

Abstract
Lugdunin, a novel thiazolidine cyclopeptide, exhibits micromolar activity against methicillin‐resistant Staphylococcus aureus (MRSA). For structure–activity relationship (SAR) studies, synthetic analogues obtained from alanine and stereo scanning as well as peptides with modified thiazolidine rings were tested for antimicrobial activity. The thiazolidine ring and the alternating d‐ and l‐amino acid backbone are essential. Notably, the non‐natural enantiomer displays equal activity, thus indicating the absence of a chiral target. The antibacterial activity strongly correlates with dissipation of the membrane potential in S. aureus. Lugdunin equalizes pH gradients in artificial membrane vesicles, thereby maintaining membrane integrity, which demonstrates that proton translocation is the mode of action (MoA). The incorporation of extra tryptophan or propargyl moieties further expands the diversity of this class of thiazolidine cyclopeptides.
Keywords: aldehyde peptide synthesis, methicillin-resistant Staphylococcus aureus, proton translocation, synthetic membrane vesicles, thiazolidine antibiotics
Infectious diseases caused by antibiotic‐resistant bacteria are an increasing health problem worldwide, especially the fast dissemination of MRSA.1 As novel antibiotic entities have rarely been discovered in the last decade, we have an urgent need to find new structures. Numerous peptides such as daptomycin add to the great structural diversity of pharmaceutical agents. Modifications of peptide antibiotics are achieved by insertion of particular moieties, for example, double bonds or heterocycles, into the backbone structure. The cyclopeptide callyaerin and one of its analogues differ by a double bond, which confers constraints that correlate directly with their activity.2 Notably, five‐ and six‐membered carbocycles within the backbone mimic conformationally restricted β‐ and γ‐amino acids.3 Naturally occurring heterocycles also determine the structural flexibility of peptide macrocycles.4 Interestingly, thiazolidine rings have not been reported as cyclopeptide components so far. During a screening approach for antibiotics using human bacterial nasal isolates,5 lugdunin (1, Scheme 1) was discovered.6 1 is a nonribosomal cyclic peptide produced by Staphylococcus lugdunensis and features a thiazolidine ring as part of the backbone. 1 shows potent antimicrobial activity against pathogenic bacteria such as MRSA with a minimum inhibitory concentration (MIC) of 3.1 μg mL−1 (3.9 μm). Furthermore, 1 mediates bactericidal effects when applied to mice after skin infection by S. aureus. However, the mode of action (MoA) of 1 has remained elusive.
Scheme 1.
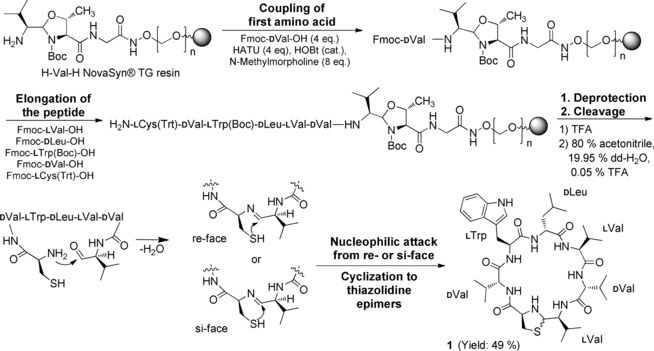
Solid‐phase aldehyde peptide synthesis of 1.
We previously established the synthesis of 1 for the structural proof of natural lugdunin.6 Now we present a comprehensive structure–activity relationship (SAR) study to identify the essential motifs of 1 for its antimicrobial activity. Derivatives of 1 were produced by solid‐phase peptide synthesis (SPPS) on an aldehyde‐generating resin. After assembly of the peptide chain and deprotection of the side chains, the linear aldehyde was released from the resin. Subsequent intramolecular heterocyclization of the C‐terminal aldehyde and the N‐terminal cysteine afforded the macrocycle 1 through in situ thiazolidine formation.7 The thiazolidine, and thus 1, exists in two interconverting and, therefore, inseparable epimeric forms (Scheme 1). The poor coupling of consecutive valine residues (Val5, Val6) was addressed by peptide elongation in acetonitrile (see Figure S1 in the Supporting Information). The optimized strategy provided access to many peptides.
All the peptides were tested as crude products for activity against the MRSA strain USA300 LAC (hereafter: USA300, Table 1). During cleavage, the aldehyde α‐carbon atom of the valine residue at position 7 (Val7, Ala7 in 8) of the peptides partially racemized, thereby leading to mixtures of l‐ and d‐valine or ‐alanine adjacent to the thiazolidine ring (see Scheme S1 and Figure S2 in the Supporting Information). Crude peptides with significant activity (MIC ≤50 μg mL−1) were purified to determine the exact MIC. We suggest terming the new class of lugdunins fibupeptides (lat. fibula, clasp) to define macrocyclic peptides with a thiazolidine moiety as an “ornament clasp”.
Table 1.
MIC values of peptides 1–25.
| AA sequence | Differences highlighted | MIC[a] | |
|---|---|---|---|
| 1 | (CVWLVVV) | lugdunin (lug) | 3.1 (3.9) |
| 2 | AVWLVVValinal[b] | 1‐Ala‐lug | ≥100 |
| 3 | (CAWLVVV) | 2‐Ala‐lug | 12.5 (16.6) |
| 4 | (CVALVVV) | 3‐Ala‐lug | ≥100 |
| 5 | (CVWAVVV) | 4‐Ala‐lug | ≥100 |
| 6 | (CVWLAVV) | 5‐Ala‐lug | 12.5 (16.6) |
| 7 | (CVWLVAV) | 6‐Ala‐lug | 25.0 (33.1) |
| 8 | (CVWLVVA) | 7‐Ala‐lug | 25.0 (33.1) |
| 9 | (CVWLVVV) | linear lug (‐COOH) | ≥100 |
| 10 | CVWLVVVcycl. | cyclized homodetic lug | ≥100 |
| 11 | (Me‐CVWLVVV) | N ‐methylthiazolidine‐lug | ≥100 |
| 12 | (Ac‐CVWLVVV) | N ‐acetylthiazolidine‐lug | ≥100 |
| 13 | (homoCVWLVVV) | 1,3‐thiazinane‐lug | ≥100 |
| 14 | PVWLVVVcycl. | 1‐Pro homodetic lug | ≥100 |
| 15 | (CVWLVVV) | 1‐d‐lug | ≥100 |
| 16 | (CVWLVVV) | 2‐l‐lug | ≥100 |
| 17 | (CVWLVVV) | 3‐d‐lug | ≥100 |
| 18 | (CVWLVVV) | 4‐l‐lug | ≥100 |
| 19 | (CVWLVVV) | 5‐d‐lug | ≥100 |
| 20 | (CVWLVVV) | 6‐l‐lug | ≥100 |
| 21 | (CVWLVVV) | 7‐d‐lug | ≥100 |
| 22 | (CVWLVVV) | enantio‐lug | 3.1 (3.9) |
| 23 | (CVWLVWV) | 6‐Trp‐lug | 1.6 (1.8) |
| 24 | (CPraWLVWV) | 2‐Pra‐6‐Trp‐lug | 3.1 (3.9) |
| 25 | VOLFPVOLFP cycl. | gramicidin S | 6.2 (5.4) |
[a] MRSA USA300 LAC (MIC in μg mL−1 (μm). For S. aureus NCTC8325 MICs, see Table S3). Single‐letter codes for amino acids, brackets indicate cyclic structure (cyclization via thiazolidine), cycl. indicates cyclization via the peptide bond, underlined letters represent d‐amino acids. [b] Detected as [M+MeOH]+ by ESI‐MS, Ala=alanine, Pro=proline, Trp=tryptophan, Pra=propargylglycine.
The importance of individual amino acids for the activity of 1 was revealed by an alanine scan,8 which yielded fibupeptides 2–8. Each amino acid of 1 was successively replaced by alanine with the same stereoconfiguration. Antimicrobially inactive peptide 2 neither carries a thiazolidine nor is cyclic because of the lack of cysteine. Mass spectrometry analysis of 2 showed the formation of an aldehyde‐methanol adduct ([M+MeOH]+, m/z 801.5232, 0.12 Δppm). This is in agreement with the finding of Enck et al. that the precursor aldehyde of tyrocidine A did not spontaneously cyclize to the imine.9
Consequently, cysteine and, hence, the thiazolidine ring is essential. Active alanine peptides 3, 6, 7, and 8 showed MIC values of 12.5 μg mL−1 (16.6 μm) to 25 μg mL−1 (33.1 μm), which is a four‐ to eightfold reduction in the antimicrobial activity. The most pronounced impact on antibiotic efficacy was detected for the exchange of Trp3 (4) and Leu4 (5) for alanine. Both derivatives showed MIC values ≥100 μg mL−1 and were regarded as inactive. Therefore, tryptophan and leucine are crucial for the antibacterial activity, whereas valine versus alanine exchanges are well‐tolerated. However, the different activities of 3, 6, 7, and 8 imply a distinct relevance of each valine.
The importance of the thiazolidine ring was investigated, starting with the linear lugdunin peptide 9 (Figure 1). Intramolecular cyclization of 9 yielded homodetic analogue 10 in which the ring is composed exclusively of normal peptide bonds. Both peptides were inactive against USA300. In addition, the role of the thiazolidine NH proton was addressed by replacing it by a tertiary amine through methylation (11) and acetylation (12). 11 and 12 are inactive as antibacterial agents, thus demonstrating the indispensability of the secondary amine of the thiazolidine ring. We speculate that the basicity of the thiazolidine amine or its three‐dimensional structure are responsible for the bioactivity of 1. Inactive 13 was prepared with homocysteine to expand the heterocycle by an additional methylene group. The six‐membered 1,3‐thiazinane affects the structure of 1 adversely in terms of activity, even though it contains a secondary amine. Since the thiazolidine resembles an unusual 2‐connected thioproline, we synthesized the proline‐containing homodetic 14, which was inactive. Thus, both, the thiazolidine ring and its secondary amine are essential for the antimicrobial activity of 1.
Figure 1.
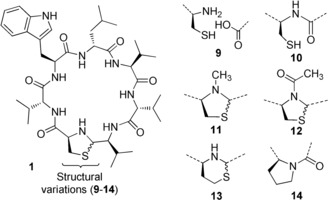
Structure of lugdunin (1) and analogues 9–14.
The intriguing d‐,l‐architecture of 1 prompted us to conduct a stereo scan, while retaining the sequence and hydrophobic character of 1. One amino acid at a time was incorporated as its enantiomer (15–21). All the diastereomers were ineffective against USA300, which demonstrates that any inversion of a stereogenic center dramatically affects the antimicrobial potency. To clarify whether the inversion of the absolute configuration of 1 also has such an impact on activity, we synthesized its enantiomer 22. Remarkably, 22 showed identical antibiotic activity as 1. This situation has been rarely discussed for natural products, for example, for the antiviral feglymycin8 or the antibiotic lysocin E.10 The insignificance of the absolute configuration of 1 suggests that the MoA does not depend on a stereospecific receptor–ligand interaction but could involve the recognition of achiral small molecules or ions.10
Together, these SAR studies revealed that an unsubstituted thiazolidine, tryptophan, leucine, and an alternating amino acid stereoconfiguration are essential structural motifs of 1. Tryptophan and leucine are abundant in peptides that interact with bacterial cell membranes such as synthetic poly‐(Trp‐Leu)‐octapeptides.11 The necessity of tryptophan and leucine and the decrease in the activity of the less hydrophobic alanine fibupeptides 3, 6, 7, and 8 pointed towards an interaction of 1 with the hydrophobic region of bacterial membranes.
A double tryptophan‐containing fibupeptide (23) was designed to intensify the presumed interaction with the bacterial membrane. d‐Tryptophan was incorporated (Trp6) within the nonpolar flank (dLeu4‐lVal5‐dVal6‐lVal7) of 1. Fibupeptide 23 showed a twofold increased activity. As could be deduced from the alanine scan, position 2 shows tolerance for side‐chain modification while retaining activity. Incorporation of d‐propargylglycine (Pra) at this position in 23 resulted in active derivative 24. 24 is suitable for 1,3‐dipolar cycloaddition to enable the production of further analogues, preferably with activity against Gram‐negative bacteria.12
With the knowledge that the enantiomer (22, Figure 2) shows activity identical to 1, we suspected that 1 might move achiral molecules or ions across lipid membranes. This concurs with our previous observation that bacterial cells exposed to 1 stopped incorporating radioactive DNA, RNA, protein, and cell‐wall precursors.6 Thus, the influence of the active fibupeptides 1, 3, 22, and 23 on the transmembrane potential of S. aureus NCTC8325 was compared to that of the less active or inactive 5, 11, and 21. When entering the cell, the green fluorescence of the dye DiOC2(3) shifts towards a red emission because of a self‐association that depends on membrane potential.13
Figure 2.
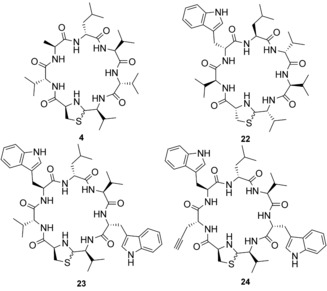
Exemplified derivatives of 1. 4 is an inactive alanine analogue. The enantiomer 22 shows identical activity as 1. 23 and 24 are specially designed analogues of 1 with twofold and equal activity, respectively.
All the tested peptides affected the transmembrane potential of S. aureus NCTC8325, in full correlation with their MIC values, and in a concentration‐dependent manner (Figure 3, see also Figure S11 and Table S3 in the Supporting Information). Partial membrane depolarization occurred at concentrations slightly below the MIC (0.125–0.5 × MIC), while inactive 11 showed no effect. This is in accordance with the parallel cessation of all biosynthetic pathways and indicates a MoA involving impairment of membrane integrity or ion leakage/transport.
Figure 3.
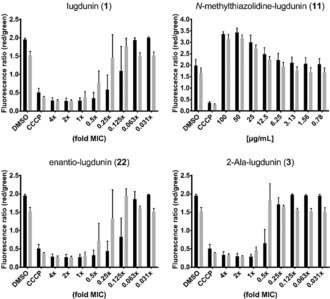
Effect of 1, 11, 22, and 3 on the S. aureus NCTC8325 membrane potential after 30 (black bars) and 60 minutes (gray bars) of treatment. The protonophore CCCP (5 μm) was used as a positive and DMSO as a negative control. Error bars represent the standard deviation (SD) of two biological replicates including two technical replicates each.
Remarkably, 1 does not tolerate amino acids with polar (Ser, Thr) or protonated (Lys) side chains without losing bioactivity—in contrast to common peptides that disrupt the membrane potential such as gramicidin A.
To analyze the effects on bacterial membranes, we treated S. aureus with 1 and subsequently added a mixture of the dyes Syto9 and propidium iodide (PI) to the cells as an indicator for pore formation. The red‐fluorescent PI can only cross the cytoplasmic membrane through large pores or lesions. Treatment with 1 up to a concentration of 30 μg mL−1 (10×MIC) did not allow for PI entry into the S. aureus cells, while nisin led to a strong influx of the dye because of its ability to form large pores (Figure 4 A).
Figure 4.
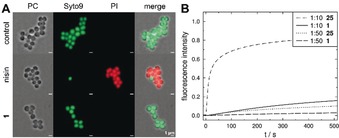
Complementary experiments excluding large pores or lesions. A) Fluorescence microscopy of S. aureus treated with pore‐forming nisin (1–2 × MIC) or 1 (10 × MIC). Scale bars: 1 μm. B) Time course of normalized CF leakage from POPC vesicles, induced by 25 and 1 at concentrations of 5 μm and 1 μm (P/L 1:10 and 1:50).
We further employed unilamellar vesicles composed of POPC (1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphocholine) as a membrane model system to assess the activity of 1. The use of artificial lipid bilayers enables the characterization of membrane activity independent of other factors such as proteins. We investigated first whether 1 impairs vesicle integrity.14 The ability of 1 to induce release of the fluorescent dye carboxyfluorescein (CF) was compared to that of the cyclic decapeptide gramicidin S (25), which can destabilize membranes.15 The dye is entrapped in vesicles in a self‐quenching concentration (100 mm) and leakage results in an increase in fluorescence. In contrast to 25, 1 induced only very slow leakage of the dye even at high concentrations (Figure 4 B). This result supports the notion that 1 does not destabilize the membrane, but rather acts by translocating ions.
We investigated the ability of 1 to transport protons by using vesicles filled with the pH‐sensitive fluorescent dye pyranine (HPTS).16 A pH gradient was established across the lipid bilayer and proton transport was observed as a change in fluorescence. As proton translocation across a membrane induces a transmembrane potential that prevents further transport, the change in the internal pH value is also dependent on charge equilibration and, therefore, transport of further ions. A control experiment with the protonophore carbonyl cyanide m‐chlorophenyl hydrazone (CCCP) and the potassium ionophore valinomycin showed that both ions have to be transported to explain rapid pH equilibration (see Figure S12 in the Supporting Information). Figure 5 shows that 1 causes rapid proton translocation at concentrations as low as 50 nm (peptide to lipid ratio (P/L) 1:1000) irrespective of the direction of the gradient. To exclude dye leakage, pyranine fluorescence was quenched outside the vesicles in a control experiment (see Figure S12 in the Supporting Information).
Figure 5.
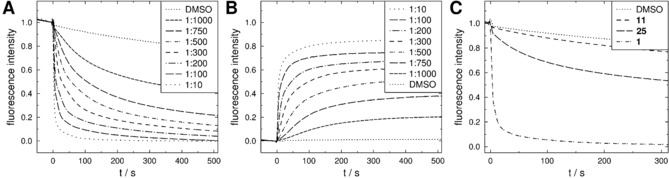
Time course of normalized pyranine fluorescence after addition of: A,B) 5 μm to 50 nm 1 (P/L 1:10 to 1:1000) with A) proton influx from pH 6.4 to 7.4, B) proton efflux from pH 7.4 to 8.4, C) after addition of 1 μm (P/L 1:50) 11, 25, and 1, proton influx from pH 6.4 to 7.4. The vesicles were composed of POPC, total lipid concentration 50 μm containing 0.5 mm pyranine.
In this pH assay, 1 demonstrated a significantly higher proton translocation capability than 25, whereas antimicrobially inactive 11 showed greatly reduced proton transport (Figure 5 C). This finding suggests a vital role of the thiazolidine moiety in proton translocation and is in agreement with the membrane depolarization data.
In summary, we established an optimized synthesis of 1 to provide access to manifold analogues. By SAR studies, we revealed the essential motifs for antimicrobial activity, notably, the alternation of d‐ and l‐amino acids, the presence of tryptophan and leucine, as well as the N‐unsubstituted thiazolidine ring. The identical activity of the enantiomer 22 suggested that chiral recognition was not relevant for the MoA of 1. Additionally, 1 and its analogues illustrate a strong correlation between membrane depolarization and MIC values with S. aureus cells. Furthermore, 1 did not induce large pores in either S. aureus cells or POPC vesicles and acts through proton translocation in synthetic membrane vesicles. In addition, the twofold more active 23 (Trp6) verified these insights.
The active analogue 24 with a propargyl function paves the way for the production of analogues with optimized bioactivity or fluorescent properties, which will contribute to elucidating the mechanistic interaction between 1 and MRSA on the molecular level. The exact role of the vital thiazolidine ring is the focus of current investigations along with the question of whether 1 translocates protons as a mobile carrier or by channel formation.
Conflict of interest
Eberhard Karls University Tübingen holds a patent (EP3072899B1) covering the compound lugdunin, derivatives thereof, and the bacterial infection prevention by lugdunin producing bacteria. The patent has also been filed in the USA (US2018/0155397A1).
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The work of N.A.S. is supported by the Institutional Strategy of the University of Tübingen (DFG, ZUK 63). We are grateful to RTG 1708 (M.C.K., A.P., H.B.‐O., S.G.) and SFB 766 (A.P., H.B.‐O., S.G.) for financial support. B.K., A.P., H.B.‐O., and A.B. acknowledge support through infrastructural funding from DZIF and Cluster of Excellence EXC 2124. We further acknowledge Jan Straetener and Jutta Gerber‐Nolte for technical support and Pascal Rath for NMR measurements.
N. A. Schilling, A. Berscheid, J. Schumacher, J. S. Saur, M. C. Konnerth, S. N. Wirtz, J. M. Beltrán-Beleña, A. Zipperer, B. Krismer, A. Peschel, H. Kalbacher, H. Brötz-Oesterhelt, C. Steinem, S. Grond, Angew. Chem. Int. Ed. 2019, 58, 9234.
Dedicated to Professor Axel Zeeck on the occasion of his 80th birthday
References
- 1. Walsh T. R., Nat. Microbiol. 2018, 3, 854–855. [DOI] [PubMed] [Google Scholar]
- 2. Zhang S., De Leon Rodriguez L. M., Leung I. K. H., Cook G. M., Harris P. W. R., Brimble M. A., Angew. Chem. Int. Ed. 2018, 57, 3631–3635; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3693–3697. [Google Scholar]
- 3. Montenegro J., Ghadiri M. R., Granja J. R., Acc. Chem. Res. 2013, 46, 2955–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kaldas S. J., Yudin A. K., Chem. Eur. J. 2018, 24, 7074–7082. [DOI] [PubMed] [Google Scholar]
- 5. Janek D., Zipperer A., Kulik A., Krismer B., Peschel A., PLoS Pathog. 2016, 12, e1005812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zipperer A., Konnerth M. C., Laux C., Berscheid A., Janek D., Weidenmaier C., Burian M., Schilling N. A., Slavetinsky C., Marschal M., Willmann M., Kalbacher H., Schittek B., Brötz-Oesterhelt H., Grond S., Peschel A., Krismer B., Nature 2016, 535, 511–516. [DOI] [PubMed] [Google Scholar]
- 7. Malins L. R., deGruyter J. N., Robbins K. J., Scola P. M., Eastgate M. D., Ghadiri M. R., Baran P. S., J. Am. Chem. Soc. 2017, 139, 5233–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dettner F., Hänchen A., Schols D., Toti L., Nußer A., Süssmuth R. D., Angew. Chem. Int. Ed. 2009, 48, 1856–1861; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 1888–1893. [Google Scholar]
- 9. Enck S., Kopp F., Marahiel M. A., Geyer A., Org. Biomol. Chem. 2010, 8, 559–563. [DOI] [PubMed] [Google Scholar]
- 10. Murai M., Kaji T., Kuranaga T., Hamamoto H., Sekimizu K., Inoue M., Angew. Chem. Int. Ed. 2015, 54, 1556–1560; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1576–1580. [Google Scholar]
- 11. Ghadiri M. R., Granja J. R., Buehler L. K., Nature 1994, 369, 301. [DOI] [PubMed] [Google Scholar]
- 12. Smith P. A., et al. Nature 2018, 561, 189–194. [DOI] [PubMed] [Google Scholar]
- 13. Novo D., Perlmutter N. G., Hunt R. H., Shapiro H. M., Cytometry 1999, 35, 55–63. [DOI] [PubMed] [Google Scholar]
- 14. Weinstein J. N., Klausner R. D., Innerarity T., Ralston E., Blumenthal R., Biochim. Biophys. Acta Biomembr. 1981, 647, 270–284. [DOI] [PubMed] [Google Scholar]
- 15. Wenzel M., Rautenbach M., Vosloo J. A., Siersma T., Aisenbrey C. H. M., Zaitseva E., Laubscher W. E., van Rensburg W., Behrends J. C., Bechinger B., Hamoen L. W., mBio 2018, 9, e00802-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kano K., Fendler J. H., Biochim. Biophys. Acta Biomembr. 1978, 509, 289–299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary