

Abstract
Background
Ontuxizumab, a humanized monoclonal antibody, targets endosialin (tumor endothelial marker 1 [TEM‐1] or CD248), which is expressed on sarcoma cells and is believed to be involved in tumor angiogenesis. This is the first trial to evaluate ontuxizumab in patients with sarcoma.
Methods
Part 1 was an open‐label, dose‐finding, safety lead‐in: 4, 6, or 8 mg/kg with gemcitabine and docetaxel (G/D; 900 mg/m2 gemcitabine on days 1 and 8 and 75 mg/m2 docetaxel on day 8). In part 2, patients were randomized in a double‐blind fashion in 2:1 ratio to ontuxizumab (8 mg/kg) or a placebo with G/D. Randomization was stratified by 4 histological cohorts.
Results
In part 2 with 209 patients, no significant difference in progression‐free survival between ontuxizumab plus G/D (4.3 months; 95% confidence interval [CI], 2.7‐6.3 months) and the placebo plus G/D (5.6 months; 95% CI, 2.6‐8.3 months) was observed (P = .67; hazard ratio [HR], 1.07; 95% CI, 0.77‐1.49). Similarly, there was no significant difference in median overall survival between the 2 groups: 18.3 months for the ontuxizumab plus G/D group (95% CI, 16.2‐21.1 months) and 21.1 months for the placebo plus G/D group (95% CI, 14.2 months to not reached; P = .32; HR, 1.23; 95% CI, 0.82‐1.82). No significant differences between the treatment groups occurred for any efficacy parameter by sarcoma cohort. The combination of ontuxizumab plus G/D was generally well tolerated.
Conclusions
Ontuxizumab plus G/D showed no enhanced activity over chemotherapy alone in soft‐tissue sarcomas, whereas the safety profile of the combination was consistent with G/D alone.
Keywords: endosialin, MORAb‐004, ontuxizumab, sarcomas, tumor endothelial marker 1 (TEM‐1)
Short abstract
Endosialin is involved in tumor blood vessel formation and is expressed on sarcoma tumor cells. This phase 1/2 randomized controlled trial shows that ontuxizumab, an endosialin‐directed monoclonal antibody, does not enhance efficacy in sarcomas when it is combined with chemotherapy (gemcitabine and docetaxel), although the combination is generally well tolerated.
Introduction
Soft‐tissue sarcomas are rare solid tumors of mesenchymal origin with more than 50 different histological subtypes, each with its own underlying biology.1 Despite optimal surgery, approximately 50% of patients will develop metastatic disease. The outcome of patients with metastatic disease is poor, with a median overall survival (OS) of 12 to 20 months and with few systemic therapy options.1 Consequently, there is an unmet need for more effective systemic therapies for advanced sarcomas.
Endosialin, or tumor endothelial marker 1 (TEM‐1), is a cell surface glycoprotein that is expressed in the stromal compartment of nearly all human tumors.2 Preclinical studies have shown that endosialin plays a key role in tumor growth and vessel formation in numerous tumor types, including sarcomas.3, 4 Endosialin expression was noted in all 9 sarcoma subtypes and in 83% of all sarcoma specimens.2 Rouleau et al2 demonstrated that endosialin was expressed by malignant, perivascular, and stromal cells in human specimens. Endosialin expression was found in human specimens of high‐grade/advanced sarcomas.5 Consequently, endosialin was considered a potential therapeutic target in sarcomas. Other studies confirmed these findings.6
Ontuxizumab is a humanized immunoglobulin G1κ antibody directed against endosialin and the first of this class to undergo clinical evaluation. Nonclinical pharmacological studies have shown that ontuxizumab has the ability to interfere with specific endosialin receptor‐ligand interactions.7 The combination of gemcitabine and docetaxel (G/D) is well established in the treatment of metastatic sarcomas.8 The aim of this trial was to assess the optimal dose of ontuxizumab in combination with G/D and to evaluate the ability of the antibody to enhance the antitumor activity of G/D.
Materials and Methods
Patients
Patients older than 18 years with histologically proven metastatic soft‐tissue sarcomas and an Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 1 were eligible. In addition, patients had to have measurable disease according to version 1.1 of the Response Evaluation Criteria in Solid Tumors (RECIST), to have been treated with 0 to 2 prior lines of systemic therapy, and to have fully recovered from all toxicities of previous treatments (apart from alopecia). Patients had to have adequate hematologic, renal, and liver parameters and no brain metastases, primary bone sarcomas, other active malignancies, or an uncontrolled medical condition.
Before enrollment was commenced, local institutional review board/ethics committee approval was obtained in accordance with the Declaration of Helsinki. Investigators obtained informed consent from each participant.
Study Design and Treatment
This was a multicenter, sequential, 2‐part trial. Part 1 was an open‐label, dose‐escalation design used to establish the safety of ontuxizumab combined with G/D and to define the recommended phase 2 dose. The dose‐escalation phase consisted of ontuxizumab (days 1 and 8 of a 21‐day cycle) in combination with gemcitabine at 900 mg/m2 (days 1 and 8) and docetaxel at 75 mg/m2 (day 8).
The recommended phase 2 dose was defined as the highest ontuxizumab dose administered in combination with G/D at which 0 of 3 patients or no more than 1 of a maximum of 6 patients in a given dose cohort experienced a dose‐limiting toxicity (DLT). Patients were treated until disease progression. A DLT was defined as treatment‐related and occurring within the first 28 days of treatment. DLTs included 1) a nonhematologic toxicity of grade 3 or higher (excluding grade 3 asthenia unless lasting longer than 3 days, nausea/vomiting unless optimally treated, and alopecia); 2) a hematologic toxicity of grade 4 neutropenia lasting longer than 7 days, grade 4 febrile neutropenia, a grade 3/4 infection with associated grade 3/4 neutropenia, a grade 4 hematologic toxicity not resolving in fewer than 14 days, or grade 3 thrombocytopenia with clinically significant bleeding; 3) delayed recovery causing a delay of the next dose longer than 28 days; and 4) an infusion‐related toxicity excluding those controlled to grade 2 or lower by management and anaphylactic reactions.
Part 2 was a randomized, double‐blind, placebo‐controlled trial of G/D with either 8 mg/kg ontuxizumab or a placebo and used the same doses and schedule used in part 1. Patients were randomly assigned in a 2:1 ratio to ontuxizumab plus G/D or the placebo plus G/D and were stratified into 4 sarcoma cohorts.
Study Assessments
The response to treatment was determined by computed tomography or magnetic resonance imaging performed at screening and every 6 weeks for the first 24 ± 1 weeks and then every 12 weeks. Patients who discontinued the study drug were followed for documentation of disease progression, any additional anticancer therapies, and survival.
The primary endpoint for part 2 was progression‐free survival (PFS). Secondary endpoints included OS, the overall response rate based on RECIST 1.1, safety, and tolerability. Exploratory objectives included the evaluation of putative predictive markers of response.
Safety was evaluated by the monitoring of adverse events (AEs; graded via Common Toxicity Criteria for Adverse Events, version 4.03), serious adverse events (SAEs), laboratory measurements, vital signs, electrocardiograms, ECOG assessments, and physical examinations.
Biomarkers, Antidrug Antibody, and Pharmacokinetic Analysis
Tumor tissue was obtained from all patients from an initial diagnostic tissue sample or an optional biopsy during screening or previous treatment. Formalin‐fixed, paraffin‐embedded slides underwent immunohistochemistry for endosialin and platelet‐derived growth factor receptor β (PDGFR‐β), as previously described,7, 9 with endosialin antibody clone 9G5 (Morphotek, Exton, Pennsylvania) and PDGFR‐β antibody 28E1 (Cell Signaling Technology, Danvers, Massachusetts). Immunohistochemistry was validated and performed by PhenoPath Laboratories (Seattle, Washington). The tumor content of slides was reviewed, and tissues controls were incorporated into each run.
Slides were evaluated by a board‐certified pathologist as percentages of cells with expression at intensities of 0 (negative), 1+ (weak), 2+ (moderate), and 3+ (strong). The level of biomarker expression in each subcompartment was assessed as the M score, which was calculated as [(% of population scoring 1+) + (2 × % of population scoring 2+) + (3 × % of population scoring 3+)]/6.
Baseline serum biomarkers were assayed to quantitate the levels of endosialin and PDGFR‐β as previously described.10 The detection of the ontuxizumab antidrug antibody in serum samples was performed as previously described.11 Serum concentrations of ontuxizumab were measured at each cycle for pharmacokinetic analysis. Ontuxizumab concentrations were measured with an endosialin antigen–based electrochemiluminescent immunoassay to capture and quantify the serum concentration of free/partially complexed ontuxizumab.11
Statistics
Parts 1 and 2 of the study were summarized and analyzed separately. The sample size was planned to be a maximum of 19 patients for part 1 and 225 patients for part 2 (120‐200 patients in part 2, with a particular sarcoma cohort to have no more than 60 patients). An independent, unblinded committee monitored the trial. Part 1 data were summarized descriptively by dose level.
In part 2, the primary analysis of PFS was conducted at the time at which 185 PFS events (progression or death) were observed; Kaplan‐Meier curves were used, with 95% confidence intervals (CIs) for medians calculated according to Brookmeyer and Crowley.12 PFS in treatment groups was compared in the intent‐to‐treat population on the basis of the log‐rank test, and the hazard ratio (HR) was estimated on the basis of the Cox proportional hazards model. These analyses were also conducted separately by sarcoma cohort. OS was summarized in a similar manner.
The overall response rate (complete or partial) was summarized with 95% CIs with the Clopper‐Pearson exact binomial CI13 for each treatment group. To identify differences between treatment groups overall and within sarcoma cohorts, a Bayesian hierarchical model was used to model PFS across the 4 strata.14 Safety data were summarized with descriptive statistics.
Cox regression modeling was used to assess the influence of baseline tissue and serum biomarkers as covariates on PFS and OS. If the univariate regression model P value (with the Wald chi‐square test) for the factor was <.2, then the factor was included as a candidate for inclusion in a stepwise selection process using a multivariate Cox regression model. Interactions between treatment and each factor were explored to assess each factor’s ability to predict a clinical response. Exposure‐response relations were evaluated with Kaplan‐Meier curves and were characterized in terms of median PFS and OS with 2‐sided 95% CIs constructed with the methodology of Brookmeyer and Crowley.12
Results
Part 1
Sixteen patients enrolled in part 1. Two dose levels of ontuxizumab (4 and 8 mg/kg) were planned in combination with G/D. No DLTs were observed in the 3 patients treated within cohort 1 (ontuxizumab at 4 mg/kg). One of the 6 patients in cohort 2 (ontuxizumab at 8 mg/kg) experienced grade 4 febrile neutropenia. An additional patient in cohort 2 experienced a non‐DLT event of grade 4 neutropenia. Both patients in cohort 2 had previously received more than 6 months of combination chemotherapy. Therefore, the G/D reduction criterion was amended to require a decrease of 25% of the starting dose for G/D for those previously treated with more than 6 months of combination chemotherapy. With this new criterion, 2 further de‐escalation dose cohorts (cohorts 3 and 4) were opened. In cohort 3, the dose was decreased to 6 mg/kg, and no DLTs were observed. Therefore, the dose was re‐escalated to 8 mg/kg in cohort 4, and no additional DLTs were observed.
In total, 3 patients received 4 mg/kg ontuxizumab, 4 patients received 6 mg/kg ontuxizumab, and 9 patients received 8 mg/kg ontuxizumab. The recommended phase 2 dose of ontuxizumab in combination with G/D was 8 mg/kg. There were no treatment‐related deaths or SAEs, and there were no AEs resulting in discontinuation from the trial in part 1.
Part 2
A total of 255 patients were screened for entry into part 2, and 209 were randomized. Among the 46 screen failures, 2 patients (0.8%) did not have measurable disease by RECIST 1.1, 1 patient (0.4%) failed to meet inclusion/exclusion criteria, and 43 patients (16.9%) were excluded for other reasons.
Patients were enrolled at 31 sites in the United States, Australia, and Europe and randomized to either ontuxizumab plus G/D (139 patients) or the placebo plus G/D (70 patients) and were included in the intent‐to‐treat population.
Of the 209 randomized patients, 2 in the ontuxizumab plus G/D arm discontinued the trial before dosing, 1 because of death from progressive disease and 1 because of complications of hypertension. A total of 207 patients received at least 1 dose of ontuxizumab plus G/D or the placebo plus G/D, and 204 of these patients (98.6%) discontinued the trial.
The baseline disease characteristics are displayed in Table 1. The study population consisted of 114 males (55%), and the median age was 56 years (range, 21‐81 years). The proportion of patients with a baseline ECOG score of 1 was higher in the ontuxizumab plus G/D group (72 of 139 patients or 52%) than the placebo plus G/D group (31 of 70 patients or 44%).
Table 1.
Baseline Characteristics for Patients Enrolled in Part 2 (Intent‐To‐Treat Population)
| Ontuxizumab at 8 mg/kg + G/D (n = 139) | Placebo + G/D (n = 70) | Total (n = 209) | |
|---|---|---|---|
| Age, mean (SD), y | 55 (13) | 54 (14) | 55 (14) |
| Sex, No. (%) | |||
| Male | 76 (55) | 38 (54) | 114 (55) |
| Female | 63 (45) | 32 (46) | 95 (46) |
| Race, No. (%) | |||
| White | 116 (83) | 57 (81) | 173 (83) |
| Black or African American | 12 (9) | 9 (13) | 21 (10) |
| Other | 11 (8) | 4 (6) | 15 (7) |
| Initial histologic diagnosis grade, No. (%) | |||
| 1 | 3 (2) | 4 (6) | 7 (3) |
| 2 | 21 (15) | 13 (19) | 34 (16) |
| 3 | 82 (59) | 38 (54) | 120 (57) |
| Unknown | 22 (16) | 9 (13) | 31 (15) |
| Missing | 11 (8) | 6 (9) | 17 (8) |
| Baseline ECOG performance status, No. (%) | |||
| 0 | 67 (48) | 39 (56) | 106 (51) |
| 1 | 72 (52) | 31 (44) | 103 (49) |
| Prior chemotherapy in the metastatic setting | 74 (53) | 40 (57) | 114 (55) |
| First line | 65 (47) | 30 (43) | 95 (45) |
| Second line | 53 (38) | 29 (41) | 82 (39) |
| Third line | 21 (15) | 11 (16) | 32 (15) |
| Histologic subtype, No. (%) | |||
| Liposarcoma | 30 (22) | 15 (21) | 45 (22) |
| Leiomyosarcoma | 41 (29) | 21 (30) | 62 (30) |
| Undifferentiated pleomorphic sarcoma or myxofibrosarcoma | 30 (22) | 15 (21) | 45 (22) |
| Other | 38 (27) | 19 (27) | 57 (27) |
| Angiosarcoma | 3 | 2 | 5 |
| Spindle cell sarcoma | 3 | 2 | 5 |
| Peripheral nerve sheath tumor | 6 | 1 | 7 |
| Synovial sarcoma | 14 | 6 | 20 |
| Miscellaneous or unclassifieda | 12 | 8 | 20 |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; G/D, gemcitabine and docetaxel; SD, standard deviation.
The miscellaneous and unclassified histologic subtypes included patients with histologic diagnoses of rhabdomyosarcoma (3), unclassified sarcoma (3), epithelioid sarcoma (3), hemangiopericytoma (2), endometrial sarcoma (2), adenosarcoma (1), clear cell sarcoma (1), fibrosarcoma (1), intimal sarcoma (1), phyllodes (1), other liposarcoma (1), and small blue round cell tumor (1).
The duration of treatment for the ontuxizumab plus G/D group was a mean of 5.1 months (range, 0.3‐21.4 months) with a mean relative dose intensity of 97%. The duration of treatment for the placebo plus G/D group was 5.4 months (range, 0.3‐21.2 months) with a mean relative dose intensity of 99.9%. Treatment delays associated with an AE occurred in 42% of the patients receiving ontuxizumab plus G/D and in 45% of the patients receiving the placebo plus G/D. Dose reductions occurred in 8% of the patients in both arms.
There was no significant difference between treatment arms for PFS. The median PFS was 4.3 months (95% CI, 2.7‐6.3 months) in the ontuxizumab plus G/D arm and 5.6 months (95% CI, 2.6‐8.3 months) in the placebo plus G/D arm (P = .67; HR, 1.07; 95% CI, 0.77‐1.49). The Kaplan‐Meier curves for PFS are displayed in Figure 1. No significant difference between treatment arms was apparent by sarcoma cohort (Table 2).
Figure 1.
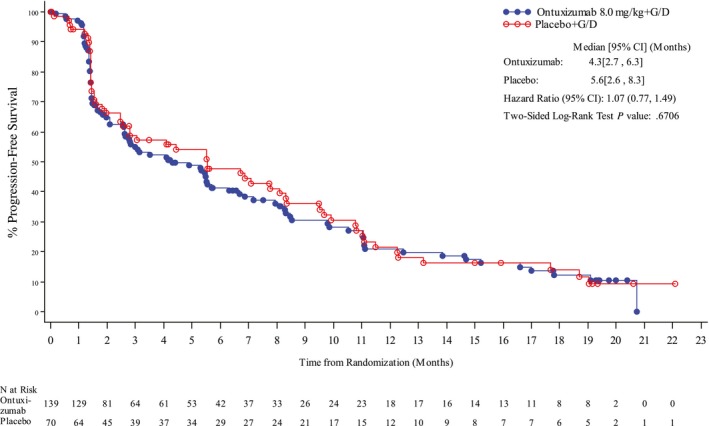
Kaplan‐Meier curves of progression‐free survival for all sarcoma subtypes in part 2 (intent‐to‐treat population). CI indicates confidence interval; G/D, gemcitabine and docetaxel.
Table 2.
PFS With the Bayesian Hierarchical Model in Part 2
| Sarcoma Type | Ontuxizumab at 8.0 mg/kg + G/D | Placebo + G/D |
|---|---|---|
| Liposarcoma | ||
| No. | 30 | 15 |
| PFS observed, median, wk | 14.6 | 24.1 |
| Hazard ratio (95% CI) | 1.12 (0.69‐1.89) | |
| OS observed, median, wk | 58.5 | 54.4 |
| Hazard ratio (95% CI) | 1.1 (0.56‐1.87) | |
| BOR (% with response) | 20/30 (67) | 12/15 (80) |
| P | .236 | |
| Leiomyosarcoma | ||
| No. | 41 | 21 |
| PFS observed, median, wk | 18.3 | 24.0 |
| Hazard ratio (95% CI) | 1.08 (0.68‐1.61) | |
| OS observed, median, wk | 64.6 | 69.1 |
| Hazard ratio (95% CI) | 1.3 (0.74‐2.28) | |
| BOR (% with response) | 27/41 (66) | 16/21 (76) |
| P | .551 | |
| UPS | ||
| No. | 30 | 15 |
| PFS observed, median, wk | 10.3 | 33.6 |
| Hazard ratio (95% CI) | 1.23 (0.73‐2.09) | |
| OS observed, median, wk | 54.1 | 55.3 |
| Hazard ratio (95% CI) | 1.2 (0.68‐2.00) | |
| BOR (% with response) | 16/30 (53) | 11/15 (73) |
| P | .174 | |
| Other | ||
| No. | 38 | 19 |
| PFS observed, median, wk | 10.3 | 6.7 |
| Hazard ratio (95% CI) | 1.01 (0.60‐1.54) | |
| OS observed, median, wk | 56.6 | 57.1 |
| Hazard ratio (95% CI) | 1.1 (0.63‐1.70) | |
| BOR (% with response) | 24/38 (63) | 8/19 (42) |
| P | .389 | |
Abbreviations: BOR, best overall response (stable disease, partial response, or complete response); CI, confidence interval; G/D, gemcitabine and docetaxel; OS, overall survival; PFS, progression‐free survival; UPS, undifferentiated pleomorphic sarcoma.
There was no significant difference in median OS between the 2 arms: 18.3 months (95% CI, 16.2‐21.1 months) in the ontuxizumab plus G/D arm and 21.1 months (95% CI, 14.2 months to not reached) in the placebo plus G/D arm (P = .32; HR, 1.23; 95% CI, 0.82‐1.83). The Kaplan‐Meier curves for OS are displayed in Figure 2.
Figure 2.
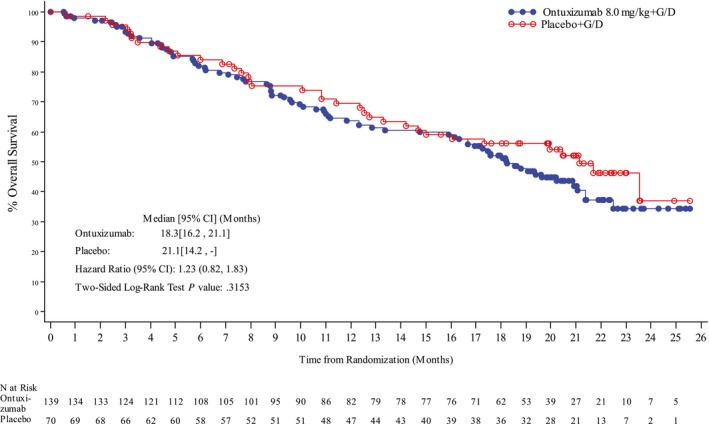
Kaplan‐Meier curves of overall survival for all sarcoma subtypes in part 2 (intent‐to‐treat population). CI indicates confidence interval; G/D, gemcitabine and docetaxel.
The only sarcoma cohort with a longer median PFS in the ontuxizumab group was the “other” cohort. The other cohort comprised at least 13 different histological subtypes and included angiosarcomas (n = 5), spindle cell sarcomas (n = 5), peripheral nerve sheath tumors (n = 7), synovial sarcomas (n = 20), and miscellaneous types (n = 20). Patients with spindle cell sarcoma treated with ontuxizumab had longer median PFS and OS than patients treated with the placebo (median PFS, 2.8 months with ontuxizumab and 1.6 months with the placebo; median OS, 10.7 months with ontuxizumab and 2.0 months with the placebo). Patients treated with ontuxizumab in the miscellaneous subcategory also had longer median PFS and OS than patients treated with the placebo. These were exploratory analyses, and no statistical comparisons were made.
There was no significant difference in the overall response rate between the 2 arms (P = 1.00) or by sarcoma cohort (P < .2 for HR <0.75). Three patients achieved a complete response: 1 (1%) was treated with ontuxizumab plus G/D, and 2 (3%) were treated with the placebo plus G/D. A partial response was achieved by 38 patients: 26 (19%) were treated with ontuxizumab plus G/D, and 12 (17%) were treated with the placebo plus G/D. Sixty patients (43%) in the ontuxizumab plus G/D arm and 33 patients (47%) in the placebo plus G/D arm had stable disease as their best response.
At trial termination, 2 patients with partial responses continued ontuxizumab. Both patients achieved a partial response at cycle 17 that continued for more than 2 years on therapy.
Safety
All patients in the ontuxizumab plus G/D arm and the placebo plus G/D arm had at least 1 treatment‐emergent AE; the most common (in ≥40% of all patients) were fatigue (74% vs 66%), anemia (61% vs 60%), nausea (56% vs 52%), diarrhea (44% vs 36%), peripheral edema (42% vs 45%), and thrombocytopenia (41% vs 43%) respectively.
A numerically higher proportion of patients in the ontuxizumab plus G/D group (86%) versus the placebo plus G/D group (76%) had treatment‐emergent AEs that were considered related to the treatment (Table 3). Treatment‐related AEs that occurred in a higher proportion of patients (>10% difference) in the ontuxizumab plus G/D arm than the placebo plus G/D arm included fatigue, headache, pyrexia, diarrhea, and vomiting. Rash occurred more frequently in the placebo plus G/D arm (18 of 67 or 27%) than the ontuxizumab plus G/D arm (16 of 140 or 11%).
Table 3.
Treatment‐Emergent Adverse Events Considered Related to the Treatment by the Investigator in ≥15% of Patients in Either Treatment Group in Part 2 (Safety Population)
| Preferred Terma | Ontuxizumab at 8 mg/kg + G/D (n = 140), No. (%) | Placebo + G/D (n = 67), No. (%) | Total (n = 207), No. (%) |
|---|---|---|---|
| Fatigue | 66 (47) | 23 (34) | 89 (43) |
| Nausea | 44 (31) | 15 (22) | 59 (29) |
| Headache | 42 (30) | 9 (13) | 51 (25) |
| Anemia | 39 (28) | 18 (27) | 57 (28) |
| Pyrexia | 35 (25) | 8 (12) | 43 (21) |
| Diarrhea | 31 (22) | 6 (9) | 37 (18) |
| Thrombocytopenia | 29 (21) | 11 (16) | 40 (19) |
| Edema, peripheral | 28 (20) | 13 (19) | 41 (20) |
| Decreased appetite | 28 (20) | 10 (15) | 38 (18) |
| Myalgia | 25 (18) | 5 (8) | 30 (15) |
| Vomiting | 24 (17) | 3 (4) | 27 (13) |
| Chills | 21 (15) | 3 (5) | 24 (12) |
| Rash | 16 (11) | 18 (27) | 34 (16) |
Abbreviation: G/D, gemcitabine and docetaxel.
Adverse events were coded with the Medical Dictionary for Drug Regulatory Activities, version 14.1.
The frequencies of patients with at least 1 SAE were similar in the 2 arms: 50% in the ontuxizumab plus G/D arm and 48% in the placebo plus G/D arm. The most frequent treatment‐related SAEs were pyrexia (3% overall, 4% in the ontuxizumab plus G/D arm, and 0% in the placebo plus G/D arm) and anemia (2% overall, 1% in the ontuxizumab plus G/D arm, and 3% in the placebo plus G/D arm). There were no differences in laboratory values, vital signs, or electrocardiogram parameters between the 2 arms.
One patient in each arm died of a treatment‐related AE (cardiac arrest in the ontuxizumab plus G/D arm and respiratory failure in the placebo plus G/D arm arm). Two patients (1%) in the ontuxizumab plus G/D arm experienced drug hypersensitivity AEs (infusion‐related reaction and pyrexia/flushing); all were nonserious and grade 1. None of the drug hypersensitivity AEs resulted in an interruption or discontinuation of ontuxizumab. One patient on ontuxizumab plus G/D developed a transient treatment‐induced antidrug antibody response.
Biomarkers and Pharmacokinetics
Baseline tumor tissue expression of endosialin and PDGFR‐β were measured in the subcompartments of arterial endothelial, capillary endothelial, cytoplasmic tumor endothelial, lymphatic endothelial, membranous tumor endothelial, nonvascular stromal, perivascular, and venous endothelial cells (Table 4). No significant difference in baseline biomarker expression between arms was observed. The highest levels of endosialin were measured in nonvascular stromal, perivascular, and venous endothelial cells. The highest levels of PDGFR‐β were measured in capillary endothelial, cytoplasmic tumor, lymphatic endothelial, nonvascular stromal, and perivascular cells.
Table 4.
Mean Baseline Biomarker Values for Patients Enrolled in Part 2 (Intent‐To‐Treat Population)
| Biomarker | Ontuxizumab at 8 mg/kg + G/D (n = 139) | Placebo + G/D (n = 70) |
|---|---|---|
| Endosialin lymphatic endothelial cell M score | 12.8 | 13.3 |
| Endosialin membranous tumor cell M score | 4.2 | 4.3 |
| Endosialin nonvascular stromal cell M score | 18.3 | 18.7 |
| Endosialin perivascular cell M score | 24.1 | 22.9 |
| Endosialin venous endothelial cell M score | 19.1 | 16.8 |
| Log plasma endosialin, ng/mL | 11.463 | 11.486 |
| PDGFR‐β arterial endothelial cell M score | 3.4 | 3.2 |
| PDGFR‐β capillary endothelial cell M score | 30.0 | 29.0 |
| PDGFR‐β cytoplasmic tumor cell M score | 22.7 | 21.5 |
| PDGFR‐β lymphatic endothelial cell M score | 22.0 | 21.7 |
| PDGFR‐β membranous tumor cell M score | 8.3 | 4.7 |
| PDGFR‐β nonvascular stromal cell M score | 21.4 | 23.4 |
| PDGFR‐β perivascular cell M score | 31.8 | 29.8 |
| PDGFR‐β venous endothelial cell M score | 3.6 | 3.5 |
| Log plasma PDGFR‐β, ng/mL | 7.830 | 7.872 |
Abbreviations: G/D, gemcitabine and docetaxel; PDGFR‐β, platelet‐derived growth factor receptor β.
A prognostic biomarker demonstrates an association with outcome, regardless of therapy. Factors considered possibly prognostic of PFS included the log serum endosialin concentration (P = .06), tissue endosialin in venous endothelial cells (P = .04), and tissue PDGFR‐β in capillary endothelial cells (P = .10). Longer PFS was associated with higher serum endosialin concentrations (HR, 0.61; 95% CI, 0.36‐1.03), lower tissue endosialin expression in venous endothelial cells (HR, 1.01; 95% CI, 1.0‐1.02), and lower tissue PDGFR‐β levels in capillary endothelial cells (HR, 0.74; 95% CI, 0.51‐1.06). Factors considered possibly prognostic of OS were endosialin cytoplasmic tumor endothelial cells (P = .09) and endosialin membranous tumor endothelial cells (P = .12). Longer OS was associated with lower tissue endosialin levels in cytoplasmic tumor endothelial cells (HR, 1.01; 95% CI, 1.0‐1.02) and higher tissue endosialin expression in membranous tumor endothelial cells (HR, 1.02; 95% CI, 1.0‐1.04).
A predictive biomarker provides information about the effect of a therapeutic intervention on clinical outcomes and can potentially be used to select patients for therapy. Tissue PDGFR‐β in capillary endothelial cells showed a significant treatment interaction (P = .02), with values below the median associated with improved PFS (HR, 0.55, 95% CI, 0.29‐1.04). No baseline biomarkers were predictive of improved OS with ontuxizumab.
Ontuxizumab had no clear exposure effect on PFS or OS.
Discussion
Ontuxizumab in combination with G/D was well tolerated in patients with soft‐tissue sarcomas. Despite promising preclinical data and some durable benefit in patients with sarcoma treated within phase 1 trials,11 the combination did not show superior activity in comparison with G/D alone in this randomized trial. This was consistent in all 4 histological cohorts studied in the randomized component of the trial.
The G/D combination was used in this study because it has proven efficacy in advanced sarcomas.8, 15, 16 A previous trial reporting a median PFS of 7.5 months with G/D and bevacizumab17 provided support for the combination of G/D with antiangiogenic agents in sarcomas.
Ontuxizumab was used in this trial to evaluate the hypothesis that blocking endosialin‐mediated tumor angiogenesis would enhance the efficacy of G/D in sarcomas. However, no improvement in PFS or OS was observed with ontuxizumab. A major difficulty in conducting trials for sarcomas is the profound heterogeneity of these diseases. One of the goals of this trial was to evaluate the benefit of ontuxizumab in all soft‐tissue sarcomas as well as specific cohorts to potentially identify subsets that might benefit from ontuxizumab. Ontuxizumab showed no additional benefit in liposarcoma, leiomyosarcoma, or undifferentiated pleomorphic sarcoma. However, in the heterogeneous “other” cohort, longer median PFS (not statistically significant) was observed with ontuxizumab. To evaluate potential benefits in specific subtypes included in the other cohort, we performed an exploratory analysis. The spindle cell and miscellaneous sarcoma subcategories showed a nonsignificantly but numerically longer median PFS with ontuxizumab treatment.
The choice of the ontuxizumab dose in this trial was based on the completed phase 1 trial of single‐agent ontuxizumab with a maximum tolerated dose of 12 mg/kg.11 One potential criticism of our trial is that the dose of ontuxizumab was not high enough. In the phase 1 study, pharmacokinetic data suggested an accumulation of ontuxizumab at 4 mg/kg, and exposures were similar between 8 and 12 mg/kg with weekly administration. For this reason, the decision was made a priori not to go above 8 mg/kg ontuxizumab in combination with G/D for the current trial. Because of the potential accumulation of ontuxizumab at doses higher than 4 mg/kg, we may not have given a high enough dose of ontuxizumab. However, pharmacokinetic analyses indicated that ontuxizumab exposure had no effect on PFS or OS.
The profile of AEs occurring more frequently in the ontuxizumab arm (fatigue, headache, pyrexia, diarrhea, and vomiting) resembles the profile of the most frequent AEs observed in the phase 1 trial.11 These results suggest that ontuxizumab did have a pharmacological effect in the current trial. Whether this dose was sufficiently high to block the angiogenic effect of endosialin is not certain.
Endosialin is believed to increase the proliferation of pericytes and result in enhanced tumor angiogenesis via a platelet‐derived growth factor receptor signaling pathway.3 In the current trial, patients were not selected on the basis of endosialin expression.
A longer PFS was associated with a higher serum endosialin concentration, a lower tissue endosialin expression in venous endothelial cells, and a lower tissue PDGFR‐β level in capillary endothelial cells at the baseline. Among ontuxizumab‐treated patients, a lower tissue PDGFR‐β expression in capillary endothelial cells was associated with improved PFS, and this indicated that it was a potential predictive indicator of improved PFS with ontuxizumab.
Although compelling evidence linked the expression of endosialin with tumor growth and progression in preclinical studies, the biomarkers measured in this trial showed no predictive association with outcome in the ontuxizumab plus G/D arm. One potential reason for the weak association between endosialin‐associated biomarkers and outcome is the low efficacy of ontuxizumab in this trial.
On the basis of these data, further trials of ontuxizumab for soft‐tissue sarcomas are not warranted. Because of the stratification by subtype, this trial provides a benchmark of the subtype‐specific efficacy of G/D. In the future, the potential use of antibody‐drug conjugates to selectively deliver cytotoxic agents to tumor sites could be evaluated. Because endosialin is highly expressed in sarcomas,2, 5 ontuxizumab could be used to target sarcoma cells and deliver cytotoxic agents linked to it. In a human endosialin‐positive sarcoma xenograft model, prolonged antitumor activity of an anti‐endosialin antibody conjugated to cytotoxic agents was observed in comparison with controls.18
Funding Support
This trial was supported by Morphotek, Inc (Exton, Pennsylvania). The University of Texas MD Anderson Cancer Center is supported by the National Institutes of Health (grant P30 CA016672).
Conflict of Interest Disclosures
Robin L. Jones reports personal fees from Adaptimmune, Blueprint, Clinigen, Eisai, Epizyme, Daiichi, Deciphera, Immune Design, Lilly, Merck, and PharmaMar outside the submitted work. Sant P. Chawla reports grants from Amgen, Roche, Threshold Pharmaceuticals, GlaxoSmithKline, CytRx, Ignyta, Immune Design, TRACON Pharmaceuticals, SARC, Karyopharm Therapeutics, and Jansen outside the submitted work. Steven Attia reports grants from Bayer, AB Science, TRACON Pharmaceuticals, CytRx, Novartis, Daiichi Sankyo, Lilly, Immune Design, Karyopharm Pharmaceuticals, Epizyme, Blueprint Medicines, Genmab, CBA Pharmaceuticals, the Desmoid Tumor Research Foundation, Merck, Deciphera, Takeda Oncology, Philogen, Gradilis, Incyte, Threshold Pharmaceuticals, Bayer, BTG, Bavarian Nordic, Advenchen Laboratories, SpringWorks, and Adaptimmune as well as travel expenses from TRACON Pharmaceuticals and Immune Design outside the submitted work. Patrick Schöffski reports institutional support from Plexxikon, Eisai, Loxo, Lilly, Blueprint Medicines, Ellipses Pharma, Deciphera, Merck, Servier, Genmab, Adaptimmune, Intellisphere, Transgene, Boehringer Ingelheim, CoBioRes NV, Exelixis, G1 Therapeutics, Novartis, and PharmaMar outside the submitted work. Bartosz Chmielowski reports personal fees from Bristol‐Myers Squibb, Merck, Genentech/Roche, Iovance, HUYA, Compugen, Array BioPharma, Regeneron, Biothera, Janssen, and Novartis outside the submitted work. Axel Le Cesne reports personal fees from PharmaMar, Pfizer, and Lilly outside the submitted work. Brian A. Van Tine reports personal fees from Epizyme, Lilly, CytRx, Janssen, Caris, Immune Design, Daiichi Sankyo, Adaptimmune, and Plexxikon and grants from Pfizer, Merck, TRACON Pharmaceuticals, and Morphotek outside the submitted work. Shreyaskumar Patel reports grants and personal fees from Janssen and personal fees from PharmaMar and MJ Hennessey/OncLive during the conduct of the study; he also reports grants from Blueprint Medicines and personal fees from Novartis Oncology, Immune Design, Epizyme, Eli Lilly, CytRx, and EMD Serono outside the submitted work. Andrew J. Wagner reports personal fees from Novartis and NanoCarrier; grants and personal fees from Daiichi‐Sankyo and Eli Lilly; and grants from Karyopharm, Aadi Bioscience, Plexxikon, and Celldex outside the submitted work. Rashmi Chugh reports grants and nonfinancial support from Morphotek during the conduct of the study. She also reports grants, personal fees, and nonfinancial support from Epizyme; personal fees from EMD Serono, Janssen, and Immune Design; and grants and nonfinancial support from AADi, Novartis, Lilly, Medivation, MabVax, Advenchen, Pfizer, and Plexxikon outside the submitted work. Kert Viele reports being an employee of Berry Consultants with multiple consulting clients. Robert G. Maki reports personal fees from Arcus, Bayer, Foundation Medicine, the American Association for Cancer Research, the American Society of Clinical Oncology, Springer, UpToDate, and Wiley; grants and personal fees from Janssen, PharmaMar, Presage Biosciences, and TRACON Pharmaceuticals; grants from Daiichi‐Sankyo, Genentech, Immune Design, Immunocor, Lilly/ImClone, Regeneron, the Sarcoma Alliance for Research Through Collaboration, and Fondazione Enrico Pallazzo; and other from AADi, Deciphera, Karyopharm, and the American Board of Internal Medicine outside the submitted work. John W. Heyburn, Susan C. Weil, and Wenquan Wang were employed by Morphotek, Inc, during the study. The other authors made no disclosures.
Author Contributions
Robin L. Jones: Conceptualization, methodology, funding acquisition, investigation, and writing–review and editing. Sant P. Chawla: Investigation and writing‐review. Steven Attia: Investigation and writing‐review. Patrick Schöffski: Investigation and writing‐review. Hans Gelderblom: Investigation and writing‐review. Bartosz Chmielowski: Investigation and writing‐review. Axel Le Cesne: Investigation and writing‐review. Brian A. Van Tine: Investigation and writing‐review. Jonathan C. Trent: Investigation and writing‐review. Shreyaskumar Patel: Investigation and writing‐review. Andrew J. Wagner: Investigation and writing‐review. Rashmi Chugh: Investigation and writing‐review. John W. Heyburn: Conceptualization, methodology, project administration, and writing–review and editing. Susan C. Weil: Conceptualization, methodology, project administration, and writing–review and editing. Wenquan Wang: Software, formal analysis, and writing–review and editing. Kert Viele, PhD: Software and formal analysis. Robert G. Maki: Conceptualization, funding acquisition, methodology, investigation, and writing–review and editing.
This study is registered at ClinicalTrials.gov (identifier NCT01574716).
References
- 1. Noujaim J, Thway K, Sheri A, et al. Histology‐driven therapy: the importance of diagnostic accuracy in guiding systemic therapy of soft tissue tumors. Int J Surg Pathol. 2016;1:5‐15. [DOI] [PubMed] [Google Scholar]
- 2. Rouleau C, Curiel M, Weber W, et al. Endosialin protein expression and therapeutic target potential in human solid tumors: sarcoma versus carcinoma. Clin Cancer Res. 2008;14:7223‐7236. [DOI] [PubMed] [Google Scholar]
- 3. Tomkowicz B, Rybinski K, Sebeck D, et al. Endosialin/TEM‐1/CD248 regulates pericyte proliferation through PDGF receptor signaling. Cancer Biol Ther. 2010;11:908‐915. [DOI] [PubMed] [Google Scholar]
- 4. Nanda A, Karim B, Peng Z, et al. Tumor endothelial marker 1 (Tem1) functions in the growth and progression of abdominal tumors. Proc Natl Acad Sci U S A. 2006;103:3351‐3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rouleau C, Smale R, Fu YS, et al. Endosialin is expressed in high grade and advanced sarcomas: evidence from clinical specimens and preclinical modeling. Int J Oncol. 2011;39:73‐89. [DOI] [PubMed] [Google Scholar]
- 6. Thway K, Robertson D, Jones RL, et al. Endosialin expression in soft tissue sarcoma as a potential marker of undifferentiated mesenchymal cells. Br J Cancer. 2016;115:473‐479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rybinski K, Imtiyaz HZ, Mittica B, et al. Targeting endosialin/CD248 through antibody‐mediated internalization results in impaired pericyte maturation and dysfunctional tumor microvasculature. Oncotarget. 2015;6:25429‐25440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maki RG, Wathen JK, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of Sarcoma Alliance for Research Through Collaboration Study 002. J Clin Oncol. 2007;25:2755‐2763. [DOI] [PubMed] [Google Scholar]
- 9. O’Shannessy DJ, Dai H, Mitchell M, et al. Endosialin and associated protein expression in soft tissue sarcomas: a potential target for anti‐endosialin therapeutic strategies. Sarcoma. 2016;2016:5213628. doi: 10.1155/2016/5213628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O’Shannessy DJ, Smith MF, Somers EB, et al. Novel antibody probes for the characterization of endosialin/TEM‐1. Oncotarget. 2016;7:69420‐69435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Diaz LA, Coughlin CM, Weil SC, et al. A first‐in‐human phase 1 study of MORAb‐004, a monoclonal antibody to endosialin in patients with advanced solid tumors. Clin Cancer Res. 2015;21:1281‐1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brookmeyer R, Crowley J. A confidence interval for the median survival time. Biometrics. 1982;38:29‐41. [Google Scholar]
- 13. Hollander M, Wolfe DA. Nonparametric Statistical Methods. New York, NY: John Wiley & Sons Inc; 1973. [Google Scholar]
- 14. Berry SM, Broglio KR, Groshen S, Berry DA. Bayesian hierarchical modeling of patient subpopulations: efficient designs of phase II oncology clinical trials. Clin Trials. 2013;10:720‐734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hensley ML, Blessing JA, Mannel R, Rose PG. Fixed‐dose rate gemcitabine plus docetaxel as first‐line therapy for metastatic uterine leiomyosarcoma: a Gynecologic Oncology Group phase II trial. Gynecol Oncol. 2008;109:329‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hensley ML, Blessing JA, Degeest K, et al. Fixed‐dose rate gemcitabine plus docetaxel as second‐line therapy for metastatic uterine leiomyosarcoma: a Gynecologic Oncology Group phase II trial. Gynecol Oncol. 2008;109:323‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dickson MA, D’Adamo DR, Keohan ML, et al. Phase II trial of gemcitabine and docetaxel with bevacizumab in soft tissue sarcoma. Sarcoma. 2015;2015:532478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rouleau C, Gianolio DA, Smale R, et al. Anti‐endosialin antibody–drug conjugate: potential in sarcoma and other malignancies. Mol Cancer Ther. 2015;14:2081‐2089. [DOI] [PubMed] [Google Scholar]