

Abstract
Guanosine penta‐ or tetraphosphate (pppGpp or ppGpp, respectively) is a nucleotide signalling molecule with a marked effect on bacterial physiology during stress. Its accumulation slows down cell metabolism and replication, supposedly leading to the formation of the antibiotic‐tolerant persister phenotype. A specifically tailored fluorescent chemosensor, PyDPA, allows the detection of (p)ppGpp in solution with high selectivity, relative to that of other nucleotides. Herein, an optimised synthetic approach is presented that improves the overall yield from 9 to 67 % over 7 steps. The simplicity and robustness of this approach will allow groups investigating the many facets of (p)ppGpp easy access to this probe.
Keywords: fluorescence, guanosine phosphates, nucleotides, sensors, synthesis design
Introduction
Molecular recognition and sensing of nucleotides has been an active research field over recent decades due to their biological significance.1 In addition to the well‐known DNA, RNA and co‐factor nucleotides NAD (nicotinamide adenine dinucleotide and FAD (flavin adenine dinucleotide), several nucleotide‐related molecules, such as cyclic diguanylate (c‐di‐GMP), cyclic adenosine monophosphate (cAMP), diadenosine tetraphosphate (Ap4A) and guanosine penta‐ or tetraphosphate (pppGpp or ppGpp, respectively), are being actively investigated for their role within second messenger signalling systems. These molecules are produced by bacteria, along with quorum sensing (QS) autoinducers, in response to environmental stimuli and, in turn, lead to a variety of phenotypic changes that allow bacteria to survive or even proliferate. In the post‐antibiotic era,2 it is of outmost importance to acquire a detailed understanding of how bacteria respond to stress to set out effective strategies not only to prevent the insurgence of antibiotic resistance, but also to eradicate recurrent and chronic infections.
Dissecting each signalling pathway requires the selective detection of each such signalling molecule in a milieu overpopulated with many other, structurally similar, compounds.
We decided to focus our attention on ppGpp or pppGpp, collectively known as (p)ppGpp, a signalling alarmone produced in response to stress conditions3 (e.g., heat shock, nutrient starvation). Discovered in 1969 by Cashel and Gallant,4 they were initially nicknamed magic spot I and II, but, even after their structures were elucidated (1; Figure 1), the nickname lingered due to the complexity of the pleiotropic effects these molecules have on bacterial physiology.5 Indeed, (p)ppGpp impacts on transcription, translation and DNA replication,3 and generates virulence factors by interfering with QS networks.6 Accumulation of (p)ppGpp is also upstream of the stringent response, a signalling cascade implicated in the formation of a dormant bacterial phenotype called persister.7 This phenotype, transiently tolerant to antibiotic treatment, is not only largely responsible for the difficulties encountered in eradicating recurrent and chronic infections, but also favours the insurgence of resistant strains.
Figure 1.

Left) Structure of the nucleotide signalling molecule (p)ppGpp and of its specific chemosensor PyDPA. Right) Structure of the 2:1 PyDPA/ppGpp complex.
In the course of our research project on small molecules able to control the onset of the persistent phenotype by interfering with the stringent response pathway, selective detection of (p)ppGpp has become a critical factor. Detection of (p)ppGpp in solution historically relied on radiolabelled compounds (either with 3H or 32P)8 or HPLC methods.9 Only in recent years has a fluorescent chemosensor (PyDPA; Figure 1)—a compound bearing a binding moiety connected to and communicating with a fluorophore10—been specifically designed to bind selectively (p)ppGpp over other nucleotides, such as ATP, GTP, UTP, TTP, cAMP and cGMP.11
From a structural point of view, PyDPA comprises two Zn2+–dipicolylamine (Zn2+‐DPA) units, which are well known for their ability to bind pyrophosphate groups in water,12 bridged to a pyrene moiety through an alkyloxy phthalate. One molecule of (p)ppGpp, with its two terminal pyrophosphate groups, is able to chelate two molecules of PyDPA, thus forcing the proximity of the corresponding pyrene units, which are exploited for their distinctive excimer emission (λ em=470 nm).13 Quantification of (p)ppGpp in solution is therefore possible with an appropriate calibration curve up to the low‐micromolar range.
The synthesis of PyDPA, as originally reported in 2008 by Rhee and co‐workers,11 was achieved in a modest yield of about 9 % over 6 steps, starting from 1‐bromopyrene (2; Scheme 1), whereas a marginal improvement to the overall yield was reported a few years later by the same group,14 starting from 1‐pyrenecarboxaldehyde (19 % over 8 steps). Herein, we report an optimised synthetic sequence that overcomes the critical steps of the original and modified synthetic approaches to increase the overall yield from 9–19 to 67 % over 7 steps.
Scheme 1.

Reported synthesis of PyDPA, according to ref. 11.
Results and Discussion
The synthesis of Rhee et al. is outlined in Scheme 1. Halogen–lithium exchange on 2 (nBuLi), followed by treatment with 1,3‐dibromopropane yields the monoarylation product 3, which is used to alkylate 5‐hydroxyisophthalic acid dimethyl ester. Ester reduction (LiAlH4) and reaction of the resulting diol with PBr3 affords dibromide 4. Alkylation of DPA with 4 affords the structure of the ligand, which is finally transformed in the Zn complex upon treatment with ZnClO4. All steps are described to proceed in good to excellent yields, except for the first one, which proceeds with a modest yield of 18 %, thus undermining the whole synthetic sequence.
To improve on these results, we envisaged exploiting the reactivity of the C−Br bond of 2 to perform a palladium‐catalysed coupling reaction with dimethyl‐5‐propargyloxy‐isophthalate (5; Scheme 2), under Sonogashira conditions. Different Pd sources ([PdCl2(PPh3)2] or [Pd(PPh3)4]), in the presence or absence of copper salts (CuI), different solvents (e.g., tertiary amines, THF, DMF) and a range of reaction temperatures (from 40 to 90 °C) were explored without success. Indeed, the high activation temperatures required to activate the C−Br bond were not compatible with the thermal instability of alkyne 5 and either no conversion of the starting material or alkyne decomposition was observed.
Scheme 2.

Palladium‐mediated coupling of 1‐iodopyrene (6) with isophthalate 5. TEA: triethylamine.
The use of more reactive 1‐iodopyrene (6)15 allowed desired product 7 to be obtained under mild conditions, although in modest yield (42 %), but it also introduced an additional step to the synthetic sequence, which reduced the overall yield to 35 % (Scheme 2).
Finally, a sequential approach was adopted to introduce the alkynyl chain first and form the ether linkage at a later stage. Although classical Sonogashira conditions are described as being effective for the reaction of propargyl alcohol (8) with 2,16 in our hands only copper‐free conditions, with [Pd(PPh3)4] as a catalyst in nBuNH2 at reflux, gave the desired product 9 in excellent yield (94 %; Scheme 3).17 It is worth noting that the same reaction conditions applied to phthalate 5 only resulted in alkyne decomposition and a mixture of unidentified by‐products. Catalytic reduction of the triple bond of 9 has been described with PtO2 (15 mol %) in THF,18 but we found that much cheaper Pd/C in methanol worked just as well, with a lower catalyst loading (5 mol %) and shorter reaction times (20 min); thus resulting in an overall dramatic decrease in the reaction cost.
Scheme 3.
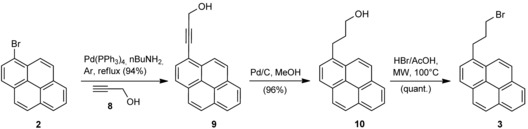
Optimised approach for the preparation of 1‐(3‐bromopropyl)pyrene (3). MW: microwave.
Finally, the reaction of alcohol 10 with HBr/AcOH under MW irradiation, as described in ref. 19 with modifications, proceeded smoothly to afford bromide 3 in quantitative yield (Scheme 3). Alternative conditions involving the use of PBr3 for functional group transformation provided lower yields and complicated the reaction workup. Thus, compared with the reported procedure, we obtained the same intermediate 3 in an overall yield of 92 %, as opposed to 18 %. Two additional steps are required, but only one chromatographic purification is involved over the three steps.
At this stage, to increase the convergence of the synthetic path and to skip the carboxymethyl ester reduction step that was described to proceed in 68 % yield (Scheme 1), triol 11 was used for the synthesis of ether 13, exploiting the lower pK a of the phenol moiety (Scheme 4). The triol could be obtained in almost quantitative yield through the reduction of commercially available dimethyl 5‐hydroxyisophthalate (12) with LiAlH4.19 Alkylation of 11 with 3 was best performed with an excess of K2CO3 and one equivalent of KI in acetonitrile at reflux. Under these conditions, ether 13 was isolated in 95 % yield, after 17 h.
Scheme 4.

Preparation of compound 13 through the alkylation of phenol 11 with compound 3 and the final steps of the synthesis of PyDPA.
The final steps of the synthetic process were reproduced as previously described, with comparable yields (Scheme 4). Diol 13 was therefore treated with PBr3 to afford dibromide 4 (89 %), which gave 14 upon reaction with excess bis(2‐picolyl)amine (88 %). Treatment of 14 with Zn(ClO4)2 ⋅6 H2O allowed chemosensor PyDPA, which was used for (p)ppGpp detection without further purification.
Spectral data
The spectral properties of PyDPA, including absorption and fluorescence spectra, in the presence of ppGpp were consistent with data reported in the literature for the PyDPA chemosensor (Figure 2).
Figure 2.

Fluorescence emission spectra of PyDPA (20 μm in 1 mm HEPES pH 7.5) alone and upon the addition of each nucleotide and ppGpp (7 μm). λ ex=344 nm.
Indeed, whereas the presence of ppGpp led to the distinctive excimer emission band at λ=470 nm, the presence of other nucleotides, such as AMP, GDP or ATP, only showed the monomer emission bands.
Conclusion
We have been able to streamline the synthesis of the PyDPA chemosensor, overcoming the critical steps of the original synthetic approach and increasing the overall yield from 9 to 67 % from the same starting material (2). We believe that this approach will be easily reproducible and useful for many groups throughout the world that are investigating the biological activity of the still puzzling nucleotide signalling molecule (p)ppGpp.
Experimental Section
General: Chemicals were purchased from commercial sources and used without further purification, unless otherwise indicated. If anhydrous conditions were required, the reactions were performed under nitrogen or argon. Anhydrous solvents were purchased from Merck. Reactions were monitored by means of analytical TLC performed on silica gel 60 F254 plates (Merck) with UV detection (λ=254 nm) and/or staining with a solution of ammonium molybdate acid or alkaline potassium permanganate. Silica gel 60 (40–63 μm; Merck) was used for flash column chromatography. NMR spectroscopy measurements were recorded on a Bruker AVANCE‐400 MHz instrument at 298 K. Chemical shifts are reported in ppm. The 1H and 13C NMR resonances of compounds were assigned with the assistance of COSY and HSQC experiments. Multiplicity is assigned as s (singlet), d (doublet), t (triplet), q (quartet), quint (quintet) and m (multiplet). Mass spectra were recorded on Apex II ICR FTMS (ESI ionisation HRMS), Waters Micromass Q‐TOF (ESI ionisation HRMS) or ThermoFischer LCQ instruments (ESI ionisation). Compound 11 was prepared as described in ref. 19.
Synthesis of 3‐Pyren‐1‐yl‐prop‐2‐yn‐1‐ol (9): 1‐Bromopyrene (2; 100 mg, 0.35 mmol) was dissolved with [Pd(PPh3)4] (13 mg, 0.0105 mmol) in nBuNH2 (12 mL) degassed with Ar. Compound 8 (102 μL, 1.78 mmol) was added to the mixture and the reaction was left to stir at reflux. After 3 h the reaction was complete (TLC: hexane/EtOAc 2:1). The solvent was evaporated under reduced pressure and the crude product was purified by means of automated flash chromatography (hexane/EtOAc gradient from 92:8 to 40:60). Product 9 was obtained as a slightly yellow solid (85.1 mg, 95 %). Spectral data matched those reported in ref. 17. 1H NMR (400 MHz, CDCl3): δ=8.55 (d, 3 J=9.2 Hz, 1 H; H3Ar), 8.25–8.19 (m, 2 H; H6Ar, H8Ar), 8.19–8.15 (m, 2 H; H5Ar, H9Ar), 8.14–8.01 (m, 4 H; H2Ar, H4Ar, H7Ar, H10Ar), 4.52 (d, 3 J=6.5 Hz, 2 H; CH2OH), 1.82 ppm (t, 1 H; OH); 13C NMR (100 MHz, CDCl3): δ=132.2 (C1), 131.5 (C5a), 131.3 (C8a), 131.1 (C3a), 129.9 (C10a), 128.5 (C2), 128.4 (C9), 127.3 (C5), 126.4 (C4), 126.2 (C7), 126.9 (C5a′), 126.7 (C3a′), 125.7 (C6), 125.5 (C8), 124.5 (C10), 124.4 (C3), 92.9 (C≡CH2OH), 52.2 (CH2OH), 84.9 ppm (CCH2OH); MS (ESI): m/z: calcd for [C19H12ONa]+: 279.08, found: 279.28.
Synthesis of 3‐(pyren‐1‐yl)propan‐1‐ol (10): Compound 9 (184 mg, 0.72 mmol) was dissolved in freshly distilled MeOH (24 mL). 10 % Pd/C (38 mg, 0.04 mmol) was added and the reaction mixture was stirred under hydrogen (1 atm) for 20 min (TLC: hexane/AcOEt 6:4). The catalyst was removed by filtration through Celite and the solvent was evaporated under reduced pressure to give pure product 10 (179 mg, 96 %). Spectral data matched those reported in ref. 20. 1H NMR (400 MHz, CDCl3): δ=8.32 (d, 3 J=9.4 Hz, 1 H; H3Ar), 8.19–8.15 (dd, 3 J=7.5 Hz, 2 H; H6Ar, H8Ar), 8.14–8.09 (dd, 3 J=7.9 Hz, 2 H; H2Ar, H9Ar), 8.06–8.01 (m, 2 H; H4Ar, H5Ar), 7.99 (t, 1 H; H7Ar), 7.90 (d, 1 H; H10Ar), 3.80 (t, 3 J=6.1 Hz, 2 H; CH 2OH), 3.50–3.43 (m, 2 H; ArCH 2), 2.18–2.10 ppm (m, 2 H; CH 2CH2OH); 13C NMR (100 MHz, CDCl3): δ=136.3 (C1), 131.6 (C5a), 131.0 (C8a), 131.1 (C3a), 128.8 (C10a), 127.7 (C2), 127.6 (C9), 127.5 (C5), 127.4 (C4), 126.2 (C7), 125.4 (C5a′), 125.3 (C3a′), 125.1 (C6), 125.0 (C8), 124.9 (C10), 123.4 (C3), 62.6 (CH2OH), 34.7 (ArCH2), 29.8 ppm (CH2‐CH2OH); MS (ESI): m/z: [C19H16ONa]+: 283.10; found: 282.96.
Synthesis of 1‐(3‐bromopropyl)pyrene (3): 33 % HBr in AcOH (0.8 mL) was added to alcohol 10 (179 mg, 0.68 mmol) in a microwave vial. The reaction was stirred under MW irradiation at 100 °C for 45 min (TLC: hexane/AcOEt 7:3). The reaction mixture was diluted with EtOAc (15 mL) and the organic phase was washed with a 50 % solution of NaHCO3 (3×15 mL) and water (1×15 mL), and then dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure to give pure product 3 as a brown viscous oil (218 mg, quant.) Spectral data matched those reported in the literature.20 1H NMR (400 MHz, CDCl3): δ=8.29 (d, 3 J=9.2 Hz, 1 H; H3Ar), 8.20–8.15 (dd, 3 J=7.6 Hz, 2 H; H6Ar, H8Ar), 8.15–8.10 (dd, 3 J=7.7 Hz, 2 H; H2Ar, H9Ar), 8.03 (s, 2 H; H4Ar, H5Ar), 7.99 (t, 1 H; H7Ar), 7.90 (d, 1 H; H10Ar), 3.56–347 (m, 4 H; CH 2OH, ArCH 2), 2.41 ppm (quint, 3 J=6.9 Hz, 2 H; CH 2CH2OH); 13C NMR (100 MHz, CDCl3): δ=134.9 (C1), 131.6 (C5a), 131.3 (C3a), 131.0 (C8a), 128.8 (C10a), 127.7 (C2), 127.6 (C9), 127.0 (C5), 126.0 (C4), 125.2 (C7), 125.1 (C6), 125.0 (C8, C5a′), 124.9 (C10), 123.4 (C3), 123.3 (C3a′), 34.7 (H2 C‐CH2Br), 33.6 (CH2Br), 31.8 ppm (ArCH2); MS (ESI): m/z: calcd for [C19H16OBr]+: 323.04; found: 323.03.
Synthesis of {5‐[3‐(pyren‐1‐yl)propoxy]‐1,3‐phenylene}dimethanol (13): Compounds 3 (100 mg, 0.31 mmol), 11 (57.2 mg, 0.37 mmol), oven‐dried K2CO3 (128 mg, 0.93 mmol) and KI (56 mg, 0.34 mmol) were dissolved in dry CH3CN (2 mL, 0.15 m). The reaction mixture was stirred at reflux under MW irradiation for 17 h (TLC: CH2Cl2/MeOH 95:5). The solvent was evaporated under reduced pressure and the resulting brown solid was dissolved in EtOAc (15 mL). The solution was washed with water (3×5 mL) and brine (1×15 mL). The organic phase was dried over anhydrous MgSO4 and the solvent was evaporated under reduced pressure. The crude was purified by means of automated flash chromatography (95:5 CH2Cl2/MeOH) to afford pure 13 (116.3 mg, 95 %). Spectral data matched those previously reported.11 1H NMR (400 MHz, CDCl3): δ=8.32 (d, 3 J=9.6 Hz, 1 H; H3Ar), 8.17 (d, 3 J=7.6 Hz, 2 H; H6Ar, H8Ar), 8.13–8.07 (dd, 3 J=7.7 Hz, 2 H; H2Ar, H9Ar), 8.03 (s, 2 H; H4Ar, H5Ar), 7.99 (t, 1 H; H7Ar), 7.90 (d, 1 H; H10Ar), 6.95 (s, 1 H; H p‐Ph), 6.87 (s, 2 H; H o‐Ph), 4.67 (s, 4 H; CH 2OH), 4.07 (t, 3 J=6.1 Hz, 2 H; CH 2OPh), 3.56 (t, 3 J=7.4 Hz, 2 H; ArCH 2), 2.35 ppm (quint, 2 H; ArCH2CH 2); 13C NMR (100 MHz, CDCl3): δ=159.7 (Cq‐OCH2), 142.9 (2×Cq), 135.9 (C1), 131.6 (C5a), 131.0 (C8a), 130.1 (C3a), 128.9 (C10a), 127.6 (C2), 127.5 (C9), 127.4 (C5), 126.8 (C4), 126.0 (C7), 125.3 (C5a′), 125.1 (C3a′), 125.0 (C6, C8), 124.9 (C10), 123.5 (C3), 117.6 (CHp‐Ph), 112.4 (2×CHo‐Ph), 67.1 (CH2OPh), 65.3 (2×CH2OH), 31.3 (ArCH2 CH2), 29.9 ppm (ArCH2); MS (ESI): m/z: calcd for [C27H24O3Na]+: 419.16; found: 419.64.
Chemosensor PyDPA: 1H NMR (400 MHz, CD3CN): δ=8.76–8.72 (d, 3 J=5.3 Hz, 4 H; CH‐o‐Py), 8.37 (d, 3 J=9.2 Hz, 1 H; H3Ar), 8.21–8.09 (m, 3 H; H6Ar, H8Ar, H9Ar), 8.07–7.90 (m, 9 H; H2Ar, H4Ar, H5Ar, H7Ar, H10Ar, CH‐p‐Py), 7.67 (t, 3 J=6.3 Hz, 4 H; CH‐m‐Py), 7.33 (d, 3 J=7.8 Hz, 4 H; CH‐m′‐Py), 6.73 (s, 2 H; H o‐Ph), 6.71 (s, 1 H; H p‐Ph), 4.16 (d, Jgem=16 Hz, 4 H; PyCH 2N), 4.02 (t, 3 J=5.5 Hz, 2 H; ArCH2CH2CH 2O), 3.82 (s, 4 H; NCH 2Ph,), 3.64 (d, 4 H; PyCH 2N), 3.61 (t, 3 J=7.3 Hz, 2 H; ArCH 2CH2CH2O), 2.36 ppm (quint, 3 J=6.9 Hz, 2 H; ArCH2CH 2CH2O); 13C NMR (100 MHz, CD3CN): δ=160.4 (Cq‐OCH2), 155.4 (Cq Py), 149.1 (Co Py), 143.0 (Cp Py), 137.3 (2x Cq Ph), 134.3 (C1), 132.3 (C5a), 131.8 (C8a), 130.9 (C3a), 129.8 (C10a), 129.0 (C2), 128.5 (C9), 128.2 (C5), 127.7 (C4), 127.4 (2xCHo‐Ph), 127.2 (C7), 126.4 (CH‐m‐Py), 126.1 (C10), 126.0 (CH‐m′‐Py), 126.0 (C6), 125.8 (C8), 125.7 (C5a′), 125.5 (C3a′), 124.7 (C3), 119.0 (CHp‐Ph), 68.0 (ArCH2CH2 CH2OPh), 56.9 (PhCH2N), 55.8 (PyCH2N), 31.9 (ArCH2 CH2CH2OPh), 29.8 ppm (ArCH2CH2CH2OPh); MS: m/z: calcd for [C51H46N6OZn2 4++3 ClO4 −]+=[M 4++3ClO4 −]+: 1187.07; found: 1187.28.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Sara Sattin obtained her PhD under the supervision of Prof. Anna Bernardi at the Università degli Studi di Milano (Italy). After a postdoctoral stay in Prof. Javier de Mendoza's group at the ICIQ in Tarragona (Spain), she joined the group of Prof. Ben G. Davis at the University of Oxford. She returned to Milan to start her independent career working on the synthesis of allosteric modulators of the chaperone protein Hsp90. Since 2018, she has been Associate Professor in Organic Chemistry at the University of Milan. Her research focuses mainly on the design and synthesis of small molecules to hamper bacterial persister formation.

Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 758108). We thank Alessio Maria Caramiello for technical assistance.
G. Conti, M. Minneci, S. Sattin, ChemBioChem 2019, 20, 1717.
References
- 1. Zhou Y., Xu Z., Yoon J., Chem. Soc. Rev. 2011, 40, 2222–2235. [DOI] [PubMed] [Google Scholar]
- 2.WHO, Antimicrobial Resistance: Global Report on Surveillance, 2014; https://www.who.int/drugresistance/documents/surveillancereport/en/.
- 3. Potrykus K., Cashel M., Annu. Rev. Microbiol. 2008, 62, 35–51. [DOI] [PubMed] [Google Scholar]
- 4. Cashel M., Gallant J., Nature 1969, 221, 838–841. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Dalebroux Z. D., Swanson M. S., Nat. Rev. Microbiol. 2012, 10, 203–212; [DOI] [PubMed] [Google Scholar]
- 5b. Dalebroux Z. D., Svensson S. L., Gaynor E. C., Swanson M. S., Microbiol. Mol. Biol. Rev. 2010, 74, 171–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fontaine B. M., Duggal Y., Weinert E. E., ACS Infect. Dis. 2018, 4, 1645–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maisonneuve E., Gerdes K., Cell 2014, 157, 539–548. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Hamagishi Y., Yoshimoto A., Oki T., Arch. Microbiol. 1981, 130, 134–137; [Google Scholar]
- 8b. Heath R. J., Jackowski S., Rock C. O., J. Biol. Chem. 1994, 269, 26584–26590. [PubMed] [Google Scholar]
- 9. Fischer M., Zimmerman T. P., Short S. A., Anal. Biochem. 1982, 121, 135–139. [DOI] [PubMed] [Google Scholar]
- 10. Wu D., Sedgwick A. C., Gunnlaugsson T., Akkaya E. U., Yoon J., James T. D., Chem. Soc. Rev. 2017, 46, 7105–7123. [DOI] [PubMed] [Google Scholar]
- 11. Rhee H.-W., Lee C.-R., Cho S.-H., Song M.-R., Cashel M., Choy H. E., Seok Y.-J., Hong J.-I., J. Am. Chem. Soc. 2008, 130, 784–785. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Lee D. H., Kim S. Y., Hong J. I., Angew. Chem. Int. Ed. 2004, 43, 4777–4780; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 4881–4884; [Google Scholar]
- 12b. Jose D. A., Mishra S., Ghosh A., Shrivastav A., Mishra S. K., Das A., Org. Lett. 2007, 9, 1979–1982; [DOI] [PubMed] [Google Scholar]
- 12c. Das P., Chandar N. B., Chourey S., Agarwalla H., Ganguly B., Das A., Inorg. Chem. 2013, 52, 11034–11041. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Cho H. K., Lee D. H., Hong J. I., Chem. Commun. 2005, 1690–1692; [DOI] [PubMed] [Google Scholar]
- 13b. Nishizawa S., Kato Y., Teramae N., J. Am. Chem. Soc. 1999, 121, 9463–9464. [Google Scholar]
- 14. Oh J., Hong J. I., Org. Lett. 2013, 15, 1210–1213. [DOI] [PubMed] [Google Scholar]
- 15. Abe H., Mawatari Y., Teraoka H., Fujimoto K., Inouye M., J. Org. Chem. 2004, 69, 495–504. [DOI] [PubMed] [Google Scholar]
- 16. Angelova P., Vieker H., Weber N. E., Matei D., Reimer O., Meier I., Kurasch S., Biskupek J., Lorbach D., Wunderlich K., Chen L., Terfort A., Klapper M., Mullen K., Kaiser U., Golzhauser A., Turchanin A., ACS Nano 2013, 7, 6489–6497. [DOI] [PubMed] [Google Scholar]
- 17. Sarathi Addy P., Saha B., Pradeep Singh N. D., Das A. K., Bush J. T., Lejeune C., Schofield C. J., Basak A., Chem. Commun. 2013, 49, 1930–1932. [DOI] [PubMed] [Google Scholar]
- 18. Mizushima T., Yoshida A., Harada A., Yoneda Y., Minatani T., Murata S., Org. Biomol. Chem. 2006, 4, 4336–4344. [DOI] [PubMed] [Google Scholar]
- 19. Rasheed O. K., Lawrence A., Quayle P., Bailey P. D., Synlett 2016, 27, 905–911. [Google Scholar]
- 20. Garms C., Francke W. in Treatment of Contaminated Soil: Fundamentals, Analysis, Applications (Eds.: R. Stegmann, G. Brunner, W. Calmano, G. Matz), Springer, Berlin, 2001, pp. 95–131. [Google Scholar]