

Abstract
Background
Previous studies have shown that alternative splicing (AS) plays a key role in carcinogenesis and prognosis of cancer. However, systematic profiles of AS signatures in head and neck cancer (HNC) have not yet been reported.
Methods
In this study, AS data, RNA‐Seq data, and corresponding clinicopathological information of 489 HNC patients were downloaded from The Cancer Genome Atlas. Univariate and multivariate Cox regression analyses were performed to screen for survival‐associated AS events. Functional and pathway enrichment analysis was also performed. The prognostic models and splicing networks were constructed using integrated bioinformatics analysis tools.
Results
Among the 42,849 alternating splicing events identified in 10,121 genes, 5,165 survival‐associated AS events in 2,419 genes were observed in univariate Cox regression analysis. Among the seven types, alternate terminator events were the most powerful prognostic factors. Multivariate Cox analysis was then used to screen for the AS genes with prognostic value. Four candidate genes (TPM1, CLASRP, PRRC1, and DNASE1L1) were found to be independent prognostic factors for HNC patients. A prognostic prediction model was built based on the four genes. The area under the receiver operating characteristic risk score curve for predicting the survival status of HNC patients was 0.704. In addition, splicing interaction network indicated that the splicing factors have significant functions in HNC.
Conclusion
A comprehensive analysis of AS events in HNC was performed. A powerful prognostic predictor for HNC patients was established based on AS events could.
Keywords: alternative splicing, head and neck cancer, The Cancer Genome Atlas
1. INTRODUCTION
Head and neck cancer (HNC) is an aggressive form of cancer with a high mortality rate and a growing global incidence rate compared with all malignancies. In 2018, an estimated 51,540 new cases of HNC were diagnosed and an estimated 10,030 deaths were recorded in the United States alone, making HNC the 10th most common type of cancer worldwide and the seventh leading cause of cancer deaths (Mehanna, Paleri, West, & Nutting, 2010; Siegel, Miller, & Jemal, 2018; Woźniak, Szyfter, Szyfter, & Florek, 2012). Thus it has been recognized as a significant component of the global cancer burden (Jemal et al., 2008; Marur & Forastiere, 2008). HNC is a heterogeneous tumor characterized by lesions in the oral cavity, pharynx, and larynx (Arantes, de Carvalho, Melendez, Carvalho, & Goloni‐Bertollo, 2014). Despite some progress in treatment approaches, the 5‐year survival rates of HNC remain very low (Carvalho et al., 2008; Siegel et al., 2018). Elucidating the underlying mechanisms in the pathogenesis of HNC could help to uncover new biomarkers and therapeutic targets.
Alternative splicing (AS) of pre‐messenger RNA (mRNA) is a key process that produces mRNA isoforms from few sets of genes (Lee & Rio, 2015; Nilsen & Graveley, 2010). AS regulates gene function by inclusion or exclusion of different exons or parts of exons in mRNA, and therefore affect the activity of protein (Narayanan, Singh, & Shukla, 2017; Salton & Misteli, 2016). Emerging evidence points to a close relationship between AS and tumor occurrence (El Marabti & Younis, 2018; Kuranaga et al., 2018; Munkley, Livermore, Rajan, & Elliott, 2017; Paschalis et al., 2018). AS is known to be involved in the mechanisms of tumorigenesis and progression and plays an important role in the regulation of gene expression, especially for specific genes during development and progression of tumors (Klinck et al., 2008; Kozlovski, Siegfried, Amar‐Schwartz, & Karni, 2017). Differences in gene expression levels of splicing regulatory factors have been observed in many cancers, and these proteins often affect the splicing patterns of many genes that function in certain cancer‐specific biological pathways, including cell cycle progression, cellular proliferation, migration, and RNA processing (David & Manley, 2010; El Marabti & Younis, 2018; Oltean & Bates, 2014).
In the current study, integrated bioinformatics analysis of the AS and gene expression profiles was conducted to identify the splicing events that have prognostic value in HNC. The Univariate Cox proportional hazards regression and multivariate Cox proportional hazards regression models were performed to screen for the potential prognostic splicing events and genes, uncovering a number of survival‐associated AS events. The regulatory AS networks in HNC were constructed to elucidate the underlying mechanisms. Four characteristic genes (TPM1, CLASRP, PRRC1, and DNASE1L1) were identified to be molecular biomarkers that predict HNC.
2. MATERIALS AND METHODS
2.1. Data acquisition of AS events
RNA sequencing data (Level 3) of The Cancer Genome Atlas (TCGA) HNC cohort were obtained from the TCGA data portal (https://tcga‐data.nci.nih.gov/tcga/). Meanwhile, AS events were retrieved from the TCGA SpliceSep. The Percent Spliced In (PSI) value, ranging from zero to one, which is commonly used in quantifying AS events (Ryan, Cleland, Kim, & Wong, 2012), was imputed for seven different types of AS events. A total of 43 adjacent nontumor tissues and 500 HNC samples were included in the AS events, while 44 adjacent nontumor control samples and 502 HNC samples were enrolled in the gene methylation data set. Only patients with complete clinicopathological information and at least 30 days of overall survival (OS) were included in this study. After the initial screen, a total of 489 samples were considered eligible in the TCGA HNC cohort.
2.2. Identification of survival‐associated AS events
Seven common patterns of AS events, including exon skip (ES), mutually exclusive exons (ME), retained intron (RI), alternate promoter (AP), alternate terminator (AT), alternate donor site (AD), and alternate acceptor site (AA) are shown in Figure 1a.
Figure 1.
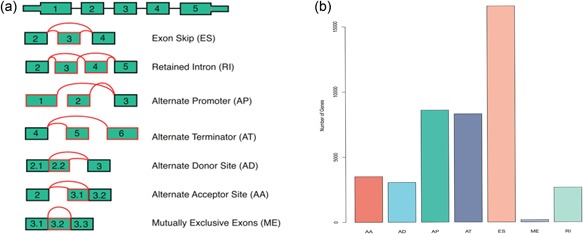
Illustrations of seven types of alternative splicing events in this study. (a) Representative model of seven types of alternative splicing, including Alternate Acceptor site (AA), Alternate Donor site (AD), Alternate Promoter (AP), Alternate Terminator (AT), Exon Skip (ES), Mutually Exclusive Exons (ME), and Retained Intron (RI). (b) Number of alternative splicing (AS) events and genes involved in 489 head and neck cancer (HNC) patients [Color figure can be viewed at wileyonlinelibrary.com]
A univariate Cox model was used to identify survival‐related AS events/mRNA using a threshold of a p < 0.05. The overlapping survival‐associated AS events were also selected for further analysis. To determine the prognostic AS events, their prognostic power was evaluated using the time‐dependent receiver operating characteristic (ROC) curves by comparing the sensitivity and specificity of the survival prediction for each model (Heagerty, Lumley, & Pepe, 2000).
2.3. UpSet plot and gene network interaction construction
For analyses of five or more sets, Upset plot (Lex, Gehlenborg, Strobelt, Vuillemot, & Pfister, 2014) was used for quantitative analysis of interactive sets between seven types of AS, rather than traditional Venn diagram, which presents the relationship between interactive sets. Moreover, gene interaction network for different types of genes in survival‐associated AS events was constructed by Cytoscape’s Reactome FI plugin (Wu, Feng, & Stein, 2010).
2.4. Functional and pathway enrichment analysis
The biological function and pathway of the prognostic spliced genes in HNC and normal samples revealed important information. Therefore, Gene Ontology (GO) analysis of the cellular component, molecular function, and biological process, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis via R package of clusterProfiler (Yu, Wang, Han, & He, 2012) were performed. p < 0.05 was considered to be statistically significant.
2.5. Construction of splicing correlation network
Splicing factors regulate AS by binding to pre‐mRNAs to affect exon selection and splicing site choice (Le, Squarize, & Castilho, 2014), whereas aberrant AS may be triggered by a few splicing factors (Kuranaga et al., 2018). This implies that survival‐associated AS events are potentially regulated by a few key splicing factors in HNC. Splicing factors were acquired from SpliceAid 2. First, splicing factors and gene expression profiles data were integrated and survival analysis was conducted to identify the prognostic splicing factors. Spearman correlation analysis was performed between splicing factor genes and the survival‐associated AS. p < 0.002 was considered to be statistically significant. A network of interaction between splicing factors and AS was generated by Cytoscape (version 3.5.1), software http://www.cytoscape.org/.
2.6. Construction of prognostic model
Multivariate Cox analysis was further used to determine whether the four prognostic genes were independent prognosis factors of a patient’s OS. The risk score model was constructed based on a linear integration of the expression level multiplied regression model (β) using the following formula: Risk score = β gene1 × exprgene1 + β gene2 × exprgene2 + ··· + β genen × exprgenen. All reported p values were two‐sided and all analysis were performed using R/Bioconductor (Version: 3.8; https://www.bioconductor.org/).
3. RESULTS
3.1. Comprehensive review of AS events in HNC cohort of TCGA
Integrated AS events were analyzed in 489 HNC patients data from TCGA. After analyzing raw data of HNC, a total of 42,849 AS events, including 3,500 AA in 2,481 genes, 3,049 AD in 2,148 genes, 8,598 AP in 3,807 genes, 8,309 AT in 3,628 genes, 16,572 ES in 6,439 genes, 174 ME in 173 genes, and 2,647 RI in 1,784 genes, were identified (see Figure 1b). Among the seven common AS events, ES was the most frequent type of AS, and accounted for more than a third of the AS events. ME was the least common type of AS. These findings demonstrate that one gene might have several AS patterns.
3.2. Identification of prognostic AS events and mRNAs
The univariate Cox regression analysis was used to screen for the survival‐associated AS events and mRNA. According to the result, a total of 5,165 AS events, including 2,419 genes, were significantly associated with OS based on the threshold/cut‐off point of the p < 0.05 (Figure 2a). Meanwhile, univariate Cox regression analysis of prognostic mRNAs revealed that there were 2,611 survival‐associated genes (Figure 2b). The common 453 genes were screen by overlapping 2,419 AS related genes and 2,611 mRNAs (Figure 2c). Because one single gene may contain two or more AS events, UpSet plot was generated to visualize the intersecting sets (Figure 3). Based on the plot, the number of survival associated with ES events was significantly reduced, while that of AP events was increased. These results suggest that most ES events are not associated with prognosis while AT events are significantly associated with survival. Based on these survival‐associated genes, gene interaction network generated by Reactome revealed important pathways including hub genes DYNLL1, SRSF2, SRSF7, and HSP90AA1 (Figure 3b).
Figure 2.
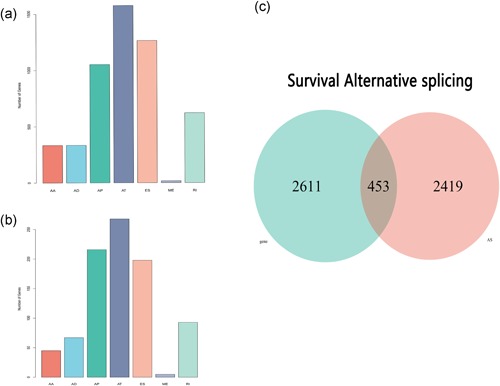
Number of survival specific alternative splicing (AS) events and genes. (a) The histogram of seven types of AS events that are significantly associated with overall survival. (b) The histogram of seven types of AS events that are significantly associated with gene expression. (c) The Venn diagram shows the intersection of survival related to AS events and messenger RNAs (mRNAs) [Color figure can be viewed at wileyonlinelibrary.com]
Figure 3.

Upset plot of different types of alternative splicing types. (a) UpSet plot of interactions between the seven types of survival‐associated alternative splicing (AS) events in head and neck cancer (HNC). (b) UpSet plot of interactions among survival‐associated genes in HNC. (c) Gene network of survival‐associated genes in HNC generated by Cytoscape [Color figure can be viewed at wileyonlinelibrary.com]
To determine the prognostic values of AS events, the top 30 genes in each of the seven AS events were chosen. Multivariate Cox regression analysis was then performed to determine the independent prognostic factors and to build prediction models. An AUC value of >0.5 was found in seven types of AS in HNC, of which AS was found to accurately predict the occurrence of HNC (AUC:0.695‐0.740; Figure 4).
Figure 4.
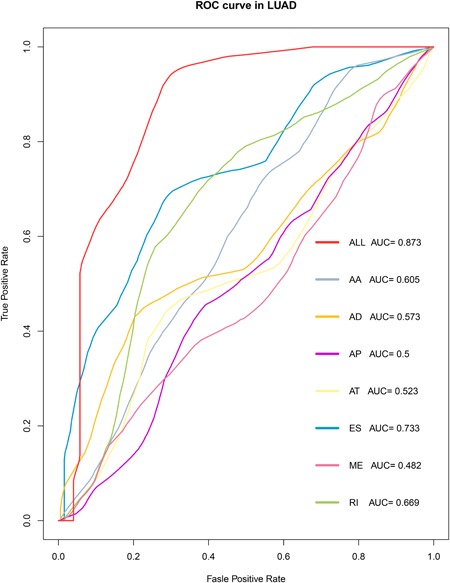
Receiver operating characteristic (ROC) curves of prognostic predictive models constructed using the seven types of alternative splicing (AS) events in head and neck cancer (HNC) patients [Color figure can be viewed at wileyonlinelibrary.com]
3.3. Functional and pathway enrichment analysis
The GO functional enrichment and KEGG pathway analysis were performed for the survival‐associated AS events. The results revealed that the AS events were an enrichment in 28 gene ontology categories. Sm‐like protein family complex, focal adhesion, spliceosomal small nuclear ribonucleoproteins complex and prespliceosome (Figure 5a) were mainly enriched. A total of 16 KEGG pathways were enriched (Figure 6b), including Valine, leucine, and isoleucine degradation (hsa00280), RNA polymerase (hsa03020), spliceosome (hsa03040), as well as Huntington disease (hsa05016; Figure 5b).
Figure 5.
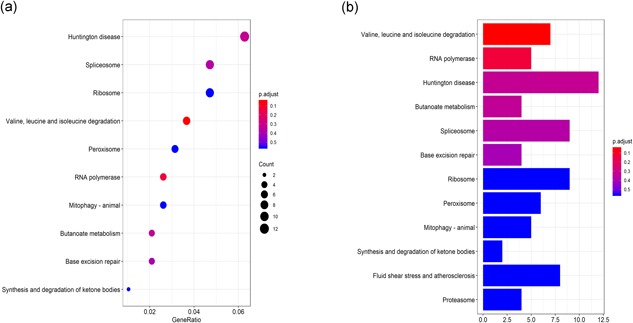
The terms of functional enrichment analyses. (a) GO, (b) KEGG. GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes [Color figure can be viewed at wileyonlinelibrary.com]
Figure 6.
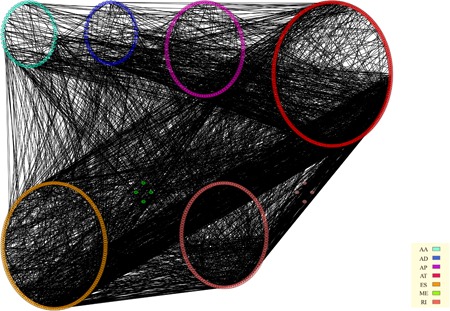
Interaction networks of KEGG enrichment results for genes in seven different AS events that are significantly associated with overall survival. AS: alternative splicing; KEGG: Kyoto Encyclopedia of Genes and Genomes [Color figure can be viewed at wileyonlinelibrary.com]
3.4. A network of survival‐associated AS events
To observe the association between different types of genes in AS events that were significantly associated with prognosis, the most significant survival‐related genes (p < 0.005) were selected and mapped to a String database using a score >0.4, to obtain interactions of these genes. The results were visualized using Cytoscape as shown in Figure 6. According to the results, most of the genes in the AS events associated with prognosis exhibited protein–protein interaction. These datasets indicate that most of the genes are involved in different biological functions.
3.5. Construction of prognostic predictor for KIRC
Nine genes with a Pearson’s correlation coefficient value >0.4 or <−0.4 between gene expression and AS event levels were identified, and these genes were associated with HNC patient’s survival. Cox multivariate analysis identified four genes including; TPM1, CLASRP, PRRC1, and DNASE1L1 (Table 1). The top four genes significantly related to AS events were further analysed to determine whether they are independent prognostic factors for HNC patients. Based on the four genes, a risk score model was constructed for each patient based on a linear combination of the mRNA expression level weighted by the regression coefficient (β) derived from the multivariate Cox regression analysis (Figure 7).
Table 1.
Four prognostic candidates for HNC patients in multivariate Cox regression analysis
| Genes | Hazard risk (HR) | Low 95% CI | High 95% CI | p value |
|---|---|---|---|---|
| TPM1 | 0.849808 | 1.1767 | 0.6874 | 0.13254 |
| CLASRP | 0.977842 | 1.0227 | 0.9541 | 0.07421 |
| PRRC1 | 0.941733 | 1.0619 | 0.8524 | 0.23766 |
| DNASE1L1 | 1.015742 | 0.9845 | 1.0054 | 0.00278 |
Note. CI: confidence interval; HNC: head and neck cancer.
Figure 7.
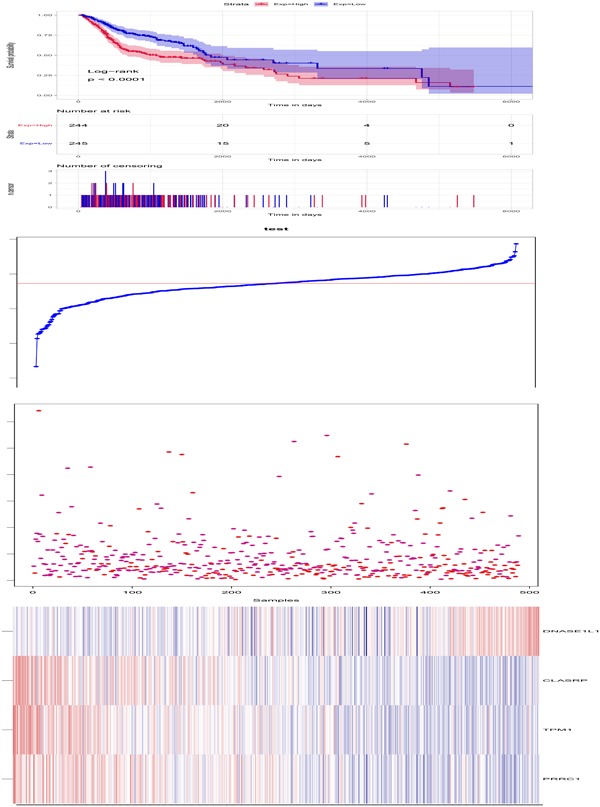
Construction of prognostic predictors based on four prognostic alternative splicing (AS) related genes. (a) Kaplan–Meier (KM) survival plot. (b) RiskScore score plot. (c) The expression heat map of four prognostic AS related genes. (d) Survival time and status of each sample for head and neck cancer (HNC) patients [Color figure can be viewed at wileyonlinelibrary.com]
3.6. Network construction of survival‐associated AS events
The most significant AS events with a p < 0.001 were considered as candidates for construction of the survival‐associated AS events. Splicing factors are defined as RNA‐binding proteins, which are characterized by cis‐regulatory elements within the pre‐mRNA that influence exon selection and splicing site choice. Using gene expression levels obtained from the TCGA, seven survival‐associated splicing factors were identified (Table 2). Meanwhile, the correlation between the gene expression of survival‐associated splicing factors and the PSI values of top significant AS events were conducted using the Spearman method. The splicing‐regulatory network was built based on the factors with significant correlations (p < 0.05). In the splicing correlation network shown in Figure 8, the expression of seven survival‐associated splicing factors (blue dots) were significantly correlated with 1,201 survival‐associated AS events, among which 497 were significantly associated with good survival in patients (green dots) while 694 were significantly associated with poor patient survival (red dots; Figure 8). Correlations relating to splicing factor CELF2, HNRNPC, TIA1, SRP54, SRSF9, HNRNPA0, and PTBP2 are shown in Figure 9. Low expression of CELF2 and HNRNPA0, HNRNPC, TIA1, SRSF9 were significantly associated with good survival in patients.
Table 2.
Prognostic survival‐associated splicing factors for HNC patients in multivariate Cox regression analysis
| Splicing factors | HR | Low 95% CI | High 95% CI | p value |
|---|---|---|---|---|
| CELF2 | 0.961693 | 0.936354 | 0.987716 | 0.004141 |
| HNRNPC | 1.003368 | 1.000895 | 1.005847 | 0.00758 |
| TIA1 | 0.98527 | 0.974261 | 0.996403 | 0.00964 |
| SRP54 | 1.007163 | 1.0012 | 1.013162 | 0.018493 |
| SRSF9 | 1.00557 | 1.000734 | 1.01043 | 0.02394 |
| HNRNPA0 | 0.949407 | 0.905967 | 0.99493 | 0.029803 |
| PTBP2 | 0.899589 | 0.810022 | 0.999059 | 0.047978 |
Note. CI: confidence interval; HNC: head and neck cancer.
Figure 8.
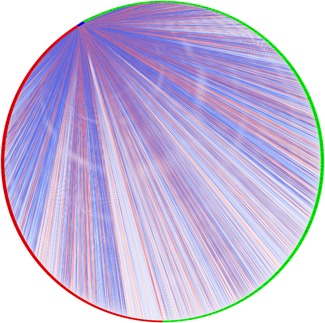
Splicing correlation network in head and neck cancer (HNC). Splicing correlation network in HNC. Expression of seven survival‐associated splicing factors (blue dots) were positively (light red line)/negatively (light blue line) correlated with the Percent Spliced In (PSI) values of 1,201 survival‐associated alternative splicing (AS) events (green dots) or adverse prognosis AS events (red dots) [Color figure can be viewed at wileyonlinelibrary.com]
Figure 9.
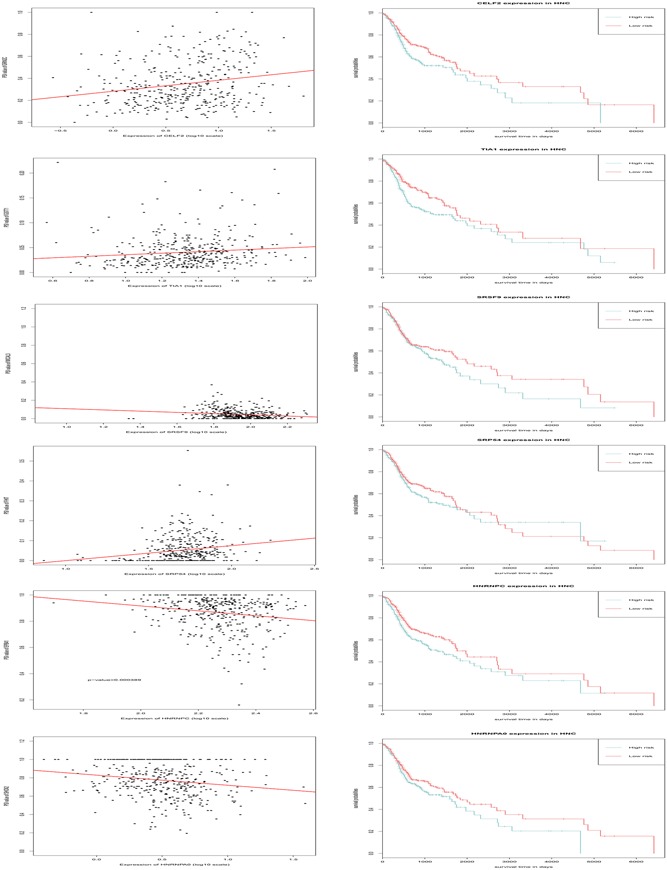
(a) Correlations between expression of splicing factors and Percent Spliced In (PSI) values of alternative splicing (AS) events (p < 0.05). (b) Kaplan–Meier (KM) survival plot for splicing factors [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
HNC is known have a rapid clinical progression and poor prognosis. In addition, its survival rates have been dismal over the last 40 years. The most common subtype is squamous cell carcinoma, which accounts for approximately 90% of all HNC malignancies (Boonkitticharoen et al., 2008). Patients with early stage HNC present with unclear symptoms and minimal physical changes; and early symptoms of this cancer are often hard to detect. Despite tremendous efforts to identify biomarkers for early detection and development of new treatments, the OS rate and prognosis remain poor. Local recurrence and metastasis are key limiting factors to the successful treatment of HNC (Le et al., 2014; Molinolo et al., 2009; Papillon‐Cavanagh et al., 2017). Therefore, there is an urgent need to improve the prognosis and decrease the mortality and morbidity of HNC. Identification of the diagnostic biomarkers is critical for early detection and risk stratification of HNC.
In recent years, several studies have explored the role of aberrant AS in HNC. Evidence has shown that AS are important in the initiation and progression of HNC. Biselli‐Chicote et al. reported that the alternatively spliced antiangiogenic family VEGFAxxxb played an important role in the development and progression of head and neck squamous cell carcinomas. A novel functional splicing variant of AKT3 was found to be associated with HPV‐positive oropharyngeal cancers (Guo et al., 2017). Yang, Jia, & Bian (2018) demonstrated that SRSF5 functions as a novel splicing factor and is upregulated by oncogene SRSF3 in oral squamous cell carcinoma. AS of TPM1 has been reported to be associated with the formation and migration of cancer cells (Huang, Zhang, Liao, Li, & Xu, 2017). Nevertheless, no studies have reported splicing variants of TPM1, CLASRP, PRRC1, and DNASE1L1, and the clinical value of the splicing events of these genes in HNC patients has not been investigated.
In this study, systematic identification and analysis of survival associated alternative events in 489 HNC patients data from TCGA was performed. A total of 48,049 AS events relating to 10,582 genes were significantly associated with HNC patients. The following genes TPM1, CLASRP, PRRC1, and DNASE1L1 were found to play important roles in cancer development. The prognostic predictors built in this study displayed high efficiency for HNC, the AUC of ROC was >0.6 at 2,000 days of OS. A multivariate Cox regression analysis was used to identify the AS events/mRNA with prognostic value. A prognostic risk model was also built on the basis of the four candidate genes. In addition, a combined survival and correlation network of the expression of splicing factors and AS events provided an avenue to address the underlying mechanism of the splicing pathway involved in patient survival. AS events were analyzed and their interaction network in HNC was constructed using TCGA data.
In summary, comprehensive profiles of AS events in HNC were explored. A series of HNC‐specific and survival‐specific AS events and genes were recognized, which may serve as targets for HNC treatment. The interaction network of splicing events of splicing factors was built, which expanded our understanding on the role of AS in tumorigenesis and prognosis of HNC.
ACKNOWLEDGEMENTS
The work was supported by Guizhou Science and Technology Cooperation Project (Qiankehe LH character [2015] 7138)
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
J. K. S., Y. X., Y. D. W., and Y. L. wrote the main manuscript text; J. K. S., Y. X., and D. Q. H. prepared Figures 1–8; J. K. S., X. H. Y., and J. G. L. contributed data analysis. All authors reviewed the manuscript.
Liang Y, Song J, He D, et al. Systematic analysis of survival‐associated alternative splicing signatures uncovers prognostic predictors for head and neck cancer. J Cell Physiol. 2019;234:15836–15846. 10.1002/jcp.28241
Contributor Information
Xinhai Yin, Email: yinxinh_cn@sina.com, Email: liujg_001@163.com.
Jianguo Liu, Email: yinxinh_cn@sina.com, Email: liujg_001@163.com.
References
REFERENCES
- Arantes, L. M. R. B. , de Carvalho, A. C. , Melendez, M. E. , Carvalho, A. L. , & Goloni‐Bertollo, E. M. (2014). Methylation as a biomarker for head and neck cancer. Oral Oncology, 50, 587–592. [DOI] [PubMed] [Google Scholar]
- Biselli‐Chicote, P. M. , Biselli, J. M. , Cunha, B. R. , Castro, R. , Maniglia, J. V. , Neto, D. S. , … Goloni‐Bertollo, E. M. (2017). Overexpression of Antiangiogenic Vascular Endothelial Growth Factor Isoform and Splicing Regulatory Factors in Oral, Laryngeal and Pharyngeal Squamous Cell Carcinomas. Asian Pacific journal of cancer prevention, 18(8), 2171–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonkitticharoen, V. , Kulapaditharom, B. , Leopairut, J. , Kraiphibul, P. , Larbcharoensub, N. , Cheewaruangroj, W. , … Pochanukul, L. (2008). Vascular endothelial growth factor a and proliferation marker in prediction of lymph node metastasis in oral and pharyngeal squamous cell carcinoma. Archives of Otolaryngology‐Head & Neck Surgery, 134, 1305–1311. [DOI] [PubMed] [Google Scholar]
- Carvalho, A. L. , Jeronimo, C. , Kim, M. M. , Henrique, R. , Zhang, Z. , Hoque, M. O. , … Califano, J. A. (2008). Evaluation of promoter hypermethylation detection in body fluids as a screening/diagnosis tool for head and neck squamous cell carcinoma. Clinical Cancer Research, 14, 97–107. [DOI] [PubMed] [Google Scholar]
- David, C. J. , & Manley, J. L. (2010). Alternative pre‐mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes & Development, 24, 2343–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Marabti, E. , & Younis, I. (2018). The cancer spliceome: Reprograming of alternative splicing in cancer. Frontiers in Molecular Biosciences, 5, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, T. , Sakai, A. , Afsari, B. , Considine, M. , Danilova, L. , Favorov, A. V. , … Califano, J. (2017). A novel functional splice variant of AKT3 defined by analysis of alternative splice expression in HPV‐positive oropharyngeal cancers. Cancer Research, 77, 5248–5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heagerty, P. J. , Lumley, T. , & Pepe, M. S. (2000). Time‐dependent ROC curves for censored survival data and a diagnostic marker. Biometrics, 56, 337–344. [DOI] [PubMed] [Google Scholar]
- Huang, G. W. , Zhang, Y. L. , Liao, L. D. , Li, E. M. , & Xu, L. Y. (2017). Natural antisense transcript TPM1‐AS regulates the alternative splicing of tropomyosin I through an interaction with RNA‐binding motif protein 4. The International Journal of Biochemistry & Cell Biology, 90, 59–67. [DOI] [PubMed] [Google Scholar]
- Jemal, A. , Siegel, R. , Ward, E. , Hao, Y. , Xu, J. , Murray, T. , & Thun, M. J. (2008). Cancer statistics, 2008. CA: Cancer Journal for Clinicians, 58, 71–96. [DOI] [PubMed] [Google Scholar]
- Klinck, R. , Bramard, A. , Inkel, L. , Dufresne‐Martin, G. , Gervais‐Bird, J. , Madden, R. , … Abou Elela, S. (2008). Multiple alternative splicing markers for ovarian cancer. Cancer Research, 68, 657–663. [DOI] [PubMed] [Google Scholar]
- Kozlovski, I. , Siegfried, Z. , Amar‐Schwartz, A. , & Karni, R. (2017). The role of RNA alternative splicing in regulating cancer metabolism. Human Genetics, 136, 1113–1127. [DOI] [PubMed] [Google Scholar]
- Kuranaga, Y. , Sugito, N. , Shinohara, H. , Tsujino, T. , Taniguchi, K. , Komura, K. , … Akao, Y. (2018). SRSF3, a splicer of the PKM gene, regulates cell growth and maintenance of cancer‐specific energy metabolism in colon cancer cells. International Journal of Molecular Sciences, 19, 3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, J. M. , Squarize, C. H. , & Castilho, R. M. (2014). Histone modifications: Targeting head and neck cancer stem cells. World Journal of Stem Cells, 6, 511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y. , & Rio, D. C. (2015). Mechanisms and regulation of alternative Pre‐mRNA splicing. Annual Review of Biochemistry, 84, 291–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lex, A. , Gehlenborg, N. , Strobelt, H. , Vuillemot, R. , & Pfister, H. (2014). UpSet: Visualization of intersecting sets. IEEE Transactions on Visualization and Computer Graphics, 20, 1983–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marur, S. , & Forastiere, A. A. (2008). Head and neck cancer: Changing epidemiology, diagnosis, and treatment. Mayo Clinic Proceedings, 83, 489–501. [DOI] [PubMed] [Google Scholar]
- Mehanna, H. , Paleri, V. , West, C. M. L. , & Nutting, C. (2010). Head and neck cancer‐‐Part 1: Epidemiology, presentation, and prevention. BMJ, 341, c4684–c4684. [DOI] [PubMed] [Google Scholar]
- Molinolo, A. A. , Amornphimoltham, P. , Squarize, C. H. , Castilho, R. M. , Patel, V. , & Gutkind, J. S. (2009). Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncology, 45, 324–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munkley, J. , Livermore, K. , Rajan, P. , & Elliott, D. J. (2017). RNA splicing and splicing regulator changes in prostate cancer pathology. Human Genetics, 136, 1143–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan, S. P. , Singh, S. , & Shukla, S. (2017). A saga of cancer epigenetics: Linking epigenetics to alternative splicing. The Biochemical Journal, 474, 885–896. [DOI] [PubMed] [Google Scholar]
- Nilsen, T. W. , & Graveley, B. R. (2010). Expansion of the eukaryotic proteome by alternative splicing. Nature, 463, 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltean, S. , & Bates, D. O. (2014). Hallmarks of alternative splicing in cancer. Oncogene, 33, 5311–5318. [DOI] [PubMed] [Google Scholar]
- Papillon‐Cavanagh, S. , Lu, C. , Gayden, T. , Mikael, L. G. , Bechet, D. , Karamboulas, C. , … Jabado, N. (2017). Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nature Genetics, 49, 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschalis, A. , Sharp, A. , Welti, J. C. , Neeb, A. , Raj, G. V. , Luo, J. , … de Bono, J. S. (2018). Alternative splicing in prostate cancer. Nature Reviews Clinical Oncology, 15, 663–675. [DOI] [PubMed] [Google Scholar]
- Ryan, M. C. , Cleland, J. , Kim, R. , Wong, W. C. , & Weinstein, J. N. (2012). SpliceSeq: A resource for analysis and visualization of RNA‐Seq data on alternative splicing and its functional impacts. Bioinformatics, 28, 2385–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salton, M. , & Misteli, T. (2016). Small molecule modulators of Pre‐mRNA splicing in cancer therapy. Trends in Molecular Medicine, 22, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel, R. L. , Miller, K. D. , & Jemal, A. (2018). Cancer statistics, 2018. CA: Cancer Journal for Clinicians, 68, 7–30. [DOI] [PubMed] [Google Scholar]
- Woźniak, A. , Szyfter, K. , Szyfter, W. , & Florek, E. (2012). Head and neck cancer‐‐history. Przeglad Lekarski, 69, 1079–1083. [PubMed] [Google Scholar]
- Wu, G. , Feng, X. , & Stein, L. (2010). A human functional protein interaction network and its application to cancer data analysis. Genome Biology, 11, R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, S. , Jia, R. , & Bian, Z. (2018). SRSF5 functions as a novel oncogenic splicing factor and is upregulated by oncogene SRSF3 in oral squamous cell carcinoma. Biochimica et Biophysica Acta, Molecular Cell Research, 1865, 1161–1172. [DOI] [PubMed] [Google Scholar]
- Yu, G. , Wang, L. G. , Han, Y. , & He, Q. Y. (2012). clusterProfiler: An R package for comparing biological themes among gene clusters. Omics: A Journal of Integrative Biology, 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]