

Abstract
Among the known liquid organic hydrogen carriers, formic acid attracts increasing interest in the context of safe and reversible storage of hydrogen. Here, the first molecularly defined cobalt pincer complex is disclosed for the dehydrogenation of formic acid in aqueous medium under mild conditions. Crucial for catalytic activity is the use of the specific complex 3. Compared to related ruthenium and manganese complexes 7 and 8, this optimal cobalt complex showed improved performance. DFT computations support an innocent non‐classical bifunctional outer‐sphere mechanism on the triplet state potential energy surface.
Keywords: cobalt, dehydrogenation, formic acid, pincer complexes, synthetic methods
With the ever‐increasing energy demand and ongoing depletion of fossil fuels, the development of renewable and environmentally friendly sources of energy is imperative. In this respect, hydrogen is considered as a promising, secure and clean energy carrier for enabling a more sustainable future.1 However, the so‐called “hydrogen economy” still faces many problems. For example, hydrogen is a highly volatile gas and consequently possesses a very low gravimetric energy density. Hence, the development of efficient hydrogen storage systems is highly desired. In this regard, liquid organic chemicals, such as methanol and formic acid (FA), constitute an attractive choice.2 Specifically, FA is interesting because it is easily obtained either from non‐edible biomass or carbon dioxide and hydrogen. Furthermore, it can undergo selective decomposition to hydrogen and carbon dioxide at mild conditions in the presence of suitable catalysts.
Thus, during the past decades, major efforts have focused on the development of catalysts for dehydrogenation of formic acid, especially in the presence of noble metals. More specifically, a number of highly active homogeneous and heterogeneous catalysts based on Ir, Ru, Au, Pd have been developed by Fukuzumi, Himeda, Laurenczy, Xu, Tsang, our group, and many others.3
Due to the limitations and price of noble metals, in recent years more and more attention has focused on the development of more abundant 3 d metal systems because of their low cost and toxicity. In 2010, our group introduced the first iron‐based catalyst for FA dehydrogenation under irradiation conditions.4a Notably, in 2011, a much more efficient iron complex has been reported, which allowed for catalyst turnover numbers of >90 000.4b Based on that work, also a water‐soluble derivative of this complex was applied by Laurenczy and co‐workers in aqueous solution.4c Milstein and co‐workers have reported another iron catalyzed dehydrogenation of FA,4d whereas Hazari and Schneider have developed a Lewis acid assisted FA dehydrogenation by using an iron pincer complex.4e In addition to iron, copper and nickel catalysts have been also disclosed to be active for this transformation.5
Alternatively, cobalt‐catalyzed decomposition of formic acid into hydrogen and carbon dioxide is relatively unknown, although it is an essential 3 d element for living organisms, which has received much attention during the past decade for its diverse activities in polymerization,6 C−H bond functionalization,7 (transfer)hydrogenation,8 hydrofunctionalization9 and dehydrogenative coupling reactions.10 For example, in 2012 Hanson and co‐workers have reported a new aliphatic PNP pincer CoII complex for the hydrogenation of olefins, ketones, aldehydes, and imines.8a Later, Milstein and co‐workers have developed another CoII system with pyridine‐based PNN pincer ligand for the hydrogenation of esters and nitriles.8c, 8d Very recently, the group of Fout has showed that CoI complexes with monoanionic bis(carbene) ligands can also promote olefin hydrogenation through a CoI/CoIII redox cycle (Figure 1).8f
Figure 1.
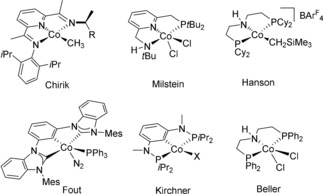
Selected cobalt pincer complexes for (transfer)hydrogenation and dehydrogenative coupling reactions.
Although cobalt catalyzed hydrogenation of carbon dioxide into formic acid/formate or alcohols, has already been reported,11 the reverse processes, that is, hydrogen generation from FA is scarcely known.12 A known nanocobalt catalyst for the selective dehydrogenation of formic acid has been reported by our group last year;13 however, the relatively harsh reaction conditions and comparably low activity under mild conditions stimulated us to look for improved molecularly‐defined systems. Based on our long standing interest in non‐noble metal catalyzed dehydrogenation reactions,14 here we report the first cobalt PNP complex for FA dehydrogenation reaction in aqueous medium.
Initially, a series of CoII complexes which were active for the hydrogenation of esters in our previous report8k was tested for the dehydrogenation of formic acid under various conditions. Unfortunately, none of these complexes showed activity for the dehydrogenation reaction (see Supporting Information). Looking at previous hydrogenation systems, we realized that CoI species might constitute the active catalyst under reductive conditions. However, these CoI species cannot be easily formed under dehydrogenative conditions. Based on this assumption, CoI complexes 1–3 were synthesized and characterized by NMR, combustion analysis and X‐ray diffraction. Indeed, when complex 1 (Figure 2) was employed, hydrogen evolution was observed from a formic acid/amine mixture at 80 °C, albeit with very low efficiency. Its bromide analogue 2 showed a similar activity. To our delight, the phenyl substituted PNP pincer complex 3 exhibited much higher activity, in which case formic acid was almost fully decomposed into hydrogen and carbon dioxide within 90 minutes, reaching a catalyst turnover number (TON) of 2260.
Figure 2.
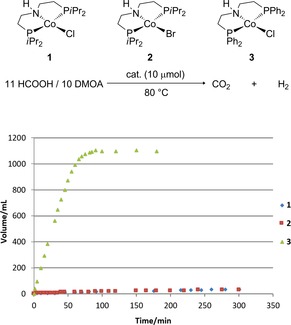
Cobalt(I) pincer complexes catalyzed dehydrogenation of formic acid. conditions: HCOOH/DMOA 11:10 (5 mL, containing 1.075 g of FA); DMOA: N,N‐dimethyloctylamine.
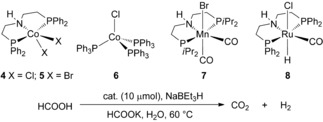
Encouraged by these results, we further tested the performance of CoI complex 3 under aqueous conditions with potassium formate as base. Under these conditions, still high activity was observed (Table 1, entry 1), whereas lowering the temperature to 60 °C significantly slowed down the gas evolution rate (Table 1, entry 2). Due to the highly air‐sensitive nature of 3, which makes the handling inconvenient, we also tried to apply the precursor complex 4 instead of 3 and performed an in situ activation by sodium triethylborohydride.15 However, treating 4 with one equivalent of sodium triethylborohydride as an activator prior to the dehydrogenation reaction, resulted in a lower activity compared to the preformed CoI complex 3 (Table 1, entries 2 and 3). This might be due to the uncompleted reduction of CoII precursor, considering that we observed a pink solution of the aqueous phase which indicates the existence of CoII species. Interestingly, when two equivalents of sodium triethylborohydride were used, a slightly higher activity than that of the preformed complex 3 was observed (Table 1, entry 4). Further optimization showed that the amount of formate can be reduced to 1:1 ratio regarding the amount of formic acid without a significant loss of the activity. Contrary, an increased FA concentration leads to a slight decrease in activity (Table 1, entries 5–8). When pure formic acid or formate were used as substrate, only very slow gas evolution was observed (Table 1, entries 9 and 10). To understand the different activity between complex 3 and the in situ activated complex 4 with two equivalents of sodium triethylborohydride, we also performed an experiment using 3 in the presence of one equivalent of sodium triethylborohydride. The result showed it exhibits the same activity as complex 4 with two equivalents of reductant (Table 1, entries 6 and 11), which suggests that the same active species is generated in both systems. The bromide analogue of complex 4, showed a slightly decreased activity. In addition, the commercially available CoI complex 6 was also tested. However, no gas evolution was observed underlining the importance of the pincer ligand in this system. Finally, we compared the activity of these new complexes with other well‐known pincer complexes, which are active in methanol and/or FA dehydrogenation.14b, 16 Surprisingly, the manganese complex 7 was completely inactive under these aqueous conditions, and even the ruthenium benchmark complex 8 gave only marginal hydrogen production. Obviously, CoI complex 3 is a superior catalyst for the dehydrogenation of formic acid under aqueous conditions.
Table 1.
Catalytic dehydrogenation of formic acid under aqueous conditions.[a]
| ||||||
|---|---|---|---|---|---|---|
| Entry | Complex | NaBEt3H [μmol] |
HCO2H [mmol] |
HCO2K [mmol] |
V
1h
[mL] |
V
3h
[mL] |
| 1[b] | 3 | – | 10 | 40 | 388 | n.d. |
| 2 | 3 | – | 10 | 40 | 131 | 396 |
| 3 | 4 | 10 | 10 | 40 | 64 | 220 |
| 4 | 4 | 20 | 10 | 40 | 134 | 426 |
| 5 | 4 | 20 | 10 | 20 | 133 | 448 |
| 6 | 4 | 20 | 10 | 10 | 125 | 436 |
| 7 | 4 | 20 | 10 | 5 | 71 | 200 |
| 8 | 4 | 20 | 20 | 20 | 102 | 292 |
| 9 | 4 | 20 | 10 | 0 | 14 | 26 |
| 10 | 4 | 20 | 0 | 10 | 12 | 22 |
| 11 | 3 | 10 | 10 | 10 | 118 | 432 |
| 12 | 5 | 20 | 10 | 10 | 100 | 300 |
| 13 | 6 | – | 10 | 10 | 0 | 0 |
| 14 | 7 | – | 10 | 10 | 0 | 0 |
| 15 | 8 | – | 10 | 10 | 6 | 18 |
[a] In all cases, the total volume of formic acid and water was 4 mL; catalyst loading was based on metal complex; gas evolution was measured by using a manual burette with correction by blank value. 100 mL gas evolution corresponds to a catalyst turnover number of 205 for CO2/H2=1:1. CO was not detected by GC measurement. [b] The reaction was performed at 80 °C. n.d.: not determined.
With the optimized conditions in hand, we further tested the stability of the system. As shown in the supporting information (Figure S14, Supporting Information), gas evolution ceased after 70 hours, and a maximum TON of 7166 was obtained.
To gain more mechanistic insight into the catalytic system, kinetic isotope effect (KIE) studies were performed. In the formic acid/amine system as well as under aqueous conditions, primary KIE values of 2.91 and 2.05 for HCOOH/DCOOH, respectively, were observed, whereas the KIE value of HCOOH/HCOOD in formic acid/amine is only 1.34. These results indicate the decarboxylation step should be involved as the rate determining step. To understand the deactivation of the system, CoI‐carbonyl complexes 9 and 10 were synthesized from complex 3 and were fully characterized (Figure 3). Neither complex 9 nor 10 showed any activity under optimized conditions, and this suggests that carbon monoxide coordination to cobalt might be responsible for the deactivation process (Figure S19). Notably, formic acid can also undergo dehydration process to give H2O and CO.17 This observation is quite different from previously reported 3 d metal catalysts, such as iron and manganese complexes, in which the catalyst needs carbonyl groups as co‐ligands to be active.
Figure 3.
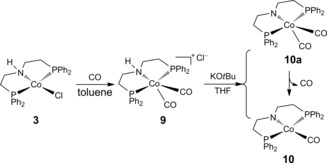
Synthesis of CoI‐carbonyl complexes 9 and 10.
Along with our experimental work, we further computed the potential energy surface to elucidate the reaction mechanism at the level of B3PW91 density functional theory. All computational details are given in the Supporting Information. On the basis of the recent study for structures and stability8k as well as of the catalytic activity of cobalt PNP pincer in acetophenone hydrogenation,18 we tested B3PW91 and M06L methods on the basis of the reaction from complex 9 to monocarbonyl complex 10 in Figure 3. It is found that M06L favours the dicarbonyl amido complex (10 a, trigonal bipyramidal) instead of the monocarbonyl complex 10 (square planar) [10 a=10+CO] both in gas phase and solution by 6.09 and 8.94 kcal mol−1, respectively, and this disagrees with the experiment. In contrast, B3PW91 prefers complex 10 instead of the dicarbonyl complex 10 a by 2.90 and 1.20 kcal mol−1 in gas phase and in solution, respectively, and this agrees with the experiment. Indeed, such monocarbonyl complex has been found for the iPr and tBu substituted counterpart.19
In agreement with the experiment, complex 10 has a square planar structure in singlet state which is more stable than the triplet state by 2.36 kcal mol−1 and stable towards CO dissociation to form 3 C (18.48 kcal mol−1). The square planar structure and the stability of the singlet state over the triplet state of complex 10 explain the amido coordination 18‐valence‐electron configuration. On the basis of this result as well as our previous results on other PNP pincer metal complexes regarding stability and catalytic activity,20 we present the B3PW91 results for discussion, while the M06L‐SCRF results are given in Supporting Information for comparison. It is noted that all species actively involved on the Gibbs free energy surface have triplet ground states and the corresponding singlet states are higher in energy and less stable. Importantly, there are no crossing points between the triplet and singlet states. Therefore, all states refer to their triplet states if no noted otherwise. The free energy potential energy surface is shown in Figure 4.
Figure 4.
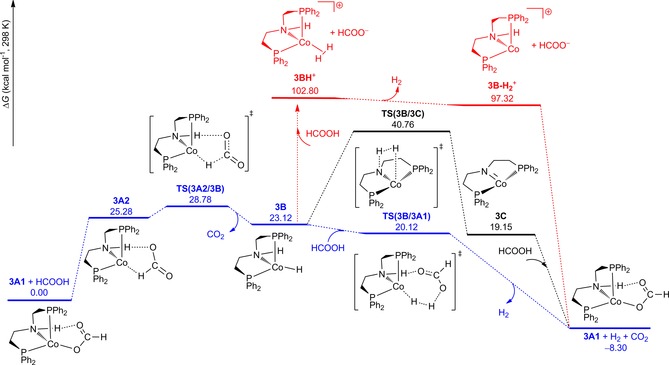
Gibbs free energy profile for formic acid dehydrogenation at B3PW91/TZVP level.
The first step of the reaction is the substitution of the chloro ligand in complex 3 by formate resulting in the formation of complex 3 A1. In 3 A1, apart from the O‐coordination (Co−O=1.983 Å), there is H‐bonding between the second O atom and the H atom of the N−H group (O−H=1.850 Å). Such coordination mode has been found also in other transition‐metal PNP pincer complexes.2, 3 Both 3 and 3 A1 have trigonal pyramidal configuration, since the sum of the two Cl‐Co‐P and one P‐Co‐P angles in 3 is 359.9° and the sum of the two O‐Co‐P and one P‐Co‐P angles in 3A1 is 359.7°. The corresponding singlet state of 3 and 3 A1 is less stable than the corresponding triplet state by 23.39 and 38.59 kcal mol−1, respectively.
Apart from the O‐coordination, we located the complex of C−H coordination 3 A2, which is responsible for the activation and dissociation of the C−H bond,20c, 20d and higher in energy than 3 A1 by 25.28 kcal mol−1. In 3 A2, the H−C distance is 1.235 Å, the Co−H distance is 1.805 Å and the H‐to‐O H‐bonding distance is 1.822 Å. Next, we located the corresponding transition state for the C−H activation and dissociation, TS (3 A2/3 B), which leads to CO2 release and the formation of the parent amine complex (3 B). In TS (3 A2/3 B), the breaking C−H distance is 1.914 Å, and the forming Co−H distance is 1.659 Å; and the distance of the N−H to O−H‐bonding distance is 2.064 Å. As expected, 3 B has trigonal pyramidal configuration on the basis of the sum of the two H‐Co‐P and one P‐Co‐P angles (359.9°). Starting from 3 A1, this step has free energy barrier of 28.78 kcal mol−1 and is endergonic by 23.12 kcal mol−1. In addition, the singlet state of 3 B is less stable than the triplet state by 33.10 kcal mol−1.
Starting from the parent amine complex 3 B, there are three possible routes for H2 release (Figure 4). The first one (black line for the classical bifunctional outer‐sphere mechanism) is the direct way from the combination of the N−H and Co−H groups via the so‐called non‐innocent mechanism with the formation of the amido complex 3 C. In the transition state TS (3 B/3 C), the forming H−H distance is 0.974 Å; and the distance of dissociating N−H and Co−H bonds is 1.451 and 1.757 Å, respectively. This step has free energy barrier of 17.64 kcal mol−1 and is exergonic by 3.97 kcal mol−1. In addition, the singlet state of 3 C is less stable than the triplet state by 24.80 kcal mol−1. Formic acid addition to the amido complex 3 C leads to the starting point of complex 3 A1. The second route (blue line for the non‐classical bifunctional outer‐sphere mechanism) involves the formic acid assisted mechanism, where H2 formation results from the Co−H group and the acid proton, whereas the N−H group simultaneously stabilizes the transition state through H‐bonding. In the transition state TS (3 B/3 A1), the forming H−H distance is 0.934 Å; and the dissociating Co−H and H−O distances are 1.699 and 1.360 Å, respectively, whereas the distance of the O−H H‐bonding is 1.753 Å. This step has negative Gibbs free energy barrier (−3.00 kcal mol−1) and is highly exergonic (−31.42 kcal mol−1). The third one (red line) involves the protonation of the Co−H group by formic acid and this step is very endergonic by 102.80 kcal mol−1 and can be discarded.
On the basis of the computed free energy potential surface, all species have formal CoI oxidation states and triplet states; and the rate‐determining step is the C−H activation accompanied by CO2 release. The simplified catalytic cycle is shown in Figure 5. Starting from complex 3 A1, the first step is C−H activation resulting in CO2 release and the formation of amine complex (3 B); and the second step is the formic acid assisted H2 formation and the regenerating of complex 3 A1 through the innocent mechanism. In contrast to the classical bifunctional outer‐sphere mechanism in which both M−H and N−H are involved directly in H2 formation, our system shows a non‐classical bifunctional outer‐sphere mechanism in which the N−H functional is not directly involved in H2 formation, and instead the N−H functional actively stabilizes the transition state for H2 formation. This is different from the proposed non‐bifunctional inner‐sphere mechanism for Hanson's CoII system.21
Figure 5.
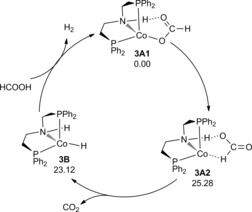
Proposed mechanism for cobalt‐catalyzed dehydrogenation of formic acid.
In summary, we have developed the first molecularly defined cobalt complex for catalytic dehydrogenation of formic acid under aqueous conditions. The system features mild conditions and good activity. Preliminary results suggested that carbonyl coordination to cobalt results in deactivation of the catalyst. On the potential energy surface, all species actively involved have formal CoI oxidation states in triplet states. The first step, which is also the rate‐determining step, is the C−H activation resulting in CO2 release and formation of the amine complex. The second step is the formic acid assisted H2 release from the amine complex through a non‐classical bifunctional outer‐sphere mechanism. We believe that these results will be interesting for the further development of non‐noble metal catalyzed hydrogenation/dehydrogenation reactions.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work is supported by the European Union under the Horizon 2020 Program as part of the NoNoMeCat project (675020‐MSCAITN‐2015‐ETN). Z.W. is grateful for the financial support of the Leibniz Foundation (Leibniz Competition, SAW‐2016‐LIKAT‐1). We thank Bianca Tannert for providing us a sample of complex 1 for initial tests, Maximilian Marx and Jacob Schneidewind for helpful discussions.
W. Zhou, Z. Wei, A. Spannenberg, H. Jiao, K. Junge, H. Junge, M. Beller, Chem. Eur. J. 2019, 25, 8459.
Dedicated to Professor Gabor Laurenczy on the occasion of his 65th birthday
References
- 1.
- 1a. Armaroli N., Balzani V., Chem. Asian J. 2011, 6, 768; [DOI] [PubMed] [Google Scholar]
- 1b. Hydrogen as a Future Energy Carrier (Eds.: A. Züttel, A. Borgschulte, L. Schlapbach), Wiley-VCH, Weinheim, 2008. [Google Scholar]
- 2.For some reviews see:
- 2a. Olah G. A., Angew. Chem. Int. Ed. 2005, 44, 2636; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 2692; [Google Scholar]
- 2b. Li Z., Xu Q., Acc. Chem. Res. 2017, 50, 1449; [DOI] [PubMed] [Google Scholar]
- 2c. Mellmann D., Sponholz P., Junge H., Beller M., Chem. Soc. Rev. 2016, 45, 3954; [DOI] [PubMed] [Google Scholar]
- 2d. Sordakis K., Tang C., Vogt L. K., Junge H., Dyson P. J., Beller M., Laurenczy G., Chem. Rev. 2018, 118, 372; [DOI] [PubMed] [Google Scholar]
- 2e. Onishi N., Laurenczy G., Beller M., Himeda Y., Coord. Chem. Rev. 2018, 373, 317. [Google Scholar]
- 3.For selected examples see:
- 3a. Loges B., Boddien A., Junge H., Beller M., Angew. Chem. Int. Ed. 2008, 47, 3962; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4026; [Google Scholar]
- 3b. Fellay C., Dyson P. J., Laurenczy G., Angew. Chem. Int. Ed. 2008, 47, 3966; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4030; [Google Scholar]
- 3c. Fukuzumi S., Kobayashi T., Suenobu T., J. Am. Chem. Soc. 2010, 132, 1496; [DOI] [PubMed] [Google Scholar]
- 3d. Himeda Y., Green Chem. 2009, 11, 2018; [Google Scholar]
- 3e. Boddien A., Loges B., Junge H., Gärtner F., Noyes J. R., Beller M., Adv. Synth. Catal. 2009, 351, 2517; [Google Scholar]
- 3f. Tanaka R., Yamashita M., Chung L. W., Morokuma K., Nozaki K., Organometallics 2011, 30, 6742; [Google Scholar]
- 3g. Tedsree K., Li T., Jones S., Chan C. W. A., Yu K. M. K., Bagot P. A. J., Marquis E. A., Smith G. D. W., Tsang S. C. E., Nat. Nanotechnol. 2011, 6, 302; [DOI] [PubMed] [Google Scholar]
- 3h. Gu X., Lu Z.-H., Jiang H.-L., Akita T., Xu Q., J. Am. Chem. Soc. 2011, 133, 11822; [DOI] [PubMed] [Google Scholar]
- 3i. Bi Q.-Y., Du X.-L., Liu Y.-M., Cao Y., He H.-Y., Fan K.-N., J. Am. Chem. Soc. 2012, 134, 8926; [DOI] [PubMed] [Google Scholar]
- 3j. Barnard J. H., Wang C., Berry N. G., Xiao J., Chem. Sci. 2013, 4, 1234; [Google Scholar]
- 3k. Jiang K., Xu K., Zou S., Cai W.-B., J. Am. Chem. Soc. 2014, 136, 4861; [DOI] [PubMed] [Google Scholar]
- 3l. Oldenhof S., de Bruin B., Lutz M., Siegler M. A., Patureau F. W., van der Vlugt J. I., Reek J. N. H., Chem. Eur. J. 2013, 19, 11507; [DOI] [PubMed] [Google Scholar]
- 3m. Wang W.-H., Ertem M. Z., Xu S., Onishi N., Manaka Y., Suna Y., Kambayashi H., Muckerman J. T., Fujita E., Himeda Y., ACS Catal. 2015, 5, 5496; [Google Scholar]
- 3n. Guerriero A., Bricout H., Sordakis K., Peruzzini M., Monflier E., Hapiot F., Laurenczy G., Gonsalvi L., ACS Catal. 2014, 4, 3002; [Google Scholar]
- 3o. Mellone I., Bertini F., Peruzzini M., Gonsalvi L., Catal. Sci. Technol. 2016, 6, 6504; [Google Scholar]
- 3p. Celaje J. J. A., Lu Z., Kedzie E. A., Terrile N. J., Lo J. N., Williams T. J., Nat. Commun. 2016, 7, 11308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Boddien A., Loges B., Gärtner F., Torborg C., Fumino K., Junge H., Ludwig R., Beller M., J. Am. Chem. Soc. 2010, 132, 8924; [DOI] [PubMed] [Google Scholar]
- 4b. Boddien A., Mellmann D., Gärtner F., Jackstell R., Junge H., Dyson P. J., Laurenczy G., Ludwig R., Beller M., Science 2011, 333, 1733; [DOI] [PubMed] [Google Scholar]
- 4c. Montandon-Clerc M., Dalebrook A. F., Laurenczy G., J. Catal. 2016, 343, 62; [Google Scholar]
- 4d. Zell T., Butschke B., Ben-David Y., Milstein D., Chem. Eur. J. 2013, 19, 8068; [DOI] [PubMed] [Google Scholar]
- 4e. Bielinski E. A., Lagaditis P. O., Zhang Y., Mercado B. Q., Würtele C., Bernskoetter W. H., Hazari N., Schneider S., J. Am. Chem. Soc. 2014, 136, 10234. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Scotti N., Psaro R., Ravasio N., Zaccheria F., RSC Adv. 2014, 4, 61514; [Google Scholar]
- 5b. Enthaler S., Brück A., Kammer A., Junge H., Irran E., Gülak S., ChemCatChem 2014, 7, 65. [Google Scholar]
- 6.
- 6a. Britovsek G. J. P., Bruce M., Gibson V. C., Kimberley B. S., Maddox P. J., Mastroianni S., McTavish S. J., Redshaw C., Solan G. A., Strömberg S., White A. J. P., Williams D. J., J. Am. Chem. Soc. 1999, 121, 8728; [Google Scholar]
- 6b. Atienza C. C. H., Milsmann C., Lobkovsky E., Chirik P. J., Angew. Chem. Int. Ed. 2011, 50, 8143; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8293; [Google Scholar]
- 6c. Chen L., Ai P., Gu J., Jie S., Li B.-G., J. Organomet. Chem. 2012, 716, 55. [Google Scholar]
- 7.
- 7a. Mo Z., Liu Y., Deng L., Angew. Chem. Int. Ed. 2013, 52, 10845; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11045; [Google Scholar]
- 7b. Palmer W. N., Obligacion J. V., Pappas I., Chirik P. J., J. Am. Chem. Soc. 2016, 138, 766; [DOI] [PubMed] [Google Scholar]
- 7c. Obligacion J. V., Semproni S. P., Pappas I., Chirik P. J., J. Am. Chem. Soc. 2016, 138, 10645. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Zhang G., Scott B. L., Hanson S. K., Angew. Chem. Int. Ed. 2012, 51, 12102; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12268; [Google Scholar]
- 8b. Monfette S., Turner Z. R., Semproni S. P., Chirik P. J., J. Am. Chem. Soc. 2012, 134, 4561; [DOI] [PubMed] [Google Scholar]
- 8c. Srimani D., Mukherjee A., Goldberg A. F. G., Ben David Y., Milstein D., Angew. Chem. Int. Ed. 2015, 54, 12357; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12534; [Google Scholar]
- 8d. Mukherjee A., Srimani D., Chakraborty S., Ben-David Y., Milstein D., J. Am. Chem. Soc. 2015, 137, 8888; [DOI] [PubMed] [Google Scholar]
- 8e. Chirik P. J., Acc. Chem. Res. 2015, 48, 1687; [DOI] [PubMed] [Google Scholar]
- 8f. Tokmic K., Markus C. R., Zhu L., Fout A. R., J. Am. Chem. Soc. 2016, 138, 11907; [DOI] [PubMed] [Google Scholar]
- 8g. Tokmic K., Jackson B. J., Salazar A., Woods T. J., Fout A. R., J. Am. Chem. Soc. 2017, 139, 13554; [DOI] [PubMed] [Google Scholar]
- 8h. Korstanje T. J., van der Vlugt J. I., Elsevier C. J., de Bruin B., Science 2015, 350, 298; [DOI] [PubMed] [Google Scholar]
- 8i. Xu R., Chakraborty S., Yuan H., Jones W. D., ACS Catal. 2015, 5, 6350; [Google Scholar]
- 8j. Zhang G., Hanson S. K., Chem. Commun. 2013, 49, 10151; [DOI] [PubMed] [Google Scholar]
- 8k. Junge K., Wendt B., Cingolani A., Spannenberg A., Wei Z., Jiao H., Beller M., Chem. Eur. J. 2018, 24, 1046; [DOI] [PubMed] [Google Scholar]
- 8l. Liu W., Sahoo B., Junge K., Beller M., Acc. Chem. Res. 2018, 51, 1858; [DOI] [PubMed] [Google Scholar]
- 8m. Junge K., Papa V., Beller M., Chem. Eur. J. 2019, 25, 122. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Zhang L., Zuo Z., Leng X., Huang Z., Angew. Chem. Int. Ed. 2014, 53, 2696; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2734; [Google Scholar]
- 9b. Obligacion J. V., Chirik P. J., J. Am. Chem. Soc. 2013, 135, 19107; [DOI] [PubMed] [Google Scholar]
- 9c. Chen C., Hecht M. B., Kavara A., Brennessel W. W., Mercado B. Q., Weix D. J., Holland P. L., J. Am. Chem. Soc. 2015, 137, 13244; [DOI] [PubMed] [Google Scholar]
- 9d. Zuo Z., Yang J., Huang Z., Angew. Chem. Int. Ed. 2016, 55, 10839; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10997. [Google Scholar]
- 10.
- 10a. Zhang G., Hanson S. K., Org. Lett. 2013, 15, 650; [DOI] [PubMed] [Google Scholar]
- 10b. Daw P., Chakraborty S., Garg J. A., Ben-David Y., Milstein D., Angew. Chem. Int. Ed. 2016, 55, 14373; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14585; [Google Scholar]
- 10c. Mastalir M., Tomsu G., Pittenauer E., Allmaier G., Kirchner K., Org. Lett. 2016, 18, 3462; [DOI] [PubMed] [Google Scholar]
- 10d. Rösler S., Ertl M., Irrgang T., Kempe R., Angew. Chem. Int. Ed. 2015, 54, 15046; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15260; [Google Scholar]
- 10e. Daw P., Ben-David Y., Milstein D., ACS Catal. 2017, 7, 7456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For homogeneous cobalt-catalyzed hydrogenation of CO2 to formic acid and derivatives see:
- 11a. Federsel C., Ziebart C., Jackstell R., Baumann W., Beller M., Chem. Eur. J. 2012, 18, 72; [DOI] [PubMed] [Google Scholar]
- 11b. Jeletic M. S., Mock M. T., Appel A. M., Linehan J. C., J. Am. Chem. Soc. 2013, 135, 11533; [DOI] [PubMed] [Google Scholar]
- 11c. Badiei Y. M., Wang W.-H., Hull J. F., Szalda D. J., Muckerman J. T., Himeda Y., Fujita E., Inorg. Chem. 2013, 52, 12576; [DOI] [PubMed] [Google Scholar]
- 11d. Spentzos A. Z., Barnes C. L., Bernskoetter W. H., Inorg. Chem. 2016, 55, 8225; [DOI] [PubMed] [Google Scholar]
- 11e. Burgess S. A., Grubel K., Appel A. M., Wiedner E. S., Linehan J. C., Inorg. Chem. 2017, 56, 8580; [DOI] [PubMed] [Google Scholar]
- 11f. Daw P., Chakraborty S., Leitus G., Diskin-Posner Y., Ben-David Y., Milstein D., ACS Catal. 2017, 7, 2500 For homogeneous cobalt-catalyzed hydrogenation of CO2 to methanol see: [Google Scholar]
- 11g. Schneidewind J., Adam R., Baumann W., Jackstell R., Beller M., Angew. Chem. Int. Ed. 2017, 56, 1890; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1916; [Google Scholar]
- 11h. Schieweck B. G., Klankermayer J., Angew. Chem. Int. Ed. 2017, 56, 10854; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10994. [Google Scholar]
- 12.Onishi has reported an early example of cobalt-catalyzed dehydrogenation under photoirradiation conditions, see: Onishi M., J. Mol. Catal. 1993, 80, 145. [Google Scholar]
- 13. Tang C., Surkus A.-E., Chen F., Pohl M.-M., Agostini G., Schneider M., Junge H., Beller M., Angew. Chem. Int. Ed. 2017, 56, 16616; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 16843. [Google Scholar]
- 14.
- 14a. Alberico E., Sponholz P., Cordes C., Nielsen M., Drexler H.-J., Baumann W., Junge H., Beller M., Angew. Chem. Int. Ed. 2013, 52, 14162; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 14412; [Google Scholar]
- 14b. Andérez-Fernández M., Vogt L. K., Fischer S., Zhou W., Jiao H., Garbe M., Elangovan S., Junge K., Junge H., Ludwig R., Beller M., Angew. Chem. Int. Ed. 2017, 56, 559; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 574. [Google Scholar]
- 15.For in situ activation of CoII complexes by NaBEt3H, see also Ref. [8c], [8d], [9a], [9d], and [19b].
- 16. Nielsen M., Alberico E., Baumann W., Drexler H.-J., Junge H., Gladiali S., Beller M., Nature 2013, 495, 85. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Mars P., Scholten J. J. F., Zwietering P. in Advances in Catalysis, Vol. 14 (Eds.: D. D. Eley, H. Pines, P. B. Weisz), Academic Press, London, 1963, p. 35; [Google Scholar]
- 17b.In our previous reported systems, low CO content was detected in gas sample, see Ref. Ref. [4a] and [14b].
- 18. Hou C., Li Y., Zhao C., Ke Z., Catal. Sci. Technol. 2019, 9, 125. [Google Scholar]
- 19.
- 19a. Rozenel S. S., Padilla R., Camp C., Arnold J., Chem. Commun. 2014, 50, 2612; [DOI] [PubMed] [Google Scholar]
- 19b. Lagaditis P. O., Schluschaß B., Demeshko S., Würtele C., Schneider S., Inorg. Chem. 2016, 55, 4529. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Wei Z., Jiao H., Adv. Inorg. Chem. 2019, 73, 323; [Google Scholar]
- 20b. Wei Z., de Aguirre A., Junge K., Beller M., Jiao H., Eur. J. Inorg. Chem. 2018, 4643; [Google Scholar]
- 20c. Wei Z., de Aguirre A., Junge K., Beller M., Jiao H., Catal. Sci. Technol. 2018, 8, 3649; [Google Scholar]
- 20d. Alberico E., Lennox A. J. J., Vogt L. K., Jiao H., Baumann W., Drexler H.-J., Nielsen M., Spannenberg A., Checinski M. P., Junge H., Beller M., J. Am. Chem. Soc. 2016, 138, 14890; [DOI] [PubMed] [Google Scholar]
- 20e. Jiao H., Junge K., Alberico E., Beller M., J. Comput. Chem. 2016, 37, 168. [DOI] [PubMed] [Google Scholar]
- 21. Huo C., Jiang J., Li Y., Zhang Z., Zhao C., Ke Z., Dalton Trans. 2015, 44, 16573. [DOI] [PubMed] [Google Scholar]
- 22. CCDC 1877384 (2), 1877385 (3), 1877386 (5), and 1877387 (10) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary