

Abstract
Solriamfetol (JZP‐110), a selective dopamine and norepinephrine reuptake inhibitor with wake‐promoting effects, is renally excreted ∼90% unchanged within 48 hours. Effects of renal impairment and hemodialysis on the pharmacokinetics and safety of 75‐mg single‐dose solriamfetol were evaluated in adults with normal renal function (n = 6); mild (n = 6), moderate (n = 6), or severe (n = 6) renal impairment; and end‐stage renal disease (ESRD) with and without hemodialysis (n = 7). Relative to normal renal function, geometric mean area under the plasma concentration–time curve from time zero to infinity increased 53%, 129%, and 339%, and mean half‐life was 1.2‐, 1.9‐, and 3.9‐fold higher with mild, moderate, and severe renal impairment, respectively. Renal excretion of unchanged solriamfetol over 48 hours was 85.8%, 80.0%, 66.4%, and 57.1% in normal, mild, moderate, and severe renal impairment groups, respectively; mean maximum concentration and time to maximum concentration did not vary substantially. Decreases in solriamfetol clearance were proportional to decreases in estimated glomerular filtration rate. Geometric mean area under the plasma concentration–time curve from time zero to time of last quantifiable concentration increased 357% and 518% vs normal in ESRD with and without hemodialysis, respectively, with half‐life >100 hours in both groups. Over the 4‐hour hemodialysis period, ∼21% of solriamfetol dose was removed. Adverse events included headache (n = 1) and nausea (n = 1). Six days after dosing, 1 participant had increased alanine and aspartate aminotransferase, leading to study discontinuation. While these adverse events were deemed study‐drug related, they were mild and resolved. Results from this study combined with population pharmacokinetic modeling/simulation suggest that solriamfetol dosage adjustments are necessary in patients with moderate or severe but not with mild renal impairment. Due to significant exposure increase/prolonged half‐life, dosing is not recommended in patients with ESRD.
Keywords: end‐stage renal disease, hemodialysis, JZP‐110, pharmacokinetics, renal impairment, solriamfetol
Solriamfetol ([R]‐2‐amino‐3‐phenylpropylcarbamate hydrochloride) is a selective dopamine and norepinephrine reuptake inhibitor. At micromolar concentrations, solriamfetol selectively binds to and inhibits reuptake at dopamine and norepinephrine transporters without promoting monoamine release.1, 2 Solriamfetol (formerly known as JZP‐110, R228060, ADX‐N05, and YKP‐10A) is a wake‐promoting agent that is being developed at daily doses of 75 mg to 300 mg for the treatment of excessive daytime sleepiness associated with narcolepsy and obstructive sleep apnea (OSA). In phase 3 trials in patients with narcolepsy and OSA, solriamfetol demonstrated reductions in excessive daytime sleepiness measured on the patient‐reported Epworth Sleepiness Scale,3 and improvement in objective assessment of wakefulness using the Maintenance of Wakefulness Test, that were statistically significantly greater than placebo.4, 5, 6 Significantly higher percentages of participants treated with solriamfetol in these trials also reported improvement on the Patient Global Improvement of Change scale relative to placebo at all evaluated time points.
Solriamfetol has high solubility and high permeability and is considered a Biopharmaceutics Classification System class 1 compound. Solriamfetol is rapidly absorbed after oral administration, with median time to maximum plasma concentration (Cmax) at 2 hours after dosing and an apparent elimination half‐life (t1/2) of ∼6 hours.7 Consumption of a high‐fat, high‐calorie meal does not change the rate and extent of solriamfetol exposure but delays time to Cmax by 1 hour, which likely has minimal clinical significance.7 The major route of solriamfetol elimination is via urinary excretion, with approximately 90% of drug excreted unchanged within 48 hours, and ∼1% of the dose excreted as the minor metabolite, N‐acetyl solriamfetol.7
Renal impairment, as well as hemodialysis in individuals with end‐stage renal disease (ESRD) could affect the pharmacokinetics (PK) of solriamfetol. Therefore, in accordance with US Food and Drug Administration guidance for PK studies in patients with impaired renal function,8 this study was designed to evaluate the PK of solriamfetol in participants with renal impairment and those with ESRD undergoing hemodialysis compared with healthy participants with normal renal function. Preliminary results from this study have previously been presented.9
Methods
Study Design
This phase 1, parallel‐group, open‐label, single‐dose study to assess the effects of renal impairment and hemodialysis on PK and safety of solriamfetol was performed at 2 sites in the United States between October and December 2015, and in accordance with US Food and Drug Administration guidance.8 The protocol was approved by the IntegReview Institutional Review Board (Austin, Texas), and the study was conducted in compliance with the protocol, the Guideline for Good Clinical Practice E6; the US Code of Federal Regulations pertaining to conduct and reporting of clinical studies; the Clinical Trials Directive of the European Medicines Agency (Directive 2001/20/EC); and the Declaration of Helsinki. Written informed consent was obtained from each subject before enrollment in the study and before performance of any study‐related procedure.
Participants
Eligible participants were men and nonpregnant, nonlactating women between the ages of 18 and 80 years, with a body mass index (BMI) ≤35 kg/m2. Women of childbearing potential were required to have used a medically accepted method of birth control for at least 2 months before the first dose of study drug, with continued use throughout the study period and for 30 days after study completion. Participants were excluded if they had a clinically significant medical abnormality (other than renal impairment or its underlying causes), or any unstable conditions including neurological or psychiatric disorder; hepatic, endocrine, cardiovascular, gastrointestinal, pulmonary, or metabolic disease; or any other abnormality that could interfere with the PK evaluation of the study drug or the participant's completion of the trial.
Eligible participants were assigned to 1 of 5 groups according to renal disease status as measured by the estimated glomerular filtration rate (eGFR) on the day before dosing, calculated using the Modification in Diet in Renal Disease equation.10 Group 1 consisted of healthy participants with normal renal function (eGFR ≥ 90 mL/min/1.73 m2), and served as the control group. Groups 2, 3, and 4 had mild, moderate, and severe renal impairment based on eGFRs of 60 to 89, 30 to 59, and <30 mL/min/1.73 m2, respectively. Group 5 consisted of participants with ESRD who required ≥3 hemodialysis treatments per week for the preceding 3 months. Every effort was made to ensure that the groups were comparable with respect to age, sex, and BMI. Group 1 was enrolled last to facilitate matching the mean age, BMI, and sex distribution of Groups 2 through 5.
Among participants with impaired renal function, continued use of medications necessary for treatment of renal function and/or coexisting disease was allowed, with the exception of monoamine oxidase inhibitors and medications with known risk for torsade de pointes.
Protocol
Groups 1 through 4 received one 75‐mg dose of solriamfetol on day 1; Group 5 received one 75‐mg dose on day 1 followed by 4‐hour hemodialysis (designated Group 5.2), and one 75‐mg dose on day 8 without hemodialysis (designated Group 5.1). All doses were administered on an empty stomach following an overnight fast except for participants in Group 5, who received a standardized snack on day 7 and breakfast early on day 8 before starting an 8‐hour fast. Participants remained fasting for 4 hours after administration, with water allowed except for 1 hour before and after dosing.
In this study, 75‐mg solriamfetol was selected as the dose for administration in participants with renal impairment, as it was considered sufficiently low and potentially safe for this population, especially considering the likelihood of higher and/or prolonged exposure to solriamfetol. The 75‐mg dose was expected to result in plasma concentrations of solriamfetol that were above the assay detection level at time points sufficient to characterize the PK profile.
Serial blood samples of ∼4 mL were collected within 30 minutes before dosing and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 24, 36, and 48 hours after dosing in Groups 1 through 5, with continued sampling at 60 and 72 hours after dosing in Groups 4 and 5. All blood samples were collected into labeled K2 ethylenediaminetetraacetic acid tubes by direct venipuncture or indwelling catheter and kept on ice until the samples were centrifuged within 30 minutes of collection at ∼2500 rpm (1315 × g) at 4°C for 10 minutes. The plasma was transferred into polypropylene tubes for freezing and storage at −70°C until analysis.
Urine samples were collected before dosing and for the time intervals of 0 to 4, 4 to 8, 8 to 12, 12 to 24, and 24 to 48 hours in Groups 1 through 5, with additional collection for the 48‐ to 72‐hour time interval in Groups 4 and 5. During the hemodialysis period on day 1 for Group 5, dialysate samples and pre‐ and postdialyzer paired blood samples were collected before dialysis (2 hours), and at 3, 4, 5, and 6 hours following dosing. Urine and dialysate samples were aliquoted into polypropylene tubes for freezing and storage at −70°C until analysis. All blood, urine, and dialysate samples were shipped on dry ice to a central bioanalytical laboratory.
Bioanalytical analyses were performed by a central laboratory (KCAS, LLC, Shawnee, Kansas) using validated proprietary methods that included extraction/derivatization and liquid chromatography–tandem mass spectrometry. Measurement of solriamfetol was over the linear range of 8.42 to 4210 ng/mL in plasma, 0.21 to 84.2 μg/mL in urine, and 1.68 to 842 ng/mL in dialysate. Assay performance was monitored by spiking blank human matrices with positive controls and internal standards to generate standard‐curve and quality control samples. After derivatization, samples were chromatographed on a C8 reversed phase analytical high‐performance liquid chromatography column, with subsequent monitoring using an API4000 liquid chromatography–tandem mass spectrometry unit (Sciex, Framingham, Massachusetts). Quantification was based on setting a calibration graph using the internal standard method. Coefficients of variation (CVs) for quality control samples at lower limits of quantitation were 3.2% to 6.0% in the plasma samples, 1.6% to 5.6% for the urine samples, and 3.5% to 7.1% for the dialysate samples.
Pharmacokinetic and Statistical Analyses
The following plasma PK parameters were evaluated using noncompartmental analysis in Phoenix® WinNonlin® Version 6.3: Cmax; time to reach Cmax following drug administration (tmax); t1/2; area under the plasma concentration‐time curve from time zero to time of last quantifiable concentration (AUCt); AUC from time zero to infinity (AUC∞); apparent total clearance of the drug from plasma after oral administration (CL/F); and apparent volume of distribution (Vd/F).
The PK parameters for solriamfetol in urine included the amount of unchanged drug excreted in urine over 48 or 72 hours, the fraction of the dose excreted unchanged in urine, and renal clearance of the drug.
For participants on hemodialysis (Group 5), the additional PK parameters included the amount of solriamfetol cleared by the 4‐hour hemodialysis (Adial); the fraction of dose removed by the 4‐hour hemodialysis; and hemodialysis clearance (CLdial) calculated as
where AUCdial is the area under the predialyzer plasma concentration‐time curve during the hemodialysis period.
PK parameters were summarized by group using descriptive statistics. To assess differences in PK between each level of renal impairment (Groups 2‐5) versus participants with normal renal function (Group 1), a linear effects model was used to compare natural log‐transformed PK parameters (Cmax, AUCt, and AUC∞). For Group 5, the participants without dialysis on day 8 (Group 5.1) and the participants who received dialysis on day 1 (Group 5.2) were analyzed and compared separately.
Point estimates and 90% confidence intervals (CIs) for differences on the natural log scale were exponentiated to obtain estimates for ratios of geometric means on the original scale. The 90%CIs around the geometric means ratios were presented for each pairwise comparison and expressed as a percentage relative to the geometric means of the reference group (Group 1). The interparticipant CV was estimated. To evaluate effects of dialysis on PK parameters for Group 5, an analysis of variance model was used that included “Day” as a fixed effect and measurements within the participant as a repeated measure. Day 8 was used as the reference for comparison. In addition, nonparametric analysis was conducted for tmax as appropriate.
All statistical analyses were conducted using SAS version 9.3 (SAS Institute, Cary, NC).
Tolerability Assessment
Tolerability was evaluated based on the occurrence of treatment‐emergent adverse events (TEAEs) regardless of causality. The identification of serious TEAEs and grading of TEAEs (mild, moderate, or severe) were according to International Conference on Harmonisation guidelines.11 TEAEs were coded using the Medical Dictionary for Regulatory Activities system version 18.0 to classify events under primary system organ class and preferred term. The relationship of TEAEs to the study drug, including categories of “related,” “suspected to be related,” or “not related,” was determined by the clinical staff. Other safety parameters included clinical laboratory testing, vital signs, electrocardiography, and physical examination.
Results
Demographics
Of the 31 participants who were enrolled and received treatment (6 participants in each of Groups 1‐4 and 7 participants in Group 5), 30 participants (97%) completed the study. One participant from Group 5 discontinued due to adverse events. Participant demographics (Table 1) show that most participants in Groups 1 through 4 were white; however, most participants in Group 5 were black. There were at least 2 participants per sex in each group, and mean age for Groups 1 through 4 were comparable, with an overlap in the range; the age range in Group 5 was lower than in the other groups. Mean BMI for Groups 1 through 5 were comparable, with an overlap in the range. Furthermore, all participants in Group 1 matched the mean age (±10 years) and BMI (±20%) of participants in Groups 2 through 5.
Table 1.
Demographic Characteristics of the Study Population
| Group 1 | Group 2 | Group 3 | Group 4 | Group 5 | |
|---|---|---|---|---|---|
| Normal Renal Function | Mild Renal Impairment | Moderate Renal Impairment | Severe Renal Impairment | End‐Stage Renal Disease | |
| Variable | (n = 6) | (n = 6) | (n = 6) | (n = 6) | (n = 7) |
| Sex, n (%) | |||||
| Female | 3 (50) | 4 (67) | 2 (33) | 2 (33) | 2 (29) |
| Male | 3 (50) | 2 (33) | 4 (67) | 4 (67) | 5 (71) |
| Race, n (%) | |||||
| White | 5 (83) | 5 (83) | 4 (67) | 5 (83) | 1 (14) |
| Black | 1 (17) | 1 (17) | 2 (33) | 1 (17) | 6 (86) |
| Ethnicity, n (%) | |||||
| Non‐Hispanic or Latino | 0 | 3 (50) | 2 (33) | 3 (50) | 6 (86) |
| Hispanic or Latino | 6 (100) | 3 (50) | 4 (67) | 3 (50) | 1 (14) |
| Age, mean (SD), y | 55.8 (3.9) | 67.8 (7.4) | 70.2 (7.7) | 59.7 (15.6) | 42.0 (7.6) |
| Weight, mean (SD), kg | 73.1 (6.8) | 67.1 (14.2) | 76.8 (11.5) | 85.5 (16.4) | 88.2 (10.5) |
| BMI, mean (SD), kg/m2 | 28.1 (2.7) | 25.1 (4.1) | 28.8 (1.9) | 29.3 (3.0) | 29.9 (3.0) |
| eGFR, mean (SD), mL/min/1.73 m2 | 111.8 (32.3) | 78.5 (8.4) | 44.2 (6.2) | 16.2 (5.8) | 7.4 (4.8) |
BMI, body mass index; eGFR, estimated glomerular filtration rate.
Plasma Pharmacokinetics
For all study groups, mean PK parameters are summarized in Table 2 and mean plasma solriamfetol concentration–time profiles are shown in Figures 1A and 1B. In general, mean Cmax and tmax were not substantially affected by renal impairment across Groups 1 through 4 (Table 2). However, solriamfetol AUC and t1/2 values increased with increasing levels of renal impairment. Solriamfetol mean ± standard deviation (SD) overall exposure (AUC∞) increased from 5273 ± 4104 ng ∙ h/mL in participants with normal renal function to 6836 ng ∙ h/mL ± 1730 in Group 2 (mild impairment), 10 470 ± 3642 in Group 3 (moderate impairment), and 23 650 ± 16 776 in Group 4 (severe impairment) (Table 2). Similarly, solriamfetol mean ± SD t1/2 was 7.6 ± 5.1 hours in participants with normal renal function and increased with greater levels of renal impairment: 9.1 ± 1.6, 14.3 ± 4.5, and 29.6 ± 14.4 hours in Groups 2, 3, and 4, respectively (Table 2). While CL/F decreased with greater levels of renal impairment, there were no substantial changes in Vd/F (Table 2). A plot of solriamfetol CL/F versus day –1 eGFR for Groups 1 through 4 is presented in Figure 2. This relationship is best described by the equation:
Table 2.
Solriamfetol Pharmacokinetic Parameters by Level of Renal Function
| Mean ± Standard Deviation (% Coefficient of Variation) | ||||||
|---|---|---|---|---|---|---|
| Normal Renal Function | Renal Impairment | End‐Stage Renal Disease (Group 5) | ||||
| Group 1 | Group 2 Mild | Group 3 Moderate | Group 4 Severe | Group 5.1 Without Hemodialysisa | Group 5.2 With Hemodialysis | |
| Variable | (n = 6) | (n = 6) | (n = 6) | (n = 6) | (n = 6) | (n = 7)b |
| Cmax, ng/mL | 499.0 ± 142.4 (28.5) | 521.8 ± 118.8 (22.8) | 517.3 ± 131.6 (25.4) | 552.8 ± 154.4 (27.9) | 474.1 ± 79.0 (16.7) | 396.4 ± 75.4 (19.0) |
| tmax, c h | 1.3 (0.5‐2.0) | 1.5 (0.5‐2.0) | 1.5 (1.0‐2.5) | 2.0 (0.5‐3.0) | 3.3 (1.0‐24.0) | 1.5 (1.5‐10.0) |
| t1/2, h | 7.6 ± 5.1 (67.7) | 9.1 ± 1.6 (18.1) | 14.3 ± 4.5 (31.4) | 29.6 ± 14.4 (48.7) | 100.5 ± 78.8 (78.4)d | 164.7 ± 81.4 (49.4)e |
| AUCt, ng ∙ h/mLf | 4849 ± 3454 (71.2) | 6613 ± 1574 (23.8) | 9230 ± 2538 (27.5) | 17 500 ± 9267 (52.9) | 25 580 ± 4544 (17.8) | 18 920 ± 3131 (16.5) |
| AUC∞, ng ∙ h/mL | 5273 ± 4104 (77.8) | 6836 ± 1730 (25.3) | 10 470 ± 3642 (34.8) | 23 650 ± 16 776 (70.9) | 64 560 ± 35 962 (55.7)d | 76 770 ± 41 993 (54.7)e |
| CL/F, L/h | 19.8 ± 10.1 (50.9) | 11.5 ± 2.5 (22.1) | 7.8 ± 2.4 (30.5) | 4.7 ± 2.8 (59.4) | 1.6 ± 1.1 (72.3)d | 1.5 ± 1.3 (91.0)e |
| Vd/F, L | 163.9 ± 23.8 (14.5) | 147.2 ± 29.1 (19.8) | 152.0 ± 32.6 (21.4) | 157.2 ± 41.2 (26.2) | 153.6 ± 45.6 (29.7)d | 231.4 ± 28.5 (12.3)e |
AUC indicates area under plasma concentration‐time curve; AUCt, AUC from time zero to time of last quantifiable concentration; AUC∞, AUC from time zero to infinity; CL/F, apparent total clearance of the drug from plasma after oral administration; Cmax, maximum concentration; tmax, time to maximum concentration; t1/2, apparent elimination half‐life, Vd/F, apparent volume of distribution.
Baseline adjusted to remove the impact of the day 1 dose on the day 8 concentration profile.
Excluding 2 concentration values: 1 participant at predose, and 1 participant at 24 hours.
For tmax, median (min‐max) is presented.
n = 3.
n = 6.
Over 48 hours for normal, mild, and moderate, and over 72 hours for severe and end‐stage renal disease.
Figure 1.

Mean (standard deviation) plasma solriamfetol concentration–time profiles following a single 75‐mg dose. (A) Participants with normal renal function and mild‐to‐severe renal impairment. (B) Participants with ESRD with and without hemodialysis. ESRD, end‐stage renal disease.
Figure 2.
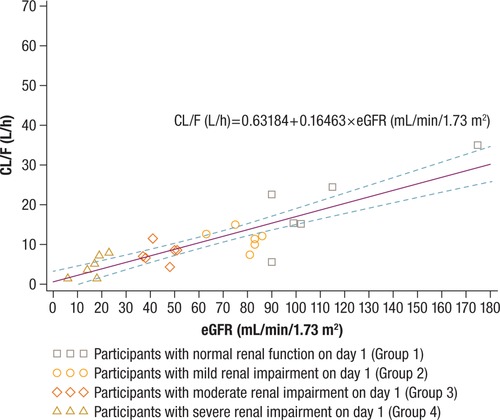
Apparent oral clearance (CL/F) versus day –1 estimated glomerular filtration rate (eGFR) for Groups 1 through 4. The broken lines represent the 90% confidence intervals.
Among participants with ESRD (Group 5), overall exposure (AUCt) was approximately 5‐fold higher for participants without dialysis on day 8 (Group 5.1; 25 580 ± 4544 ng ∙ h/mL) and about 4‐fold higher among participants with dialysis on day 1 (Group 5.2; 18 920 ± 3131) relative to Group 1 (4849 ± 3454) (Table 2). Mean t1/2 values exceeded 100 hours in both Group 5.1 (100.5 hours) and Group 5.2 (164.7 hours) (Table 2), and compared with Group 1, Cmax values were slightly lower and tmax values differed significantly (P ≤ .05 for both).
Ratios of geometric means and their associated 90% CIs for the pairwise comparisons of solriamfetol plasma PK parameters for Groups 2 through 5 vs Group 1 are presented in Table 3. As shown, small increases were observed in Cmax, which was approximately 6%, 4%, and 11% higher in Groups 2, 3, and 4, respectively, versus Group 1. However, total solriamfetol exposure (AUC∞) in Groups 2, 3, and 4 was 53%, 129%, and 339% higher, respectively, relative to Group 1. In participants with ESRD, Cmax was approximately 3% and 19% lower in Groups 5.1 (ESRD without hemodialysis) and 5.2 (ESRD with hemodialysis), respectively, versus Group 1, and exposure was approximately 518% and 357% higher in the 2 groups versus Group 1.
Table 3.
Comparisons of Solriamfetol Plasma PK Parameters
| Group 1 Normal | Group 2 Mild | Group 3 Moderate | Group 4 Severe | Group 5.1 Without Hemodialysis | Group 5.2 With Hemodialysis | |
|---|---|---|---|---|---|---|
| PK Parameter | (n = 6) | (n = 6) | (n = 6) | (n = 6) | (n = 6) | (n = 7)a |
| Geometric LS mean | ||||||
| Cmax, ng/mL | 482.3 | 510.5 | 503.2 | 533.0 | 468.8 | 389.9 |
| AUCt, ng ∙ h/mLb | 4087.3 | 6469.6 | 8960.2 | 15 549 | 25 253 | 18 689 |
| AUC∞, ng ∙ h/mL | 4363.9 | 6672.4 | 10 002 | 19 140 | 56 319c | 65 306d |
| Percent ratio (90% confidence interval) of geometric mean relative to Group 1 | ||||||
| Cmax | — | 105.9 (80.6‐139.0) | 104.3 (78.4‐138.9) | 110.5 (81.1‐150.6) | 97.2 (76.1‐124.1) | 80.9 (63.4‐103.1) |
| AUCt | — | 158.3 (97.5‐256.9) | 219.2 (133.7‐359.6) | 380.4 (208.4‐694.4) | 617.8 (385.3‐990.8) | 457.2 (296.6‐704.9) |
| AUC∞ | — | 152.9 (92.9‐251.7) | 229.2 (135.6‐387.4) | 438.6 (217.3‐885.3) | 1290.6 (542.8‐3068.5) | 1496.5 (748.7‐2991.2) |
Notes: Parameters were Ln‐transformed prior to analysis. Geometric least squares means (LSMs) are calculated by exponentiating the LSMs from the analysis of variance. % mean ratio = 100 × (test/reference).
AUC indicates area under plasma concentration‐time curve; AUCt, AUC from time zero to time of last quantifiable concentration; AUC∞, AUC from time zero to infinity; Cmax, maximum concentration; ESRD, end‐stage renal disease; LS, least squares; PK, pharmacokinetics.
Excluding 2 concentration values: 1 participant at predose, and 1 participant at 24 hours.
Over 48 hours for Groups 1 through 3 and over 72 hours for Groups 4 and 5.
n = 3.
n = 6.
Urinary Excretion
Renal clearance and the cumulative amount of solriamfetol excreted in urine decreased as renal impairment increased (Table 4). In Group 1, the mean ± SD percentage of solriamfetol recovered unchanged in urine over 48 hours was 85.8% ± 7.7% and decreased to 80.0% ± 9.0%, 66.4% ± 12.8%, and 57.1% ± 18.6% in Groups 2, 3, and 4, respectively. Mean solriamfetol renal clearance also decreased with renal impairment, from 17.0 ± 7.7 L/h in the normal renal function group to 9.3 ± 1.6 L/h in Group 2, 5.8 ± 2.0 L/h in Group 3, and 3.8 ± 2.6 L/h in Group 4. Only 1 participant made urine and was able to provide data
Table 4.
Urinary Excretion of Solriamfetol
| Mean ± Standard Deviation (% Coefficient of Variation) | ||||
|---|---|---|---|---|
| Group 1 | Group 2 | Group 3 | Group 4 | |
| Normal Renal Function | Mild | Moderate | Severe | |
| PK Parameter | (n = 6) | (n = 6) | (n = 6) | (n = 6) |
| Fe(0‐48), % | 85.8 ± 7.7 (9.0) | 80.0 ± 9.0 (11.2) | 66.4 ± 12.8 (19.2) | 57.1 ± 18.6 (32.5) |
| CLR, L/h | 17.0 ± 7.7 (45.4) | 9.3 ± 1.6 (17.1) | 5.8 ± 2.0 (34.1) | 3.8 ± 2.6 (68.0) |
CLR indicates renal clearance; Fe(0‐48), fraction of the dose excreted unchanged in urine in 48 hours.
in Group 5, and the cumulative amount of solriamfetol excreted in urine was lower with hemodialysis, 42.1%, compared with 52.9% without hemodialysis.
Dialysate Parameters
Over the 4‐hour hemodialysis period on day 1 for participants with ESRD, the mean ± SD cumulative fraction of the 75‐mg solriamfetol dose removed was 20.6% ± 1.7% (range 19.2% to 24.1%), and the hemodialysis clearance was 12.4 ± 1.5 L/h (range, 11.3‐15.9 L/h).
Tolerability
There were no deaths or other serious adverse events during this study. A total of 4 participants (13%), 1 each in Groups 2 and 3, and 2 in Group 5 (1 with and 1 without hemodialysis), reported 5 TEAEs (Table 5), including single events of nausea, skin abrasion, and headache in 1 participant each, and an increase in alanine aminotransferase (ALT; to 144 IU/L; reference range, 8‐54 IU/L) and aspartate aminotransferase (AST; to 66 IU/L; reference range, 8‐40 IU/L) observed 6 days after dosing in 1 participant that led to discontinuation. All TEAEs were considered by the investigator to be mild, and all but the skin abrasion were considered to be related to study drug. All TEAEs resolved, including the increased ALT and AST, which resolved on day 11. No other abnormal laboratory findings were considered clinically meaningful. No clinically significant abnormal findings were observed in vital sign and electrocardiographic measurements.
Table 5.
Number (%) of Participants with Treatment‐Emergent Adverse Events
| Normal Renal Function | Renal Impairment | End‐Stage Renal Disease (Group 5) | ||||
|---|---|---|---|---|---|---|
| Adverse Event | Group 1 Normal (n = 6) | Group 2 Mild (n = 6) | Group 3 Moderate (n = 6) | Group 4 Severe (n = 6) | Group 5.1 Without Hemodialysisa (n = 6) | Group 5.2 With Hemodialysis (n = 7) |
| Any TEAE | 0 | 1 (17%) | 1 (17%) | 0 | 1 (17%) | 1 (14%) |
| Nausea | 0 | 0 | 0 | 0 | 1 (17%) | 0 |
| Skin abrasion | 0 | 1 (17%) | 0 | 0 | 0 | 0 |
| ALT increased | 0 | 0 | 0 | 0 | 0 | 1 (14%) |
| AST increased | 0 | 0 | 0 | 0 | 0 | 1 (14%) |
| Headache | 0 | 0 | 1 (17%) | 0 | 0 | 0 |
ALT indicates alanine aminotransferase; AST, aspartate aminotransferase; TEAE, treatment‐emergent adverse event.
One participant from Group 5 discontinued the study before day 8 due to adverse events of mild elevated ALT and AST.
Discussion
This study showed that consistent with renal excretion of unchanged drug being the primary route of elimination, renal impairment increases overall exposure to solriamfetol, with the magnitude of the increase reflecting the level of impairment. The incremental decreases in CL/F with worsening renal function resulted in corresponding increases in overall solriamfetol exposure that was 53% for mild, 129% for moderate, and 339% for severe impairment relative to normal renal function. Increasing renal impairment was also associated with decreasing cumulative percentage of solriamfetol excreted in urine. Additionally, as there were no substantial changes in Vd/F, the decreases in solriamfetol CL/F resulted in increased t1/2 by approximately 1.2‐, 1.9‐, and 3.9‐fold in participants with mild, moderate, and severe renal impairment, respectively, compared with participants with normal renal function.
In this regard, it should also be noted that while Cmax values were not substantially affected by renal impairment, the observed increases in t1/2 associated with renal impairment are expected to translate to changes in steady‐state Cmax that are not fully accounted for by the single‐dose regimen evaluated in the current study, due to accumulation.
Consistent with the inability of ESRD participants requiring hemodialysis to eliminate solriamfetol via renal excretion, these participants had increased overall exposure to solriamfetol (≥4‐fold), longer t1/2 values (≥13‐fold), and slightly lower Cmax values (≤19%), relative to participants with normal renal function. Furthermore, ESRD participants had lower solriamfetol Cmax and AUCt values after undergoing a 4‐hour hemodialysis session, with 20.6% of the solriamfetol dose removed as unchanged drug. Notably, the solriamfetol hemodialysis clearance of 12.4 L/h estimated from solriamfetol recovered in the dialysate was approximately 30% lower than solriamfetol renal clearance in participants with normal renal function.
This analysis and the results reported may be especially relevant to the OSA patient population which has relatively high rates of impaired renal function.12, 13, 14, 15 Conversely, studies in participants with chronic kidney disease have reported high rates of comorbid OSA determined by polysomnography, ranging from 16% to 65%.16, 17, 18, 19, 20, 21 Reviews of study data on the relationship between OSA and chronic kidney disease/ESRD suggest that both conditions may mutually increase risks of their concomitant occurrence, possibly through other comorbidities and risk factors common to each, such as hypertension, obesity, cardiovascular disease, metabolic disorders, and diabetes.22, 23, 24 However, some data also suggest that OSA may be a direct, independent risk factor for chronic kidney disease, possibly through pathogenic mechanisms such as hypoxia and renin‐angiotensin system activation.22, 23, 24 Therefore, clinicians treating patients with excessive daytime sleepiness who have OSA should take into consideration potential risk factors for impaired renal function.
Dose adjustment recommendations in patients with renal impairment are based on combined analyses of renal impairment study data and population PK modeling/simulation. The goal is to adjust the dose in renally impaired patients to achieve similar steady‐state exposures as in participants with normal renal function. These analyses suggest that adjustments to solriamfetol dosage are not needed in patients with mild renal impairment but are necessary in patients with moderate or severe renal impairment. Dosing is not recommended in patients with ESRD due to significant exposure increase and t1/2 prolongation in this population.
Despite the disparities in exposure to solriamfetol, the 5 TEAEs that were reported showed no distinct pattern. However, the TEAEs of increased ALT and AST that led to discontinuation (and resolved 5 days after onset) occurred in a Group 5 participant. All TEAEs were judged mild in severity, all were considered treatment related, and all resolved.
Conclusions
Solriamfetol exposure and t1/2 increased, and urinary recovery of solriamfetol decreased with increasing levels of renal impairment, while Cmax was essentially unchanged. The apparent oral clearance of solriamfetol was approximately proportional to renal function as measured by eGFR. In participants with ESRD requiring hemodialysis, solriamfetol AUC was lower in participants who underwent 4‐hour hemodialysis 2 hours after dosing compared with participants without hemodialysis but was still nearly 4‐fold higher than in participants with normal renal function. The mean cumulative fraction of the solriamfetol 75‐mg dose removed by hemodialysis was 20.6%. These results show that renal impairment affects the PK of solriamfetol in a manner consistent with current knowledge of its PK properties. The safety profile of solriamfetol in this study was consistent with clinical research in larger patient populations. Combined analyses based on the current study data and population PK modeling/simulation suggest that adjustments to solriamfetol dosage are not needed in patients with mild renal impairment but are necessary in patients with moderate or severe renal impairment. Dosing is not recommended in patients with ESRD due to significant exposure increase and t1/2 prolongation in this population.
Funding
This research was funded by Jazz Pharmaceuticals.
Declaration of Conflicting Interests
K.Z., D.C., and L.L. are employees of Jazz Pharmaceuticals who, in the course of this employment, have received stock options exercisable for, and other stock awards of, ordinary shares of Jazz Pharmaceuticals plc. T.M. is an employee of a research organization that provided services to Jazz Pharmaceuticals plc. K.L. declares no conflict of interest.
Data Sharing Statement
All summary data that consolidates information for all study subjects are provided within the manuscript and supporting files. Researchers with additional questions or requests for the full data set as part of a comprehensive research proposal may contact Katie Zomorodi at Katie.Zomorodi@jazzpharma.com.
Acknowledgments
In 2014, Jazz Pharmaceuticals acquired a license to develop and commercialize solriamfetol from Aerial Biopharma. Jazz Pharmaceuticals has worldwide development, manufacturing, and commercialization rights to solriamfetol, excluding certain jurisdictions in Asia. SK Biopharmaceuticals, the discoverer of the compound (also known as SKL‐N05), maintains rights in 12 Asian markets, including Korea, China, and Japan. Under the direction of the authors, E. Jay Bienen, PhD, and Larry Deblinger, employees of The Curry Rockefeller Group, LLC (CRG), provided medical writing assistance for this publication. Editorial assistance in formatting, proofreading, copy editing, and fact checking was also provided by CRG. Jazz Pharmaceuticals provided funding to CRG for support in writing and editing of this manuscript.
References
- 1. Carter L, Baladi M, Black J. JZP‐110: a dopamine‐norepinephrine reuptake inhibitor (DNRI) with robust wake‐promoting effects and low abuse potential. Poster presented at: Winter Conference on Brain Research; January 23‐28, 2016; Breckenridge, Colorado. Poster Su23.
- 2. Baladi MG, Forster MJ, Gatch MB, et al. Characterization of the neurochemical and behavioral effects of solriamfetol (JZP‐110), a selective dopamine and norepinephrine reuptake inhibitor. J Pharmacol Exp Ther. 2018;366:367‐376. [DOI] [PubMed] [Google Scholar]
- 3. Johns MW. A new method for measuring daytime sleepiness: the Epworth Sleepiness Scale. Sleep. 1991;14(6):540‐545. [DOI] [PubMed] [Google Scholar]
- 4. Thorpy MJ, Shapiro C, Mayer G, et al. A randomized study of solriamfetol for excessive sleepiness in narcolepsy. Ann Neurol. 2019;85:359‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schweitzer PK, Rosenberg R, Zammit GK, et al. Solriamfetol for excessive sleepiness in obstructive sleep apnea (TONES 3): a randomized controlled trial [published online ahead of print December 6, 2018]. Am J Respir Crit Care Med. 10.1164/rccm.201806-1100OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Strollo PJ Jr, Hedner J, Collop N, et al. Solriamfetol for the treatment of excessive sleepiness in obstructive sleep apnea: a placebo‐controlled randomized‐withdrawal study. Chest. 2019;155(2):364‐374. [DOI] [PubMed] [Google Scholar]
- 7. Zomorodi K, Kankam M, Lu Y. A phase 1, randomized, crossover, open‐label study of the pharmacokinetics of solriamfetol (JZP‐110) in healthy adult subjects with and without food [published online ahead of print December 28, 2018]. Clin Ther. 10.1016/j.clinthera.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 8. U.S. Food and Drug Administration . Draft Guidance for Industry. Pharmacokinetics in patients with impaired renal function—study design, data analysis, and impact on dosing and labeling. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm204959.pdf. Published 2010. Accessed January 11, 2019.
- 9. Zomorodi K, Chen D, Lee L, Lasseter K, Marbury T. An open‐label, single‐dose, phase 1 study of the pharmacokinetics and safety of JZP‐110 in subjects with normal or impaired renal function and with end‐stage renal disease requiring hemodialysis [abstract]. Sleep. 2017;40(suppl):A382‐A383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Levey AS, Coresh J, Greene T, et al. Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate. Ann Intern Med. 2006;145(4):247‐254. [DOI] [PubMed] [Google Scholar]
- 11. International Conference on Harmonisation . Guideline for Industry. Clinical safety data management: definitions and standards for expedited reporting. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E2A/Step4/E2A_Guideline.pdf. Published 1994. Accessed January 11, 2019.
- 12. Iseki K, Tohyama K, Matsumoto T, Nakamura H. High prevalence of chronic kidney disease among patients with sleep related breathing disorder (SRBD). Hypertens Res. 2008;31(2):249‐255. [DOI] [PubMed] [Google Scholar]
- 13. Chou YT, Lee PH, Yang CT, et al. Obstructive sleep apnea: a stand‐alone risk factor for chronic kidney disease. Nephrol Dial Transplant. 2011;26(7):2244‐2250. [DOI] [PubMed] [Google Scholar]
- 14. Kanbay A, Buyukoglan H, Ozdogan N, et al. Obstructive sleep apnea syndrome is related to the progression of chronic kidney disease. Int Urol Nephrol. 2012;44(2):535‐539. [DOI] [PubMed] [Google Scholar]
- 15. Marrone O, Battaglia S, Steiropoulos P, et al. Chronic kidney disease in European patients with obstructive sleep apnea: the ESADA cohort study. J Sleep Res. 2016;25(6):739‐745. [DOI] [PubMed] [Google Scholar]
- 16. Sakaguchi Y, Shoji T, Kawabata H, et al. High prevalence of obstructive sleep apnea and its association with renal function among nondialysis chronic kidney disease patients in Japan: a cross‐sectional study. Clin J Am Soc Nephrol. 2011;6(5):995‐1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nicholl DD, Ahmed SB, Loewen AH, et al. Clinical presentation of obstructive sleep apnea in patients with chronic kidney disease. J Clin Sleep Med. 2012;8(4):381‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuhlmann U, Becker HF, Birkhahn M, et al. Sleep‐apnea in patients with end‐stage renal disease and objective results. Clin Nephrol. 2000;53(6):460‐466. [PubMed] [Google Scholar]
- 19. Miskowiec I, Klawe JJ, Tafil‐Klawe M, et al. Prevalence of sleep apnea syndrome in hemodialyzed patients with end‐stage renal disease. J Physiol Pharmacol. 2006;57(suppl 4):207‐211. [PubMed] [Google Scholar]
- 20. Markou N, Kanakaki M, Myrianthefs P, et al. Sleep‐disordered breathing in nondialyzed patients with chronic renal failure. Lung. 2006;184(1):43‐49. [DOI] [PubMed] [Google Scholar]
- 21. de Oliveira Rodrigues CJ, Marson O, Tufic S, et al. Relationship among end‐stage renal disease, hypertension, and sleep apnea in nondiabetic dialysis patients. Am J Hypertens. 2005;18(2 Pt 1):152‐157. [DOI] [PubMed] [Google Scholar]
- 22. Hanly PJ, Ahmed SB. Sleep apnea and the kidney: is sleep apnea a risk factor for chronic kidney disease? Chest. 2014;146(4):1114‐1122. [DOI] [PubMed] [Google Scholar]
- 23. Abuyassin B, Sharma K, Ayas NT, Laher I. Obstructive sleep apnea and kidney disease: a potential bidirectional relationship? J Clin Sleep Med. 2015;11(8):915‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gildeh N, Drakatos P, Higgins S, Rosenzweig I, Kent BD. Emerging co‐morbidities of obstructive sleep apnea: cognition, kidney disease, and cancer. J Thorac Dis. 2016;8(9):E901‐E917. [DOI] [PMC free article] [PubMed] [Google Scholar]