

Abstract
Although thousands of long noncoding RNAs (lncRNAs) have been annotated, only a limited number of them have been functionally characterized. Here, we identified an oncogenic lncRNA, named lnc‐UCID (lncRNA up‐regulating CDK6 by interacting with DHX9). Lnc‐UCID was up‐regulated in hepatocellular carcinoma (HCC), and a higher lnc‐UCID level was correlated with shorter recurrence‐free survival of HCC patients. Both gain‐of‐function and loss‐of function studies revealed that lnc‐UCID enhanced cyclin‐dependent kinase 6 (CDK6) expression and thereby promoted G1/S transition and cell proliferation. Studies from mouse xenograft models revealed that tumors derived from lnc‐UCID‐silenced HCC cells had a much smaller size than those from control cells, and intratumoral injection of lnc‐UCID small interfering RNA suppressed xenograft growth. Mechanistically, the 850‐1030‐nt domain of lnc‐UCID interacted physically with DEAH (Asp‐Glu‐Ala‐His) box helicase 9 (DHX9), an RNA helicase. On the other hand, DHX9 post‐transcriptionally suppressed CDK6 expression by binding to the 3′‐untranslated region (3′UTR) of CDK6 mRNA. Further investigation disclosed that lnc‐UCID enhanced CDK6 expression by competitively binding to DHX9 and sequestering DHX9 from CDK6‐3′UTR. In an attempt to explore the mechanisms responsible for lnc‐UCID up‐regulation in HCC, we found that the lnc‐UCID gene was frequently amplified in HCC. Furthermore, miR‐148a, whose down‐regulation was associated with an increase of lnc‐UCID in HCC, could bind lnc‐UCID and inhibit its expression. Conclusion: Up‐regulation of lnc‐UCID, which may result from amplification of its gene locus and down‐regulation of miR‐148a, can promote HCC growth by preventing the interaction of DHX9 with CDK6 and subsequently enhancing CDK6 expression. These findings provide insights into the biological functions of lncRNAs, the regulatory network of cell cycle control, and the mechanisms of HCC development, which may be exploited for anticancer therapy.
Abbreviations
- 3′UTR
3′‐untranslated region
- CCA
cholangiocarcinoma
- CDK6
cyclin‐dependent kinase 6
- DBE
DHX9‐binding element
- DHX9
DEAH (Asp‐Glu‐Ala‐His) box helicase 9
- EMSA
electrophoretic mobility shift assay
- FACS
fluorescence‐activated cell sorting
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- HCC
hepatocellular carcinoma
- IgG
immunoglobulin G
- lncRNA
long noncoding RNA
- lnc‐UCID
lncRNA up‐regulating CDK6 by interacting with DHX9
- NC
negative control
- ns
not significant
- nt
nucleotide
- pRb
retinoblastoma protein
- RACE
rapid amplification of cDNA ends
- RIP
RNA immunoprecipitation
- SF
skin fibroblast
- siRNA
small interfering RNA
Long noncoding RNAs (lncRNAs) belong to a class of noncoding transcripts greater than 200 nucleotides in length,1 and they may regulate cell activities by interacting with DNA, RNA, and proteins.2, 3 It has been shown that lncRNAs play important roles in diverse biological processes and that their dysfunction contributes to pathological conditions, such as cancer.4, 5 Although thousands of lncRNAs have been annotated, only a limited number of them have been characterized functionally.
Uncontrolled cell proliferation, one of the most important hallmarks of cancer, results from defects in the control of the cell cycle, particularly at the G1 to S phase (G1/S) transition. The G1/S transition is the critical point that determines whether cells proceed to proliferation; this checkpoint is tightly controlled by the retinoblastoma protein (pRb) pathway, which involves primarily pRb, D‐type cyclins, E‐type cyclins, cyclin‐dependent kinase (CDK)4/6, CDK inhibitors, and E2F.6, 7 Dysfunction of the pRb pathway is detected in every type of cancer. To date, a few lncRNAs have been identified to regulate the pRb pathway and G1/S transition, and their deregulation has been shown to affect tumor growth.8, 9, 10, 11, 12 Obviously, it is highly worthwhile to identify more lncRNAs that regulate the pRb pathway.
Hepatocellular carcinoma (HCC) is one of the most common malignancies worldwide.13 Emerging evidence indicates that deregulation of lncRNAs is associated with uncontrolled cell proliferation, apoptosis resistance, active angiogenesis, and metastasis of HCC,14, 15, 16 and it also affects the stemness of HCC cells.17, 18 Genomic gain represents an important mechanism in the activation of oncogenes,19 but lncRNAs on the amplified chromosome region of HCC remain to be explored. In an attempt to identify oncogenic lncRNAs in HCC, we screened for the lncRNAs that were up‐regulated in HCC and located on the chromosome regions with frequent amplification. One lncRNA, which we named lnc‐UCID (lncRNA up‐regulating CDK6 by interacting with DHX9), was found to be located on chromosome 1q22 and was frequently amplified and up‐regulated in HCC tissues. Lnc‐UCID enhanced CDK6 expression and promoted tumor growth of HCC cells. Mechanism analyses revealed that lnc‐UCID was associated with DEAH (Asp‐Glu‐Ala‐His) box helicase 9 (DHX9) and prevented the DHX9‐mediated down‐regulation of CDK6, which thus promoted G1/S transition and cell proliferation. Our findings reveal the fundamental roles of lnc‐UCID and DHX9 in cell cycle control and tumorigenesis, and provide the lnc‐UCID‐DHX9‐CDK6 axis as a potential target for anticancer therapy.
Materials and Methods
More details are provided in the Supporting Information.
Tissue Specimens and Cell Lines
Human HCC and adjacent nontumor liver tissues were collected from patients who underwent radical tumour resection at Sun Yat‐sen University Cancer Center, China. Both tumor and nontumor tissues were confirmed histologically. No local or systemic treatment had been conducted before surgery. After surgical resection, no other anticancer therapy was administered before relapse. Informed consent was obtained from each patient, and the protocol was approved by the Institutional Research Ethics Committee. Tissues were immediately snap‐frozen in liquid nitrogen until use. Relevant characteristics of the 139 subjects included are summarized in Supporting Table S1.
HEK293T cells, immortalized human liver cell line LO2, and two hepatoma cell lines (HepG2 and QGY‐7703) were cultured in Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (FBS, Gibco). Primary skin fibroblasts (SFs) were isolated from human neonatal foreskin and maintained in RPMI 1640 medium (Gibco) supplemented with 10% FBS (Gibco). Isolation of SF cells was performed as previously described.20
RNA Oligoribonucleotides and Vectors
All RNA oligonucleotides were purchased from GenePharma (Shanghai, China). The small interfering RNAs (siRNAs) targeting the human lnc‐UCID (Ensembl transcript ID: ENST00000497831.1), DHX9 (GeneBank accession No. NM_001357.4), and CDK6 (NM_001259) transcripts were designated as siUCID, siDHX9 and siCDK6, respectively. The negative control (NC) RNA duplex for both miR‐148a and siRNAs was nonhomologous to any human genome sequence. The miR‐148a inhibitor (anti‐miR‐148a) with a sequence complementary to the mature miR‐148a and its negative control (anti‐NC) consisted of 2′‐O‐methyl‐modified RNA oligonucleotides.
The expression vectors pc3‐puro‐UCID, pc3‐puro‐UCID‐Δcore, pc3‐gab‐UCID, pc3‐gab‐CDK6, pCDH‐Flag‐DHX9, and luciferase reporter vectors psi‐CDK6‐DHX9‐binding element (DBE) 1, psi‐CDK6‐DBE2, psi‐UCID‐WT, and psi‐UCID‐MUT were generated as described in the Supporting Information.
All oligonucleotide sequences are listed in Supporting Table S2.
Analysis of Gene Expression
The expression level of target genes was analyzed by quantitative real‐time quantitative PCR or western blotting.
Cell Transfection
RNA oligos were transfected using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA). A final concentration of 50 nM duplex was used. Plasmid DNAs were transfected with Lipofectamine 3000 (Invitrogen).
Cell Counting Assay
Cell counting assay was used to evaluate cell growth. Twenty‐four hours after the last transfection, 1 × 104 viable cells were reseeded in a 24‐well plate and cultured for 4 days before cell counting.
Colony Formation Assay
Twenty‐four hours after transfection, aliquots of the viable transfected cells (500 for the HepG2 cells, and 150 for the QGY‐7703 cells) were placed in a 6‐well plate and maintained in complete medium for 18 (HepG2) or 10 (QGY‐7703) days. Colonies were fixed in methanol and stained with a 0.1% crystal violet solution in 20% methanol for 15 minutes.
Mouse Xenograft Models
All mouse experiments were approved by the Institutional Animal Care and Use Committee at the School of Life Sciences, Sun Yat‐sen University. Experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication No. 80‐23, revised 1996) and according to the institutional ethical guidelines for animal experiments. For tumorigenicity assay, QGY‐7703 cells (2 × 106) transfected with NC (50 nM) or siUCID (25 nM each of siUCID‐1 and siUCID‐2) were suspended in 100 μL phosphate‐buffered saline (PBS)/Matrigel (1:1) and then injected subcutaneously into the posterior flanks of 4‐5‐week‐old female NSG (NOD.Cg‐Prkdcscid Il2rgtm1Wjl/SzJ) mice (6 mice for NC group and 7 mice for siUCID group). The experiments ended 22 days after tumor cell inoculation. To evaluate the therapeutic effect of siUCID in vivo, 7 days after QGY‐7703 cell (2 × 106) implantation, 5 nmol cholesterol‐conjugated NC or siUCID‐1 (Ribobio, Guangzhou, China) in 0.1 mL 1 × PBS was intratumorally injected 3 times per week for 2 weeks (7 mice for each group). Tumor volume (V) was monitored by measuring the length (L) and width (W) with calipers and calculated with the following formula: (L × W2) × 0.5.
Cell Cycle Analysis
For loss‐of‐function experiments, cell cycle analyses were performed using a detergent‐containing hypotonic solution (Krishan's reagent) and fluorescence‐activated cell sorting (FACS) (Becton Dickinson, San Jose, CA). For the gain‐of‐function and rescue experiment, the DNA content of live cells was stained with Hoechst 33342 and analyzed by FACS.
Bromodeoxyuridine Incorporation Assay
DNA replication was examined by bromodeoxyuridine (BrdU) incorporation assay. The BrdU incorporation rate is expressed as the percentage of BrdU‐stained cells/total number of cells. At least 500 cells were counted for each sample.
Polysome Analysis
Cells were treated with cycloheximide (cat. 2112, Cell Signaling Technology, CST, Beverly, MA) and then lysed. The ribosomes were separated by ultracentrifugation in 10%‐50% sucrose gradient. The collected fractions were subjected to quantitative real‐time PCR analysis.
Rapid Amplification of Complementary DNA Ends
To amplify the 3′‐end of lnc‐UCID, the total RNA from normal liver tissues was subjected to reverse transcription with a 3′RACE (rapid amplification of complementary DNA ends)‐adaptor primer, followed by nested PCR using gene‐specific primers and 3′RACE‐adaptor primer. The 5′‐end of the lnc‐UCID transcript was characterized using a 5′‐Full RACE Kit (D315; Takara, Kyoto, Japan).
RNA Pulldown Assay
RNA pulldown assay was performed using in vitro transcribed biotinylated RNA and streptavidin Dynabeads (Invitrogen). The retrieved proteins were resolved on a sodium dodecyl sulfate‐polyacrylamide gel, and the specific band was excised and analysed by mass spectrometry (Beijing Protein Innovation, Beijing, China).
RNA‐Immunoprecipitation Assay
The DHX9‐RNA complex was immunoprecipitated by anti‐DHX9 antibody, and the isotype‐matched immunoglobulin G (IgG) was used as a negative control. RNA was extracted from the precipitates by TRIzol reagent (Invitrogen) and detected by quantitative real‐time PCR.
RNA Electrophoretic Mobility Shift Assay
Flag‐DHX9 protein was obtained from the pCDH‐Flag‐DHX9‐transfected HEK293T cells by immunoprecipitation with Anti‐FLAG M2 magnetic beads (M8823, Sigma‐Aldrich, St. Louis, MO, USA). A biotin‐labelled RNA probe was generated as described in the RNA pulldown assay and incubated with the Flag‐DHX9 protein or Flag peptide (negative control, B23111, Bimake, Houston, TX, USA) at RT for 20 minutes in RNA–electrophoretic mobility shift assay (EMSA) binding buffer (10 mM HEPES at pH 7.3, 20 mM KCl, 1 mM MgCl2, 1 mM DTT, 5% glycerol, 100 μg/mL yeast transfer RNA) and subjected to native polyacrylamide gel electrophoresis (PAGE) analysis. Biotinylated RNA was measured in the blots with a chemiluminescent EMSA kit (Beyotime).
Luciferase Reporter Assay
For loss‐of‐function studies and the validation of miR‐148a target, HepG2 cells in a 48‐well plate were transfected with 50 nM RNA duplex for 24 hours and then transfected with 10 ng psi‐CDK6‐DBE1, psi‐CDK6‐DBE2, psi‐UCID‐WT, or psi‐UCID‐MUT. For gain‐of‐function studies, HepG2 cells were seeded in a 48‐well plate for 24 hours, followed by the co‐transfection of psi‐CDK6‐DBE1 with pc3‐puro‐UCID alone or together with pCDH‐Flag‐DHX9. Forty‐eight hours after transfection, the cell lysates were subjected to dual luciferase assays as described.21 Constitutively expressed Firefly luciferase from the psiCHECK2 vector (Promega) was used as a control to correct for the differences in both the transfection and harvest efficiencies.
Statistical Analysis
The differences in lnc‐UCID, DHX9, CDK6, and miR‐148a expression levels between the paired HCC tissues and adjacent nontumor liver tissues were compared by paired t test. The correlations between the RNA levels of lnc‐UCID and CDK6, DHX9 or miR‐148a were explored with Pearson's correlation coefficient. Recurrence‐free survival was calculated from the date of HCC resection to the time of first recurrence. Patients who were lost to follow‐up or who died from causes unrelated to HCC were treated as censored events. Kaplan‐Meier survival curves and Cox proportional hazard regression analyses were performed using SPSS version 13.0 (SPSS Inc., Chicago, IL) to identify prognostic factors.
The data are expressed as the mean ± SEM from at least three independent experiments. The differences between groups were analyzed using an unpaired t test when only two groups were compared and one‐way analysis of variance when more than two groups were compared. A P value of less than 0.05 was considered statistically significant. All statistical tests were two‐sided and were performed using GraphPad Prism (GraphPad Software Inc., San Diego, CA).
Results
LNC‐UCID is Up‐Regulated in HCC Tissues, and Silencing LNC‐UCID Inhibits Tumor Growth in Vitro and in Vivo
To identify oncogenic lncRNAs, two criteria were used to select candidate lncRNAs (Supporting Fig. S1A): (1) the RNA level of lncRNA displayed more than 2‐fold increase in HCC tissues compared with adjacent nontumor tissues; and (2) the gene locus of lncRNA was located within the chromosome regions known to be frequently amplified in HCC.22 Among eight candidate lncRNAs (Supporting Table S3), the functions of ENST00000497831.1, ENST00000621798.4, TMED10P1, and ENST00000441932.1 have not yet been reported. These lncRNAs were therefore examined in our study cohort by quantitative real‐time PCR. Compared with nontumor liver tissues, more than 50% increases in the RNA levels of ENST00000497831.1, TMED10P1, and ENST00000621798.4 were observed in 68.8%, 53.1%, and 46.9% HCC samples, respectively (Fig. 1A and Supporting Fig. S1B). ENST00000497831.1, the lncRNA with most significant up‐regulation in HCC, was selected for further exploration of clinical significance and biological function, and was then named lnc‐UCID. The analysis on the genomic DNA of lnc‐UCID in paired HCC and adjacent nontumor liver tissues disclosed that the lnc‐UCID gene was amplified in 38.7% of HCC tissues, and the RNA levels of lnc‐UCID were positively correlated with its genomic DNA contents (r = 0.402, P = 0.025) (Fig. 1B). Furthermore, the Kaplan‐Meier survival analysis revealed an association of a higher lnc‐UCID level with a shorter recurrence‐free survival (P = 0.005; Fig. 1C), and both univariate and multivariate analyses confirmed the up‐regulation of lnc‐UCID as an independent risk factor for shorter recurrence‐free survival (hazard ratio = 2.052, P = 0.021; Supporting Table S1).
Figure 1.
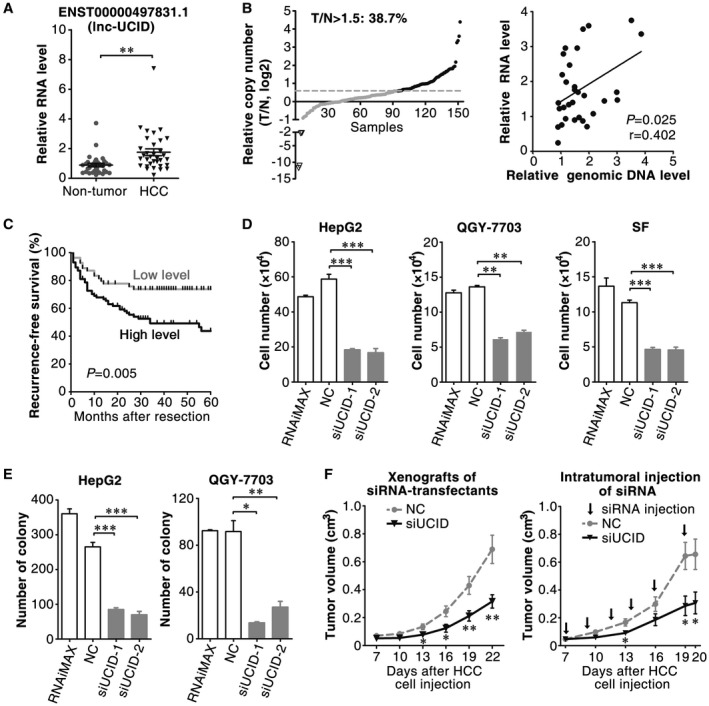
Lnc‐UCID is up‐regulated in HCC tissues, and silencing lnc‐UCID expression inhibits tumor cell growth in vitro and in vivo. (A) Lnc‐UCID expression was significantly increased in HCC tissues. Lnc‐UCID expression was assessed by quantitative real‐time PCR in 32 paired HCC and adjacent nontumor liver tissues. (B) Lnc‐UCID was frequently amplified in HCC tissues, and its RNA level was positively correlated with genomic DNA content. The genomic level of the lnc‐UCID gene was analyzed by quantitative real‐time PCR in 150 paired HCC (T) and adjacent nontumor (N) tissues. The lnc‐UCID locus was considered to be amplified when T/N > 1.5. (C) A Kaplan‐Meier plot revealed the association between a higher lnc‐UCID level and a shorter recurrence‐free survival. Based on the minimum P‐value approach, the 39th percentile of the lnc‐UCID level in 139 HCC tissues was chosen as the cut‐off value for separating the lnc‐UCID low‐level group (n = 54) from the lnc‐UCID high‐level group (n = 85). (D) Lnc‐UCID knockdown inhibited cell growth in vitro. (E) Lnc‐UCID knockdown suppressed the colony formation of hepatoma cells. (F) Silencing lnc‐UCID suppressed tumor growth in vivo. Left panel, QGY‐7703 cells were transfected with NC or mixture of siUCID‐1 and siUCID‐2, then injected into NSG mice. Right panel, QGY‐7703 cells were implanted into NSG mice for 7 days; cholesterol‐conjugated NC or siUCID‐1 was injected intratumorally. *P < 0.05; **P < 0.01; ***P < 0.001.
We next evaluated whether lnc‐UCID regulated cell growth. Silencing lnc‐UCID (Supporting Fig. S2A) significantly reduced the number of HepG2, QGY‐7703, and SF cells (Fig. 1D). Consistently, siUCID‐transfected hepatoma cells displayed fewer and smaller colonies than the NC‐transfectants (Fig. 1E and Supporting Fig. S2B). Further investigations using an in vivo xenograft model showed that the size of xenografts derived from siUCID‐transfected tumor cells significantly decreased (Fig. 1F, left panel). Most importantly, intratumoral injection of siUCID suppressed the growth of HCC xenografts (Fig. 1F, right panel). These results suggest that the up‐regulation of lnc‐UCID may promote tumor cell growth, and targeting lnc‐UCID may represent a potential therapeutic strategy.
LNC‐UCID Promotes G1/S Transition by Up‐Regulating CDK6
The function of lnc‐UCID in cell‐cycle progression was examined next. As shown, lnc‐UCID knockdown caused the marked accumulation of the G1 population in nocodazole‐synchronized cells (Fig. 2A). Serum starvation–stimulation assays further revealed that most of the siUCID transfectants remained in the G1 phase, whereas a large proportion of the NC transfectants had already entered the S phase after serum re‐addition (Fig. 2B). Consistently, silencing lnc‐UCID decreased the mRNA levels of the S phase genes (Fig. 2C) that are transactivated by E2F family members and essential for S‐phase onset, and subsequently abrogated DNA replication (Fig. 2D). These results suggest that lnc‐UCID may promote G1/S transition and cell proliferation.
Figure 2.
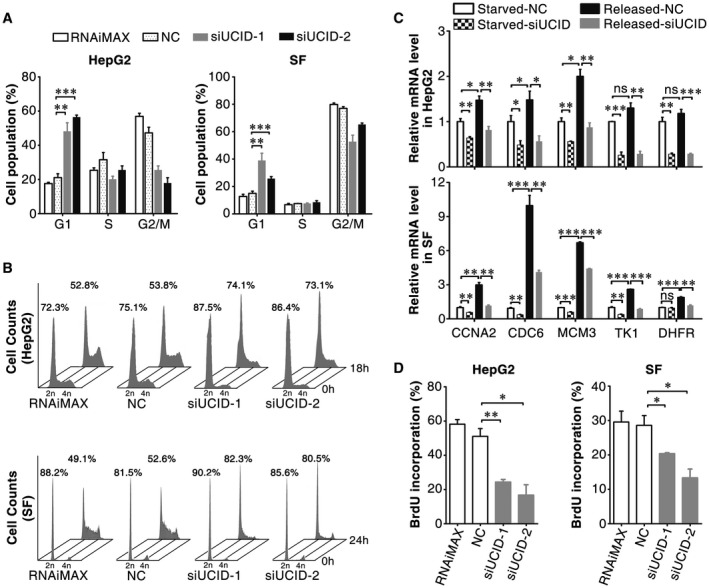
Lnc‐UCID knockdown inhibits G1/S transition. (A) Lnc‐UCID silencing induced a substantial increase in the G1 population. NC‐transfected or siUCID‐transfected cells were synchronized with nocodazole before FACS analysis. (B) Lnc‐UCID silencing blocked S‐phase entry after serum stimulation. NC‐transfectants or siUCID‐transfectants were serum‐deprived for 48 hours, followed by serum re‐addition; then, the cells were harvested at the indicated time points. The time point when serum was re‐added was set as 0 hours. (C) Lnc‐UCID knockdown inhibited the expression of S‐phase genes. NC‐transfectants or siUCID‐transfectants were serum‐deprived (Starved) for 48 hours and then maintained in culture medium with 15% FBS (Released) for 16 (HepG2) or 12 (SF) hours. (D) Lnc‐UCID knockdown reduced DNA replication. For (A), (C) and (D), error bars represent mean ± SEM from three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
The pRb pathway is the master controller of G1/S transition. Lnc‐UCID knockdown caused a prominent decrease in phosphorylated pRb protein level (Fig. 3A). The levels of the key regulators of pRb phosphorylation, including cyclin D, cyclin E, cyclin A, CDK4, CDK6, E2F1, p15 and p16, were therefore examined after lnc‐UCID silencing. CDK6 and cyclin A2 protein levels were reduced in siUCID transfectants (Fig. 3B and Supporting Fig. S3A). Cyclin A was a downstream target of CDK6‐pRB‐E2Fs axis23, we therefore focused on the regulation of CDK6 by lnc‐UCID. Both the protein (Fig. 3B) and mRNA (Fig. 3C) levels of CDK6 were reduced in siUCID transfectants. Lnc‐UCID knockdown shortened the half‐life of CDK6 mRNA (Fig. 3D) and reduced the proportion of CDK6 mRNA present in the polyribosomal fraction (Fig. 3E and Supporting Fig. S3B), indicating the posttranscriptional regulation of CDK6 by lnc‐UCID. Furthermore, CDK6 was also up‐regulated in HCC (Supporting Fig. S3C), and the CDK6 level was positively correlated with the lnc‐UCID level (Fig. 3F). As expected, CDK6 silencing increased the G1 population, reduced DNA replication, and inhibited cell growth (Supporting Fig. S4A‐D), which phenocopied the outcome of lnc‐UCID silencing.
Figure 3.
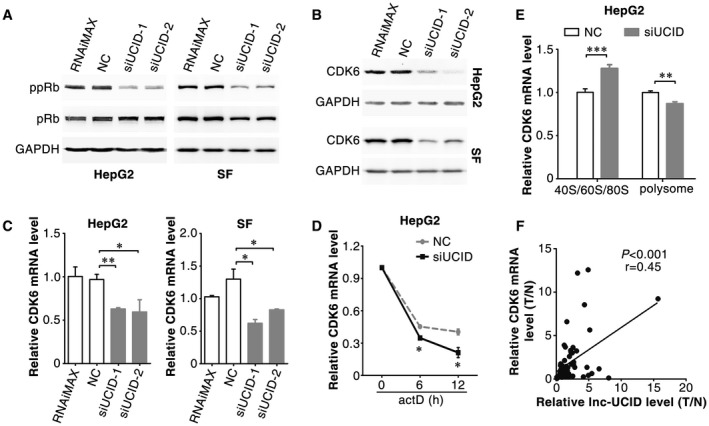
Silencing lnc‐UCID post‐transcriptionally down‐regulates CDK6 expression. (A) Lnc‐UCID knockdown reduced the level of phosphorylated pRb. (B,C) Lnc‐UCID knockdown reduced the protein and mRNA levels of CDK6. Forty‐eight hours after transfection with NC or siUCID, the protein (A,B) and mRNA (C) levels of genes were assessed by western blotting and quantitative real‐time PCR, respectively. (D) Lnc‐UCID knockdown reduced the stability of CDK6 mRNA. NC‐transfected or siUCID‐transfected cells were treated with 5 μg/mL actinomycin D for the indicated times. (E) Lnc‐UCID knockdown reduced the proportion of CDK6 mRNA in the polysome fraction. For (C) and (E), error bars represent mean ± SEM from three independent experiments. (F) CDK6 levels were positively correlated with lnc‐UCID levels. The RNA levels of CDK6 and lnc‐UCID were detected in 62 paired HCC and nontumor liver tissues by quantitative real‐time PCR. *P < 0.05; **P < 0.01; ***P < 0.001. Abbreviation: ppRb, S780‐phosphorylated pRb.
To perform gain‐of‐function studies, the full‐length transcript of lnc‐UCID was determined by 5′‐RACE and 3′‐RACE. As shown, lnc‐UCID was a polyadenylated RNA containing a 5′‐cap structure (Supporting Fig. S5A,B) and 1201 nucleotides (Supporting Fig. S5C) and had no protein‐coding capacity (Supporting Fig. S5D,E). Lnc‐UCID overexpression increased the protein (Fig. 4A) and mRNA (Fig. 4B) levels of CDK6, reduced the G1 population (Fig. 4C), and increased DNA replication and cell numbers (Fig. 4D); these effects phenocopied CDK6 overexpression. Furthermore, silencing CDK6 abrogated the stimulatory effect of lnc‐UCID overexpression on DNA replication and cell growth (Fig. 4E), whereas overexpressing CDK6 rescued the siUCID‐induced reductions in DNA replication and cell growth (Fig. 4F). These data suggest that lnc‐UCID may promote G1/S transition and cell proliferation by enhancing CDK6 expression.
Figure 4.
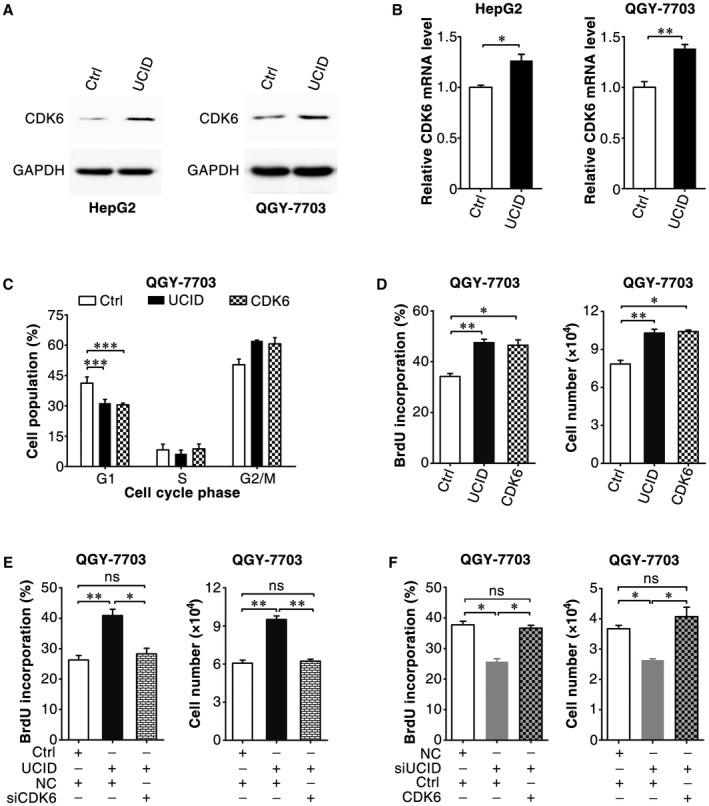
Lnc‐UCID overexpression promotes G1/S transition by enhancing CDK6 expression. (A,B) Ectopic lnc‐UCID expression increased the protein and mRNA levels of CDK6. HepG2 or QGY‐7703 cells were transfected with the pc3‐puro (Ctrl) or pc3‐puro‐UCID plasmid for 24 hours, followed by culture with puromycin for 48 hours; then, the cells were subjected to western blotting (A) or quantitative real‐time PCR (B) analysis. (C) Ectopic lnc‐UCID and CDK6 expression promoted G1/S transition. pc3‐gab (Ctrl), pc3‐gab‐UCID, and pc3‐gab‐CDK6‐transfected cells were synchronized with nocodazole before FACS analysis. (D) Ectopic lnc‐UCID and CDK6 expression increased DNA replication and promoted cell growth. (E) Silencing CDK6 abrogated the lnc‐UCID‐promoted DNA replication and cell growth. (F) CDK6 expression rescued the siUCID‐induced reductions in DNA replication and cell growth. For (D)‐(F), QGY‐7703 cells were transfected twice at 24‐hour intervals with the indicated plasmids. Twenty‐four hours after the last transfection, the cells were reseeded, transfected with the indicated siRNA duplex (E,F), and then subjected to BrdU incorporation or cell counting assays (D‐F). For cell counting assay, 1 × 104 transfected QGY‐7703 cells were seeded on day 0 and counted on day 4. For (B)‐(F), error bars represent mean ± SEM from three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
LNC‐UCID is Associated With DHX9 Through its 3′‐End
To explore how lnc‐UCID up‐regulated CDK6, RNA pulldown assays were used to identify lnc‐UCID‐associated proteins. DHX9, an RNA helicase with RNA‐binding activity, was identified in the protein complexes pulled down by lnc‐UCID but not in those pulled down by the antisense (AS) RNA of lnc‐UCID (lnc‐UCID‐AS was used as a negative control; Fig. 5A,B and Supporting Fig. S6A). RNA immunoprecipitation (RIP) assay was performed to further confirm the in vivo interaction between lnc‐UCID and DHX9. Compared with the IgG‐control complex, considerably more lnc‐UCID, but not the negative control GAPDH (glyceraldehyde 3‐phosphate dehydrogenase), was detected in the anti‐DHX9‐precipitated complex (Fig. 5C and Supporting Fig. S6B). Subsequent RNA pulldown assays using different fragments of lnc‐UCID as probes showed that DHX9 was pulled down by the fragments containing the 850‐1030‐nucleotide (nt) region of lnc‐UCID, but not by those without this domain; these results revealed 850‐1030‐nt to be the core sequence for lnc‐UCID binding to DHX9 (designated as lnc‐UCID‐core, Fig. 5D). The association between DHX9 and lnc‐UCID‐core was further verified by RNA‐EMSA. A shifted band was detected when biotin‐labeled lnc‐UCID‐core was used as an RNA probe and incubated with purified Flag‐tagged DHX9 protein, whereas no shifted band was observed when biotin‐labeled 1‐184‐nt of the lnc‐UCID was added as a probe (Fig. 5E). These results indicate an association between DHX9 and the 850‐1030‐nt domain of lnc‐UCID.
Figure 5.
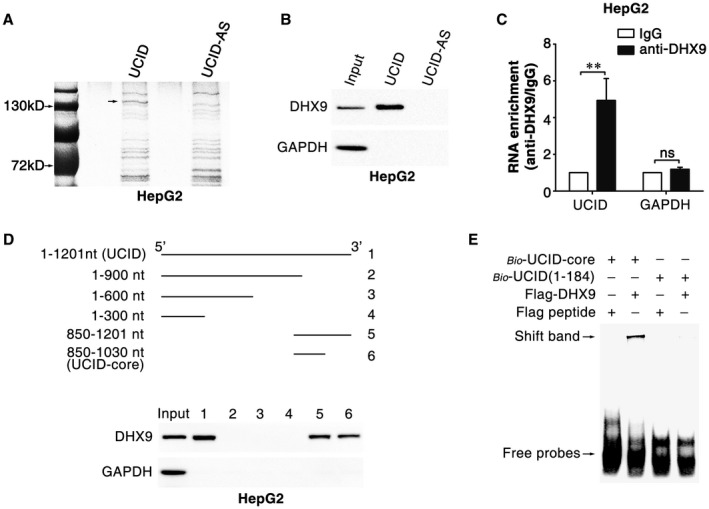
The 850‐1030‐nt region of lnc‐UCID is associated with DHX9. (A) Identification of the lnc‐UCID‐associated proteins by RNA pulldown assay. The proteins pulled down by lnc‐UCID or the antisense RNA of lnc‐UCID (lnc‐UCID‐AS, used as a NC) were resolved by SDS‐PAGE (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) and subjected to silver staining. A specific band was identified in the lnc‐UCID group and is marked with an arrow. (B) DHX9 was enriched in the protein complexes pulled down by lnc‐UCID. HepG2 cell proteins were pulled down with lnc‐UCID or lnc‐UCID‐AS and then subjected to western blotting. (C) Lnc‐UCID was associated with DHX9 in vivo. HepG2 cells were subjected to RIP assays with a DHX9 antibody or isotype‐matched control IgG, and the amount of lnc‐UCID in the precipitates was analyzed by quantitative real‐time PCR analysis. RNA enrichment indicates the RNA levels of lnc‐UCID or GAPDH in the anti‐DHX9 precipitates relative to those in the IgG precipitates. Error bars represent mean ± SEM from three independent experiments. (D) DHX9 bound to the 850‐1030‐nt region of lnc‐UCID. HepG2 cell proteins were pulled down with a series of truncated lnc‐UCID mutants and then subjected to western blotting. For (B)‐(D), GAPDH was used as a negative control. (E) The interaction between DHX9 and lnc‐UCID‐core was verified by RNA‐EMSA assays. Flag peptide and the probe containing the 1‐184‐nt sequence of lnc‐UCID were used as negative controls. **P < 0.01.
DHX9 Binds to the CDK6‐3′‐Untranslated Region and Decreases CDK6 Expression
DHX9 has been shown to regulate the expression of genes at the transcriptional and translational levels. We found that DHX9 knockdown significantly increased the protein and mRNA levels of CDK6 (Fig. 6A), but had no impact on the level of CDK6 precursor mRNA (Supporting Fig. S7). Furthermore, silencing DHX9 dramatically prolonged the half‐life of CDK6 mRNA (Fig. 6B) and increased the proportion of CDK6 mRNA present in the polyribosomal fraction (Fig. 6C), indicating that DHX9 may negatively regulate CDK6 expression through a post‐transcriptional regulatory mechanism.
Figure 6.
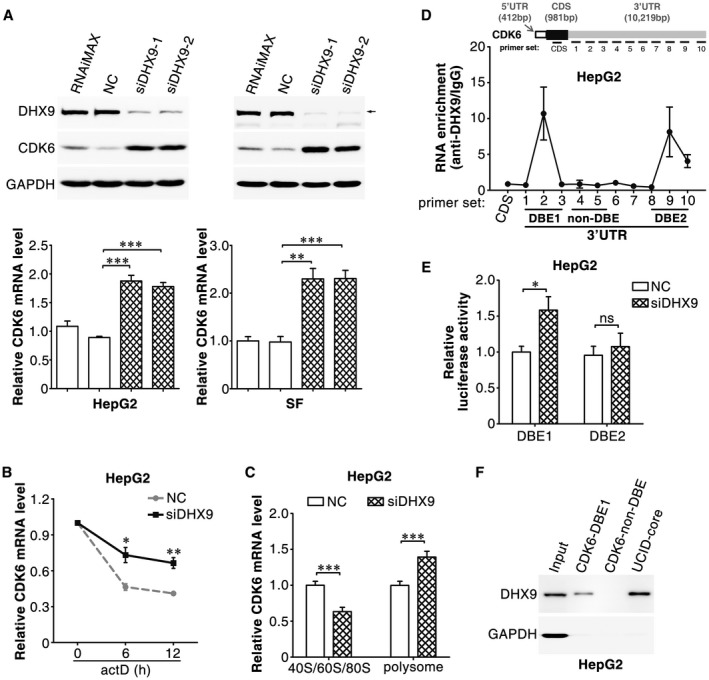
DHX9 post‐transcriptionally inhibits CDK6 expression by binding to the CDK6‐3′UTR. (A) DHX9 knockdown increased the protein and mRNA levels of CDK6. Forty‐eight hours after transfection with NC or siDHX9, the protein (upper panel) and mRNA (lower panel) levels of cellular CDK6 were assessed by western blotting and quantitative real‐time PCR, respectively. (B) DHX9 knockdown increased the stability of CDK6 mRNA. NC‐transfected or siDHX9‐transfected HepG2 cells were treated with 5 μg/mL actinomycin D for the indicated times. (C) DHX9 knockdown increased the proportion of CDK6 mRNA in the polysome fraction. (D) DHX9 was associated with the CDK6‐3′UTR. RIP assays were conducted with a DHX9 antibody or isotype‐matched control IgG, and the precipitated RNAs were detected by quantitative real‐time PCR using a series of primer sets covering the entire CDK6 mRNA. RNA enrichment indicates the levels of the indicated CDK6 mRNA fragments in the anti‐DHX9 precipitates relative to those in the IgG precipitates. (E) DHX9 knockdown enhanced the activity of the luciferase containing CDK6‐DBE1. NC or siDHX9 was co‐transfected with psi‐CDK6‐DBE1 or psi‐CDK6‐DBE2. For (A)‐(E), error bars represent mean ± SEM from three independent experiments. (F) DHX9 was associated with CDK6‐DBE1. RNA pulldown assays were performed with the indicated RNA probes. CDK6‐non‐DBE, a sequence between primer sets 4 and 5 that was not associated with DHX9 in the RIP assays, was used as a NC. UCID‐core, the 850‐1030‐nt region of lnc‐UCID that was associated with DHX9 in the RIP assays, was used as a positive control. *P < 0.05; **P < 0.01; ***P < 0.001.
To explore whether DHX9 could bind directly to CDK6 mRNA, RIP assays with a DHX9 antibody were performed and the amount of CDK6 mRNA was detected by quantitative real‐time PCR with a series of primers covering the entire CDK6 transcript. Compared with those from the IgG control, three fragments within the 3′‐untranslated region (UTR) of CDK6 mRNA, which were respectively detected by primer sets 2, 9 and 10, were significantly enriched in the anti‐DHX9 immunoprecipitates (Fig. 6D), suggesting that two regions that are located between primer sets 1‐3 (designated as DHX9‐binding element 1, CDK6‐DBE1) and between primer sets 8‐10 (CDK6‐DBE2) in the CDK6‐3′UTR may be associated with DHX9. Luciferase reporter assays were then used to validate whether DHX9 down‐regulated the expression of CDK6 by binding to these two elements. Notably, DHX9 knockdown increased the activity of the luciferase reporter containing the CDK6‐DBE1, but not the CDK6‐DBE2 element downstream of the Renilla luciferase gene (Fig. 6E). Consistently, RNA pulldown assays revealed that DHX9 was pulled down by CDK6‐DBE1 and lnc‐UCID‐core (Fig. 6F), but not by CDK6‐non‐DBE, which was located between primer sets 4 and 5 in the CDK6‐3′UTR (Fig. 6D). These results indicate that DHX9 may decrease the mRNA stability of CDK6 and inhibit CDK6 translation by binding to the CDK6‐3′UTR.
LNC‐UCID Promotes CDK6 Expression by Sequestering DHX9 From the CDK6‐3′UTR
These results showed that DHX9 interacted with lnc‐UCID and CDK6 mRNA, and that both lnc‐UCID and DHX9 regulated CDK6 expression. We therefore explored whether lnc‐UCID enhanced CDK6 expression by binding to DHX9. Significantly, DHX9 knockdown abrogated the siUCID‐induced CDK6 down‐regulation (Fig. 7A) and in turn abolished the siUCID‐induced G1 population accumulation (Fig. 7B). Moreover, the lnc‐UCID‐induced increases in CDK6 levels, DNA replication, and cell number were attenuated when its 850‐1030‐nt sequence, the core domain interacting with DHX9, was deleted (Fig. 7C,D and Supporting Fig. S8). These results suggest that lnc‐UCID may promote CDK6 expression and cell proliferation by associating with DHX9 through the 850‐1030‐nt at the 3′‐end.
Figure 7.
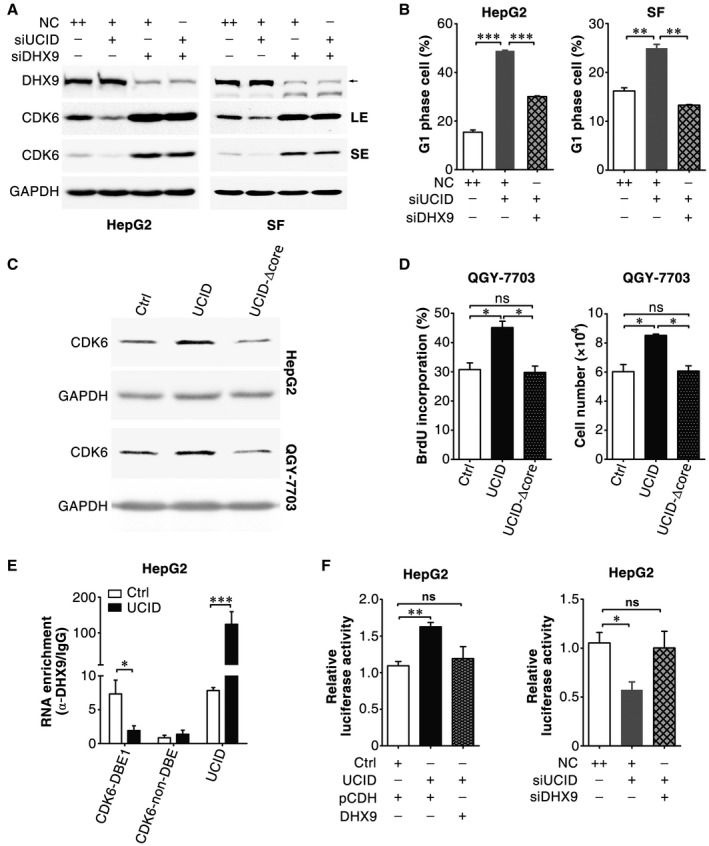
Lnc‐UCID promotes CDK6 expression and cell proliferation by sequestering DHX9 from the CDK6‐3′UTR. (A) DHX9 knockdown abrogated the siUCID‐induced down‐regulation of CDK6. HepG2 or SF cells were co‐transfected with the indicated siRNAs for 48 hours. (B) DHX9 knockdown abrogated the siUCID‐induced G1 population accumulation. The siRNA‐transfected cells were synchronized with nocodazole before FACS analysis. (C) The lnc‐UCID‐induced up‐regulation of CDK6 was attenuated when its 850‐1030‐nt sequence was deleted. HepG2 or QGY‐7703 cells were transfected with the indicated plasmid for 24 hours, cultured with puromycin for 48 hours, and then subjected to western blotting. (D) The lnc‐UCID‐induced increases in DNA replication and cell growth were abrogated when the 850‐1030‐nt sequence was deleted. QGY‐7703 cells were transfected twice with the indicated plasmids at 24‐hour intervals. Twenty‐four hours after the last transfection, the cells were subjected to BrdU incorporation or cell counting assays. (E) Lnc‐UCID overexpression diminished the association of DHX9 with CDK6‐DBE1. HepG2 cells were transfected twice at 24‐hour intervals with the indicated plasmids and then subjected to RIP assays 48 hours after the last transfection. RIP assays were conducted with a DHX9 antibody or isotype‐matched control IgG, and the precipitated RNAs were detected by quantitative real‐time PCR. RNA enrichment indicates the levels of CDK6‐DBE1, CDK6‐non‐DBE, or lnc‐UCID in the anti‐DHX9 precipitates relative to those in the IgG precipitates. (F) Expression of DHX9 abolished the lnc‐UCID‐increased luciferase activity of the CDK6‐DBE1‐containing reporter, and silencing DHX9 rescued the siUCID‐reduced luciferase activity of this reporter. HepG2 cells were co‐transfected with psi‐CDK6‐DBE1 and the indicated plasmids or siRNA. For (C)‐(F), Ctrl, pc3‐puro was used as a NC. *P < 0.05; ***P < 0.01; ***P < 0.001. Abbreviations: LE, long exposure; SE, short exposure; UCID, pc3‐puro‐lnc‐UCID containing the full‐length lnc‐UCID; and UCID‐Δcore, pc3‐puro‐lnc‐UCID‐Δcore containing mutant lnc‐UCID with the 850‐1030‐nt deletion.
Lnc‐UCID and DHX9 were present in both the cytoplasm and nucleus (Supporting Fig. S9A,B). Neither the subcellular localization (Supporting Fig. S9B) nor the protein level (Supporting Fig. S9C) of DHX9 was changed when lnc‐UCID was knocked down. We next investigated whether lnc‐UCID bound competitively to DHX9 and sequestered it from CDK6 mRNA. RIP assays showed that CDK6‐DBE1 and lnc‐UCID, but not CDK6‐non‐DBE, were significantly enriched in the anti‐DHX9‐precipitated complexes, whereas lnc‐UCID overexpression significantly decreased the enrichment of CDK6‐DBE1 in the DHX9 precipitates (Fig. 7E). Furthermore, expression of DHX9 abolished the lnc‐UCID‐increased luciferase activity of the CDK6‐DBE1‐containing reporter (Fig. 7F, left panel), whereas silencing DHX9 rescued the siUCID‐reduced luciferase activity of this reporter (Fig. 7F, right panel). These results suggest that lnc‐UCID may disrupt the association between DHX9 and the CDK6‐3′UTR.
MIR‐148A Directly Binds and Down‐Regulates LNC‐UCID
As shown above, gene amplification may represent one of the mechanisms responsible for lnc‐UCID up‐regulation in some HCC tissues (Fig. 1A,B). We further explored whether other mechanisms exist. miR‐148a, a tumor‐suppressive microRNA,24, 25 was predicted to bind to the 407‐428‐nt region of lnc‐UCID by using the TargetScan database (Supporting Fig. S10). Restoration of miR‐148a reduced the lnc‐UCID level (Fig. 8A, left panel), whereas antagonism of miR‐148a increased the lnc‐UCID expression (Fig. 8A, right panel). A dual‐luciferase reporter assay showed that miR‐148a expression significantly attenuated the luciferase activity of the reporter with wild‐type lnc‐UCID, but not that of the mutant reporter (Fig. 8B and Supporting Fig. S10). Furthermore, the expression of miR‐148a was frequently down‐regulated (Fig. 8C) and was negatively correlated with lnc‐UCID level in HCC tissues (Fig. 8D). Consistently, overexpression of miR‐148a reduced DNA replication and cell growth of hepatoma cells (Supporting Fig. S11A,B), which phenocopied the effect of lnc‐UCID silencing. These results suggest that up‐regulation of lnc‐UCID may also result from down‐regulation of miR‐148a in HCC.
Figure 8.
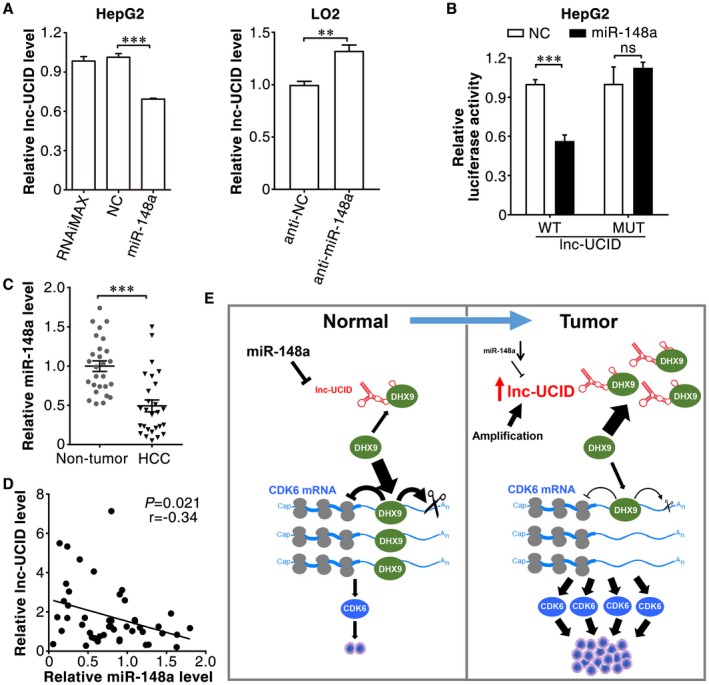
miR‐148a directly binds and down‐regulates lnc‐UCID. (A) Ectopic expression of miR‐148a or antagonism of cellular miR‐148a affected lnc‐UCID expression. Forty‐eight hours after transfection with the indicated RNA oligoribonucleotides, HepG2 cells were analyzed by quantitative real‐time PCR. (B) miR‐148a suppressed Renilla luciferase activity of the reporters containing the wild‐type but not mutant lnc‐UCID. WT, psi‐UCID‐WT; MUT, psi‐UCID‐MUT. (C) miR‐148a expression was significantly decreased in HCC tissues. miR‐148a expression was analyzed in 27 paired HCC and adjacent nontumor liver tissues by quantitative real‐time PCR. (D) Expression of miR‐148a was negatively correlated with lnc‐UCID level. Statistical analysis was performed using Pearson's correlation coefficient. (E) Model of the miR‐148a/lnc‐UCID/DHX9/CDK6 axis and its role in cell proliferation and tumorigenesis. **P < 0.01; ***P < 0.001. Abbreviations: MUT, mutant; and WT, wild type.
Taken together, amplification of the lnc‐UCID gene and down‐regulation of miR‐148a may lead to increased expression of lnc‐UCID, which binds to DHX9 and abrogates its inhibition on CDK6 expression, resulting in up‐regulation of CDK6 and in turn uncontrolled cell proliferation (Fig. 8E).
Discussion
LncRNAs belong to a class of identified RNA molecules, and their functions remain largely unknown. In this study, we identified an oncogenic lncRNA that was frequently up‐regulated in HCC tissues and promoted G1/S transition and tumor growth by up‐regulating CDK6 expression.
The deregulation of cell cycle progression, particularly the G1/S transition, is a common feature of cancer cells, and targeting cell cycle pathways has been considered a promising strategy for cancer therapy.6, 26 CDK6 is the key driver of G1/S transition and cell division,7 and its expression and activity are tightly controlled. Cyclin D and CDK inhibitors have been identified as critical regulators of CDK6 activity,6, 7 and the expression of CDK6 is regulated by transcription factors such as SP1 and PU.1,27, 28 and by miRNAs like miR‐195,29 miR‐26,20 and miR‐107.30 Recent studies indicate that lncRNAs may also regulate CDK6 expression. It has been shown that a lncRNA from Chinese hamster cells called gadd7 binds to TAR DNA‐binding protein (TDP‐43) and disrupts the interaction between TDP‐43 and CDK6 mRNA, thus resulting in the degradation of CDK6 mRNA31; the lncRNA MYU forms a complex with hnRNPK and stabilizes CDK6 expression in colon cancer cells.32 Here, we identified lnc‐UCID as a CDK6 regulator, which physically bound to DHX9 and sequestered DHX9 from CDK6 3′UTR, thereby preventing DHX9‐mediated CDK6 down‐regulation.
DHX9 is a helicase that binds to double‐stranded nucleic acids and unwinds the complex structure of DNA/RNA. It also interacts with proteins to function as a molecular scaffold or transcription coactivator.33, 34 DHX9 appears to play a central role in many cellular processes, including the regulation of DNA replication, gene transcription and translation, microRNA biogenesis, RNA processing and transport, and genome stability maintenance.35, 36, 37 It has been reported that DHX9 regulates gene expression by binding to 5′UTR to unwind its structure.38 Here, we found that DHX9 negatively regulated CDK6 expression by binding to a CDK6‐DBE1 region in the CDK6‐3′UTR, resulting in the decreased stability and reduced polyribosome incorporation of CDK6 mRNA. We supposed that DHX9 might unwind the structure of CDK6‐3′UTR, which led to mRNA instability or made CDK6‐3′UTR easier to be targeted by other regulatory molecules, such as microRNAs.
DHX9 is overexpressed in different types of cancer, including HCC (Supporting Fig. S12A). However, whether DHX9 possesses growth stimulating or inhibiting activity remains controversial. DHX9 can stimulate the expression of oncogenes, including CCND139 and IGF2,40 but it also enhances the expression of tumor suppressors, such as p1641 and p53.42 We found that DHX9 knockdown increased CDK6 levels but reduced IGF2 levels (Supporting Fig. S12B). Nevertheless, lnc‐UCID had no impact on the expression of p16, CCND1, and IGF2 (Supporting Fig. S3A, S12C) in our cell models. Hence, lnc‐UCID up‐regulation may facilitate HCC development by specifically abolishing the inhibitory effects of DHX9 on CDK6 expression. Consistently, the expression of lnc‐UCID was positively correlated with DHX9 level in HCC tissues (Supporting Fig. S12D), indicating that the co‐up‐regulation of lnc‐UCID may be essential for maintaining a high level of CDK6 in the DHX9‐highly expressed HCC cells. The regulatory network of DHX9 appears to be complex and may be highly dependent on the cellular context. Further research is warranted to delineate the mechanistic consequences of DHX9 in tumorigenesis.
Amplification of chromosome 1q is one of the most frequent genetic events in HCC.19, 43 In particular, the gain of chromosome 1q21‐23 has been associated with the early development of HCC.44 Few oncogenic protein‐coding genes on this region, including chromodomain helicase/ATPase DNA‐binding protein 1‐like, CDC28 protein kinase regulatory subunit 1B, SHC‐transforming protein 1, and jumping translocation breakpoint, have been identified to contribute to hepatocarcinogenesis.19, 43 In this study, we identified lnc‐UCID as a potential oncogene that was located at chromosome 1q22 and frequently amplified in HCC. Furthermore, miR‐148a, whose down‐regulation was associated with the increase of lnc‐UCID in HCC tissues, directly bound and down‐regulated lnc‐UCID. Therefore, up‐regulation of lnc‐UCID in HCC may result from amplification of its gene locus and down‐regulation of miR‐148a.
The levels of lnc‐UCID were similar, regardless of the hepatitis B virus (HBV) infection and cirrhosis status of HCC patients, suggesting that HBV infection and cirrhosis were not responsible for lnc‐UCID up‐regulation in HCC (Supporting Fig. S13A). Interestingly, lnc‐UCID was also increased in colorectal cancer tissues (Supporting Fig. S13B). However, its level was significantly lower in cholangiocarcinoma (CCA) tissues, compared with nontumor livers (Supporting Fig. S13C). Moreover, lnc‐UCID expression in two CCA cell lines was lower than all four HCC cell lines examined (Supporting Fig. S13D). These results indicate that the expression and function of lnc‐UCID may be tissue or cancer type specific. Consistent with our findings, the gene‐expression profiles of HCC and CCA are largely different,45 and it has been shown that fatty acid synthase, glypican‐3, miR‐221, and miR‐222 increase in HCC but decrease in CCA.46, 47, 48
In summary, we characterize lnc‐UCID as an oncogenic lncRNA that is up‐regulated in HCC and functions as a molecular sponge of DHX9 to up‐regulate CDK6 expression, and in turn promotes G1/S transition, consequently resulting in HCC development. We also detail the mechanisms of lnc‐UCID up‐regulation and identify a function of DHX9 in regulating CDK6 expression. Our findings suggest the pivotal role of lnc‐UCID in cell cycle control and tumorigenesis, and implicate the miR‐148a‐lnc‐UCID‐DHX9‐CDK6 axis as a potential target for anticancer therapy.
Supporting information
Supported by the National Key R&D Program of China (2017YFA0504402); National Natural Science Foundation of China (91440205, 81572400, 31701256, and 31771554); Natural Science Foundation of Guangdong Province (2014A030311031); and Fundamental Research Funds for the Central Universities (171gjc32).
Potential conflict of interest: Nothing to report.
Contributor Information
Ying Zhu, Email: zhuy68@mail.sysu.edu.cn.
Shi‐Mei Zhuang, Email: zhuangshimei@163.com.
References
Author names in bold designate shared co‐first authorship.
- 1. St. Laurent G, Wahlestedt C, Kapranov P. The landscape of long noncoding RNA classification. Trends Genet 2015;31:239‐251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell 2009;136:629‐641. [DOI] [PubMed] [Google Scholar]
- 3. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell 2011;43:904‐914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Song Y, Liu C, Liu X, Trottier J, Beaudoin M, Zhang L, et al. H19 promotes cholestatic liver fibrosis by preventing ZEB1‐mediated inhibition of epithelial cell adhesion molecule. Hepatology 2017;66:1183‐1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell 2016;29:452‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer 2017;17:93‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 2013;14:518‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tseng YY, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H, et al. PVT1 dependence in cancer with MYC copy‐number increase. Nature 2014;512:82‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fu WM, Zhu X, Wang WM, Lu YF, Hu BG, Wang H, et al. Hotair mediates hepatocarcinogenesis through suppressing miRNA‐218 expression and activating P14 and P16 signaling. J Hepatol 2015;63:886‐895. [DOI] [PubMed] [Google Scholar]
- 10. Lu Y, Hu Z, Mangala LS, Stine ZE, Hu X, Jiang D, et al. MYC targeted long noncoding RNA DANCR promotes cancer in part by reducing p21 levels. Cancer Res 2018;78:64‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tang J, Xie Y, Xu X, Yin Y, Jiang R, Deng L, et al. Bidirectional transcription of Linc00441 and RB1 via H3K27 modification‐dependent way promotes hepatocellular carcinoma. Cell Death Dis 2017;8:e2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cao C, Sun J, Zhang D, Guo X, Xie L, Li X, et al. The long intergenic noncoding RNA UFC1, a target of MicroRNA 34a, interacts with the mRNA stabilizing protein HuR to increase levels of beta‐catenin in HCC cells. Gastroenterology 2015;148:415‐426 .e418. [DOI] [PubMed] [Google Scholar]
- 13. Heimbach JK, Kulik LM, Finn RS, Sirlin CB, Abecassis MM, Roberts LR, et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018;67:358‐380. [DOI] [PubMed] [Google Scholar]
- 14. Wong CM, Tsang FH, Ng IO. Non‐coding RNAs in hepatocellular carcinoma: molecular functions and pathological implications. Nat Rev Gastroenterol Hepatol 2018;15:137‐151. [DOI] [PubMed] [Google Scholar]
- 15. Zhang J, Li Z, Liu L, Wang Q, Li S, Chen D, et al. Long noncoding RNA TSLNC8 is a tumor suppressor that inactivates the interleukin‐6/STAT3 signaling pathway. Hepatology 2018;67:171‐187. [DOI] [PubMed] [Google Scholar]
- 16. Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF, et al. A long noncoding RNA activated by TGF‐beta promotes the invasion‐metastasis cascade in hepatocellular carcinoma. Cancer Cell 2014;25:666‐681. [DOI] [PubMed] [Google Scholar]
- 17. Wang Y, He L, Du Y, Zhu P, Huang G, Luo J, et al. The long noncoding RNA lncTCF7 promotes self‐renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell 2015;16:413‐425. [DOI] [PubMed] [Google Scholar]
- 18. Yuan SX, Wang J, Yang F, Tao QF, Zhang J, Wang LL, et al. Long noncoding RNA DANCR increases stemness features of hepatocellular carcinoma by derepression of CTNNB1. Hepatology 2016;63:499‐511. [DOI] [PubMed] [Google Scholar]
- 19. Shibata T, Aburatani H. Exploration of liver cancer genomes. Nat Rev Gastroenterol Hepatol 2014;11:340‐349. [DOI] [PubMed] [Google Scholar]
- 20. Zhu Y, Lu Y, Zhang Q, Liu JJ, Li TJ, Yang JR, et al. MicroRNA‐26a/b and their host genes cooperate to inhibit the G1/S transition by activating the pRb protein. Nucleic Acids Res 2012;40:4615‐4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Su H, Yang JR, Xu T, Huang J, Xu L, Yuan Y, et al. MicroRNA‐101, down‐regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Res 2009;69:1135‐1142. [DOI] [PubMed] [Google Scholar]
- 22. Ding J, Huang S, Wu S, Zhao Y, Liang L, Yan M, et al. Gain of miR‐151 on chromosome 8q24.3 facilitates tumour cell migration and spreading through downregulating RhoGDIA. Nat Cell Biol 2010;12:390‐399. [DOI] [PubMed] [Google Scholar]
- 23. Bracken AP, Ciro M, Cocito A, Helin K. E2F target genes: unraveling the biology. Trends Biochem Sci 2004;29:409‐417. [DOI] [PubMed] [Google Scholar]
- 24. Li L, Liu Y, Guo Y, Liu B, Zhao Y, Li P, et al. Regulatory MiR‐148a‐ACVR1/BMP circuit defines a cancer stem cell‐like aggressive subtype of hepatocellular carcinoma. Hepatology 2015;61:574‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang JP, Zeng C, Xu L, Gong J, Fang JH, Zhuang SM. MicroRNA‐148a suppresses the epithelial‐mesenchymal transition and metastasis of hepatoma cells by targeting Met/Snail signaling. Oncogene 2014;33:4069‐4076. [DOI] [PubMed] [Google Scholar]
- 26. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009;9:153‐166. [DOI] [PubMed] [Google Scholar]
- 27. Choe KS, Ujhelly O, Wontakal SN, Skoultchi AI. PU.1 directly regulates cdk6 gene expression, linking the cell proliferation and differentiation programs in erythroid cells. J Biol Chem 2010;285:3044‐3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cram EJ, Liu BD, Bjeldanes LF, Firestone GL. Indole‐3‐carbinol inhibits CDK6 expression in human MCF‐7 breast cancer cells by disrupting Sp1 transcription factor interactions with a composite element in the CDK6 gene promoter. J Biol Chem 2001;276:22332‐22340. [DOI] [PubMed] [Google Scholar]
- 29. Xu T, Zhu Y, Xiong Y, Ge YY, Yun JP, Zhuang SM. MicroRNA‐195 suppresses tumorigenicity and regulates G1/S transition of human hepatocellular carcinoma cells. Hepatology 2009;50:113‐121. [DOI] [PubMed] [Google Scholar]
- 30. Bohlig L, Friedrich M, Engeland K. p53 activates the PANK1/miRNA‐107 gene leading to downregulation of CDK6 and p130 cell cycle proteins. Nucleic Acids Res 2011;39:440‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu X, Li D, Zhang W, Guo M, Zhan Q. Long non‐coding RNA gadd7 interacts with TDP‐43 and regulates Cdk6 mRNA decay. EMBO J 2012;31:4415‐4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kawasaki Y, Komiya M, Matsumura K, Negishi L, Suda S, Okuno M, et al. MYU, a target lncRNA for Wnt/c‐Myc signaling, mediates induction of CDK6 to promote cell cycle progression. Cell Rep 2016;16:2554‐2564. [DOI] [PubMed] [Google Scholar]
- 33. Anderson SF, Schlegel BP, Nakajima T, Wolpin ES, Parvin JD. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat Genet 1998;19:254‐256. [DOI] [PubMed] [Google Scholar]
- 34. Nakajima T, Uchida C, Anderson SF, Lee CG, Hurwitz J, Parvin JD, et al. RNA helicase A mediates association of CBP with RNA polymerase II. Cell 1997;90:1107‐1112. [DOI] [PubMed] [Google Scholar]
- 35. Jain A, Bacolla A, Del Mundo IM, Zhao J, Wang G, Vasquez KM. DHX9 helicase is involved in preventing genomic instability induced by alternatively structured DNA in human cells. Nucleic Acids Res 2013;41:10345‐10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Robb GB, Rana TM. RNA helicase A interacts with RISC in human cells and functions in RISC loading. Mol Cell 2007;26:523‐537. [DOI] [PubMed] [Google Scholar]
- 37. Chen ZX, Wallis K, Fell SM, Sobrado VR, Hemmer MC, Ramskold D, et al. RNA helicase A is a downstream mediator of KIF1Bbeta tumor‐suppressor function in neuroblastoma. Cancer Discov 2014;4:434‐451. [DOI] [PubMed] [Google Scholar]
- 38. Hartman TR, Qian S, Bolinger C, Fernandez S, Schoenberg DR, Boris‐Lawrie K. RNA helicase A is necessary for translation of selected messenger RNAs. Nat Struct Mol Biol 2006;13:509‐516. [DOI] [PubMed] [Google Scholar]
- 39. Huo L, Wang YN, Xia W, Hsu SC, Lai CC, Li LY, et al. RNA helicase A is a DNA‐binding partner for EGFR‐mediated transcriptional activation in the nucleus. Proc Natl Acad Sci U S A 2010;107:16125‐16130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu M, Roth A, Yu M, Morris R, Bersani F, Rivera MN, et al. The IGF2 intronic miR‐483 selectively enhances transcription from IGF2 fetal promoters and enhances tumorigenesis. Genes Dev 2013;27:2543‐2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Myohanen S, Baylin SB. Sequence‐specific DNA binding activity of RNA helicase A to the p16INK4a promoter. J Biol Chem 2001;276:1634‐1642. [DOI] [PubMed] [Google Scholar]
- 42. Halaby MJ, Harris BR, Miskimins WK, Cleary MP, Yang DQ. Deregulation of internal ribosome entry site‐mediated p53 translation in cancer cells with defective p53 response to DNA damage. Mol Cell Biol 2015;35:4006‐4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen L, Chan TH, Guan XY. Chromosome 1q21 amplification and oncogenes in hepatocellular carcinoma. Acta Pharmacol Sin 2010;31:1165‐1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Midorikawa Y, Yamamoto S, Tsuji S, Kamimura N, Ishikawa S, Igarashi H, et al. Allelic imbalances and homozygous deletion on 8p23.2 for stepwise progression of hepatocarcinogenesis. Hepatology 2009;49:513‐522. [DOI] [PubMed] [Google Scholar]
- 45. Chaisaingmongkol J, Budhu A, Dang H, Rabibhadana S, Pupacdi B, Kwon SM, et al. Common molecular subtypes among Asian hepatocellular carcinoma and cholangiocarcinoma. Cancer Cell 2017;32:57‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li L, Che L, Tharp KM, Park HM, Pilo MG, Cao D, et al. Differential requirement for de novo lipogenesis in cholangiocarcinoma and hepatocellular carcinoma of mice and humans. Hepatology 2016;63:1900‐1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Man XB, Tang L, Zhang BH, Li SJ, Qiu XH, Wu MC, et al. Upregulation of Glypican‐3 expression in hepatocellular carcinoma but downregulation in cholangiocarcinoma indicates its differential diagnosis value in primary liver cancers. Liver Int 2005;25:962‐966. [DOI] [PubMed] [Google Scholar]
- 48. Karakatsanis A, Papaconstantinou I, Gazouli M, Lyberopoulou A, Polymeneas G, Voros D. Expression of microRNAs, miR‐21, miR‐31, miR‐122, miR‐145, miR‐146a, miR‐200c, miR‐221, miR‐222, and miR‐223 in patients with hepatocellular carcinoma or intrahepatic cholangiocarcinoma and its prognostic significance. Mol Carcinog 2013;52:297‐303. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials