~9.5 months compared to saline-injected controls at ~2.7 months. Forelimb strength, mouse activity, and muscle fiber count were significantly improved yet still significantly less than wild-type. Laminin-111 therapy can also improve the regenerative capacity of dyW/dyW cardiotoxin-injured muscles.83 For this kind of therapy, however, an in-depth study of the pharmacokinetic characteristics of laminin-111 is recommended to ensure delivery and bioavailability. Excitingly for the field, laminin-111 has proven highly beneficial for the treatment of Duchenne muscular dystrophy (DMD), a related disorder caused by lack of the dystrophin protein and subsequent disruption of the DGC. With promising results in a large animal model,84 it is likely that clinical trials testing laminin-111 for DMD treatment will soon be underway. This shows that therapies aimed at ECM restoration may not necessarily be limited to treating a single neuromuscular disorder, given the often-shared molecular pathophysiology of this group of diseases.
Use of linker proteins
Restoring interactions between laminins and cell surface receptors contributes substantially to the therapeutic efficacy, since these interactions mediate signaling between the ECM and adjacent cells, as well as help maintain membrane integrity. To treat LAMA2 MD, certain groups have instead focused on restoring or strengthening these interactions through linker proteins. The most studied linker protein for LAMA2 MD therapy is the miniaturized form of agrin or mini-agrin. Agrin is a heparan sulfate proteoglycan whose muscle-specific isoform has N- and C-terminal domains that bind laminins and α-dystroglycan, respectively.85 Agrin is thought to be important for helping transmit forces between the basement membrane and the cortical cytoskeleton of muscle cells via the DGC.85 Mini-agrin is composed of these N- and C-terminal domains, connected by one follistatin-like domain.86 Laminin-α4 is upregulated in LAMA2 MD and forms laminin-411 with the β1 and γ1 chains, but only weakly binds α-dystroglycan.86,87 It is expected that mini-agrin will help make laminin-411 a substitute for laminin-211.
Moll et al (2001) created transgenic dyW/dyW mice with a chick mini-agrin cDNA construct driven by the mouse MCK promoter.86 Mini-agrin was correctly localized in basement membranes at high levels in skeletal muscles, but only minimally in the heart. Transgenic mice had generally improved health, with wild-type-like body weight and growth, as well as improved performance in the open field and rotarod tests. Survival was increased to at least 40 weeks. Myopathy was mostly non-evident in 4-week-old transgenic mice, but became more apparent in 16-week-old mice. CK activity was also significantly reduced in transgenic mice, yet still about thrice the levels observed in wild-type. Late-onset expression of the mini-agrin transgene had a similar effect.88 Transgenic dy3K/dy3K Lama2-null mice with mini-agrin were made in a different study, and while improvements in muscle morphology and regeneration were observed, these were not as good as those displayed by transgenic dyW/dyW mice.89
Mini-agrin treatment does not completely ameliorate LAMA2 MD pathology. This can be due to a number of reasons, one being insufficient delivery to target tissues. Many studies have investigated potential means of delivery including using adeno-associated viruses (AAVs) with serotypes 1, 2,90 and 9,91 or using a combined cell- and gene-therapy approach with mesoangioblasts, which are mesodermal, blood vessel-associated progenitor cells.92 Thus far, use of AAV9 seems to be most promising in terms of treating both muscular and neurological phenotypes of LAMA2 MD.
Another reason for the reduced efficacy could be that laminin polymerization, which is not addressed by mini-agrin, is required for complete therapy. To remedy this, a fusion protein was made that consisted of the laminin α1 LN polymerization domain at the N-terminus, and the nidogen-1 G2 and G3 domains at the C-terminus. The protein, called αLNNd, can direct laminin polymerization through the LN domain, bind laminin γ1 via the G3 domain, and bind collagen IV via the G2 and G3 domains.93 Studies showed that αLNNd can rescue the polymerization of mutant, N-terminal truncated laminins, such as those in dy2J/dy2J mice, which significantly ameliorated fibrosis and myofiber morphology, as well as improved forelimb grip strength.94 Mice transgenic in both mini-agrin and αLNNd had more continuous basement membranes, less fibrosis, and more and bigger muscle fibers than single transgenic mice.95 Muscle function, body weight, and survival were also better in double transgenic mice, yet there is room for improvement to reach wild-type levels. Given such findings, exploration of other linker proteins, eg, those with both polymerization and cell surface receptor binding functions, would be worth looking into for therapy.
Adjusting integrin expression and activity
Approaches have also been developed to modify integrin expression for LAMA2 MD, as integrin dysregulation is a feature of the disease. Overexpression of the α7 integrin subunit was done by Doe et al (2011) in dyW/dyW mice by transgene introduction.96 This led to enhanced localization of the α7β1 integrin at skeletal muscle cell membranes and generally ameliorated muscle histology. Lifespan was prolonged 2.4-fold compared to non-transgenic dyW/dyW mice, accompanied by improvements in muscle function. Body weight, however, was not significantly increased by treatment.
A different group showed that a seemingly opposite strategy, ie, β1 integrin inhibition, may be beneficial for LAMA2 MD treatment. Using a lama2−/- zebrafish model of LAMA2 MD, Wood et al (2018) reported that treatment with RGD peptide, a β1 integrin receptor antagonist, significantly increased collagen deposition at the ECM and enhanced muscle fiber stability.97 However, this did not lead to functional improvement as evaluated by a swimming test.
These studies highlight the complexity surrounding the role of the α7β1 integrin in LAMA2 MD pathogenesis. More studies are needed to understand LAMA2 MD biology in this respect. Whichever approach is used, modifying integrin expression or activity does not appear to result in considerable alleviation of LAMA2 MD symptoms. The restoration of other laminin interactions at the basement membrane may be more important for treatment, or perhaps there are key regulators of integrin expression or alternative integrin isoforms, eg, αV and α5,98 that have to be targeted for therapy. While challenging, combinational therapy of the various basement membrane treatment approaches is also an option and will likely result in increased efficacy.
Modulating downstream cellular events
Here, we primarily discuss treatments targeting cell growth and death, the immune response and fibrosis, as well as intracellular systems of regulation. Most of these approaches make use of pharmacological agents, which may have broad ranges of effect. The cellular events listed earlier also exhibit some degree of interdependence with each other. Thus, while we attempt to categorize these treatments for ease of reading, their effects may not be limited to the group we place them in.
Targeting cell growth and death
LAMA2 MD is characterized by muscle wasting and, at the cellular level, reduced myofiber size and number. Treatment with insulin-like growth factor 1 (IGF-1) has been explored as a way to counter this issue. IGF-1 initiates pathways that promote cell growth, differentiation, and survival, eg, MAPK and PI3K signaling.99 Lynch et al (2001) subcutaneously treated dy2J/dy2J mice with 2 mg/kg IGF-1 for 4 weeks and found that it significantly increased the cross-sectional area and mass of the extensor digitorum longus (EDL) and soleus (SOL) muscles.100 Treated mice had significantly higher body mass than dy2J/dy2J controls; however, it did not reach wild-type levels. Kumar et al (2011) conducted a more in-depth study of the restorative effects of IGF-1 treatment by overexpressing it in dyW/dyW mice under control of the myosin light chain (MLC) promoter.101 Besides beneficial effects on growth and survival, the transgene also improved muscle regeneration, decreased apoptosis, and increased mouse activity. Additionally, systemic IGF-1 administration with human mesenchymal stromal cells has been shown to treat dy2J/dy2J mice well.102 Clenbuterol, a β-adrenergic receptor agonist and muscle anabolic agent, also ameliorated disease in the dy/dy model.103
Occurring with reduced muscle growth in LAMA2 MD is increased muscle death. Studies show that interfering with the expression of genes involved in apoptosis can help treat LAMA2 MD. For instance, dyW/dyW mice that were either null for the pro-apoptotic Bax gene or overexpressing the anti-apoptotic Bcl2 gene showed improvements in lifespan, growth, and muscle histology.104,105 Small molecules have also been used to inhibit apoptosis, including the (-)-deprenyl analog omigapil and the antibiotic doxycycline. Omigapil inhibits the GAPDH-Siah1-CBP/p300 apoptotic pathway, which is activated in dyW/dyW mice.106 Omigapil treatment decreased the expression of apoptosis-related genes and the number of apoptotic nuclei in skeletal muscles. Treatment also led to improvements in overall health of the treated mice, and decreased the severity of skeletal defects. Omigapil also decreased fibrosis and improved respiration in dy2J/dy2J mice, but appreciable effects were not observed for muscle function.107 Santhera Pharmaceuticals recently completed a Phase I open-label clinical trial (NCT01805024) in 2018 testing the pharmacokinetic properties, safety, and tolerability of omigapil in CMD patients, including those with LAMA2 MD.108 Results from the dose escalation study have not been published yet; however, the outcome seems favorable.
On the other hand, doxycycline ameliorated both muscle and nerve pathologies in dyW/dyW mice.109,110 Optimization of doxycycline therapy, given its adverse effect on angiogenesis and other cellular processes, remains to be achieved. Studies show that inhibiting apoptosis in tandem with other approaches, eg, IGF-1 supplementation111 or mini-agrin introduction,112 give enhanced benefit in LAMA2 MD mice. Combinational therapy can be an avenue to explore for LAMA2 MD, not only with treatments targeting apoptosis but for other treatments described in this review as well.
Targeting the immune response and fibrosis
The most-studied drug in this category is losartan, an antifibrotic agent. Losartan blocks the angiotensin II receptor, which inhibits activation of the transforming growth factor β1 (TGF-β1) pathway that promotes fibrosis.113,114 Angiotensin II also activates fibrotic pathways independent of TGF-β1,114 which are inhibited by losartan. Losartan and its derivative L-158,809 have been shown to improve muscle strength, regeneration, and histopathology in LAMA2 MD mice.113,115 Interestingly, losartan also exerts effects on the MAPK and NFκB pathways,113,116 as well as reverses dysregulation of the αV and α5 integrins in LAMA2 MD mice.98 Combinational treatment of losartan with IGF-1 further enhanced its therapeutic efficacy.117 In 2016, the US Food and Drug Administration granted Orphan Drug status for TXA127 (Tarix Orphan) for LAMA2 MD treatment.118 TXA127 is a pharmaceutical equivalent of angiotensin (1–7), a naturally occurring peptide derived from angiotensin II cleavage that counteracts angiotensin II.118,119 Studies on TXA127 are yet to be published.
Chronic inflammation is typical in muscular dystrophies and poses numerous harmful effects besides fibrosis such as increased immune cell infiltration and hyperactivity, decreased muscle regeneration, and propagation of muscle death.120,121 Modulators of these processes have been investigated for their potential to treat LAMA2 MD, eg, prednisolone,122 glatiramer acetate,123 halofuginone,124 and farnesylthiosalicyclic acid.125 All have been found to ameliorate LAMA2 MD pathology to different extents. LAMA2 MD mice deficient in the expression of complement 3, galectin-3, and osteopontin—genes that promote inflammation and/or fibrosis—have also been generated and examined. A spectrum of outcomes was observed: complement 3 deficiency proved beneficial for LAMA2 MD treatment,122 whereas loss of galectin-3 and osteopontin had negligible or surprisingly deleterious effects on disease progression, respectively.126
Targeting intracellular systems of regulation
LAMA2 MD is also characterized by increased proteasomal activity, autophagy, and intracellular calcium levels. Inhibiting these processes has proven useful for treating LAMA2 MD. Systemic treatment of dy3K/dy3K mice with the proteasome inhibitor MG-132 significantly reduced fibrosis and apoptosis, as well as significantly increased overall muscle fiber size, mouse activity, and lifespan.127 Systemic treatment with bortezomib, a more selective proteasome inhibitor, likewise resulted in significant improvements in the same model.128 However, proteasome inhibition in general only achieves partial recovery at best and is not particularly useful for treating partial laminin-α2 deficiency.129 The same can be said for the therapeutic efficacy of 3-methyladenine, an inhibitor of autophagy.130 Early studies on handling high intracellular calcium levels in LAMA2 MD have been done and yielded positive results, eg, using caldecrin to reduce serum calcium concentrations131 and decreasing cyclophilin D expression to inhibit activation of calcium-mediated apoptosis.132 Besides these agents, metformin has also been found to reduce pathology and improve health in dy2J/dy2J mice, being generally more beneficial for females than males.133 The mechanisms of action of metformin are uncertain, but it is suggested that it targets metabolism. Finally, overexpression of the cytotoxic T cell GalNAc transferase was also found to reduce dystrophic pathology in dyW/dyW mice, likely by influencing protein glycosylation or by promoting agrin expression in skeletal muscles.134
Corrective genetic approaches to treat LAMA2 MD
Two corrective genetic strategies have been tested for LAMA2 MD: exon skipping and CRISPR/Cas9. A third with a somewhat similar purpose, premature stop codon readthrough using antibiotics, was attempted but did not restore laminin-α2 expression.135
Exon skipping uses antisense oligonucleotides (AOs) to exclude selected exons from the final mRNA product of a gene.136 AOs bind pre-mRNA sequences via base-pairing at chosen sites to influence splicing. Target sites are usually splice sites or exonic splicing enhancers, the masking of which will cause the splicing machinery to skip certain exons. Exon skipping can restore the reading frame of mutant mRNAs created by out-of-frame deletions, as well as exclude out in-frame exons with nonsense mutations. Ultimately, this restores translation of truncated but partially functional proteins, as extensively shown for DMD.137 To test this approach for LAMA2 MD, Aoki et al (2013) intramuscularly injected AOs of the phosphorodiamidate morpholino oligomer (PMO) chemistry into the TAs of dy3K/dy3K mice to skip Lama2 exon 4.138 Two PMOs were used (400 µg/kg), both targeting exon 4, which contained the disruptive neomycin cassette in the dy3K/dy3K model. Exon 4 skipping was induced, and up to 20% laminin-α2-positive fibers (presumably N-terminal truncated forms) were observed in the TAs of treated mice. Intraperitoneal injections of the same PMOs, at a total dose of 150 mg/kg, also resulted in exon 4 skipping and an improvement in lifespan compared to saline-treated controls. It is unclear if exon skipping therapy can lead to other forms of improvement, eg, muscle function, or if its efficacy for treating LAMA2 MD can be improved through enhancing oligonucleotide delivery.
On the other hand, CRISPR/Cas9 induces a more permanent form of correction, through targeted gene editing. The CRISPR/Cas9 system is essentially composed of the Cas9 endonuclease that can make double-stranded DNA breaks and a guide RNA (gRNA) that can direct Cas9 to where it can induce DNA cleavage.139 CRISPR/Cas9 has been tested if it can correct the Lama2 mutation in dy2J/dy2J mice. Kemaladewi et al (2017) designed gRNAs with Cas9 derived from Staphylococcus aureus (SaCas9) to delete the Lama2 intron 2 region containing the splice site mutation through nonhomologous end-joining (NHEJ).140 This leads to use of a different splice site downstream in the intron, which results in exon 2 inclusion and eventual successful translation of laminin-α2. Intramuscular, intraperitoneal, and temporal vein injections of these CRISPR components all led to Lama2 exon 2 inclusion and restored laminin-α2 protein synthesis. Systemic treatment significantly reduced fibrosis and CNF count, and temporal vein injection in particular significantly ameliorated both muscular and neurological phenotypes to nearly wild-type status.
A variant of the CRISPR/Cas9 system can also be used to induce gene expression. Catalytically inactive Cas9 fused to a transcription activation domain such as VP160 can be directed to promoters of selected genes and enhance gene transcription. Perrin et al (2017) showed that this increases laminin-α1 expression in vitro and in vivo.141 Whether this leads to functional improvement, however, remains to be determined.
Conclusion
Extensive progress has been made in understanding the multi-faceted, complex pathophysiology of LAMA2 MD since its initial characterization. Advances in research on laminin-α2 and its role in muscle have contributed largely to this development. We are also now beginning to more appreciate the functions of laminin-α2 in the heart, with cardiac involvement becoming increasingly represented as a feature of LAMA2 MD in the literature. ECM integrity is recognized as a vital contributor to heart structure and function, with pathological alterations to the ECM linked to cardiovascular disease.142 As laminin-211 is a major component of the myocardial ECM, it is likely that its loss will have important implications for cardiac physiology. Furthermore, while we only touched on it briefly here, LAMA2 MD also has a neurological component that primarily concerns the peripheral nerves. Studies developing therapies for LAMA2 MD are increasingly taking this into consideration in treatment or drug design, ensuring that both muscles and nerves benefit from the restorative effects of an approach.
On the subject of treatment, it is reassuring that numerous strategies have been and are being investigated for LAMA2 MD therapy. While some of these are moving closer to the clinic, eg, omigapil and TXA127, much work still needs to be done to improve therapeutic efficacy. It is often the case with these therapies that partial amelioration of LAMA2 MD pathology is achieved; rarely is the effect consistent, complete, and long-lived. More treatment optimization and research are recommended. As mentioned earlier, combinational therapy is an option. Other targets can also be selected for therapy. Various microRNAs are dysregulated in LAMA2 MD, and their expression can certainly be modulated as an approach.143,144 Polyamines,145 decorin,146 Ku70,147 p53, and sirtuin148 are also starting to be recognized as potential therapeutic targets. Managing diet149 or bone marrow transplantation150 may also be options, but follow-up studies into these are lacking. Therapies for other muscular dystrophies can also be adapted and tested for LAMA2 MD, as have been done for many of the strategies described. Overall, a combination of basic, translational, and clinical research efforts are needed to ensure that we not only understand LAMA2 MD in its entirety but also know how to treat it, so as to provide patients with a cure as soon as possible.
Acknowledgments
This work was supported by Muscular Dystrophy Canada, The Friends of Garrett Cumming Research Fund, the HM Toupin Neurological Science Research Fund, Canadian Institutes of Health Research (CIHR) – 132574 and 143251, Alberta Innovates: Health Solutions (AIHS), Canada Foundation for Innovation (CFI), Alberta Advanced Education and Technology, and the Women and Children’s Health Research Institute (WCHRI).
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Quijano-Roy S, Sparks SE, Rutkowski A. LAMA2-Related Muscular Dystrophy. 2012. Jun 7. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. [Google Scholar]
- 2.Durbeej M. Laminin-α2 chain-deficient congenital muscular dystrophy. Curr Top Membr. 2015;31–60. doi: 10.1016/bs.ctm.2015.05.002 [DOI] [PubMed] [Google Scholar]
- 3.Geranmayeh F, Clement E, Feng LH, et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord. 2010;20(4):241–250. doi: 10.1016/j.nmd.2010.02.001 [DOI] [PubMed] [Google Scholar]
- 4.Allamand V, Guicheney P. Merosin-deficient congenital muscular dystrophy, autosomal recessive (MDC1A, MIM#156225, LAMA2 gene coding for α2 chain of laminin). Eur J Hum Genet. 2002;10(2):91–94. doi: 10.1038/sj.ejhg.5200743 [DOI] [PubMed] [Google Scholar]
- 5.Graziano A, Bianco F, D’Amico A, et al. Prevalence of congenital muscular dystrophy in Italy: a population study. Neurology. 2015;84(9):904–911. doi: 10.1212/WNL.0000000000001303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mostacciuolo ML, Miorin M, Martinello F, Angelini C, Perini P, Trevisan CP. Genetic epidemiology of congenital muscular dystrophy in a sample from north-east Italy. Hum Genet. 1996;97(3):277–279. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/8786062. [DOI] [PubMed] [Google Scholar]
- 7.Løkken N, Born AP, Duno M, Vissing J. LAMA2-related myopathy: frequency among congenital and limb-girdle muscular dystrophies. Muscle Nerve. 2015;52(4):547–553. doi: 10.1002/mus.24588 [DOI] [PubMed] [Google Scholar]
- 8.Tomé FM, Evangelista T, Leclerc A, et al. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci III. 1994;317(4):351–357. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/8000914. [PubMed] [Google Scholar]
- 9.Helbling-Leclerc A, Zhang X, Topaloglu H, et al. Mutations in the laminin α2–chain gene (LAMA2) cause merosin–deficient congenital muscular dystrophy. Nat Genet. 1995;11(2):216–218. doi: 10.1038/ng1095-216 [DOI] [PubMed] [Google Scholar]
- 10.Patton BL, Miner JH, Chiu AY, Sanes JR. Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J Cell Biol. 1997. doi: 10.1083/jcb.139.6.1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones KJ, Morgan G, Johnston H, et al. The expanding phenotype of laminin alpha2 chain (merosin) abnormalities: case series and review. J Med Genet. 2001;38(10):649–657. doi: 10.1136/jmg.38.10.649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilhuis HJ, Ten Donkelaar HJ, Tanke RB, et al. Nonmuscular involvement in merosin-negative congenital muscular dystrophy. Pediatr Neurol. 2002;26(1):30–36. doi: 10.1016/S0887-8994(01)00352-6 [DOI] [PubMed] [Google Scholar]
- 13.Bentley G, Haddad F, Bull TM, Seingry D. The treatment of scoliosis in muscular dystrophy using modified Luque and Harrington-Luque instrumentation. J Bone Joint Surg Br. 2001;83(1):22–28. doi: 10.1302/0301-620X.83B1.10029 [DOI] [PubMed] [Google Scholar]
- 14.He Y, Jones KJ, Vignier N, et al. Congenital muscular dystrophy with primary partial laminin α2 chain deficiency: molecular study. Neurology. 2001. doi: 10.1212/WNL.57.7.1319 [DOI] [PubMed] [Google Scholar]
- 15.Wallgren-Pettersson C, Bushby K, Mellies U, Simonds A. ENMC. 117th ENMC workshop: ventilatory support in congenital neuromuscular disorders – congenital myopathies, congenital muscular dystrophies, congenital myotonic dystrophy and SMA (II) 4-6 April 2003, Naarden, The Netherlands. Neuromuscul Disord. 2004;14(1):56–69. doi: 10.1016/j.nmd.2003.09.003 [DOI] [PubMed] [Google Scholar]
- 16.Philpot J, Bagnall A, King C, Dubowitz V, Muntoni F. Feeding problems in merosin deficient congenital muscular dystrophy. Arch Dis Child. 1999;80(6):542–547. doi: 10.1136/adc.80.6.542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Philpot J, Cowan F, Pennock J, et al. Merosin-deficient congenital muscular dystrophy: the spectrum of brain involvement on magnetic resonance imaging. Neuromuscul Disord. 1999;9(2):81–85. doi: 10.1016/S0960-8966(98)00110-2 [DOI] [PubMed] [Google Scholar]
- 18.Sunada Y, Edgar TS, Lotz BP, Rust RS, Campbell KP. Merosin-negative congenital muscular dystrophy associated with extensive brain abnormalities. Neurology. 1995. doi: 10.1212/WNL.45.11.2084 [DOI] [PubMed] [Google Scholar]
- 19.Pini A, Merlini L, Fms T, Chevallay M, Gobbi G. Merosin-negative congenital muscular dystrophy, occipital epilepsy with periodic spasms and focal cortical dysplasia. Report of three Italian cases in two families. Brain Dev. 1996;18(4):316–322. doi: 10.1016/0387-7604(96)00028-9 [DOI] [PubMed] [Google Scholar]
- 20.Di Muzio A, De Angelis MV, Di Fulvio P, et al. Dysmyelinating sensory-motor neuropathy in merosin-deficient congenital muscular dystrophy. Muscle Nerve. 2003;27(4):500–506. doi: 10.1002/mus.10326 [DOI] [PubMed] [Google Scholar]
- 21.Sasaki T, Giltay R, Talts U, Timpl R, Talts JF. Expression and distribution of laminin α1 and α2 chains in embryonic and adult mouse tissues: an immunochemical approach. Exp Cell Res. 2002. doi: 10.1006/excr.2002.5499 [DOI] [PubMed] [Google Scholar]
- 22.Marques J, Duarte ST, Costa S, et al. A typical phenotype in two patients with LAMA2 mutations. Neuromuscul Disord. 2014;24(5):419–424. doi: 10.1016/j.nmd.2014.01.004 [DOI] [PubMed] [Google Scholar]
- 23.Carboni N, Marrosu G, Porcu M, et al. Dilated cardiomyopathy with conduction defects in a patient with partial merosin deficiency due to mutations in the laminin-α2-chain gene: a chance association or a novel phenotype? Muscle Nerve. 2011;44(5):826–828. doi: 10.1002/mus.22228 [DOI] [PubMed] [Google Scholar]
- 24.Spyrou N, Philpot J, Foale R, Camici PG, Muntoni F. Evidence of left ventricular dysfunction in children with merosin-deficient congenital muscular dystrophy. Am Heart J. 1998;136(3):474–476. doi: 10.1016/S0002-8703(98)70222-4 [DOI] [PubMed] [Google Scholar]
- 25.Bönnemann CG, Wang CH, Quijano-Roy S, et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul Disord. 2014;24(4):289–311. doi: 10.1016/j.nmd.2013.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ehrig K, Leivo I, Argraves WS, Ruoslahti E, Engvall E. Merosin, a tissue-specific basement membrane protein, is a laminin-like protein. Proc Natl Acad Sci U S A. 1990;87(9):3264–3268. doi: 10.1073/pnas.87.9.3264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vuolteenaho R, Nissinen M, Sainio K, et al. Human laminin M chain (merosin): complete primary structure, chromosomal assignment, and expression of the M and A chain in human fetal tissues. J Cell Biol. 1994. doi: 10.1083/jcb.124.3.381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smirnov SP, McDearmon EL, Li S, Ervasti JM, Tryggvason K, Yurchenco PD. Contributions of the LG modules and furin processing to laminin-2 functions. J Biol Chem. 2002. doi: 10.1074/jbc.M201880200 [DOI] [PubMed] [Google Scholar]
- 29.Talts JF, Mann K, Yamada Y, Timpl R. Structural analysis and proteolytic processing of recombinant G domain of mouse laminin α2 chain. FEBS Lett. 1998. doi: 10.1016/S0014-5793(98)00312-3 [DOI] [PubMed] [Google Scholar]
- 30.Aumailley M, Bruckner-Tuderman L, Carter WG, et al. A simplified laminin nomenclature. Matrix Biol. 2005. doi: 10.1016/j.matbio.2005.05.006 [DOI] [PubMed] [Google Scholar]
- 31.Leivo I, Engvall E. Merosin, a protein specific for basement membranes of Schwann cells, striated muscle, and trophoblast, is expressed late in nerve and muscle development. Proc Natl Acad Sci. 1988. doi: 10.1073/pnas.85.5.1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res. 2004;94(8):1023–1031. doi: 10.1161/01.RES.0000126574.61061.25 [DOI] [PubMed] [Google Scholar]
- 33.Ervasti JM. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta - Mol Basis Dis. 2007;1772(2):108–117. doi: 10.1016/j.bbadis.2006.05.010 [DOI] [PubMed] [Google Scholar]
- 34.Yurchenco PD, Quan Y, Colognato H, et al. The alpha chain of laminin-1 is independently secreted and drives secretion of its beta- and gamma-chain partners. Proc Natl Acad Sci U S A. 1997. doi: 10.1073/pnas.94.19.10189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Colognato H, Yurchenco PD. The laminin α2 expressed by dystrophic dy2J mice is defective in its ability to form polymers. Curr Biol. 1999. doi: 10.1016/S0960-9822(00)80056-1 [DOI] [PubMed] [Google Scholar]
- 36.Hopf M, Göhring W, Mann K, Timpl R. Mapping of binding sites for nidogens, fibulin-2, fibronectin and heparin to different IG modules of perlecan. J Mol Biol. 2001. doi: 10.1006/jmbi.2001.4878 [DOI] [PubMed] [Google Scholar]
- 37.Campbell KP, Kahl SD. Association of dystrophin and an integral membrane glycoprotein. Nature. 1989. doi: 10.1038/338259a0 [DOI] [PubMed] [Google Scholar]
- 38.Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992. doi: 10.1038/355696a0 [DOI] [PubMed] [Google Scholar]
- 39.Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122(4):809–823. doi: 10.1083/jcb.122.4.809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tisi D, Talts JF, Timpl R, Hohenester E. Structure of the C-terminal laminin G-like domain pair of the laminin alpha2 chain harbouring binding sites for alpha-dystroglycan and heparin. Embo J. 2000;19(7):1432–1440. doi: 10.1093/emboj/19.7.1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wizemann H, Garbe JHO, Friedrich MVK, Timpl R, Sasaki T, Hohenester E. Distinct requirements for heparin and α-dystroglycan binding revealed by structure-based mutagenesis of the laminin α2 LG4-LG5 domain pair. J Mol Biol. 2003. doi: 10.1016/S0022-2836(03)00848-9 [DOI] [PubMed] [Google Scholar]
- 42.Von der Mark H, Durr J, Sonnenberg A, Von der Mark K, Deutzmann R, Goodman SL. Skeletal myoblasts utilize a novel β1-series integrin and not α6β1 for binding to the E8 and T8 fragments of laminin. J Biol Chem. 1991;266(35):23593-601. [PubMed] [Google Scholar]
- 43.Mayer U. Integrins: redundant or important players in skeletal muscle? J Biol Chem. 2003. doi: 10.1074/jbc.R200022200 [DOI] [PubMed] [Google Scholar]
- 44.Song WK, Wang W, Foster RF, Bielser DA, Kaufman SJ. H36-alpha 7 is a novel integrin alpha chain that is developmentally regulated during skeletal myogenesis [published erratum appears in J Cell Biol 1992 Jul;118(1):213]. J Cell Biol. 1992. doi: 10.1083/jcb.117.3.643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vachon PH, Xu H, Liu L, et al. Integrins (α7β1) in muscle function and survival disrupted expression in merosin-deficient congenital muscular dystrophy. J Clin Invest. 1997. doi: 10.1172/JCI119716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang X, Vuolteenaho R, Tryggvason K. Structure of the human laminin alpha2-chain gene (LAMA2), which is affected in congenital muscular dystrophy. J Biol Chem. 1996;271(44):27664–27669. doi: 10.1074/jbc.271.44.27664 [DOI] [PubMed] [Google Scholar]
- 47.Oliveira J, Gruber A, Cardoso M, et al. LAMA2 gene mutation update: toward a more comprehensive picture of the laminin-α2 variome and its related phenotypes. Hum Mutat. 2018;39(10):1314–1337. doi: 10.1002/humu.23599 [DOI] [PubMed] [Google Scholar]
- 48.Oliveira J, Gonçalves A, Oliveira ME, et al. Reviewing large LAMA2 deletions and duplications in congenital muscular dystrophy patients. J Neuromuscul Dis. 2014. doi: 10.3233/JND-140031 [DOI] [PubMed] [Google Scholar]
- 49.Oliveira J, Santos R, Soares-Silva I, et al. LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin Genet. 2008. doi: 10.1111/j.1399-0004.2008.01068.x [DOI] [PubMed] [Google Scholar]
- 50.Mann CJ, Perdiguero E, Kharraz Y, et al. Aberrant repair and fibrosis development in skeletal muscle. Skelet Muscle. 2011;1(1):21. doi: 10.1186/2044-5040-1-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gawlik KI, Holmberg J, Durbeej M. Loss of dystrophin and β-sarcoglycan significantly exacerbates the phenotype of laminin α2 chain-deficient animals. Am J Pathol. 2014. doi: 10.1016/j.ajpath.2013.11.017 [DOI] [PubMed] [Google Scholar]
- 52.Pegoraro E, Mancias P, Swerdlow SH, et al. Congenital muscular dystrophy with primary laminin alpha2 (Merosin) deficiency presenting as inflammatory myopathy. Ann Neurol. 1996. doi: 10.1002/ana.410400515 [DOI] [PubMed] [Google Scholar]
- 53.Wardrop KE, Dominov JA. Proinflammatory signals and the loss of lymphatic vessel hyaluronan receptor-1 (LYVE-1) in the early pathogenesis of laminin alpha2-deficient skeletal muscle. J Histochem Cytochem. 2011. doi: 10.1369/jhc.2010.956672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taniguchi M, Kurahashi H, Noguchi S, et al. Expression profiling of muscles from Fukuyama-type congenital muscular dystrophy and laminin-alpha 2 deficient congenital muscular dystrophy; is congenital muscular dystrophy a primary fibrotic disease? Biochem Biophys Res Commun. 2006;342(2):489–502. doi: 10.1016/j.bbrc.2005.12.224 [DOI] [PubMed] [Google Scholar]
- 55.Häger M, Bigotti MG, Meszaros R, et al. Cib2 binds integrin a7BB1D and is reduced in laminin a2 chain-deficient muscular dystrophy. J Biol Chem. 2008. doi: 10.1074/jbc.M801166200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Lunteren E. Gene expression profiling of diaphragm muscle in 2-laminin (merosin)-deficient dy/dy dystrophic mice. Physiol Genomics. 2006. doi: 10.1152/physiolgenomics.00226.2005 [DOI] [PubMed] [Google Scholar]
- 57.Cohn RD, Mayer U, Saher G, et al. Secondary reduction of alpha7B integrin in laminin alpha2 deficient congenital muscular dystrophy supports an additional transmembrane link in skeletal muscle. J Neurol Sci. 1999;163(2):140–152. doi: 10.1016/S0022-510X(99)00012-X [DOI] [PubMed] [Google Scholar]
- 58.Jimenez-Mallebrera C, Torelli S, Feng L, et al. A comparative study of α-dystroglycan glycosylation in dystroglycanopathies suggests that the hypoglycosylation of α-dystroglycan does not consistently correlate with clinical severity. Brain Pathol. 2009. doi: 10.1111/j.1750-3639.2008.00198.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gawlik KI, Mayer U, Blomberg K, Sonnenberg A, Ekblom P, Durbeej M. Laminin α1 chain mediated reduction of laminin α2 chain deficient muscular dystrophy involves integrin α7β1 and dystroglycan. FEBS Lett. 2006. doi: 10.1016/j.febslet.2006.02.027 [DOI] [PubMed] [Google Scholar]
- 60.Hodges BL, Hayashi YK, Nonaka I, Wang W, Arahata K, Kaufman SJ. Altered expression of the alpha7beta1 integrin in human and murine muscular dystrophies. J Cell Sci. 1997;110(Pt 2):2873–2881. doi: 10.1103/PhysRevE.86.011907 [DOI] [PubMed] [Google Scholar]
- 61.Rooney JE, Gurpur PB, Yablonka-Reuveni Z, Burkin DJ. Laminin-111 restores regenerative capacity in a mouse model for α7 integrin congenital myopathy. Am J Pathol. 2009. doi: 10.2353/ajpath.2009.080522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y, Li H, Lian Z, Li N. Myofibroblasts protect myoblasts from intrinsic apoptosis associated with differentiation via β1 integrin-PI3KAkt pathway. Dev Growth Differ. 2010. doi: 10.1111/j.1440-169X.2010.01209.x [DOI] [PubMed] [Google Scholar]
- 63.Gawlik KI, Durbeej M. Skeletal muscle laminin and MDC1A: pathogenesis and treatment strategies. Skelet Muscle. 2011;1(1):9. doi: 10.1186/2044-5040-1-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vilquin JT, Kinoshita I, Roy B, et al. Partial laminin alpha2 chain restoration in alpha2 chain-deficient dy/dy mouse by primary muscle cell culture transplantation. J Cell Biol. 1996;133(1):185–197. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/8601607. doi: 10.1083/jcb.133.1.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sunada Y, Bernier SM, Kozak CA, Yamada Y, Campbell KP. Deficiency of merosin in dystrophic dy mice and genetic linkage of laminin M chain gene to dy locus. J Biol Chem. 1994;269(19):13729–13732. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/8188645. [PubMed] [Google Scholar]
- 66.Xu H, Christmas P, Wu XR, Wewer UM, Engvall E. Defective muscle basement membrane and lack of M-laminin in the dystrophic dy/dy mouse. Proc Natl Acad Sci. 1994;91(12):5572–5576. doi: 10.1073/pnas.91.12.5572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Michelson AM, Russell ES, Harman PJ. Dystrophia Muscularis: a hereditary primary myopathy in the house mouse. Proc Natl Acad Sci U S A. 1955;41(12):1079–1084. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/16589799. doi: 10.1073/pnas.41.12.1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vilquin J-T, Guérette B, Puymirat J, et al. Myoblast transplantations lead to the expression of the laminin α2 chain in normal and dystrophic (dy/dy) mouse muscles. Gene Ther. 1999;6(5):792–800. doi: 10.1038/sj.gt.3300889 [DOI] [PubMed] [Google Scholar]
- 69.Kuang W, Xu H, Vachon PH, et al. Merosin-deficient congenital muscular dystrophy. Partial genetic correction in two mouse models. J Clin Invest. 1998;102(4):844–852. doi: 10.1172/JCI3705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu H, Wu XR, Wewer UM, Engvall E. Murine muscular dystrophy caused by a mutation in the laminin alpha 2 (Lama2) gene. Nat Genet. 1994;8(3):297–302. doi: 10.1038/ng1194-297 [DOI] [PubMed] [Google Scholar]
- 71.Willmann R, Gordish-Dressman H, Meinen S, et al. Improving reproducibility of phenotypic assessments in the dyw mouse model of laminin-α2 related congenital muscular dystrophy. J Neuromuscul Dis. 2017;4(2):115–126. doi: 10.3233/JND-170217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Domogatskaya A, Rodin S, Tryggvason K. Functional Diversity of Laminins. Annu Rev Cell Dev Biol. 2012;28(1):523–553. doi: 10.1146/annurev-cellbio-101011-155750 [DOI] [PubMed] [Google Scholar]
- 73.Colognato H, Yurchenco PD. Form and function: the laminin family of heterotrimers. Dev Dyn. 2000;218(2):213–234. 10.1002/(SICI)1097-0177(200006)218:2<213::AID-DVDY1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 74.Gawlik K. Laminin 1 chain reduces muscular dystrophy in laminin 2 chain deficient mice. Hum Mol Genet. 2004;13(16):1775–1784. doi: 10.1093/hmg/ddh190 [DOI] [PubMed] [Google Scholar]
- 75.Miyagoe Y, Hanaoka K, Nonaka I, et al. Laminin alpha2 chain-null mutant mice by targeted disruption of the Lama2 gene: a new model of merosin (laminin 2)-deficient congenital muscular dystrophy. FEBS Lett. 1997;415(1):33–39. http://www.ncbi.nlm.nih.gov/pubmed/9326364. [DOI] [PubMed] [Google Scholar]
- 76.Gawlik KI, Durbeej M. Transgenic overexpression of laminin α1 chain in laminin α2 chain-deficient mice rescues the disease throughout the lifespan. Muscle Nerve. 2010;42(1):30–37. doi: 10.1002/mus.21616 [DOI] [PubMed] [Google Scholar]
- 77.Gawlik KI, Li J-Y, Petersén Å, Durbeej M. Laminin α1 chain improves laminin α2 chain deficient peripheral neuropathy. Hum Mol Genet. 2006;15(18):2690–2700. doi: 10.1093/hmg/ddl201 [DOI] [PubMed] [Google Scholar]
- 78.Gawlik KI, Harandi VM, Cheong RY, Petersén Å, Durbeej M. Laminin α1 reduces muscular dystrophy in dy 2J mice. Matrix Biol. 2018;70:36–49. doi: 10.1016/j.matbio.2018.02.024 [DOI] [PubMed] [Google Scholar]
- 79.Häger M, Gawlik K, Nyström A, Sasaki T, Durbeej M. Laminin {alpha}1 chain corrects male infertility caused by absence of laminin {alpha}2 chain. Am J Pathol. 2005;167(3):823–833. http://www.ncbi.nlm.nih.gov/pubmed/16127160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ehrke-Schulz E, Zhang W, Schiwon M, et al. Cloning and large-scale production of high-capacity adenoviral vectors based on the human adenovirus type 5. J Vis Exp. 2016;(107):e52894. doi: 10.3791/52894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vilquin JT, Kennel PF, Paturneau-Jouas M, et al. Electrotransfer of naked DNA in the skeletal muscles of animal models of muscular dystrophies. Gene Ther. 2001;8(14):1097–1107. doi: 10.1038/sj.gt.3301484 [DOI] [PubMed] [Google Scholar]
- 82.Rooney JE, Knapp JR, Hodges BL, Wuebbles RD, Burkin DJ. Laminin-111 protein therapy reduces muscle pathology and improves viability of a mouse model of merosin-deficient congenital muscular dystrophy. Am J Pathol. 2012;180(4):1593–1602. doi: 10.1016/j.ajpath.2011.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Van Ry PM, Minogue P, Hodges BL, Burkin DJ. Laminin-111 improves muscle repair in a mouse model of merosin-deficient congenital muscular dystrophy. Hum Mol Genet. 2014;23(2):383–396. doi: 10.1093/hmg/ddt428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barraza-Flores P, Fontelonga TM, Wuebbles RD, et al. Laminin-111 protein therapy enhances muscle regeneration and repair in the GRMD dog model of duchenne muscular dystrophy. Hum Mol Genet. 2019. doi: 10.1093/hmg/ddz086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bezakova G, Ruegg MA. New insights into the roles of agrin. Nat Rev Mol Cell Biol. 2003;4(4):295–309. doi: 10.1038/nrm1074 [DOI] [PubMed] [Google Scholar]
- 86.Moll J, Barzaghi P, Lin S, et al. An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature. 2001;413(6853):302–307. doi: 10.1038/35095054 [DOI] [PubMed] [Google Scholar]
- 87.Ringelmann B, Röder C, Hallmann R, et al. Expression of laminin α1, α2, α4, and α5 chains, fibronectin, and tenascin-c in skeletal muscle of dystrophic 129ReJdy/dyMice. Exp Cell Res. 1999;246(1):165–182. doi: 10.1006/excr.1998.4244 [DOI] [PubMed] [Google Scholar]
- 88.Meinen S, Barzaghi P, Lin S, Lochmüller H, Ruegg MA. Linker molecules between laminins and dystroglycan ameliorate laminin-α2–deficient muscular dystrophy at all disease stages. J Cell Biol. 2007;176(7):979–993. doi: 10.1083/jcb.200611152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bentzinger CF, Barzaghi P, Lin S, Ruegg MA. Overexpression of mini-agrin in skeletal muscle increases muscle integrity and regenerative capacity in laminin-α2-deficient mice. Faseb J. 2005;19(8):934–942. doi: 10.1096/fj.04-3376com [DOI] [PubMed] [Google Scholar]
- 90.Qiao C, Li J, Zhu T, et al. Amelioration of laminin- 2-deficient congenital muscular dystrophy by somatic gene transfer of miniagrin. Proc Natl Acad Sci. 2005;102(34):11999–12004. doi: 10.1073/pnas.0502137102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Qiao C, Dai Y, Nikolova VD, et al. Amelioration of muscle and nerve pathology in LAMA2 muscular dystrophy by aav9-mini-agrin. Mol Ther - Methods Clin Dev. 2018;9:47–56. doi: 10.1016/j.omtm.2018.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Domi T, Porrello E, Velardo D, et al. Mesoangioblast delivery of miniagrin ameliorates murine model of merosin-deficient congenital muscular dystrophy type 1A. Skelet Muscle. 2015;5(1):30. doi: 10.1186/s13395-015-0055-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McKee KK, Capizzi S, Yurchenco PD. Scaffold-forming and adhesive contributions of synthetic laminin-binding proteins to basement membrane assembly. J Biol Chem. 2009;284(13):8984–8994. doi: 10.1074/jbc.M809719200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McKee KK, Crosson SC, Meinen S, Reinhard JR, Rüegg MA, Yurchenco PD. Chimeric protein repair of laminin polymerization ameliorates muscular dystrophy phenotype. J Clin Invest. 2017;127(3):1075–1089. doi: 10.1172/JCI90854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reinhard JR, Lin S, McKee KK, et al. Linker proteins restore basement membrane and correct LAMA2 -related muscular dystrophy in mice. Sci Transl Med. 2017;9(396):eaal4649. doi: 10.1126/scitranslmed.aal4649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Doe JA, Wuebbles RD, Allred ET, Rooney JE, Elorza M, Burkin DJ. Transgenic overexpression of the α7 integrin reduces muscle pathology and improves viability in the dyW mouse model of merosin-deficient congenital muscular dystrophy type 1A. J Cell Sci. 2011;124(13):2287–2297. doi: 10.1242/jcs.083311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wood AJ, Cohen N, Joshi V, et al. RGD inhibition of itgb1 ameliorates laminin- α 2-deficient zebrafish fibre pathology. Hum Mol Genet. 2018. doi: 10.1093/hmg/ddy426 [DOI] [PubMed] [Google Scholar]
- 98.Accorsi A, Mehuron T, Kumar A, Rhee Y, Girgenrath M. Integrin dysregulation as a possible driver of matrix remodeling in Laminin-deficient congenital muscular dystrophy (MDC1A). J Neuromuscul Dis. 2015;2(1):51–61. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28198706. [PubMed] [Google Scholar]
- 99.Hakuno F, Takahashi S-I. 40 years of IGF1: IGF1 receptor signaling pathways. J Mol Endocrinol. 2018;61(1):T69–T86. doi: 10.1530/JME-17-0311 [DOI] [PubMed] [Google Scholar]
- 100.Lynch GS, Cuffe SA, Plant DR, Gregorevic P. IGF-I treatment improves the functional properties of fast- and slow-twitch skeletal muscles from dystrophic mice. Neuromuscul Disord. 2001;11(3):260–268. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/11297941. [DOI] [PubMed] [Google Scholar]
- 101.Kumar A, Yamauchi J, Girgenrath T, Girgenrath M. Muscle-specific expression of insulin-like growth factor 1 improves outcome in Lama2Dy-w mice, a model for congenital muscular dystrophy type 1A. Hum Mol Genet. 2011;20(12):2333–2343. doi: 10.1093/hmg/ddr126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Secco M, Bueno C, Vieira NM, et al. Systemic delivery of human mesenchymal stromal cells combined with IGF-1 enhances muscle functional recovery in LAMA2 dy/2j dystrophic mice. Stem Cell Rev Reports. 2013;9(1):93–109. doi: 10.1007/s12015-012-9380-9 [DOI] [PubMed] [Google Scholar]
- 103.Hayes A, Williams DA. Examining potential drug therapies for muscular dystrophy utilising the dy/dy mouse: I. Clenbuterol. J Neurol Sci. 1998;157(2):122–128. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/9619633. [DOI] [PubMed] [Google Scholar]
- 104.Girgenrath M, Dominov JA, Kostek CA, Boone Miller J. Inhibition of apoptosis improves outcome in a model of congenital muscular dystrophy. J Clin Invest. 2004;114(11):1635–1639. doi: 10.1172/JCI22928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dominov JA, Kravetz AJ, Ardelt M, Kostek CA, Lou BM, Miller JB. Muscle-specific BCL2 expression ameliorates muscle disease in laminin α2-deficient, but not in dystrophin-deficient, mice. Hum Mol Genet. 2005;14(8):1029–1040. doi: 10.1093/hmg/ddi095 [DOI] [PubMed] [Google Scholar]
- 106.Erb M, Meinen S, Barzaghi P, et al. Omigapil ameliorates the pathology of muscle dystrophy caused by laminin- 2 deficiency. J Pharmacol Exp Ther. 2009;331(3):787–795. doi: 10.1124/jpet.109.160754 [DOI] [PubMed] [Google Scholar]
- 107.Yu Q, Sali A, Van der Meulen J, et al. Omigapil treatment decreases fibrosis and improves respiratory rate in dy2J mouse model of congenital muscular dystrophy. Gillingwater TH, ed. PLoS One. 2013;8(6):e65468. doi: 10.1371/journal.pone.0065468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Santhera Pharmaceuticals. Santhera announces successful completion of first clinical trial with omigapil in patients with congenital muscular dystrophy. Cure CMD. Available from: https://www.curecmd.org/single-post/2018/04/04/Santhera-Announces-Successful-Completion-of-First-Clinical-Trial-with-Omigapil-in-Patients-with-Congenital-Muscular-Dystrophy. Published 2018. Accessed February13, 2019.
- 109.Girgenrath M, Lou BM, Vishnudas VK, Homma S, Miller JB. Pathology is alleviated by doxycycline in a laminin-α2-null model of congenital muscular dystrophy. Ann Neurol. 2008;65(1):47–56. doi: 10.1002/ana.21523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Homma S, Beermann ML, Miller JB. Peripheral nerve pathology, including aberrant Schwann cell differentiation, is ameliorated by doxycycline in a laminin- 2-deficient mouse model of congenital muscular dystrophy. Hum Mol Genet. 2011;20(13):2662–2672. doi: 10.1093/hmg/ddr168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yamauchi J, Kumar A, Duarte L, Mehuron T, Girgenrath M. Triggering regeneration and tackling apoptosis: a combinatorial approach to treating congenital muscular dystrophy type 1 A. Hum Mol Genet. 2013;22(21):4306–4317. doi: 10.1093/hmg/ddt280 [DOI] [PubMed] [Google Scholar]
- 112.Meinen S, Lin S, Thurnherr R, Erb M, Meier T, Rüegg MA. Apoptosis inhibitors and mini-agrin have additive benefits in congenital muscular dystrophy mice. EMBO Mol Med. 2011;3(8):465–479. doi: 10.1002/emmm.201100151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Elbaz M, Yanay N, Aga-Mizrachi S, et al. Losartan, a therapeutic candidate in congenital muscular dystrophy: studies in the dy2J/dy2J mouse. Ann Neurol. 2012;71(5):699–708. doi: 10.1002/ana.22694 [DOI] [PubMed] [Google Scholar]
- 114.Murphy AM, Wong AL, Bezuhly M. Modulation of angiotensin II signaling in the prevention of fibrosis. Fibrogenesis Tissue Repair. 2015;8(1):7. doi: 10.1186/s13069-015-0023-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Meinen S, Lin S, Ruegg MA. Angiotensin II type 1 receptor antagonists alleviate muscle pathology in the mouse model for laminin-α2-deficient congenital muscular dystrophy (MDC1A). Skelet Muscle. 2012;2(1):18. doi: 10.1186/2044-5040-2-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Elbaz M, Yanay N, Laban S, Rabie M, Mitrani-Rosenbaum S, Nevo Y. Life or death by NFκB, Losartan promotes survival in dy2J/dy2J mouse of MDC1A. Cell Death Dis. 2015;6(3):e1690. doi: 10.1038/cddis.2015.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Accorsi A, Kumar A, Rhee Y, Miller A, Girgenrath M. IGF-1/GH axis enhances losartan treatment in Lama2-related muscular dystrophy. Hum Mol Genet. 2016;ddw291. doi: 10.1093/hmg/ddw291 [DOI] [PubMed] [Google Scholar]
- 118.Henriques C LAMA2 muscular dystrophy drug candidate TXA127 Is granted orphan drug status. Muscular Dystrophy News. Available from: https://musculardystrophynews.com/2016/02/18/tarix-orphan-granted-orphan-drug-status-for-txa127-as-potential-treatment-for-congenital-muscular-dystrophy-mdc1a/. Published 2016. Accessed February13, 2019.
- 119.Machado-Silva A, Passos-Silva D, Santos RA, Sinisterra RD. Therapeutic uses for Angiotensin-(1-7). Expert Opin Ther Pat. 2016;26(6):669–678. doi: 10.1080/13543776.2016.1179283 [DOI] [PubMed] [Google Scholar]
- 120.Nitahara-Kasahara Y, Takeda S, Okada T. Inflammatory predisposition predicts disease phenotypes in muscular dystrophy. Inflamm Regen. 2016;36(1):14. doi: 10.1186/s41232-016-0019-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mack M. Inflammation and fibrosis. Matrix Biol. 2018;68-69:106–121. doi: 10.1016/j.matbio.2017.11.010 [DOI] [PubMed] [Google Scholar]
- 122.Connolly AM, Keeling RM, Streif EM, Pestronk A, Mehta S. Complement 3 deficiency and oral prednisolone improve strength and prolong survival of laminin α2-deficient mice. J Neuroimmunol. 2002;127(1–2):80–87. doi: 10.1016/S0165-5728(02)00104-2 [DOI] [PubMed] [Google Scholar]
- 123.Dadush O, Aga-Mizrachi S, Ettinger K, et al. Improved muscle strength and mobility in the dy2J/dy2J mouse with merosin deficient congenital muscular dystrophy treated with Glatiramer acetate. Neuromuscul Disord. 2010;20(4):267–272. doi: 10.1016/j.nmd.2010.02.002 [DOI] [PubMed] [Google Scholar]
- 124.Nevo Y, Halevy O, Genin O, et al. Fibrosis inhibition and muscle histopathology improvement in laminin-α2-deficient mice. Muscle Nerve. 2010;42(2):218–229. doi: 10.1002/mus.21706 [DOI] [PubMed] [Google Scholar]
- 125.Nevo Y, Aga-Mizrachi S, Elmakayes E, et al. The ras antagonist, farnesylthiosalicylic acid (FTS), decreases fibrosis and improves muscle strength in dy2j/dy2j mouse model of muscular dystrophy. Lamitina T, ed. PLoS One. 2011;6(3):e18049. doi: 10.1371/journal.pone.0018049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gawlik KI, Holmberg J, Svensson M, et al. Potent pro-inflammatory and pro-fibrotic molecules, osteopontin and galectin-3, are not major disease modulators of laminin α2 chain-deficient muscular dystrophy. Sci Rep. 2017;7(1):44059. doi: 10.1038/srep44059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Carmignac V, Quéré R, Durbeej M. Proteasome inhibition improves the muscle of laminin α2 chain-deficient mice. Hum Mol Genet. 2011;20(3):541–552. doi: 10.1093/hmg/ddq499 [DOI] [PubMed] [Google Scholar]
- 128.Körner Z, Fontes-Oliveira CC, Holmberg J, Carmignac V, Durbeej M. Bortezomib partially improves laminin α2 chain–deficient muscular dystrophy. Am J Pathol. 2014;184(5):1518–1528. doi: 10.1016/j.ajpath.2014.01.019 [DOI] [PubMed] [Google Scholar]
- 129.Körner Z, Durbeej M. Bortezomib does not reduce muscular dystrophy in the dy2J/dy2J mouse model of laminin α2 chain-deficient muscular dystrophy. Fraidenraich D, ed. PLoS One. 2016;11(1):e0146471. doi: 10.1371/journal.pone.0146471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Carmignac V, Svensson M, Körner Z, et al. Autophagy is increased in laminin α2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum Mol Genet. 2011;20(24):4891–4902. doi: 10.1093/hmg/ddr427 [DOI] [PubMed] [Google Scholar]
- 131.Tomomura M, Fujii T, Sakagami H, Tomomura A. Serum calcium-decreasing factor, caldecrin, ameliorates muscular dystrophy in dy/dy mice. In Vivo. 2011;25(2):157–163. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/21471529. [PubMed] [Google Scholar]
- 132.Millay DP, Sargent MA, Osinska H, et al. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med. 2008;14(4):442–447. doi: 10.1038/nm1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fontes-Oliveira CC, Soares OB, Körner Z, Harandi V, Durbeej M. Effects of metformin on congenital muscular dystrophy type 1A disease progression in mice: a gender impact study. Sci Rep. 2018;8(1):16302. doi: 10.1038/s41598-018-34362-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xu R, Chandrasekharan K, Yoon JH, Camboni M, Martin PT. Overexpression of the Cytotoxic T Cell (CT) Carbohydrate inhibits muscular dystrophy in the dyw mouse model of congenital muscular dystrophy 1A. Am J Pathol. 2007;171(1):181–199. doi: 10.2353/ajpath.2007.060927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Allamand V, Bidou L, Arakawa M, et al. Drug-induced readthrough of premature stop codons leads to the stabilization of laminin α2 chain mRNA in CMD myotubes. J Gene Med. 2008;10(2):217–224. doi: 10.1002/jgm.1140 [DOI] [PubMed] [Google Scholar]
- 136.Lim KRQ, Yokota T. Invention and Early History of Exon Skipping and Splice Modulation. Methods Mol Biol. 2018;3–30. doi: 10.1007/978-1-4939-8651-4_1 [DOI] [PubMed] [Google Scholar]
- 137.Kole R, Krieg AM. Exon skipping therapy for Duchenne muscular dystrophy. Adv Drug Deliv Rev. 2015;87:104–107. doi: 10.1016/j.addr.2015.05.008 [DOI] [PubMed] [Google Scholar]
- 138.Aoki Y, Nagata T, Yokota T, et al. Highly efficient in vivo delivery of PMO into regenerating myotubes and rescue in laminin-α2 chain-null congenital muscular dystrophy mice. Hum Mol Genet. 2013;22(24):4914–4928. doi: 10.1093/hmg/ddt341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jiang F, Doudna JA. CRISPR–cas9 structures and mechanisms. Annu Rev Biophys. 2017;46(1):505–529. doi: 10.1146/annurev-biophys-062215-010822 [DOI] [PubMed] [Google Scholar]
- 140.Kemaladewi DU, Maino E, Hyatt E, et al. Correction of a splicing defect in a mouse model of congenital muscular dystrophy type 1A using a homology-directed-repair-independent mechanism. Nat Med. 2017. doi: 10.1038/nm.4367 [DOI] [PubMed] [Google Scholar]
- 141.Perrin A, Rousseau J, Tremblay JP. Increased expression of laminin subunit alpha 1 chain by dCas9-VP160. Mol Ther - Nucleic Acids. 2017;6:68–79. doi: 10.1016/j.omtn.2016.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Takawale A, Sakamuri SSVP, Kassiri Z. Extracellular matrix communication and turnover in cardiac physiology and pathology Compr Physiol. 2015;5(2):687–719. doi: 10.1002/cphy.c140045 [DOI] [PubMed] [Google Scholar]
- 143.Holmberg J, Alajbegovic A, Gawlik KI, Elowsson L, Durbeej M. Laminin α2 chain-deficiency is associated with microrna deregulation in skeletal muscle and plasma. Front Aging Neurosci. 2014;6 10.3389/fnagi.2014.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Moreira Soares Oliveira B, Gawlik KI, Durbeej-Hjalt M, Holmberg J. Exploratory profiling of urine microRNAs in the dy2J/dy2J mouse model of LAMA2-CMD: relation to disease progression. PLoS Curr. 2018. doi: 10.1371/currents.md.d0c203c018bc024f2f4c9791ecb05f88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kemaladewi DU, Benjamin JS, Hyatt E, Ivakine EA, Cohn RD. Increased polyamines as protective disease modifiers in congenital muscular dystrophy. Hum Mol Genet. 2018;27(11):1905–1912. doi: 10.1093/hmg/ddy097 [DOI] [PubMed] [Google Scholar]
- 146.Zanotti S, Negri T, Cappelletti C, et al. Decorin and biglycan expression is differentially altered in several muscular dystrophies. Brain. 2005;128(11):2546–2555. doi: 10.1093/brain/awh635 [DOI] [PubMed] [Google Scholar]
- 147.Vishnudas VK, Miller JB. Ku70 regulates Bax-mediated pathogenesis in laminin- 2-deficient human muscle cells and mouse models of congenital muscular dystrophy. Hum Mol Genet. 2009;18(23):4467–4477. doi: 10.1093/hmg/ddp399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yoon S, Lou BM, Yu B, Shao D, Bachschmid M, Miller JB. Aberrant caspase activation in laminin-α2-deficient human myogenic cells is mediated by p53 and sirtuin activity. J Neuromuscul Dis. 2018;5(1):59–73. doi: 10.3233/JND-170262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zdanowicz MM, Slonim AE, Bilaniuk I, O’Connor MM, Moyse J, Teichberg S. High protein diet has beneficial effects in murine muscular dystrophy. J Nutr. 1995;125(5):1150–1158. doi: 10.1093/jn/125.5.1150 [DOI] [PubMed] [Google Scholar]
- 150.Hagiwara H, Ohsawa Y, Asakura S, Murakami T, Teshima T, Sunada Y. Bone marrow transplantation improves outcome in a mouse model of congenital muscular dystrophy. FEBS Lett. 2006;580(18):4463–4468. doi: 10.1016/j.febslet.2006.07.015 [DOI] [PubMed] [Google Scholar]