

Abstract
We present SymPortal (SymPortal.org), a novel analytical framework and platform for genetically resolving the algal symbionts of reef corals using next‐generation sequencing (NGS) data of the ITS2 rDNA. Although the ITS2 marker is widely used to genetically characterize taxa within the family Symbiodiniaceae (formerly the genus Symbiodinium), the multicopy nature of the marker complicates its use. Commonly, the intragenomic diversity resultant from this multicopy nature is collapsed by analytical approaches, thereby focusing on only the most abundant sequences. In contrast, SymPortal employs logic to identify within‐sample informative intragenomic sequences, which we have termed ‘defining intragenomic variants' (DIVs), to identify ITS2‐type profiles representative of putative Symbiodiniaceae taxa. By making use of this intragenomic ITS2 diversity, SymPortal is able to resolve genetic delineations using the ITS2 marker at a level that was previously only possible by using additional genetic markers. We demonstrate this by comparing this novel approach to the most commonly used alternative approach for NGS ITS2 data, the 97% similarity clustering to operational taxonomic units (OTUs). The SymPortal platform accepts NGS raw sequencing data as input to provide an easy‐to‐use, standardization‐enforced, and community‐driven framework that integrates with a database to gain resolving power with increased use. We consider that SymPortal, in conjunction with ongoing large‐scale sampling and sequencing efforts, should play an instrumental role in making future sampling efforts more comparable and in maximizing their efficacy in working towards the classification of the global Symbiodiniaceae diversity.
Keywords: ITS2, molecular ecology, multicopy, phylogentics, Symbiodiniaceae, SymPortal
1. INTRODUCTION
Corals and the ecosystems they construct have been among the most susceptible to climate change, but corals have been shown to exhibit variation in their responses to stress (Hughes et al., 2018; Norstrom et al., 2016). One important factor that contributes to reef resilience is the genetic identity of the algal symbionts harboured by the reef‐building corals (Hume et al., 2016; Kemp, Hernandez‐Pech, Iglesias‐Prieto, Fitt, & Schmidt, 2014; LaJeunesse et al., 2010; Rowan, 2004; Rowan, Knowlton, Baker, & Jara, 1997; Silverstein, Cunning, & Baker, 2015; Thornhill, Howells, Wham, Steury, & Santos, 2017). Effective characterization of these algal symbionts, family Symbiodiniaceae (formerly genus Symbiodinium containing Clades A–I, now equivalent to family Symbiodiniaceae, currently with seven named genera; LaJeunesse et al., 2018), is therefore important.
Despite the multitude of markers available for assessing Symbiodiniaceae diversity, the internal transcribed spacer 2 (ITS2) of the rRNA gene shows an uninterrupted popularity and remains the most commonly used marker (Cunning, Gates, & Edmunds, 2017; Fujise et al., 2018; Pochon, Putnam, & Gates, 2014; Smith, Ketchum, & Burt, 2017b; Varasteh, Shokri, Rajabi‐Maham, Behzadi, & Hume, 2018). However, this marker is multicopy in nature, which complicates its use. A single Symbiodiniaceae cell usually contains hundreds to thousands of rRNA gene copies (Arif et al., 2014; LaJeunesse, 2002; Thornhill, Lajeunesse, & Santos, 2007). On the one hand, diversity of ITS2 sequences can come from sequence variations among these gene copies, giving rise to intragenomic diversity. On the other hand, sample sequence diversity may also be due to hosts associating with multiple genotypes of Symbiodiniaceae, referred to as intergenomic diversity (Sampayo, Dove, & Lajeunesse, 2009). Without the use of additional genetic markers, differentiating among these sources of variation can be challenging (Thornhill et al., 2007).
Historically, analyses have primarily used one of two techniques to characterize ITS2 sequence diversity from PCR amplicons: denaturing gradient gel electrophoresis (DGGE) or molecular cloning‐and‐sequencing. Regardless of the technique used, the responsibility of characterizing different sequences as being due to intra‐ or intergenomic sources of variance lays with the researcher. Most analyses successfully incorporated intragenomic theory, leading to the core understanding that specific combinations of intragenomic sequences (commonly known as ITS2 profiles) could differentiate, and therefore define, taxa (often referred to as so‐called ‘types’; LaJeunesse, 2002). An example is the identification of both the D1 and D4 sequences to characterize Durusdinium trenchii, formerly Symbiodinium trenchii (LaJeunesse, Wham, & Pettay, 2014). However, analyses interpreting every sequence as representative of a distinct Symbiodiniaceae genotype leads to erroneous inflation of Symbiodiniaceae diversity estimates (Apprill & Gates, 2007; Thornhill et al., 2007).
Whilst DGGE and to a lesser extent molecular cloning‐and‐sequencing approaches are still used to analyse the state of rDNA variation (Hume, D’Angelo, Burt, & Wiedenmann, 2018; Smith, Hume, Delaney, Wiedenmann, & Burt, 2017a; Varasteh et al., 2018; Wham, Ning, & LaJeunesse, 2017), contemporary analyses are starkly shifting towards employing next‐generation sequencing (NGS) technologies (Cunning et al., 2017; Hume et al., 2016; Hume, Ziegler et al., 2018; Smith, Ketchum et al., 2017b; Ziegler et al., 2017). These NGS approaches afford a greater sequencing depth, but yield sequence diversities orders of magnitude larger than previous methodologies. NGS approaches also overcome limitations associated with the gel‐based DGGE methodologies, such as, but not limited to, poor resolution between sequences and a sequence detection limit reliant on the effectiveness of the staining technique employed.
Despite the advances offered by NGS approaches, there is no consensus on how best to exploit their increased sequencing depth and differentiate between intragenomic and intergenomic sequence diversity. Initial analyses have used operational taxonomic unit (OTU) approaches to collapse sequence diversity at a 97% similarity threshold (Arif et al., 2014; Cunning et al., 2017). Yet other approaches have combined OTU analyses with searches for key ITS2 type‐defining sequences in attempts to better exploit the data (Ziegler et al., 2017). Most recently, a minimum entropy decomposition (MED)‐based approach (referred to as the metahaplotype approach) has been used to consolidate the high diversity to a smaller set of core sequence nodes, based on biologically informative sequence positions rather than the fixed similarity thresholds of the OTU approach (Eren et al., 2015; Smith, Ketchum et al., 2017b).
However, these above‐mentioned approaches are ultimately limited in their ability to resolve taxa from NGS ITS2 data. As numerous Symbiodiniaceae species share the same most abundant ITS2 sequence, and a 1‐bp difference in this most abundant sequence may relate to evolutionary divergences of >10 million years (Thornhill, Lewis, Wham, & LaJeunesse, 2014), the fixed similarity threshold clustering of OTU approaches greatly limits their ability to maintain informative intragenomic structure and thus, resolve Symbiodiniaceae taxa. In contrast, the metahaplotype approach (Smith, Ketchum et al., 2017b) does retain intragenomic diversity information. However, it does not differentiate between intra‐ and intergenomic diversity and is thus limited to scenarios where samples only contain a single symbiont taxon (Smith, Ketchum et al., 2017b). To verify this single symbiont assumption, the method is reliant on an additional less conserved marker, the chloroplastic psbA noncoding region (psbA ncr), a marker often used to differentiate between closely related Symbiodiniaceae taxa (LaJeunesse & Thornhill, 2011). While this marker may appear to be an attractive alternative to the ITS2 region due to its power to resolve closely related taxa, its highly variable nature severely limits the taxonomic range over which returned sequences can be aligned, and therefore it is effective only for reconstructing fine‐level phylogenies among closely related taxa (Thornhill et al., 2014).
To maximize biological inferences obtained from NGS ITS2 data sets, we present SymPortal, a new analytical framework for resolving Symbiodiniaceae taxa using NGS data of the ITS2 marker gene. Our methodology augments tried‐and‐tested intragenomic resolution theory with the power of NGS to resolve between symbiont taxa at a level far surpassing alternative methodologies using this marker. We employ novel logic to identify within‐sample informative intragenomic sequences, which we have termed 'defining intragenomic variants' (DIVs), and use combinations of these DIVs to identify ITS2 type profiles representative of putative Symbiodiniaceae taxa. This approach means that SymPortal is better able to differentiate between intragenomic and intergenomic sources of ITS2 variation without the need for more resolute markers, such as the psbA ncr. Here we give an overview of the SymPortal analytical framework and demonstrate its ability to achieve superior ITS2‐based taxonomic resolutions within the family Symbiodiniaceae. We analyse NGS ITS2 data in the SymPortal framework and compare the results with the most commonly used approach, the 97% similarity OTU analysis.
2. MATERIALS AND METHODS
This section provides an overview of the SymPortal analytical framework before detailing the approach used in the methodological comparison of the SymPortal and 97% similarity OTU analyses. More detailed and continually updated documentation of the SymPortal framework can be found on GitHub (https://github.com/didillysquat/SymPortal_framework).
2.1. SymPortal and the reorganization of the former genus Symbiodinium
The former genus Symbiodinium has recently undergone a revision in systematics to the taxonomic level of family and named Symbiodiniaceae (LaJeunesse et al., 2018). The phylogenetic groupings commonly referred to as ‘Clades’ are now recognized as different genera. Currently seven, about half of the total, are given formal names. The group designated Clade A retains the genus name Symbiodinium. Thus, any mention of Symbiodinium refers to taxa in Clade A. As pertains to this research, Clade C is now the genus Cladocopium.
Of note, the SymPortal framework was written before this taxonomic reorganization and all analyses to date have been completed according to the old organizational system/taxonomy. Given the need for backwards comparability to previous studies, the SymPortal framework will maintain its use of the clade systematics whilst working towards the additional incorporation of the Symbiodiniaceae systematics to ensure forward comparability. The term and taxonomic level of ‘clade’ will therefore still be referenced in the explanation of the SymPortal framework below, and it should be clear that this refers to the phylogenetic groupings of the former genus Symbiodinium (Clades A–I).
2.2. Overview of the SymPortal analytical framework
The SymPortal analytical framework consists of two parts: the SymPortal analysis and an SQL database with which the analysis is integrated. The analysis may be run either remotely via submission to SymPortal.org or locally. The main difference between these two modes of operation is the database used for the analysis. If running the analysis via submission to SymPortal.org, the remotely hosted SymPortal database is used, while analyses run locally make use of a user‐constructed database. Please see Sections 2.3.4: Accessing the SymPortal analytical framework and 2.3.5: The SymPortal analytical framework's database below for further details.
Within the SymPortal framework, the SymPortal analysis works by employing the usually substantial ITS2 intragenomic diversity harboured within every Symbiodiniaceae genome, and captured by NGS ITS2 sequencing, to resolve between genetically differentiated taxa (Figure 1). The analysis does this by identifying specific sets of ‘defining intragenomic [ITS2 sequence] variants’ (DIVs) that represent the taxonomic unit of SymPortal, the ‘ITS2 type profile’ (a term derived from the ‘ITS2 types’ defined by ‘ITS2 profiles’ in DGGE‐based methodologies). The database with which the SymPortal analysis is integrated stores sequencing information from every previously conducted SymPortal analysis. With continued use, the SymPortal framework will therefore accrue a catalog of ITS2 type profiles representative of identified putative Symbiodiniaceae taxa.
Figure 1.
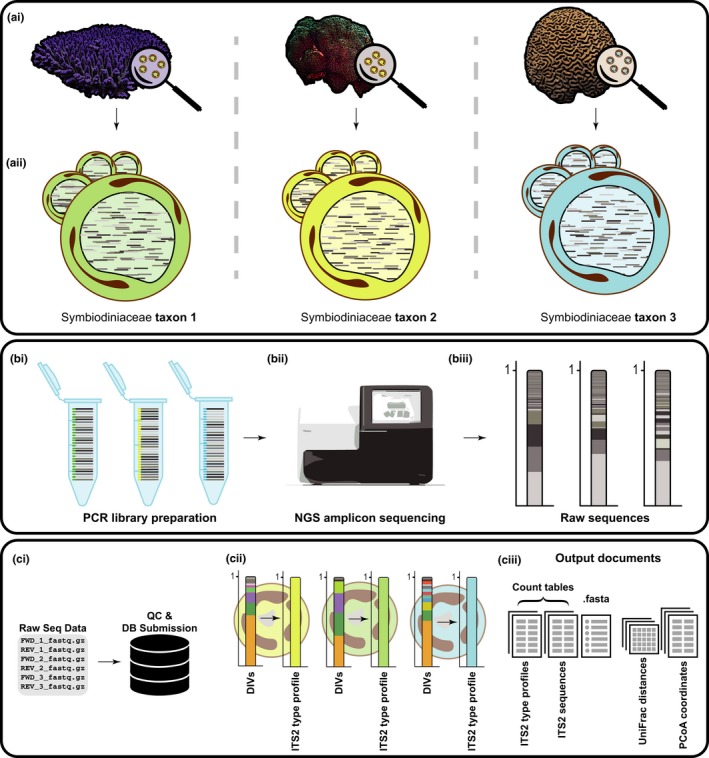
Overview of the typical workflow employing the SymPortal analytical framework. Sampling: (ai) Field sampling: corals containing populations of Symbiodiniaceae are sampled. (aii) Each Symbiodiniaceae cell contains hundreds of copies of the ITS2 gene representing many unique sequences (intragenomic sequence diversity). In this example, three corals are sampled, each containing a single genetically differentiated population of Symbiodiniaceae. PCR library preparation and sequencing: (bi) genomic DNA is extracted from each sample, the ITS2 region is PCR‐amplified and PCR libraries are prepared for sequencing through clean‐up and adapter ligation. (bii) Prepared libraries are sequenced on the Illumina platform producing, (biii) raw sequencing reads in paired.fastq.gz format. SymPortal analysis: (ci) paired.fastq.gz files are quality control processed and loaded into the SymPortal framework's database before being run through the SymPortal analysis. (cii) During the analysis, re‐occurring sets of ITS2 sequences are identified as ‘defining intragenomic [ITS2 sequence] variants’ (DIVs) that are used to define novel or pre‐existing, taxa‐representative, ITS2 type profiles. (ciii) tab‐delimited count tables are output detailing the abundance of predicted ITS2 type profiles and post‐quality control sequences found in each sample. A .fasta is also provided detailing nucleotide sequence information. Finally, clade‐separated UniFrac distance matrices and calculated principal coordinate analysis (PCoA) coordinates are output for between‐sample and between‐ITS2 type profile pairwise comparisons [Colour figure can be viewed at wileyonlinelibrary.com]
Whilst several different Symbiodiniaceae taxa may be harboured by an individual coral, commonly only a single Symbiodiniaceae taxon is found to be predominant (Baums, Devlin‐Durante, & Lajeunesse, 2014; Goulet & Coffroth, 2003; Pettay, Wham, Pinzon, & Lajeunesse, 2011; Thornhill, Xiang, Fitt, & Santos, 2009). In cases where multiple Symbiodiniaceae are harboured by a single coral, these taxa are usually from different genera. Nevertheless, although rare, individual corals may harbour multiple Symbiodiniaceae taxa from the same genera. SymPortal must therefore be able to differentiate between intra‐ and intergenomic sources of ITS2 sequence variants returned from sample amplicon libraries (Arif et al., 2014; Cunning et al., 2017; Thornhill et al., 2007). To achieve this, SymPortal applies a central principle: the probability that a given set of ITS2 sequences found in a single coral sample are representative of a single Symbiodiniaceae genotype increases with the number of samples that set is found in (Figure 1c). Key to this principle is that although distinct coral colonies may contain DNA from millions of individual Symbiodiniaceae cells, when considered on a genus by genus basis (ITS2 sequences from Symbiodiniaceae taxa of different genera are genetically diverse and mostly unalignable), the vast majority of the cells are usually of a single genotype (Baums et al., 2014; Goulet & Coffroth, 2003; Pettay et al., 2011; Thornhill et al., 2009). On the rare occasions when multiple taxa belonging to the same genus are found within a single sample, these taxa may be successfully resolved using SymPortal when each of them is present in other samples as standalone taxa. This principle has been used in DGGE‐based methodologies to successfully identify genetically distinct Symbiodiniaceae taxa that have been verified by additional genetic markers (LaJeunesse & Thornhill, 2011; LaJeunesse et al., 2014; Sampayo et al., 2009).
Given the above approach to differentiating between intra‐ and intergenomic sources of variation, SymPortal's ability to accurately identify DIVs and therefore ITS2 type profiles representative of Symbiodiniaceae taxa will increase as more samples are incorporated into the analysis. Therefore, to maximize the confidence with which each analysis identifies ITS2 type profiles indicative of putative taxa, SymPortal makes use of the sequencing information stored in its database from previously run analyses. By having more samples at its disposal, from which it may search for re‐occurring sets of ITS2 sequences, SymPortal's power to resolve within Symbiodiniaceae increases with use.
2.3. The SymPortal workflow
The processes underlying data submission and analysis in the SymPortal analytical framework are documented in detail online at the GitHub wiki (https://github.com/didillysquat/SymPortal_framework/wiki). Below, we provide an overview of these processes alongside an introduction to the general principles underlying the operation of SymPortal.
2.3.1. Sample input
A data submission to SymPortal is typically in the form of a batch of samples in which each sample's sequencing information is represented by a pair of demultiplexed .fastq.gz files (one file containing the forward read and one file containing the reverse read; Figure 1). Amplicon libraries must have been amplified with Symbiodiniaceae ITS2‐specific primers. We recommend use of the SYM_VAR primer pair (SYM_VAR_REV/SYM_VAR_5.8S2; Hume et al., 2013; Hume et al., 2015, respectively; Hume, Ziegler et al., 2018), but any amplicon is appropriate as long as the amplicon produced by the SYM_VAR primer pair is nested within it (e.g. ITSintfor2/ITS‐Reverse as used in Arif et al., 2014; Coleman, Suarez, & Goff, 1994; LaJeunesse, 2002; Ziegler et al., 2017). For a comparison of commonly used primer pairs, please refer to Hume, Ziegler et al. (2018).
To maintain standardization between data submissions, all sequence quality control (QC) is done within the SymPortal framework. This is currently conducted using mothur 1.39.5 (Schloss et al., 2009), the blast+ suite of executables (Camacho et al., 2009), minimum entropy decomposition (MED; Eren et al., 2015) and custom functions written in Python to minimize the incorporation of artefactual and non‐Symbiodiniaceae sequences, while increasing the informative potential of each library.
Post‐QC, the sequencing information from each sample is loaded into the SymPortal database, at which point it is available for subsequent analyses (see Section 2.3.5: The SymPortal analytical framework's database below).
2.3.2. An analysis: Searching for ITS2 type profiles and identifying DIVs
Each new SymPortal analysis makes use of the sequencing information associated with not only the new samples, but all previously analysed samples as well. During an analysis, each sample's collection of ITS2 sequences is algorithmically searched in a genus‐/clade‐separated manner (Figure 2) to identify the largest set of ITS2 sequences that also occur in other samples in the analysis. To minimize the effects of sequencing depth artefacts, these genera‐separated collections of sequences will only be searched if they contain more than 200 sequences. When a set of ITS2 sequences is found to re‐occur in a sufficient number of samples, each of these sequences is considered to be a DIV and these DIVs are used to characterize an ITS2 type profile (Figures 1 and 2). If for any given sample a re‐occurring set of DIVs cannot be found, then an ITS2 type profile will be assigned to this sample that is defined simply by the sample's most abundant ITS2 sequence.
Figure 2.
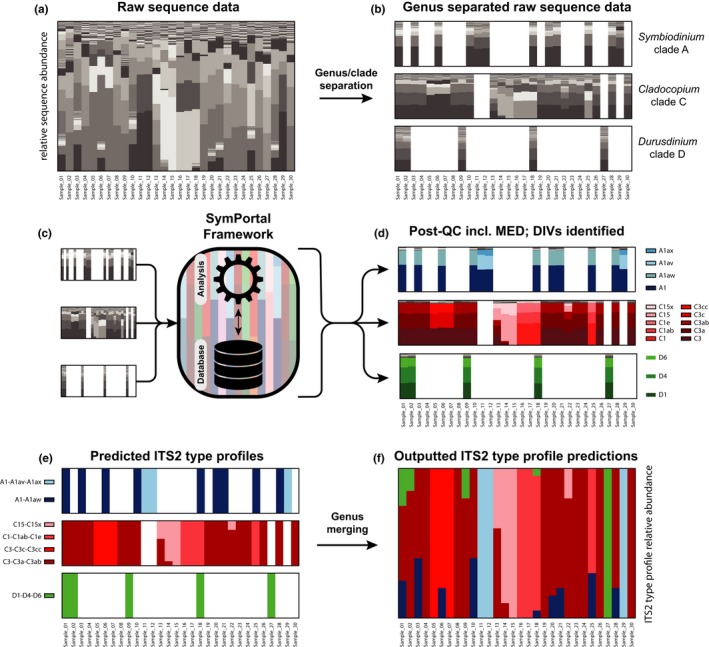
Determination of ITS2 type profiles in samples containing mixed Symbiodiniaceae communities. In all subplots, each stacked bar represents sequence data or predicted ITS2 type profiles for a single given sample with individual bars of the stack representing relative abundances within the sample. Coloured bars represent specific ITS2 sequences or ITS2 type profiles. Grey scale bars represent unidentified ITS2 sequences. (a) Samples that contain ITS2 sequence diversity originating from intergenomic and intragenomic variation are submitted to SymPortal. (b) Raw sequences are separated by genus as part of the standard quality control pipeline. (c–e) The remaining analysis is conducted on the genus‐separated sequence data, but only genus‐separated collections of more than 200 sequences (per sample) are searched for ITS2 type profiles to minimize sequencing depth artefacts. Multiple ITS2 type profiles of the same genus (e.g. C15‐C15x and C3‐C3a‐C3ab) may be predicted within individual samples, if each of the ITS2 type profiles are found in isolation in the current or previously analysed data sets. (f) Before output, the genus‐separated predicted ITS2 type profiles are merged and abundances are adjusted relative to preseparation proportions. The data used in this figure have been generated for the purpose of illustration and are not based on actual data. The code used to generate the data can be found at https://github.com/didillysquat/sp_ms_figure_creation [Colour figure can be viewed at wileyonlinelibrary.com]
When identifying ITS2 type profiles, the presence as well as the abundance of characterizing DIVs are considered. For each set of supported ITS2 sequences that is found in the algorithmic search, each constituent ITS2 sequence is tested to see whether it has a multimodal distribution across the samples in which the ITS2 set of sequences was found (i.e. whether there are two or more distinctly different abundances of the sequence). If a multimodal distribution is identified, the collection of samples to which the ITS2 set of sequences were associated is split according to the distribution of the sequences between the different modes. Once no further multimodal distributions are identified, the ITS2 sequences of the separated sets of ITS2 sequences are considered DIVs. In this way, it is possible for different ITS2 type profiles to contain exactly the same collection of DIVs but at different abundances. At this point, the relative abundances of the ITS2 type profile's DIVs within all the samples they were found in are then used to characterize the ITS2 type profile. Therefore, when searching for ITS2 type profiles within samples, a match is only considered if all of the characterizing DIVs are found, and these have relative abundances that fall within the abundance ranges determined during characterization of the given ITS2 type profile.
2.3.3. Mixed Symbiodiniaceae community samples
Individual samples may contain taxa from more than one genus in the family Symbiodiniaceae (formerly Clades A–G; LaJeunesse et al., 2018). Between these genera, ITS2 sequences are mostly unalignable. Therefore, as a precursor to all analyses, sequence data are separated by genus/clade before subsequent analysis of ITS2 type profiles. The genus‐/clade‐separated data are then merged back together before final output with abundances of predicted ITS2 type profiles adjusted relative to the preseparation proportions (Figure 2).
Samples may also contain multiple Symbiodiniaceae populations from the same genus, and each can make up a considerable proportion of the host's algal symbiont complement. SymPortal is able to identify multiple ITS2 type profiles of the same genus within a given sample as long as each of the ITS2 type profiles has been found as the sole ITS2 type profile in another sample, such as C3‐C3a‐C3ab and C15‐C15x in Figure 2 (see Section 2.3.6, SymPortal output, for an explanation of ITS2 type profile systematics).
2.3.4. Accessing the SymPortal analytical framework
The SymPortal analytical framework may be run either remotely, through submission of data to a version of the framework hosted remotely at SymPortal.org, or locally, by running the Python scripts housed on the GitHub repository (https://github.com/didillysquat/SymPortal_framework). Instructions for submitting data to the remote instance of the framework are detailed at the GitHub wiki and at SymPortal.org. Additionally, policies regarding data ownership and accessibility when submitting to the remote instance of SymPortal are also hosted on SymPortal's GitHub wiki. Analyses run remotely through SymPortal.org will have access to the latest version of the SymPortal database. This remotely hosted PostgreSQL database contains sequencing information from all samples previously analysed that were submitted via SymPortal.org. Analyses run via SymPortal.org will therefore have access to the greatest possible resolving power and enable comparability to other data sets previously submitted to SymPortal.org. If running the SymPortal framework locally, it will be necessary for the user to populate the local database with which the local SymPortal analysis will integrate. As such, when running analyses locally, the ability to resolve ITS2 type profiles will be contingent on the extent of sequencing information housed in the local database. Further documentation may be found at the SymPortal GitHub repository.
2.3.5. The SymPortal analytical framework's database
Irrespective of whether analyses are run locally or remotely via SymPortal.org, the sequencing information of every data set analysed by SymPortal is stored in the associated database. This sequencing information is stored when a data set is loaded into a SymPortal database instance (Table 1) and when analyses are run (Table 2). Remotely run analyses will use the remote SymPortal database, while local analyses will use a locally defined database. All database instances are integrated into the Python‐scripted analysis through the Django API (https://www.djangoproject.com/). In this way, the sequencing information from all previously analysed samples is accessible for use in future analyses.
Table 1.
Database tables populated upon loading of a new data set into the SymPortal framework
| Table name | Table description |
|---|---|
| DataSet | Equivalent to a set of samples loaded into, and analysed on, the SymPortal framework. Contains fields to store basic information such as: the submitting user, the working directory, or the data set's unique name |
| DataSetSample | Equivalent to a single sample from a specific data set. Has a foreign key relation to the DataSet table. Contains fields that store quality control statistics, as well as basic sample information, for example name of samples, number of reads after making contiguous sequences, or number of nodes after minimum entropy decomposition |
| CladeCollection | An abstract object that is a collection of all DataSetSampleSequence instances for a specific DataSetSample that are all of the same clade and have a total abundance > 200. Has a foreign key relationship to DataSetSample. Contains fields that store basic information on the DataSetSampleSequence instances in the collection and of the cladea |
| DataSetSampleSequence | Equivalent to a single unique post‐QC sequencing read returned from a given data set sample. Contains a field for abundance. Has foreign key relationships to ReferenceSequence, CladeCollection and DataSetSample |
| ReferenceSequence | Equivalent to a single unique post‐QC sequencing read, independent of any one sample. Contains fields for name, sequence, clade and accession number |
The clade system of taxonomic naming within the former genus Symbiodinium has now been superseded by new family‐ and genera‐level descriptions (LaJeunesse et al., 2018). In the interest of maintaining reverse comparability to previous studies, SymPortal will continue to relate its outputs to the clade divisions. In time, SymPortal will also be updated to include the new Symbiodiniaceae organization.
Table 2.
Database tables populated during a SymPortal analysis
| Table name | Table description |
|---|---|
| DataAnalysis | Equivalent to a single SymPortal analysis. Contains fields to store high‐level information about the analysis, such as DataSet objects included in the analysis, name, description and parameters used within the analysis |
| AnalysisType | Equivalent to an ITS2 type profile. Contains fields relating to ITS2 type profile features, for example the ReferenceSequence objects that characterize the AnalysisType (DIVs) listed in order of abundance, the ReferenceSequence objects that are found as the most abundant sequences in each of the samples in which the AnalysisType is found, a list of the CladeCollection objects in which the AnalysisType is found, or the maximum and minimum relative abundances of the DIVs that characterize the AnalysisType. Has a foreign key relationship to DataAnalysis |
| CladeCollectionType | An abstract table used to link the database tables associated with data analyses to the database tables associated with data set loading. Has foreign key relationships to CladeCollection and AnalysisTypea |
The clade system of taxonomic division within the former genus Symbiodinium has now been superseded by new family‐ and genera‐level descriptions (LaJeunesse et al., 2018). In the interest of maintaining reverse comparability to previous studies, SymPortal will continue to relate its outputs to the clade divisions. In time SymPortal will also be updated to include the new Symbiodiniaceae organization.
In addition to storing detailed sequencing information for each submitted data set, the database also stores details of all completed analyses (Table 2). Analysis details, such as which ITS2 type profiles were found and which DIVs characterize those profiles, are associated with the respective sequencing information, e.g. which samples the ITS2 type profiles were found in, and which of the samples' sequences were identified as DIVs. The remotely hosted SymPortal database therefore represents a powerful analytical and reference resource, effectively accruing a catalogue of global Symbiodiniaceae diversity as the number of samples analysed grows.
With every new data set submitted to the SymPortal framework's database, the amount of sequencing information available for use in the SymPortal analysis will increase. Effectively, each sequential analysis will have access to a more resource‐rich version of the database. In this way, subtle differences between the identities of the ITS2 type profiles discovered in consecutive analyses may occur as SymPortal's power to resolve improves. As such, when comparing outputs from two different SymPortal analyses, it will be helpful to consider which data sets from the SymPortal database were included in the given analyses. For this reason, every analysis count table output will provide the unique database identifier (UID) of the analysis it is related to. From this information, a list of the data sets that were incorporated into the analysis may be generated. However, changes that occur between analyses run against sequential database versions are likely to be small. Samples from different analyses that contain similar ITS2 sequence diversities will still have comparable ITS2 type profile designations. At the time of writing, the latest analysis (DataAnalysis UID 44) contained 7,173 samples, representing the sampling efforts of 35 studies (35 DataSet objects). Within these samples, 771 different ITS2 type profiles were identified, representing 10,539 unique ITS2 type profile/sample associations. Importantly, 50% of these unique associations are represented by the 92 most abundant ITS2 type profiles (Figure S1). A summary of the database tables held in the SymPortal framework's database is given in Tables 1 and 2.
2.3.6. SymPortal output
The SymPortal output consists of five core output files. Two tab‐delimited count tables each report on post‐QC sequences, ITS2 type profile abundances identified in the output's samples (each reported in absolute and relative abundances) and a .fasta file containing nucleotide sequence information for every ITS2 sequence reported in the count table. In addition, a set of genus‐separated Bray–Curtis‐ or UniFrac‐based distance matrices for between‐sample and between‐ITS2 type profile comparisons, and principal coordinate analysis (PCoA) coordinates for each of these distance matrices, are output. To give an overview of data submissions and data analyses, plots of sequences and ITS2 type profile abundances across samples as well as graphical representations of the PCoA results are additionally generated for the user (see Figures S2 and S3 as an example).
For both count tables (i.e. sequences and IT2 type profiles), the format resembles the OTU count tables commonly used in 16S analyses. Therefore, the format of these tables is ideally suited to be used as direct input to commonly implemented statistical analyses for sequencing diversity and abundance data. The SymPortal outputs for the data analysed in this study is provided as Files S1–S11.
The sequence output table lists the sequences found in order of their abundances across all the samples included in the output (Files S1 and S2). For each sequence, its abundance in each of the samples is reported. A GenBank accession number (if available) or alternatively a UID is also provided for each ITS2 sequence that allows it to be identified in the SymPortal database. In this way, a single definitive source for the ITS2 sequence is always available. The nucleotide sequence for each of the ITS2 sequences reported in the count table is available in a .fasta file (File S3). Finally, for each sample, the number of sequences retained at each step of QC is reported.
The ITS2 type profile abundances are output in decreasing order of the number of samples they were found in for the current analysis output (Files S4 and S5). For each ITS2 type profile, several features are reported as summarized in Table 3.
Table 3.
ITS2 type profile count table output features
| Count table feature (table header name) | Description |
|---|---|
| ITS2 type profile UID | A unique database identifier number that uniquely identifies each ITS2 type profile in the SymPortal database |
| Clade | The clade (A–I) of the ITS2 type profilea |
| Majority ITS2 sequence | The sequence(s) that was(were) identified as the most abundant in each of the samples that the type profile was found in |
| Associated species | A list of any Symbiodiniaceae species descriptions that the given ITS2 type profile could relate to |
| ITS2 type profile abundance local | The number of samples in the output that the given ITS2 type profile was found in |
| ITS2 type profile abundance DB | The number of samples in the entire database that contained the ITS2 type profile in question |
| ITS2 type profile | The name of the ITS2 type profile in question |
| Sequence accession/SymPortal UID | Either an accession number or SymPortal UID for each of the DIV sequences that define the ITS2 type profile in question |
| Counts | Number of sequence reads for DIVs of a given ITS2 type profile in a given sample |
The clade system of taxonomic naming within the former genus Symbiodinium has now been superseded by new family‐ and genera‐level descriptions (LaJeunesse et al., 2018). In the interest of maintaining reverse comparability to previous studies, SymPortal will continue to relate its outputs to the former clade divisions. In time, SymPortal will also be updated to include the new Symbiodiniaceae organization.
To aid in relating the determined ITS2 type profiles to species descriptions, a list of associated Symbiodiniaceae species is reported for each ITS2 type profile where applicable (Table 3). The purpose of this list is to aid the researcher in identifying potential Symbiodiniaceae species that the ITS2 type profile could represent. For a species to be associated with an ITS2 type profile, the ITS2 sequence(s) characteristic of the species, as detailed in its description, must all be found in the list of DIVs that define the ITS2 type profile in question. New species information will be added to the SymPortal database, and therefore associated with outputs, as new descriptions are made. Importantly, this is not a list of species that the ITS2 type profile definitely represents, and it remains the responsibility of the researcher to carefully consider the species descriptions to assess whether the queried sample may be the species in question. Depending on the description, this may require the use of further genetic, morphological, or physiological evidence.
The naming scheme of the ITS2 type profiles is informative. It denotes which underlying DIVs the ITS2 type profiles are composed of in decreasing order of abundance. Hence, the ITS2 type profile ‘C3‐C3gulf‐C3c‐C3aq’ contains DIVs that are ITS2 sequences denoted as C3, C3gulf, C3c and C3aq. This nomenclature also provides information on which of the DIVs were the most abundant in each of the samples in which the ITS2 type profile was found. In the case of ‘C3‐C3gulf‐C3c‐C3aq,’ the C3 sequence was the most abundant sequence in all of the samples in which this ITS2 type profile was found, as this DIV is listed first in the name. Interestingly, a Symbiodiniaceae taxon represented by an ITS2 type profile is sometimes characterized by more than one most abundant sequence. For example, in the type ‘C3/C3c‐C3gulf’ some of the samples in which this ITS2 type profile was found contained the C3 sequence as the most abundant DIV, but in others the C3c sequence was most abundant. This comajority abundance is denoted by the ‘/’ in the ITS2 type profile name.
Distance matrices of pairwise comparisons are output from SymPortal to aid in the quantification of similarity or relatedness between study samples (Files S8 and S10) and ITS2 type profiles (File S6), respectively. Given the inability to align some ITS2 sequences from different genera within the Symbiodiniaceae, pairwise differences are calculated for genus‐separated groupings of ITS2 sequences. Distance matrices are calculated based on either a Bray–Curtis method (sensu Smith, Vaughan, Ketchum, McParland, & Burt, 2017c) or using the weighted UniFrac method (Lozupone, Lladser, Knights, Stombaugh, & Knight, 2011). UniFrac is often used to compare bacterial communities by assessing similarity between sets of 16S rDNA sequences representative of bacterial taxa (Lozupone et al., 2011). As such, it is well suited to comparing similarities between sets of ITS2 sequences where closely related ITS2 type profiles (those sharing a most abundant ITS2 sequence) are likely to be more closely related, the more similar their ITS2 sequence complements are. To compare similarity between two sets of sequences, UniFrac calculates the proportion of sequences from each set that share a branch on a precomputed phylogenetic tree containing all sequences from both sequence sets. Such a phylogenetic tree is therefore required for the analysis and computed as detailed in the following.
Weighted UniFrac distance matrices are calculated in mothur 1.39.5 (Schloss et al., 2009). A bootstrapped neighbour‐joining (NJ) tree is used as input to the unifrac.weighted command and generated using a combination of: mafft (Katoh & Standley, 2013) to create a multiple sequence alignment, seqboot from phylip (http://evolution.genetics.washington.edu/phylip/) to generate multiple alignments by resampling, mothur's implementation of clearcut (Sheneman, Evans, & Foster, 2006) to generate multiple trees, and sumtrees (http://dendropy.org/programs/sumtrees.html) to generate a 50% majority rule consensus tree.
2.4. Comparison of SymPortal's resolution to that of a 97% similarity cutoff clustering approach
To compare the ability of SymPortal to resolve putative Symbiodiniaceae taxa with that of the most commonly used ITS2 NGS analysis approach, the 97% similarity OTU approach, we reanalysed data from a previous study with both methods (Figure 3).
Figure 3.
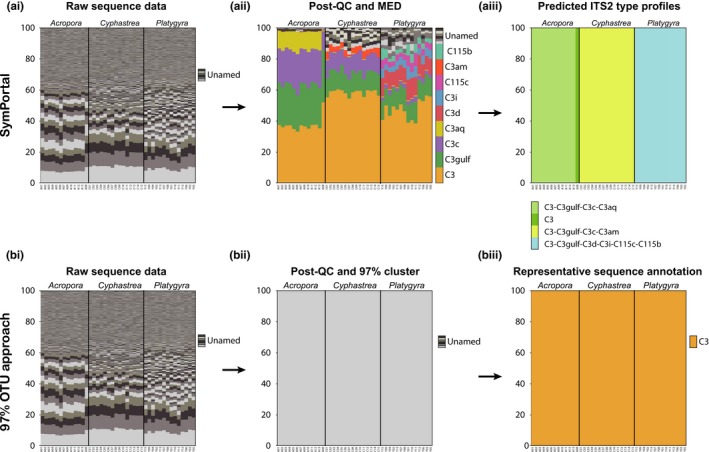
Comparison of the SymPortal analytical framework (top) with operational taxonomic unit (OTU) generation based on 97% similarity cutoff clustering (bottom) from the Smith, Ketchum et al. (2017b) data set (three coral species with 13–15 samples for each coral species). The comparison demonstrates the ability of both methods to resolve between closely related (i.e. the same most common ITS2 sequence) Symbiodiniaceae taxa. From left to right, plots represent progress in each analysis: (ai, bi) raw sequence data, (aii) post‐quality control (QC) and minimum entropy decomposition analyses (MED), (bii) post‐QC and 97% similarity clustering, (aiii) determination of ITS2 type profiles through the identification of defining intragenomic sequence variants (DIVs), and (biii) identification and annotation of representative OTUs. In all plots, each stacked bar column represents a single sample with each individual bar representing either the proportion of a single ITS2 sequence relative to the total abundance of sequences in that sample or the relative abundance of ITS2 type profiles identified in each sample (plot aiii only). Notably, SymPortal ITS2 type profiles determine distinct Symbiodiniaceae taxa associated with distinct hosts, whereas a 97% OTU approach suggests the same symbiont is associated with all hosts. Coloured bars denote DIV sequences (see coloured key) as opposed to grey scale bars that represent non‐DIV sequences [Colour figure can be viewed at wileyonlinelibrary.com]
2.4.1. The test data set
The test data set was collected as part of Smith, Ketchum et al. (2017b). It consisted of Symbiodiniaceae ITS2 and psbA ncr NGS amplicon data from 13, 15 and 14 coral samples from each of three coral species: Acropora downingi, Cyphastrea microphthalma and Platygyra daedalea, respectively. All samples, from each of the coral species, were found to contain Symbiodiniaceae taxa of the species Cladocopium thermophilum (Hume et al., 2015; LaJeunesse et al., 2018) by Smith, Ketchum et al. (2017b). Briefly, corals were collected from Saadiyat reef (24°35′56.4″N, 54°25′17.4″E) in the southern Persian/Arabian Gulf in February 2016. DNA was extracted through: sample lysis in an sodium dodecylsulphate‐based lysis buffer, binding of DNA on carboxylated modified SeraMag beads (GE Healthcare Life Sciences), washing in 80% ethanol and subsequent dissolution in molecular‐grade water. Amplicon libraries were generated using Symbiodiniaceae‐specific primer pairs for ITS2, SYM_VAR_5.8S2 and SYM_VAR_REV (Hume et al., 2013, 2015; Hume, Ziegler et al., 2018) and for psbA ncr, PSBA_NC_F and PSBA_NC_R (Smith, Ketchum et al., 2017b). Amplicon libraries were prepared for, and sequenced on, the Illumina MiSeq platform. For the sake of clarity in demonstrating each approach's analysis, only sequences of the genus Cladocopium (formerly Clade C; LaJeunesse et al., 2018) were considered in the comparison (although samples contained some sequences from the genus Symbiodinium, formerly Clade A). Of note, the figures automatically generated during the SymPortal data analysis conducted for this study that contain genus Cladocopium and Symbiodinium information are provided in Figures S2 and S3.
The availability of both ITS2 and psbA ncr NGS amplicon data in this data set makes it ideal for testing ITS2‐based genetic resolutions. ITS2‐resolved genetic delineations can be verified against phylogenies made from the highly variable, but less intragenomically diverse psbA ncr marker. Smith, Ketchum et al. (2017b) have used the metahaplotype approach to resolve the samples in this data set into three groups that corresponded to the respective coral host origin. Critically, however, the metahaplotype approach has no means of differentiating between within‐sample intra‐ and intergenomic sources of ITS2 diversity. Rather, it is reliant on the psbA ncr marker to verify that only a single dominant taxon is present in each sample prior to application of the ITS2‐based ordination resolution approach (Smith, Ketchum et al., 2017b).
2.4.2. Sample analysis using generation of OTUs by 97% similarity cutoff clustering
Clustering to OTUs at the 97% similarity cutoff threshold is an analytical approach originating from the analysis of 16S rDNA in bacterial ecological studies (Stackebrandt & Goebel, 1994; Vetrovsky & Baldrian, 2013). This approach was applied to Symbiodiniaceae ITS2 DNA to collapse intragenomic sequence variability and to be able to resolve ecologically discrete entities (Arif et al., 2014). The 97% threshold was applied to Symbiodiniaceae due to its documented ability to collapse all ITS2 diversity in monoclonal cultures of Symbiodiniaceae (Arif et al., 2014). In the approach described by Arif et al. (2014), sequences recovered from all samples are split by genus, pooled and clustered into OTUs. Representative sequences are then associated with each of the OTUs. More recently, a modification to this method was proposed by Cunning et al. (2017) in which sequences are still clustered at a 97% similarity cutoff but on a sample‐by‐sample basis rather than across all samples. This more recent approach was developed to address the fact that the most abundant ITS2 sequences from different Symbiodiniaceae species may be more similar to one another than the intragenomic variants found within those species (Arif et al., 2014; Cunning et al., 2017). The representative OTUs returned from this approach will usually be equivalent to the most abundant sequence in each sample per genus. To maintain the maximum potential for resolution between the samples in our test data set, we undertook both analyses, namely within and across sample clustering approaches.
For both clustering methods, the initial QC of sample sequences was the same as the mothur‐based component of QC used in SymPortal. Briefly, mothur_1.39.5 (Schloss et al., 2009) was used to create contigs from paired forward and reverse demultiplexed .fastq.gz files using the make.contigs command. On a sample‐by‐sample basis, the following sequence of commands was then applied. The screen.seqs command was used (maxambig = 0, maxhomop = 5) to discard sequences putatively generated from sequencing errors. The resultant .fasta file was used as an input for blastn with the max_target_seqs argument set to 1 and an output format string of ‘6 qseqid sseqid evalue pident gcovs’ (Camacho et al., 2009). A custom blast database was used that contained a single representative sequence for each of the nine clades of the former genus Symbiodinium (Clades A–I; LaJeunesse et al., 2018; File S12). Any sequences not returning the Clade C entry as the closest match were disregarded from the analyses (see above). The unique.seqs command was used to create a nonredundant collection of sequences represented by a .name and a .fasta file. The remainder of the QC was performed using both the .name and the .fasta files produced. Next, split.abund was run (cutoff = 2) to discard sequences that were not found at an abundance > 2 in each sequenced sample – again, to reduce incorporating sequences with sequencing errors. The pcr.seqs command (pdiffs = 2, rdiffs = 2) was used to trim the primer sequence regions from the returned sequences. In addition, this command was used to discard sequences in which the specific primer pairs could not be found – indicative of poor sequencing quality – allowing a deviation of ≤2‐nucleotide differences in either of the forward or the reverse primer sequences. Sequences were again made nonredundant using the unique.seqs command, then sequences <184 bp and >310 bp in length were removed using the screen.seqs minlength and maxlength parameters. Sequences were again made nonredundant.
To perform clustering on pooled sequences across all samples, for each of the .fasta and .name file pairs created above (one pair per sample), a single redundant .fasta file was created using the deunique.seqs command. Each of these deuniqued .fasta files was then combined to create a single redundant .fasta file containing all post‐QC sequences from the sequencing effort. A single master nonredundant .fasta and .name file set was then created using the unique.seqs command. This .fasta file was then aligned using mafft (Katoh & Standley, 2013) with the ‐‐auto flag active. A phylip style distance matrix was created using mothur's dist.seqs command with countends = F and output = lt. The sequences were then clustered at a 3% dissimilarity cutoff using the average‐neighbour algorithm, the phylip distance matrix and the .name file using the cluster command (method = average, cutoff = 0.03) to produce OTUs. Finally, the get.oturep command was used to return the most abundant sequence as a representative sequence for each OTU generated by the clustering.
To perform clustering on sequences within samples: The same process as above was used to align, cluster and generate representative OTU sequences, only working on a sample‐by‐sample basis with the respective pairs of nonredundant .fasta and .name files.
2.4.3. Sample analysis using SymPortal's framework
Paired .fastq.gz files for each sample were run through a SymPortal analysis in one batch. The abundances of the ITS2 type profiles belonging to Clade C, as reported in the SymPortal output count table, were directly used to create Figure 3. Output count tables are provided as Files [Link], [Link], [Link], [Link], [Link].
2.4.4. Plotting of the psbA ncr maximum‐likelihood (ML) phylogeny
To create a psbA ncr phylogeny, the preprocessed psbA ncr sequences from the data associated with Smith, Ketchum et al. (2017b) were used to generate a single consensus sequence for each sample. Generation of these consensus sequences is detailed at https://github.com/didillysquat/sp_ms_figure_creation/blob/master/SP_MS_figure_making.ipynb. Unfortunately, psbA ncr marker amplicons were not available for one of the Platygyra samples, Y11, due to an indexing error (Y11 was not included in the original study; Smith, Vaughan et al., 2017c). A multiple sequence alignment was created from these sequences (File S13). A best nucleotide substitution model was selected in mega7 (Tamura, Stecher, Peterson, Filipski, & Kumar, 2013) and used to plot a bootstrapped (100 replications) ML phylogeny.
2.5. Figure creation
The largely programmatic process used to make figures 2–5 is documented here: https://github.com/didillysquat/sp_ms_figure_creation.
3. RESULTS
3.1. ITS2 type profile determination with SymPortal
Running the 42 samples from three coral species (Acropora downingi, Cyphastrea microphthalma and Platygyra daedalea) from the Persian/Arabian Gulf (see Methods) generated four Cladocopium ITS2 type profiles characterized by between one and six DIVs (output count tables for sequences and ITS2 type profiles are provided in Files S1–S5). Notably, 41 of the 42 samples were represented by three of the four ITS2 type profiles, which correlated to host genus (Figure 3): A. downingi, C3‐C3gulf‐C3c‐C3aq; C. microphthalma, C3‐C3gulf‐C3c‐C3am; and P. daedalea, C3‐C3gulf‐C3d‐C3i‐C115c‐C115b. The fourth ITS2 type profile, denoted as ‘C3’ in the SymPortal analysis (Files S4 and S5), was found in only one sample that was collected from an A. downingi coral, sample A08. This ITS2 type profile was most closely related to the ITS2 type profile found in the other samples from A. downingi corals (denoted as C3‐C3gulf‐C3c‐C3aq in the SymPortal analysis, Files S4 and S5). Despite the C3, C3gulf, C3c and C3aq sequences being present, the A08 sample was not assigned to C3‐C3gulf‐C3c‐C3aq due to the C3aq sequence being at a relative abundance that was lower than the defining abundances for the ITS2 type profile. As no other ITS2 type profile could be assigned to the sample, it was conservatively assigned the C3 ITS2 type profile based only on the sample's most abundant ITS2 sequence from the clade in question (Clade C). Plotting the within‐genus UniFrac distance‐based PCoA coordinates for pairwise sample comparisons (provided as part of the SymPortal output; File S11) also illustrates a clustering of samples according to host species (Figure 4). One Acropora sp. sample, A08, falls outside of the main Acropora sp. grouping. The cause of this divergence is the difference in ITS2 diversity (abundance and presence/absence of ITS2 sequences) contained in this sample compared to the other samples of this host species, as can be seen in Figure 3(aii).
Figure 4.
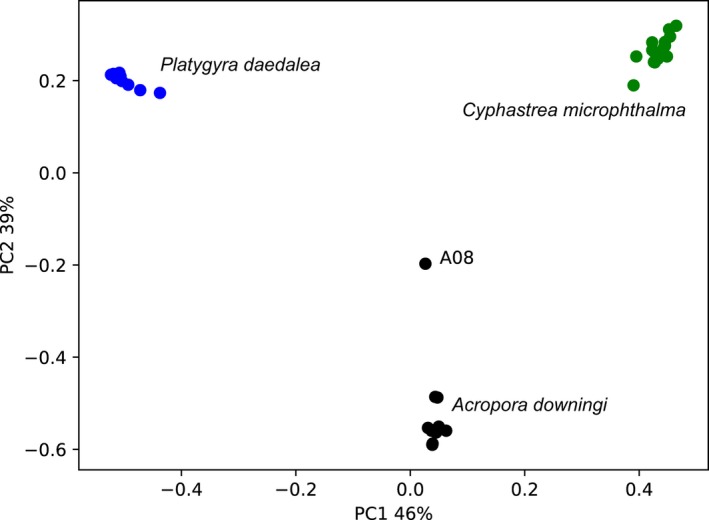
Principal coordinate analysis (PCoA) plot based on UniFrac distances between Cladocopium (formerly Clade C; LaJeunesse et al., 2018) ITS2 sequences found in samples from this study. The plot is made directly from the principal component coordinates output as part of the SymPortal analysis (File S11). Samples are coloured according to host species, as annotated. Sample A08 is separately annotated given its dissimilarity from the main Acropora spp. cluster [Colour figure can be viewed at wileyonlinelibrary.com]
The host‐correlated resolution of the SymPortal analysis is in accordance with the results from the ordination‐based metahaplotype approach of Smith, Ketchum et al. (2017b) that also split the samples into three groups.
3.2. ITS2 OTU generation by 97% similarity cutoff clustering, within and across samples
The within‐ and across‐sample clustering approaches provided identical results. All sequences clustered into a single OTU, which was represented by the C3 sequence (Figure 3; nucleotides 5–267 of sequence KX815267). Clustering into this single OTU was concluded at >98% dissimilarity (i.e. before the 97% threshold could be reached), illustrating the much lower sequence and taxon diversity in comparison to 16S rDNA amplicon studies (Arif et al., 2014; Ziegler et al., 2017).
3.3. Agreement between the ITS2 and psbA ncr genetic markers
Construction of an unrooted ML phylogeny from the psbA ncr sequences associated with the Smith, Ketchum et al. (2017b) data set demonstrated a host‐correlated resolution of the study's samples (Figure 5). This resolution is in accordance with that of the SymPortal results but undetected by the OTU approach. Although not detected in the ML phylogeny, the psbA ncr sequence of sample A08 differed from all other Acropora‐derived sequences in four positions (three separate indels). All other Acropora‐derived sequences were identical. This difference is also realized in the ITS2 sequence and type profiles recovered and designated for A08 by the SymPortal analytical framework. Of the 13 Platygyra sequences for which psbA ncr sequences were available, four showed a divergence from the consensus (three at a single position and one at two positions; samples Y03, Y04, Y05 and Y06; File S13). This divergence from the consensus was not correlated with a separate ITS2 type profile designation and when the ITS2 data for the Platygyra samples is ordinated (PCoA from UniFrac‐based distances), separation of these samples from the other Platygyra samples is questionable (Figure S4). Only one Cyphastrea sample differed from the psbA ncr consensus sequence (one single nucleotide indel).
Figure 5.
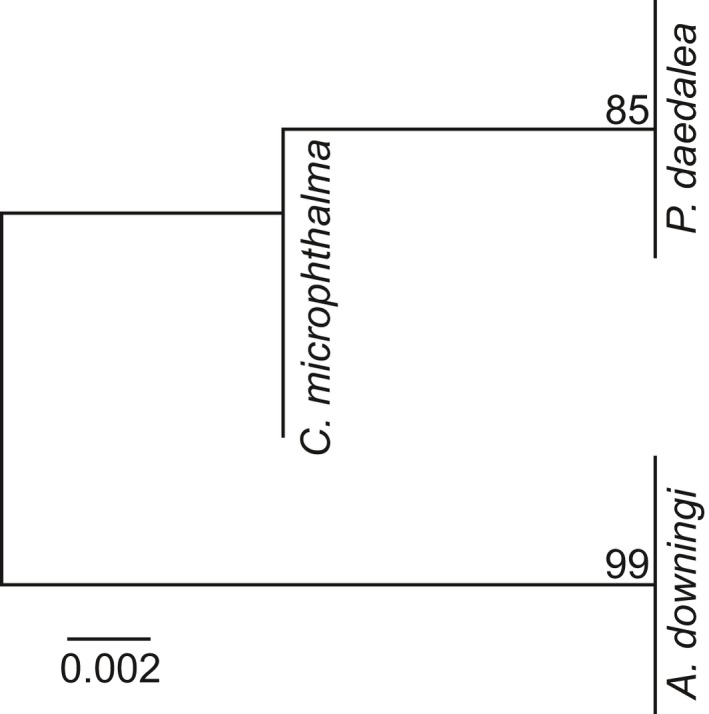
Unrooted maximum likelihood phylogeny based on the psbA ncr sequences from all the samples associated with the Smith, Ketchum et al. (2017b) study. Samples resolved in three positions on the tree according to host species, as annotated. Bootstrap support values < 100 (100 replicates) are annotated
4. DISCUSSION
The multicopy ITS2 sequence diversity harboured within every Symbiodiniaceae genome represents a wealth of information that is currently underutilized in many NGS analytical approaches. Here, we demonstrate that by exploiting this diversity, the SymPortal analytical framework is able to resolve between closely related Symbiodiniaceae genotypes at a resolution never before achievable with the ITS2 marker alone (Figure 3). Our results demonstrate that while all sequence diversity was effectively collapsed using a 97% similarity clustering method (as commonly used in current Symbiodiniaceae ITS2 approaches using NGS data), the SymPortal framework was able to identify four putative and distinct symbiont taxa that were harboured in multiple samples across each of the three coral host species investigated (Figure 3).
While intragenomic ITS2 sequence analysis has been used in characterizing Symbiodiniaceae taxa in more traditional approaches such as DGGE for some time (LaJeunesse, 2002), the uptake of NGS technologies has afforded researchers the power to interrogate PCR amplicon libraries never previously possible (Arif et al., 2014). In particular, the improvement in sequencing depth and accuracy afforded by NGS makes it the perfect platform for characterizing multicopy marker amplicon diversity. Indeed, we see from the example data set analysed here that the introduction of the concept of using ‘defining intragenomic variants’ (so‐called DIVs in our SymPortal approach) for NGS within the SymPortal analysis is (a) effective in resolving between intra‐ and intergenomic sources of ITS2 sequence variants (more so than any a priori clustering cutoff method or most‐abundant sequence method) and (b) does so at a resolution surpassing classical DGGE approaches. For example, DIVs used to differentiate between different ITS2 type profiles may be found at relatively low abundances (~<5%; Figure 3). Such sequences would previously have been below detection limits. As such, taxonomic inferences afforded by these DIVs would not have been achievable (Hume et al., 2015; Smith, Hume et al., 2017a).
The informative nature of the intragenomic diversity found within Symbiodiniaceae samples may be put into context by comparing the ITS2 type profile outputs from the SymPortal analysis, the ordination‐based ITS2 analytical approaches (the metahaplotype approach of Smith et al. and the UniFrac‐based PCoA groupings of the SymPortal output), and the psbA ncr alignment and phylogeny (Figures 4 and 5; File S13 and Figure S4). The only sample to return an ITS2 type profile designation different from other samples of the same host origin was sample A08. The visually different ITS2 sequence profile of this sample (in the ordination and sequence profiles), as well as the conservative C3 ITS2 type profile designation of SymPortal, are in agreement with its disparate psbA ncr sequence (differing from the consensus by three indels across four nucleotides). Within the other two host species, the maximum divergence from the psbA ncr consensus sequence was by two separate single nucleotide indels. Whilst only one of the Cyphastrea samples differed from the consensus, four of the Platygyra samples returned different psbA ncr sequences, three of which had the same indel. Although none of these samples were designated as unique ITS2 type profiles, ordination of the ITS2 profiles did suggest some grouping of the samples that returned the same psbA ncr indel (Figure S4). A greater number of samples would be required to elucidate the robustness of this resolution, but it would appear that divergence in the psbA ncr (even as small as a single indel) may also be reflected in the ITS2 marker. Given the hypervariable character of the psbA ncr region in general, and the fact that even closely related taxa may have considerable differences between their most abundant psbA ncr sequence (as evidenced by the between‐host species differences seen here), it would appear that exploiting intragenomic ITS2 diversity in phylogenetic resolutions within Symbiodiniaceae enables a resolution comparable to that of psbA ncr (at least when considering the short read sequencing‐derived psbA ncr amplicons of the Smith et al. data). Furthermore, because any two ITS2 sequences from the same Symbiodiniaceae clade (A–I) may be aligned, the taxonomic breadth over which comparisons may be made with ITS2 greatly exceeds that of psbA ncr (psbA ncr sequences from the C3 radiation and the C1 radiation are largely unalignable).
As our ability to resolve Symbiodiniaceae improves, taxonomic descriptions may need to be revisited and updated. The majority of Symbiodiniaceae taxa descriptions to date include information related to ITS2 sequence characterization (for example, but not limited to, Hume et al., 2015; Jeong et al., 2014; LaJeunesse, 2017; Lajeunesse, Parkinson, & Reimer, 2012). In many cases, this information relates only to the most common ITS2 sequence associated with the taxa (e.g. C1, A1). However, more recent descriptions contain information relating to additional ITS2 sequence variants that are instrumental in identifying the taxa in question using the ITS2 marker (e.g. Hume et al., 2015; Hume et al., 2016; LaJeunesse et al., 2014). Indeed, we see that ITS2 sequences referred to in such descriptions (e.g. Cladocopium thermophilum : C3, C3‐gulf; Durusdinium trenchii [formerly Symbiodinium trenchii]: D1, D4) are correspondingly being identified as DIVs in the SymPortal framework. Just as these more recent descriptions have exploited improvements in scientific understanding and sequencing power to offer more fine‐scale resolution, the improved resolution of the SymPortal framework should also be incorporated into taxonomic descriptions where appropriate. Where a unique ITS2 type profile is able to identify a discrete Symbiodiniaceae taxon, incorporating information relating to its characterizing DIVs into its description will increase the ease with which taxa may be identified. Of course, further subdivisions of Symbiodiniaceae taxa proposed by differentiated ITS2 type profiles will need to be corroborated with additional supporting evidence. This may take the form of fine‐scale genetic markers such as the chloroplastic psbA ncr and microsatellite flanking sequences, biogeographical data such as specific host‐species fidelities or physical distributions, or correlations of genetic divisions to host or symbiont physiological measurements.
Aside from considering how taxonomic descriptions may be advanced, it is equally important to consider how the outputted resolutions of SymPortal may be evaluated in relation to previous analyses that used alternative methods of analysis in conjunction with the ITS2 marker. By reporting all levels of resolution from the coarser to the finer scale, namely clade, most abundant sequence, putative species and ITS2 type profile, the SymPortal output enables researchers to work at the most appropriate level for their investigation, while maintaining the ability to compare with previous studies. For example, many early analyses reported only to the clade (genus) level, which may be compared directly to SymPortal's ‘clade’ output (see Section 2.3.6 for further details). More recently, samples were often assigned to a ‘type,’ a somewhat ambiguous term, often related to the most abundant sequence, or sequences, found in a sample. Such findings will be comparable using the ‘Majority ITS2 sequence’ output of SymPortal. Previously used OTU clustering approaches may also be compared: those approaches that cluster within samples may be approximated to identifying the most abundant sequences of samples (therefore comparable again to the ‘Majority ITS2 sequence’ output), and those that cluster across samples may be compared by searching for OTU representative sequences amongst ITS2 type profile DIVs. With time, the new Symbiodiniaceae systematics will also be fully integrated into the SymPortal outputs to ensure both forward and reverse comparability of studies analysed through the SymPortal framework.
While we have demonstrated SymPortal's ability to resolve between closely related Symbiodiniaceae taxa, it is also important to consider whether this level of resolution is biologically pertinent. In the analysed data set, we have demonstrated SymPortal's ability to differentiate subtaxa within Cladocopium thermophilum that associate with specific scleractinian coral hosts (Figure 3). Analyses conducted since the initial description of C. thermophilum have returned a larger diversity of chloroplastic psbA ncr marker sequences from genotypes belonging to this species, than were found in the initial description (Hume et al., 2016). This additional diversity has led to the hypothesis that the C. thermophilum taxon probably contains multiple subtaxa that may be species in their own right. With the incorporation of ITS2 intragenomic sequence variants into analyses, delineation of specific geographical and host‐specific populations has now been made possible (Hume, D’Angelo, Burt et al., 2018; Smith, Ketchum et al., 2017b). In particular, the analyses conducted with SymPortal here allow for the identification of these host‐specific subtaxa by their specific combinations of DIVs. The fidelity of a given algal symbiont to a specific coral host can be viewed as being representative of a complex and complementary suite of physiological mechanisms that result in the specific host–symbiont association (D'Angelo et al., 2015; Davy, Allemand, & Weis, 2012; Ochsenkuhn, Rothig, D'Angelo, Wiedenmann, & Voolstra, 2017; Voolstra et al., 2009). As such it would appear that the resolution offered by SymPortal is probably biologically significant. Importantly, it should be noted that Symbiodiniaceae taxa to date have been classified at a coarser resolution than is used here, either with alternative markers (e.g. Picciani, de Lossio e Seiblitz, de Paiva, e Castro, & Zilberberg, 2016; Thomas, Kendrick, Kennington, Richards, & Stat, 2014) or with alternative means of analysis using the ITS2 marker (Howells, Abrego, Meyer, Kirk, & Burt, 2016; Hume et al., 2013). Given the biologically meaningful finer scale resolutions presented through the use of the SymPortal framework, generalizations based on these coarser resolutions should be exercised with care, as exceptions to these generalizations may simply be unresolved, if derived from more traditional classification approaches (e.g. Symbiodiniaceae containing the C3 sequence as their majority sequence are generally considered to be thermally sensitive). Yet, C. thermophilum, the predominant algal symbiont harboured by corals surviving in the world's hottest sea, the Persian/Arabian Gulf, also has a most abundant sequence of C3 (Howells et al., 2016; Hume et al., 2013; Smith, Vaughan et al., 2017c). However, only by undertaking further fine‐scale analyses and correlating these results with associated metadata will we be able to better assess at what level phylogenetic resolutions correlate with meaningful biological divisions.
The central analytical principle that the SymPortal and DGGE‐based analyses are built on has some inherent limitations. The principle relies on identifying re‐occurring sets (found in multiple samples) of ITS2 sequences with the understanding that the more commonly a set of sequences occurs, the more likely that set of sequences is representative of a single taxon (LaJeunesse, 2001, 2002). However, although rare, it is possible that Symbiodiniaceae taxa co‐occur in a sufficient number of samples that the ITS2 sequences returned from several taxa may be documented as being representative of a single taxon. In the analyses that have already been completed, such ‘super types’ have been a very rare occurrence with only one detected case so far. In this study (unpublished), an ITS2 type profile was generated by SymPortal that was co‐dominant for the C15 and C3 sequences. This ITS2 type profile was easily diagnosed as a ‘super type’ due to its profile being a combination of two ITS2 type profiles also identified in the same study (one C3 radiation ITS2 type profile, and one C15 radiation ITS2 type profile). In cases where these super types have been identified, mitigations to their specific instances have been incorporated into the SymPortal algorithms directly to prevent their propagation and re‐occurrence. In the future, however, a median joining network‐based approach will probably be used to assess whether co‐occurring sets of sequences may represent several taxa. Sequence collections will be assessed to see whether they form contiguous networks characteristic of a single taxon's profile, or whether multiple discrete radiations exist, probably characteristic of multiple taxa. Another limitation occurs when trying to quantify the abundances of closely related symbiont taxa (taxa that share the same most abundant ITS2 sequence) found in the same sample. In such cases, where several taxa in a single sample have some of the same DIVs in common, it is extremely difficult to partition what proportions of each of the DIVs are representative of which taxa (as it would be for any other approach given the nature of the problem). Currently, SymPortal uses a conservative approach and does not aim to partition the DIVs, but instead assigns only one of the taxa in question to the sample (the ITS2 type profile containing the greatest abundance of DIVs from the sample). However, given that the majority of coral samples typically harbour one dominant Symbiodiniaceae taxon, especially within a single genus, we anticipate this to be an issue restricted to a small number of cases.
Free‐living or environmental assemblages of Symbiodiniaceae may be structured very differently to those assemblages associated with an animal host. Outside of the host, Symbiodiniaceae populations are free from any host‐selective influence and might therefore be more evenly distributed. Therefore, it cannot be assumed that a single Symbiodiniaceae taxon will be prevalent in a sample from the environment, an assumption required for SymPortal to be able to successfully identify ITS2 type profiles of putative taxa (Thornhill et al., 2017, and references therein). However, as mentioned previously, with continued use, SymPortal will accrue a catalogue of ITS2 type profiles representative of identified putative Symbiodiniaceae taxa. This catalogue of defining ITS2 type profiles may be applied to ITS2 amplicon libraries from environmental samples to predict the presence of Symbiodiniaceae that have already been identified in animal hosts. However, given the breakdown of the aforementioned assumption in these free‐living environments, it is unlikely to be straightforward to identify previously unaccounted putative taxa de novo from environmental samples. Notably, many of the free‐living taxa may not form stable associations with hosts (Thornhill et al., 2017). Besides attempting to use additional markers to attain a finer scale resolution in free‐living samples, another option would be to extract, culture, sequence and characterize monoclonal strains found in such samples (sensu Arif et al., 2014). While host‐associating Symbiodiniaceae can be notoriously difficult to culture, free‐living Symbiodiniaceae are often more readily cultured (Krueger & Gates, 2012; LaJeunesse, 2002; Santos, Taylor, & Coffroth, 2001). Once cultured, monoclonal lines may be sequenced and their ITS2 type profiles directly submitted to the SymPortal database to enable their identification in subsequent analyses.
Basin‐ and global‐scale coral reef ecosystem sampling efforts (e.g. the Tara Oceans or Tara Pacific expeditions https://oceans.taraexpeditions.org/en/, or the Global Coral Microbiome Project http://coralmicrobes.org/) are bringing us closer to having a complete catalog of Symbiodiniaceae diversity (Decelle et al., 2018). Thanks to the wide use of the ITS2 marker in many previously conducted analyses we can already observe closely related Symbiodiniaceae taxa (having the same most abundant ITS2 sequence in common) harboured in a range of species found across geographical ranges that span ocean basins (e.g. Pettay, Wham, Smith, Iglesias‐Prieto, & LaJeunesse, 2015). As sampling efforts intensify, and sequencing and informatic power becomes more accessible, we may be able to document and catalogue such a global convergence of Symbiodiniaceae taxa but at an even finer scale, unlocking the biological inferences associated with this increased resolution. However, standardization between future sampling initiatives (i.e. use of methodologies that incorporate comparable approaches to resolution and nomenclature) will be critical in achieving this goal. To this end, SymPortal enforces standardization through consistent quality control parameters built‐in to the analytical framework and encourages integration and data curation through the offered ability to run SymPortal against the centralized SymPortal database via SymPortal.org. SymPortal is also a community‐driven platform, in that its power to resolve grows with the addition of more sequence data. By embracing this standardized and user‐driven approach, we hope that SymPortal will be widely adopted by the research community as a tool that can generate better results from data that are already being produced. We consider that SymPortal should play an instrumental role in making future sampling efforts comparable and in maximizing their efficacy in working towards the classification of the global Symbiodiniaceae diversity.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
The SymPortal framework was conceived by B.C.C.H., C.R.V., J.W., E.G.S. and M.Z. The framework was built by B.C.C.H. with significant input from: C.R.V. and J.W. for funding and provision of computational resources; C.R.V., E.G.S., M.Z. and T.L.J. for developing analysis theory; E.G.S. and J.B. for providing example data sets; H.J.M.W. for informatic support; and C.R.V. for sourcing additional data to aid in development of the framework prior to public release. B.C.C.H. conducted data analyses and created the figures with significant input from C.R.V., M.Z. and E.G.S. B.C.C.H. and C.R.V. wrote the manuscript; all authors commented and approved the final manuscript.
Supporting information
ACKNOWLEDGEMENTS
This work was supported by KAUST baseline funding to C.R.V., Red Sea Research Center funding (FCC/1/1973‐22‐01) to C.R.V. and B.C.C.H., Natural Environment Research Council Grant NE/K00641X/1 to J.W., and the European Research Council under the European Union's Seventh Framework Programme Grant FP7/2007‐013/ERC Grant Agreement 311179 to J.W. The NYU Abu Dhabi Institute is acknowledged for funding coral sample collections. Alex Mercière is thanked for his help in preparing Figure 1. Finally, three anonymous reviewers are thanked for their considerable contributions in improving the manuscript and the accessibility of the SymPortal framework in general.
Hume BCC, Smith EG, Ziegler M, et al. SymPortal: A novel analytical framework and platform for coral algal symbiont next‐generation sequencing ITS2 profiling. Mol Ecol Resour. 2019;19:1063–1080. 10.1111/1755-0998.13004
Contributor Information
Benjamin C. C. Hume, Email: benjamin.hume@kaust.edu.sa.
Christian R. Voolstra, Email: christian.voolstra@kaust.edu.sa.
DATA ACCESSIBILITY
Data from the original Smith, Vaughan et al. (2017c) study are deposited in the Dryad Digital Repository (http://dx.doi.org/10.5061/dryad.h6s54). The data used to create Figure 3 are available through the SymPortal wiki hosted on GitHub: https://github.com/didillysquat/SymPortal_framework/wiki. The largely programmatic process used to make figures 2–5 is documented here: https://github.com/didillysquat/sp_ms_figure_creation. Version 0.2.2 of the SymPortal framework's source code has been archived at https://doi.org/10.5281/zenodo.2552178.
REFERENCES
- Apprill, A. M. , & Gates, R. D. (2007). Recognizing diversity in coral symbiotic dinoflagellate communities. Molecular Ecology, 16, 1127–1134. 10.1111/j.1365-294X.2006.03214.x [DOI] [PubMed] [Google Scholar]
- Arif, C. , Daniels, C. , Bayer, T. , Banguera‐Hinestroza, E. , Barbrook, A. , Howe, C. J. , … Voolstra, C. R. (2014). Assessing Symbiodinium diversity in scleractinian corals via next‐generation sequencing‐based genotyping of the ITS2 rDNA region. Molecular Ecology, 23, 4418–4433. 10.1111/mec.12869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baums, I. B. , Devlin‐Durante, M. K. , & Lajeunesse, T. C. (2014). New insights into the dynamics between reef corals and their associated dinoflagellate endosymbionts from population genetic studies. Molecular Ecology, 23, 4203–4215. 10.1111/mec.12788 [DOI] [PubMed] [Google Scholar]
- Camacho, C. , Coulouris, G. , Avagyan, V. , Ma, N. , Papadopoulos, J. , Bealer, K. , & Madden, T. L. (2009). BLAST+: Architecture and applications. BMC Bioinformatics, 10, 421 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman, A. W. , Suarez, A. , & Goff, L. J. (1994). Molecular delineation of species and syngens in volvocacean green‐algae (Chlorophyta). Journal of Phycology, 30, 80–90. 10.1111/j.0022-3646.1994.00080.x [DOI] [Google Scholar]
- Cunning, R. , Gates, R. D. , & Edmunds, P. J. (2017). Using high‐throughput sequencing of ITS2 to describe Symbiodinium metacommunities in St. John, US Virgin Islands. Peerj, 5, e3472 10.7717/peerj.3472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angelo, C. , Hume, B. C. C. , Burt, J. , Smith, E. G. , Achterberg, E. P. , & Wiedenmann, J. (2015). Local adaptation constrains the distribution potential of heat‐tolerant Symbiodinium from the Persian/Arabian Gulf. The ISME Journal, 9, 2551–2560. 10.1038/ismej.2015.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davy, S. K. , Allemand, D. , & Weis, V. M. (2012). Cell biology of cnidarian‐dinoflagellate symbiosis. Microbiology and Molecular Biology Reviews, 76, 229–261. 10.1128/MMBR.05014-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decelle, J. , Carradec, Q. , Pochon, X. , Henry, N. , Romac, S. , Mahé, F. , … de Vargas, C. (2018). Worldwide occurrence and activity of the reef-building coral symbiont symbiodinium in the open Ocean. Current Biology, 28(22), 3625–3633.e3. [DOI] [PubMed] [Google Scholar]
- Eren, A. M. , Morrison, H. G. , Lescault, P. J. , Reveillaud, J. , Vineis, J. H. , & Sogin, M. L. (2015). Minimum entropy decomposition: Unsupervised oligotyping for sensitive partitioning of high‐throughput marker gene sequences. The ISME Journal, 9, 968 10.1038/ismej.2014.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujise, L. , Nitschke, M. R. , Frommlet, J. C. , Serôdio, J. , Woodcock, S. , Ralph, P. J. , & Suggett, D. J. (2018). Cell cycle dynamics of cultured coral endosymbiotic microalgae (Symbiodinium) across different types (species) under alternate light and temperature conditions. Journal of Eukaryotic Microbiology, 65, 505–517. 10.1111/jeu.12497 [DOI] [PubMed] [Google Scholar]
- Goulet, T. L. , & Coffroth, M. A. (2003). Stability of an octocoral‐algal symbiosis over time and space. Marine Ecology Progress Series, 250, 117–124. 10.3354/meps250117 [DOI] [Google Scholar]
- Howells, E. J. , Abrego, D. , Meyer, E. , Kirk, N. L. , & Burt, J. A. (2016). Host adaptation and unexpected symbiont partners enable reef‐building corals to tolerate extreme temperatures. Global Change Biology, 22, 2702–2714. 10.1111/gcb.13250 [DOI] [PubMed] [Google Scholar]
- Hughes, T. P. , Anderson, K. D. , Connolly, S. R. , Heron, S. F. , Kerry, J. T. , Lough, J. M. , … Wilson, S. K. (2018). Spatial and temporal patterns of mass bleaching of corals in the Anthropocene. Science, 359, 80–83. 10.1126/science.aan8048 [DOI] [PubMed] [Google Scholar]
- Hume, B. C. C. , D’Angelo, C. , Burt, J. A. , & Wiedenmann, J. (2018) Fine‐scale biogeographical boundary delineation and sub‐population resolution in the Symbiodinium thermophilum coral symbiont group from the Persian/Arabian Gulf and Gulf of Oman. Frontiers in Marine Science, 5 10.3389/fmars.2018.00138 [DOI] [Google Scholar]
- Hume, B. , D’Angelo, C. , Burt, J. , Baker, A. C. , Riegl, B. , & Wiedenmann, J. (2013). Corals from the Persian/Arabian Gulf as models for thermotolerant reef‐builders: Prevalence of clade C3 Symbiodinium, host fluorescence and ex situ temperature tolerance. Marine Pollution Bulletin, 72, 313–322. 10.1016/j.marpolbul.2012.11.032 [DOI] [PubMed] [Google Scholar]
- Hume, B. C. C. , D'Angelo, C. , Smith, E. g. , Stevens, J. r. , Burt, J. , & Wiedenmann, J. (2015). Symbiodinium thermophilum sp nov., a thermotolerant symbiotic alga prevalent in corals of the world's hottest sea, the Persian/Arabian. Gulf. Scientific Reports, 5, 8562 10.1038/srep08562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume, B. C. C. , Voolstra, C. R. , Arif, C. , D’Angelo, C. , Burt, J. A. , Eyal, G. , … Wiedenmann, J. (2016). Ancestral genetic diversity associated with the rapid spread of stress‐tolerant coral symbionts in response to Holocene climate change. Proceedings of the National Academy of Sciences of the United States of America, 113, 4416–4421. 10.1073/pnas.1601910113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume, B. C. C. , Ziegler, M. , Poulain, J. , Pochon, X. , Romac, S. , Boissin, E. , … Voolstra, C. R. (2018). An improved primer set and amplification protocol with increased specificity and sensitivity targeting the Symbiodinium ITS2 region. Peerj, 6, e4816 10.7717/peerj.4816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong, H. J. , Lee, S. Y. , Kang, N. S. , Yoo, Y. D. , Lim, A. S. , Lee, M. J. , … LaJeunesse, T. C. (2014). Genetics and morphology characterize the dinoflagellate Symbiodinium voratum, n. sp., (Dinophyceae) as the sole representative of Symbiodinium Clade E. Journal of Eukaryotic Microbiology, 61, 75–94. 10.1111/jeu.12088 [DOI] [PubMed] [Google Scholar]
- Katoh, K. , & Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution, 30, 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp, D. W. , Hernandez‐Pech, X. , Iglesias‐Prieto, R. , Fitt, W. K. , & Schmidt, G. W. (2014). Community dynamics and physiology of Symbiodinium spp. before, during, and after a coral bleaching event. Limnology and Oceanography, 59, 788–797. 10.4319/lo.2014.59.3.0788 [DOI] [Google Scholar]
- Krueger, T. , & Gates, R. D. (2012). Cultivating endosymbionts – Host environmental mimics support the survival of Symbiodinium C15 ex hospite . Journal of Experimental Marine Biology and Ecology, 413, 169–176. 10.1016/j.jembe.2011.12.002 [DOI] [Google Scholar]
- LaJeunesse, T. C. (2001). Investigating the biodiversity, ecology, and phylogeny of endosymbiotic dinoflagellates in the genus Symbiodinium using the its region: In search of a "species" level marker. Journal of Phycology, 37, 866–880. 10.1046/j.1529-8817.2001.01031.x [DOI] [Google Scholar]
- LaJeunesse, T. C. (2002). Diversity and community structure of symbiotic dinoflagellates from Caribbean coral reefs. Marine Biology, 141, 387–400. 10.1007/s00227-002-0829-2 [DOI] [Google Scholar]
- LaJeunesse, T. C. (2017). Validation and description of Symbiodinium microadriaticum, the type species of Symbiodinium (Dinophyta). Journal of Phycology, 53, 1109–1114. 10.1111/jpy.12570 [DOI] [PubMed] [Google Scholar]
- LaJeunesse, T. C. , Parkinson, J. E. , Gabrielson, P. W. , Jeong, H. J. , Reimer, J. D. , Voolstra, C. R. , & Santos, S. R. (2018). Systematic revision of Symbiodiniaceae highlights the antiquity and diversity of coral endosymbionts. Current Biology, 28, 2570–2580. 10.1016/j.cub.2018.07.008 [DOI] [PubMed] [Google Scholar]
- Lajeunesse, T. C. , Parkinson, J. E. , & Reimer, J. D. (2012). A genetics‐based description of Symbiodinium minutum sp. nov. and S. psygmophilum sp. nov. (Dinophyceae), two dinoflagellates symbiotic with cnidaria. Journal of Phycology, 48, 1380–1391. 10.1111/j.1529-8817.2012.01217.x [DOI] [PubMed] [Google Scholar]
- LaJeunesse, T. C. , Pettay, D. T. , Sampayo, E. M. , Phongsuwan, N. , Brown, B. , Obura, D. O. , … Fitt, W. K. (2010). Long‐standing environmental conditions, geographic isolation and host‐symbiont specificity influence the relative ecological dominance and genetic diversification of coral endosymbionts in the genus Symbiodinium . Journal of Biogeography, 37, 785–800. 1111/j.1365-2699.2010.02273.x [Google Scholar]
- LaJeunesse, T. C. , & Thornhill, D. J. (2011). Improved resolution of reef‐coral endosymbiont (Symbiodinium) species diversity, ecology, and evolution through psbA non‐coding region genotyping. PLoS ONE, 6, e29013 10.1371/journal.pone.0029013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaJeunesse, T. C. , Wham, D. C. , Pettay, D. T. , et al. (2014). Ecologically differentiated stress‐tolerant endosymbionts in the dinoflagellate genus Symbiodinium (Dinophyceae) Clade D are different species. Phycologia, 53, 305–319. 10.2216/13-186.1 [DOI] [Google Scholar]
- Lozupone, C. , Lladser, M. E. , Knights, D. , Stombaugh, J. , & Knight, R. (2011). UniFrac: An effective distance metric for microbial community comparison. ISME Journal, 5, 169–172. 10.1038/ismej.2010.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norström, A. V. , Nyström, M. , Jouffray, J.‐B. , Folke, C. , Graham, N. A. J. , Moberg, F. , … Williams, G. J. (2016). Guiding coral reef futures in the Anthropocene. Frontiers in Ecology and the Environment, 14, 490–498. 10.1002/fee.1427 [DOI] [Google Scholar]
- Ochsenkuhn, M. A. , Rothig, T. , D’Angelo, C. , Wiedenmann, J. , & Voolstra, C. R. (2017). The role of floridoside in osmoadaptation of coral‐associated algal endosymbionts to high‐salinity conditions. Science Advances, 3, e1602047 10.1126/sciadv.1602047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettay, D. T. , Wham, D. C. , Pinzon, J. H. , & Lajeunesse, T. C. (2011). Genotypic diversity and spatial‐temporal distribution of Symbiodinium clones in an abundant reef coral. Molecular Ecology, 20, 5197–5212. 10.1111/j.1365-294X.2011.05357.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettay, D. T. , Wham, D. C. , Smith, R. T. , Iglesias‐Prieto, R. , & LaJeunesse, T. C. (2015). Microbial invasion of the Caribbean by an Indo‐Pacific coral zooxanthella. Proceedings of the National Academy of Sciences, 112, 7513–7518. 10.1073/pnas.1502283112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciani, N. , de Lossio e Seiblitz, I. G. , de Paiva, P. C. , e Castro, C. B. , & Zilberberg, C. (2016). Geographic patterns of Symbiodinium diversity associated with the coral Mussismilia hispida (Cnidaria, Scleractinia) correlate with major reef regions in the Southwestern Atlantic Ocean. Marine Biology, 163, 236 10.1007/s00227-016-3010-z [DOI] [Google Scholar]
- Pochon, X. , Putnam, H. M. , & Gates, R. D. (2014) Multi‐gene analysis of Symbiodinium dinoflagellates: A perspective on rarity, symbiosis, and evolution. Peerj, 2, e394 10.7717/peerj.394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan, R. (2004). Coral bleaching – Thermal adaptation in reef coral symbionts. Nature, 430, 742–742. 10.1038/430742a [DOI] [PubMed] [Google Scholar]
- Rowan, R. , Knowlton, N. , Baker, A. , & Jara, J. (1997). Landscape ecology of algal symbionts creates variation in episodes of coral bleaching. Nature, 388, 265–269. 10.1038/40843 [DOI] [PubMed] [Google Scholar]
- Sampayo, E. M. , Dove, S. , & Lajeunesse, T. C. (2009). Cohesive molecular genetic data delineate species diversity in the dinoflagellate genus Symbiodinium . Molecular Ecology, 18, 500–519. [DOI] [PubMed] [Google Scholar]
- Santos, S. R. , Taylor, D. J. , & Coffroth, M. A. (2001). Genetic comparisons of freshly isolated versus cultured symbiotic dinoflagellates: Implications for extrapolating to the intact symbiosis. Journal of Phycology, 37, 900–912. 10.1046/j.1529-8817.2001.00194.x [DOI] [Google Scholar]
- Schloss, P. D. , Westcott, S. L. , Ryabin, T. , Hall, J. R. , Hartmann, M. , Hollister, E. B. , … Weber, C. F. (2009). Introducing mothur: Open‐source, platform‐independent, community‐supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75, 7537–7541. 10.1128/AEM.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheneman, L. , Evans, J. , & Foster, J. A. (2006). Clearcut: A fast implementation of relaxed neighbor joining. Bioinformatics, 22, 2823–2824. 10.1093/bioinformatics/btl478 [DOI] [PubMed] [Google Scholar]
- Silverstein, R. N. , Cunning, R. , & Baker, A. C. (2015). Change in algal symbiont communities after bleaching, not prior heat exposure, increases heat tolerance of reef corals. Global Change Biology, 21, 236–249. 10.1111/gcb.12706 [DOI] [PubMed] [Google Scholar]
- Smith, E. G. , Hume, B. C. C. , Delaney, P. , Wiedenmann, J. , & Burt, J. A. (2017a). Genetic structure of coral‐Symbiodinium symbioses on the world’s warmest reefs. PLoS ONE, 12, e0180169 10.1371/journal.pone.0180169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, E. G. , Ketchum, R. N. , & Burt, J. A. (2017b). Host specificity of Symbiodinium variants revealed by an ITS2 metahaplotype approach. The ISME Journal, 11, 1500–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, E. G. , Vaughan, G. O. , Ketchum, R. N. , McParland, D. , & Burt, J. A. (2017c). Symbiont community stability through severe coral bleaching in a thermally extreme lagoon. Scientific Reports, 7, 2428 10.1038/s41598-017-01569-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stackebrandt, E. , & Goebel, B. M. (1994). A place for DNA‐DNA reassociation and 16s ribosomal‐RNA sequence‐analysis in the present species definition in bacteriology. International Journal of Systematic Bacteriology, 44, 846–849. 10.1099/00207713-44-4-846 [DOI] [Google Scholar]
- Tamura, K. , Stecher, G. , Peterson, D. , Filipski, A. , & Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 30, 2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, L. , Kendrick, G. A. , Kennington, W. J. , Richards, Z. T. , & Stat, M. (2014). Exploring Symbiodinium diversity and host specificity in Acropora corals from geographical extremes of Western Australia with 454 amplicon pyrosequencing. Molecular Ecology, 23, 3113–3126. 10.1111/mec.12801 [DOI] [PubMed] [Google Scholar]
- Thornhill, D. J. , Howells, E. J. , Wham, D. C. , Steury, T. D. , & Santos, S. R. (2017). Population genetics of reef coral endosymbionts (Symbiodinium, Dinophyceae). Molecular Ecology, 26, 2640–2659. 10.1111/mec.14055 [DOI] [PubMed] [Google Scholar]
- Thornhill, D. J. , Lajeunesse, T. C. , & Santos, S. R. (2007). Measuring rDNA diversity in eukaryotic microbial systems: How intragenomic variation, pseudogenes, and PCR artifacts confound biodiversity estimates. Molecular Ecology, 16, 5326–5340. 10.1111/j.1365-294X.2007.03576.x [DOI] [PubMed] [Google Scholar]
- Thornhill, D. J. , Lewis, A. M. , Wham, D. C. , & LaJeunesse, T. C. (2014). Host‐specialist lineages dominate the adaptive radiation of reef coral endosymbionts. Evolution, 68, 352–367. 10.1111/evo.12270 [DOI] [PubMed] [Google Scholar]
- Thornhill, D. J. , Xiang, Y. , Fitt, W. K. , & Santos, S. R. (2009). Reef endemism, host specificity and temporal stability in populations of symbiotic dinoflagellates from two ecologically dominant Caribbean corals. PLoS ONE, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varasteh, T. , Shokri, M. R. , Rajabi‐Maham, H. , Behzadi, S. , & Hume, B. C. (2018). Symbiodinium thermophilum symbionts in Porites harrisoni and Cyphastrea microphthalma in the northern Persian Gulf, Iran. Journal of the Marine Biological Association of the United Kingdom, 98, 2067–2073. 10.1017/s0025315417001746 [DOI] [Google Scholar]
- Vetrovsky, T. , & Baldrian, P. (2013). The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS ONE, 8, e57923 10.1371/journal.pone.0057923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voolstra, C. R. , Schwarz, J. A. , Schnetzer, J. , Sunagawa, S. , Desalvo, M. K. , Szmant, A. M. , … Medina, M. (2009). The host transcriptome remains unaltered during the establishment of coral‐algal symbioses. Molecular Ecology, 18, 1823–1833. 10.1111/j.1365-294X.2009.04167.x [DOI] [PubMed] [Google Scholar]
- Wham, D. C. , Ning, G. , & LaJeunesse, T. C. (2017). Symbiodinium glynnii sp nov., a species of stress‐tolerant symbiotic dinoflagellates from pocilloporid and montiporid corals in the Pacific Ocean. Phycologia, 56, 396–409. 10.2216/16-86.1 [DOI] [Google Scholar]
- Ziegler, M. , Arif, C. , Burt, J. A. , Dobretsov, S. , Roder, C. , LaJeunesse, T. C. , & Voolstra, C. R. (2017). Biogeography and molecular diversity of coral symbionts in the genus Symbiodinium around the Arabian Peninsula. Journal of Biogeography, 44, 674–686. 10.1111/jbi.12913 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data from the original Smith, Vaughan et al. (2017c) study are deposited in the Dryad Digital Repository (http://dx.doi.org/10.5061/dryad.h6s54). The data used to create Figure 3 are available through the SymPortal wiki hosted on GitHub: https://github.com/didillysquat/SymPortal_framework/wiki. The largely programmatic process used to make figures 2–5 is documented here: https://github.com/didillysquat/sp_ms_figure_creation. Version 0.2.2 of the SymPortal framework's source code has been archived at https://doi.org/10.5281/zenodo.2552178.