

Abstract
The application of modeling and simulation (M&S) tools to biological, physiological, and clinical data has great potential to enhance drug development and regulatory decision making. The strategic development of multidisciplinary projects aimed at integrating methodologies from different disciplines may bridge between preclinical and clinical drug development as well as between academic curiosity and clinical practice. Herein we review the history and present the state of M&S approaches as well as our vision for future challenges and applications.
History and Present State
M&S tools have long been used in engineering and aerospace industries to develop products that would be prohibitively expensive to optimize through iterative improvement of prototypes. Modern drug development is now adapting and integrating analogous tools based on information from all phases of the drug‐development process. The adoption of these tools has gained traction, particularly because it is neither cost‐effective nor time‐efficient to tackle all open questions experimentally. As a result, an increasing number of decisions in drug development are now based on M&S. As such, M&S has evolved from a research nicety in the 1970s1 to a mechanistic science for the prediction and extrapolation aimed at improving the quality, efficiency, and cost effectiveness of decision making during drug discovery, development, regulatory assessment, and life‐cycle management.2
This wide range of applications allows the use of M&S approaches at various levels of physiological and temporal complexity. The choice of the approach to be used should be governed by the complexity of the question to be answered (i.e., fit for purpose), not the other way around. For example, although systems pharmacology models have been increasingly employed in drug discovery and early development for target identification and lead optimization, population pharmacokinetic/pharmacodynamic (pharmacometric) models are typically used for dose selection and identification of intrinsic and extrinsic factors that warrant dose adjustment in patient subgroups and the optimization of trial design during clinical development.
All of these M&S approaches, irrespective of their complexity, are intended to reduce the uncertainty about the benefits and risks of a drug during the development and evaluation processes. Sheiner3 outlined in his learn‐and‐confirm paradigm the following three key questions that are intended to guide the iterative reduction of uncertainty: The only author listed in the reference list for this reference is Sheiner.
What do we want to know?
How confident do we want to be?
What are we willing to assume?
The application of this paradigm has had a lasting effect on the way that today's clinical trials are conducted. It has also fostered the development of standards and methodologies that are now routinely employed in drug development and regulatory decision making. These standards are most evolved for pharmacometric approaches, but regulatory guidances are either forthcoming or recently put in place for physiologically based and systems pharmacology approaches.4, 5 However, there is still a disconnection between (pre)clinical and real‐world patient scenarios, which can lead to unexpected adverse events or even the withdrawal of approved drugs from the market. This is, for example, because of the fact that some low‐frequency adverse events, even if serious, are hard if not impossible to study within the sample‐size constraints of clinical trials but surface in clinical practice. Other variables that are difficult to study in controlled trials include patient adherence, polypharmacy, or changes in the exposure–response relationship over time.
Challenges and Opportunities for the Future
In the authors’ opinion, the gap between bench, trial, and bedside can be bridged using combined physiologically based, pharmacometric, and pharmacoepidemiologic approaches. Overall, this integrated approach is based on an iterative learning process that alternates population‐based observational research and mechanistic modeling that essentially provide answers to the following three questions:
What are the clinically relevant sources of variability?
How much of this variability can be captured in controlled clinical trials?
How can the lessons learned be implemented into clinical practice in a cost‐effective fashion?
We would like to use the following section of the manuscript to illustrate the concept and to highlight the value added by this joint M&S and epidemiologic approach to make causal inferences using two examples.
The first example is a collaborative research project between the University of Florida's Center for Pharmacometrics and Systems Pharmacology, the Center for Drug Evaluation and Safety and the U.S. Food and Drug Administration's Office of Generic Drugs. The objective of this project was to develop a mechanism‐based and risk‐based strategy that systematically investigated postmarketing concerns of therapeutic inequivalence following the switch between brand and generic products. The jointly developed research strategy rested on the following three integrated pillars: (i) pharmacoepidemiologic methods applied to real‐world data, (ii) physiologically‐based absorption models, and (iii) population pharmacokinetic/pharmacodynamic models. The pharmacoepidemiologic approach highlighted clinically significant associations between certain generics and scenarios of reduced efficacy or adverse events. The results of the pharmacoepidemiologic analyses showed that signals indicating potential formulation issues altered overall utilization of generic medications, such as discontinuation and treatment switching as well as increased clinical event rates associated with both increased and decreased pharmacologic actions.6 Although methods to establish causal inferences were used, unmeasured confounding factors may produce bias, thus requiring a theoretical basis (biological plausibility). The hypothesis generated by the pharmacoepidemiologic approach was then evaluated by integrated physiologically‐based absorption and population pharmacokinetic/pharmacodynamic models to evaluate the biological, physiological, and drug‐related and/or formulation‐related causes of the observed reduced efficacy or adverse event–drug pair report. Physiologically‐based absorption modeling coupled with population pharmacokinetic/pharmacodynamic modeling can be used to investigate if realistic changes in pharmaceutical variables (e.g., variables affecting dissolution rate, intestinal motility, or primary and secondary active transport across the intestinal membrane) may result in the observed purported reduced efficacy or adverse events arising from generic substitution.7, 8 This strategy was applied retrospectively to a historic case example of extended‐release metoprolol as well as to antiepileptic drugs to establish the scientific approach (Figure 1) followed by a prospective application to direct acting oral anticoagulants to rank order the anticipated bleeding risk associated with soon‐to‐be marketed generic dabigatran, edoxaban, rivaroxaban, and apixaban.
Figure 1.
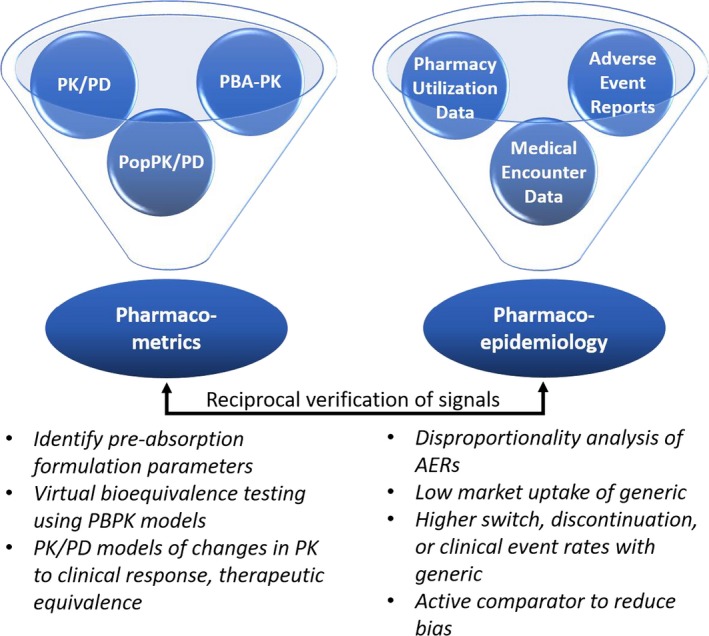
Integrated evidence pillars between pharmacometrics and pharmacoepidemiology for generic vs. brand‐name drug comparisons. Signals and parameters for bioequivalence can be identified and confirmed in a bidirectional workflow. Figure and legend modified with permission from Brown, J.D. et al.6 Real‐world data approaches for early detection of potential safety and effectiveness signals for generic substitution: a metoprolol extended‐release case study. J. Clin. Pharmacol. (2019). https://doi.org/doi10.1002/jcph.1436. AERs, adverse event reporting systems; PBA‐PK, physiologically‐based absorption pharmacokinetic; PBPK, physiologically‐based pharmacokinetics; PK, pharmacokinetics; PK/PD, pharmacokinetic/pharmacodynamic; PopPK/PD, population pharmacokinetics/pharmacodynamics.
The second example is another collaborative project of the Center for Pharmacometrics and Systems Pharmacology and the Center for Drug Evaluation and Safety in collaboration with the Bill & Melinda Gates Foundation with the objective to evaluate the impact of drug–drug interactions on the efficacy and safety of hormonal contraceptive agents.9 The employed research strategy rests on the integration of exposure data from physiologically based drug–drug interaction models, dose–response relationships derived from model‐based meta‐analysis, and real‐world pharmacoepidemiologic analyses that are informed by the drug–drug interaction models. Based on the projected strengths of the interactions between types (e.g., active ingredient and route) of hormonal contraceptives and select perpetrators (various enzyme inducers or inhibitors), pharmacoepidemiologic analyses allow the real‐world validation of these model‐based projections based on the observed comparative risk for unplanned pregnancy or venous‐thromboembolic events. These pharmacoepidemiologic studies are in turn enhanced by the integration of mechanistic pathways, thus increasing the biological plausibility of observed differences in effects and thus decreasing the chance for spurious findings (Figure 2). It is complemented by a pharmacoeconomic analysis to determine the associated costs.
Figure 2.
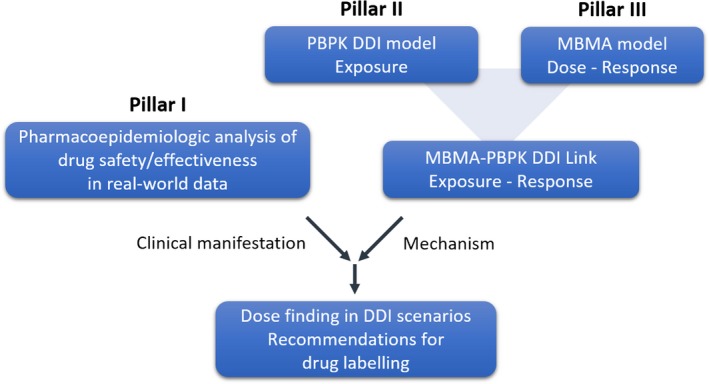
Integrated workflow for establishing a multidisciplinary framework to study DDIs of hormonal contraceptives. Signals related to altered efficacy (e.g., unintended pregnancies) or safety (e.g., venous thromboembolisms) can be detected by pharmacoepidemiologic analysis (pillar I) using real‐world data. The mechanistic plausibility of these signals is subsequently evaluated via physiologically‐based DDI models (pillar II). To be able to provide optimal dosing and labeling recommendations, an exposure–response analysis will be conducted by linking exposure in the presence and absence of DDIs to dose–response data derived from a model‐based meta‐analysis (pillar III). DDI, drug–drug interaction; PBPK, physiologically‐based pharmacokinetics; MBMA, model‐based meta‐analysis.
In both examples, a reciprocal research framework is established in which M&S and real‐world outcome approaches are used in an iterative learning process. That is, M&S approaches may help direct, confirm, and inspire the pharmacoepidemiologic design of drug safety or effectiveness evaluations in population‐level databases while the findings of such pharmacoepidemiologic studies can spark investigations of novel mechanistic pathways by M&S‐based scientists. Real‐world data have the potential to provide a conduit of external validation and supplementation of pharmacometric models to focus on specific populations or specific disease states or to identify high‐value drug targets for further study. The nature of these databases facilitates a shift to what may be more important considerations in the real‐world, and successful collaborations with pharmacoepidemiologists must consider the nature of the data being used. For example, prototypical perpetrators in drug–drug interaction studies (e.g., midazolam, rifampin) will not be prevalent enough in the real world to be studied and may be considered less clinically impactful from a population‐based perspective. Rather, perpetrators that are highly prevalent but perhaps with weaker drug–drug interaction profiles may be prioritized as they represent an overall larger absolute impact. Likewise, the extension of pharmacokinetic/pharmacodynamic and physiologically‐based pharmacokinetic models must consider the measurement of relevant population‐level clinical outcomes rather than more discrete pharmacological changes that are not captured in these databases, e.g., increased rate of heart attacks vs. increased heart rate variability.7, 8 By using a combined approach, the strength of evidence is greatly increased and elevates its impact on regulatory and clinical decision‐making with much greater certainty.
In summary, inasmuch as M&S approaches have strong internal validity through their mechanistic basis, they lack proven clinical validation, which in turn can be realized with real‐world pharmacoepidemiological analyses. In contrast, although pharmacoepidemiology has strong external validity, i.e., generalizability to the population effects, it is more prone to biases that threaten internal validity but that can be tested against mechanistic models. Similar to the collaborative research environment, the integration of real‐world evidence into part of the regulatory process offers many opportunities and challenges. One major obstacle that needs to be overcome for these highly collaborative projects, and potential subsequent regulatory activities, is the establishment of a clear understanding of how the individual objectives tie in to the overall research strategy. This must acknowledge that the measurement of exposures, outcomes, and the extrapolation of findings between these distinct sciences will not always be ideal, but lessons learned from each can strengthen and expand the other approach. In creating this collaborative environment, it allows us to emphasize the “pharmacology” in pharmacoepidemiology and brings a real‐world application to M&S approaches. Consequently, frequent and appropriate communication between stakeholders is key to success, and it is prime time for pharmaceutical scientists and pharmacoepidemiologists to come to the table for collaboration.
Funding
No funding was received for this work.
Conflict of Interest
The authors declared no competing interests for this work.
References
- 1. Sheiner, L.B. Computer‐aided long‐term anticoagulation therapy. Comput. Biomed. Res. 2, 507–518 (1969). [DOI] [PubMed] [Google Scholar]
- 2. Marshall, S.F. et al Good practices in model‐informed drug discovery and development: practice, application, and documentation. CPT Pharmacometrics Syst. Pharmacol. 5, 93–122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sheiner, L.B. Learning versus confirming in clinical drug development. Clin. Pharmacol. Ther. 61, 275–291 (1997). [DOI] [PubMed] [Google Scholar]
- 4. U.S. Food and Drug Administration. Guidance for Industry Population Pharmacokinetics (U.S. Department of Health and Human Services, U.S. Food and Drug Administration, Silver Spring, MD, 1999). [Google Scholar]
- 5. U.S. Food and Drug Administration. Physiologically Based Pharmacokinetic Analyses–Format and Content Guidance for Industry (U.S. Department of Health and Human Services, U.S. Food and Drug Administration, Silver Spring, MD, 2018). [Google Scholar]
- 6. Brown, J.D. et al Real‐world data approaches for early detection of potential safety and effectiveness signals for generic substitution: a metoprolol extended‐release case study. J. Clin. Pharmacol. 10.1002/jcph.1436 [DOI] [PubMed] [Google Scholar]
- 7. Basu, S. et al Physiologically‐based pharmacokinetic modeling to evaluate formulation factors influencing bioequivalence of metoprolol extended‐release products. J. Clin. Pharmacol. 10.1002/jcph.1017 [DOI] [PubMed] [Google Scholar]
- 8. Kim, S. et al Evaluating the clinical impact of formulation variability: a metoprolol extended‐release case study. J. Clin. Pharmacol. 10.1002/jcph.1433 [DOI] [PubMed] [Google Scholar]
- 9. Lesko, L.J. et al Establishing a multidisciplinary framework to study drug‐drug interactions of hormonal contraceptives: an invitation to collaborate. CPT Pharmacometrics Syst. Pharmacol. 7, 706–708 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]