

Abstract
Chronic infection of hepatitis B virus (HBV) is associated with an increased incidence of hepatocellular carcinoma (HCC). HBV encodes an oncoprotein, hepatitis B x protein (HBx), that is crucial for viral replication and interferes with multiple cellular activities including gene expression, histone modifications, and genomic stability. To date, it remains unclear how disruption of these activities contributes to hepatocarcinogenesis. Here, we report that HBV exhibits antiresection activity by disrupting DNA end resection, thus impairing the initial steps of homologous recombination (HR). This antiresection activity occurs in primary human hepatocytes undergoing a natural viral infection–replication cycle as well as in cells with integrated HBV genomes. Among the seven HBV‐encoded proteins, we identified HBx as the sole viral factor that inhibits resection. By disrupting an evolutionarily conserved Cullin4A–damage‐specific DNA binding protein 1–RING type of E3 ligase, CRL4WDR70, through its H‐box, we show that HBx inhibits H2B monoubiquitylation at lysine 120 at double‐strand breaks, thus reducing the efficiency of long‐range resection. We further show that directly impairing H2B monoubiquitylation elicited tumorigenesis upon engraftment of deficient cells in athymic mice, confirming that the impairment of CRL4WDR70 function by HBx is sufficient to promote carcinogenesis. Finally, we demonstrate that lack of H2B monoubiquitylation is manifest in human HBV‐associated HCC when compared with HBV‐free HCC, implying corresponding defects of epigenetic regulation and end resection. Conclusion: The antiresection activity of HBx induces an HR defect and genomic instability and contributes to tumorigenesis of host hepatocytes.
Abbreviations
- BrdU
bromodeoxyuridine
- ChIP
chromatin immunoprecipitation
- CPT
camptothecin
- CRISPR
clustered regularly interspaced short palindromic repeats
- CRL4WDR70
Cullin4A‐DDB1‐RING type of E3 ligase
- DCAF
DDB1 and CUL4–associated factor
- DDB1
damage‐specific DNA binding protein 1
- DSB
double‐strand break
- EXO1
exonuclease 1
- gRNA
genomic RNA
- HBcAg
hepatitis B core antigen
- HBV
hepatitis B virus
- HBVHCC
HBV‐associated HCC
- HBx
hepatitis B x protein
- HCC
hepatocellular carcinoma
- HR
homologous recombination
- IHC
immunohistochemical
- IR
ionizing radiation
- IRIF
IR‐induced foci
- NBS1
Nijmegen breakage syndrome 1
- PHH
primary human hepatocyte
- PPP1R12C/p84
protein phosphatase 1 regulatory subunit 12C
- RPA
replication protein A
- si
small interfering
- ssDNA
single‐stranded DNA
- SV5‐V
protein encoded by simian virus 5
- uH2B
H2B monoubiquitylation at lysine 120
- USP22
ubiquitin‐specific peptidase 22
- WDR70
WD repeat domain 70
Worldwide, chronic hepatitis B virus (HBV) infection and its associated diseases such as cirrhosis account for about 50% of all cases of human hepatocellular carcinoma (HCC), and the risk of carcinogenesis is elevated with increasing viral load.1, 2 The vast majority of HBV‐associated HCC (HBVHCC) cases harbor chromosomally integrated HBV genomes,3, 4 although site‐specific integration of the viral genome into a known oncogene seems to be a rare event. This strongly suggests that HBV encodes a viral element that intrinsically promotes oncogenic transformation. During HBV infection, the small 3.2‐kb HBV genome expresses seven genes (preCore [hepatitis B e antigen, HBeAg], Core, Pol, X, and three envelope proteins, L, M, and S) for completion of the viral replication.5 The X gene encoding a 154‐residue pleiotropic transcriptional activator (hepatitis B x protein [HBx]) serves as a stimulator of viral replication and gene expression.6, 7 A plethora of evidence suggests that HBx has the capacity to induce HCC in vivo, behaving as a prominent oncogenic driver for HBVHCC.8 However, the precise oncogenic targets and/or signal transduction pathways influenced by HBx remain enigmatic.
HBx lacks DNA binding activity and thus likely carries out its functions through protein–protein interactions with cellular factors. One such HBx‐interacting factor is damage‐specific DNA binding protein 1 (DDB1), a core component of the Cullin‐4 ring ligase (CRL4) family of ubiquitin ligases.9 The core of CRL4 complexes (CUL4‐DDB1–ring of cullins) is associated with a family of WD40 proteins, which act as substrate‐targeting subunits and are known as DDB1 and CUL4–associated factor (DCAFs).10 Two short peptide stretches of DCAFs, the DDB1‐binding WD40 (DWD) and H‐box motifs, are required for the docking of DCAFs to CRL4s through DDB1.11, 12, 13 Intriguingly, the C terminus of HBx resembles the H‐box of a subset of cellular DCAFs (including DDB2 and DCAF9/WD and tetratricopeptide repeats 1 [WDTC1]) and can selectively disengage DCAFs from CRL4.12, 14 Recent studies have shown that HBx hijacks the E3 ligase activity of CRL4 to form viral CRL4HBx and remove specific cellular factors (i.e., structural maintenance of chromosomes 5/6 [SMC5/6]) that are restrictive to viral replication.15
Impairment of the CRL4 core complex activity by tissue‐specific knockout of DDB1 in hepatocytes has been shown to induce liver regeneration and tumorigenesis,16 suggesting a restriction of HCC by CRL4. This could be attributed to the potential of HBx to interfere with DNA damage responses: it compromises cell cycle checkpoint control by its interaction with p539 and inhibits the ultraviolet light–induced nucleotide excision repair (NER) pathway by disrupting the CRL4DDB2 complex.17 However, no role has been established for disabled NER in promoting human HBVHCC, and removal of known DCAF factors does not account for the oncogenic activity of HBx. Thus, there exists a mechanistic gap in HBx‐triggered hepatocarcinogenesis, implying an uncharacterized mechanism underlying HBVHCC.
Here, we present evidence that HBV infection inhibits DNA double‐strand break (DSB) repair in both HBV‐infected and HBV‐integrated cells. We show that the disassembly of a recently identified CRL4 subcomplex, CRL4WDR70, by the HBx H‐box motif compromises DNA end resection. The resulting loss of CRL4WDR70‐dependent histone H2B monoubiquitylation prevents the association of replication protein A (RPA) and RAD51 with DSBs and prevents efficient homologous recombination (HR) of DSBs. We demonstrate that the loss of WD repeat domain 70 (WDR70) function phenocopies the impact of HBx on HR and that the epigenetic and repair defects caused by HBx can directly trigger tumorigenesis. Thus, this study provides compelling evidence that HBx possesses an antiresection activity that inhibits CRL4WDR70‐mediated HR repair. This results in genomic instability of host cells and potentially drives the carcinogenesis of HBVHCC.
Materials and Methods
Cell Lines
L02, T43,18 HepG2, HepG2.2.15, HEK293T, and their derived cells were cultured in standard conditions. Primary human hepatocyte (PHH) culture and infection with HBV were performed as described.19, 20 Additional details are in the Supporting Information.
Biochemical, Immunological, and Molecular Assays
Details of immunoassays, measurement of DNA damage responses, real‐time quantitative PCR, and chromatin immunoprecipitation (ChIP) analysis are available in the Supporting Information.
Statistics
All histograms are presented as means ± standard deviation. For quantitative analysis, including the ChIP assay and image analysis, at least three independent experiments, each containing three parallel samples, were performed. Statistical analysis between two groups was performed by t test in GraphPad Prism 5.
Results
HBV Infection Induces a Resection Defect in Host Cells
HBVHCC genomes are remarkably unstable21 (data not shown). We therefore examined whether HBV impacts DSB repair, the malfunction of which often causes genome‐wide structural variations. Congenic hepatocarcinoma cell lines (parental HepG2 and derived HepG2.2.15 that harbors a functional HBV genome22) were challenged with DSB‐generating agents, and DSB responses were evaluated by indirect immunofluorescence. Upon camptothecin (CPT) treatment, HepG2.2.15 cells, when compared to control HepG2 cells, showed reduced foci formation for Nijmegen breakage syndrome 1 (NBS1) and RPA32, the latter of which is a replication protein A subunit. RPA is a prominent marker for the single‐stranded DNA (ssDNA) that arises from 5′‐3′ DSB resection23 (Supporting Fig. S1A). Phosphorylation of RPA32 (phosphorylated serines 33 and 4/8) was also shown to be diminished by immunoblotting (Fig. 1A; Supporting Fig. S1B). Because RPA constitutes a platform to attract HR factors onto processed DNA ends and is a prerequisite for HR (see Supporting Fig. S1C), these results suggest that DNA end resection, and consequently HR itself, is compromised.24
Figure 1.
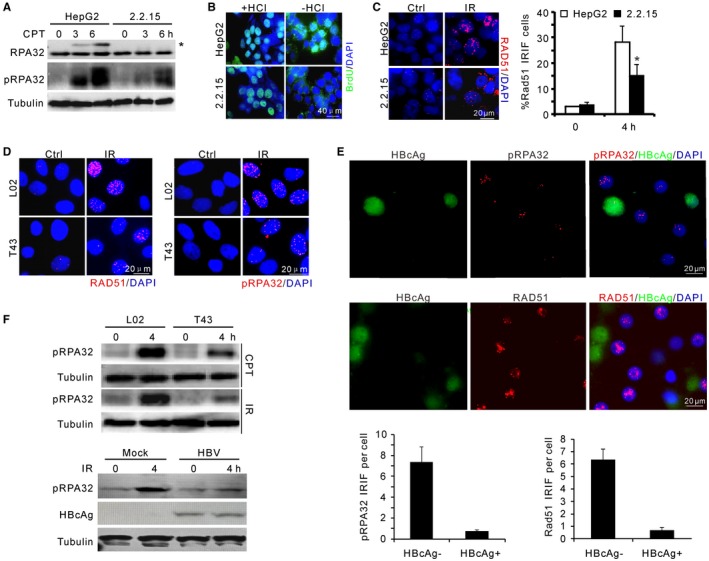
HBV attenuates DNA end resection upon genotoxic insults. (A) Immunoblotting for RPA32 phosphorylation in HepG2 and HepG2.2.15 cells upon CPT treatment. pRPA32 indicates phosphorylated serine 33 of RPA32. *Band shift of phosphorylated RPA32 detected by α‐RPA32 serum. (B) Non‐(−HCl) and denaturing‐(+HCl) indirect immunofluorescence showing ssDNA at resected DSBs and equal incorporation of BrdU after CPT treatment, respectively. Scale bar, 40 μm. (C) Representative image and percentage of RAD51 IRIF‐positive cells after 5‐Gy irradiation in HepG2 and HepG2.2.15 cells. (D) Representative images for IRIF of RAD51 and phosphorylated RPA32 in L02 and T43 cells after IR treatment. (E) Images (top) and quantification (bottom) of indicated IRIF in mock or HBV‐infected PHHs at day 7. Cells were fixed for immunostaining 4 hours after 1‐Gy irradiation. HBcAg (green) marks HBV‐infected PHHs. (F) Immunoblotting for RPA32 phosphorylation in L02/T43 cells or HBV‐infected PHHs at indicated times. Genotoxic treatments: 2 μM CPT or 5‐Gy irradiation. Abbreviation: DAPI, 4′,6‐diamidino‐2‐phenylindole.
To confirm that DSB resection is attenuated in HepG2.2.15 cells, ssDNA was visualized directly using bromodeoxyuridine (BrdU) incorporation and subsequent α‐BrdU antibody staining under nondenaturing conditions (Fig. 1B). Because the generation of sufficient ssDNA from DNA end resection is required for the recruitment of Rad51 recombinase to mediate homology search and downstream HR events,25 we next visualized RAD51 foci. In HepG2.2.15 cells, the recruitment of RAD51 to DSBs after ionizing radiation (IR) was dramatically decreased in comparison with HepG2 controls (Fig. 1C). Thus, HBV is associated with resection and HR defects in a hepatocarcinoma cell line with integrated viral genomes.
Resection Defect in HBV‐Infected Cells
Although HepG2.2.15 is widely used as a model system for studying HBV replication, the hepatocarcinoma origin of this cell line and random integration of the viral genome may result in genetic variation of the host genome and therefore unfaithfully represent the cellular impact of HBV. To exclude this possibility and extend our observations to the circumstances of natural viral infection, we present two lines of evidence to establish that the HBV‐induced resection and HR defects represent a generalized phenomenon. First, we show that the defects in RPA phosphorylation and RAD51 IR‐induced foci (IRIF) hold true in an independently derived HBV‐positive cell line (T43) that was generated by integrating viral genomes into normal hepatic L02 cells18 (Fig. 1D,F; Supporting Fig. S1D).
Second, we employed HBV‐infected PHH cultures to investigate the impact of natural viral infection on resection activity.19, 20 The application of concentrated supernatants from HepG2.2.15 cells or high‐titer HBV patient serum to PHHs resulted in efficient infection and viral replication as shown by increased hepatitis B surface antigen/HBeAg titres and the positivity for nucleocapsid antigen (HBcAg) during days 3‐7 postinoculation (Supporting Fig. S1E,F). As a consequence of HBV infection, IR‐induced RPA32 phosphorylation was reduced significantly in PHHs, coincidental with reduced RPA32/RAD51 IRIF in HBcAg‐positive cells (Fig. 1E,F). Taken together, these data demonstrate that DNA end resection is impeded during the course of HBV infection and maintained after integration of viral genomes.
HBx Counteracts CUL4A‐DDB1‐Dependent Resection through its H‐box Motif
Genes involved in homology‐dependent repair are frequently mutated in breast and ovarian cancers.26, 27 However, no human genetic abnormalities involving HR have been described for HBVHCC. To consolidate our observation that HBV‐induced HR defects are directly attributable to a viral element, as opposed to genetic variation in the host genomes, we performed a screen for the impact of HBV viral genes on resection (Supporting Fig. S2A). By examining CPT‐induced RPA and NBS1 phosphorylation, HBx was identified as the sole viral factor that renders cells defective in resection (Fig. 2A; Supporting Fig. S2B). HBx expression in L02 cells also perturbed RPA32 and RAD51 foci formation (Fig. 2B; Supporting Fig. S2C). In corresponding experiments, we also showed that the defects in RPA phosphorylation and DSB loading were significantly restored in T43 cells upon silencing of HBx by small interfering (si) RNA (Fig. 2C; Supporting Fig. S2D,E). siHBx could also rescue the defective IRIF of RPA and RAD51 in PHH cells (Supporting Fig. S2F).
Figure 2.
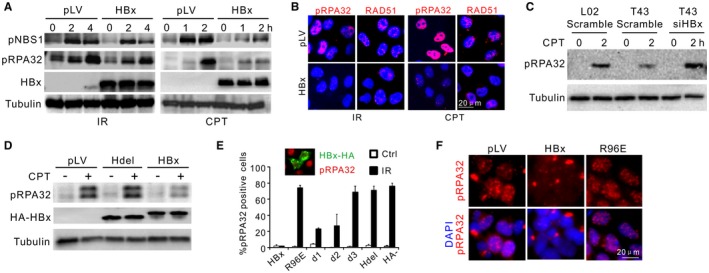
HBx impairs DNA end resection. (A) Immunoblotting for IR‐induced or CPT‐induced RPA32 and NBS1 phosphorylation in the presence or not of HBx. L02 cells were preinfected with 2 multiplicities of infection/cell of pLV (mock infection) or pLV‐HA‐HBx lentivirus. (B) pLV‐transfected or pLV‐HBx‐transfected L02 cells were treated as in (A), followed by immunostaining of RAD51 and pRPA32 foci. (C) Monitoring of RPA32 phosphorylation in indicated cells treated or not with siHBx. (D) Immunoblotting for pRPA32 in CPT‐treated L02 cells transfected with indicated expression plasmids. HBx and Hdel (HBx with H‐box deleted: amino acids 88‐98) were monitored by α‐HA. (E) Percentage of pRPA32 IRIF‐positive L02 cells pretransfected with different HA‐HBx constructs. pRPA32 foci were monitored 4 hours after irradiation. Cells with double‐positive staining of HA and phosphorylated serine 33 were judged as positive except in “HBx−” cells (no HA‐HBx transfection) where pRPA32 were counted in random cells. Inset: Costaining of HA‐HBx (green) and phosphorylated serine 33 (red). n = 3 biological repeats. Error bars = SD. (F) Immunofluorescence for pRPA32 in L02 cells infected with HBx wild‐type or R96E lentivirus (2 multiplicities of infection/cell) followed by IR treatment. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; HA, hemagglutinin; LV, lentivirus.
To exclude the possibility that overexpression of HBx produces off‐target effects not seen in physiological conditions, mRNA levels of HBx in T43 cells, HepG2.2.15 cells, and PHHs were quantified (Supporting Fig. S2G). T43 cells express HBx at a comparable level to infected PHHs but at significantly lower levels than that observed in HepG2.2.15 cells. This likely reflects the relative replicative activities, as indicated by the covalently closed circular DNA copies (Supporting Fig. S2H). Thus, we conclude that HBx confers on HBV an antiresection activity under physiological conditions.
The majority of HBx protein is in a complex with DDB1 that is mediated by its H‐box motif (residues 88‐98).28 This results in the redirection of CRL4 activity to support the viral life cycle.15, 29 We therefore tested the hypothesis that HBx interferes with resection by disrupting the function of CRL4 complexes. Indeed, the formation of RPA32 and RAD51 IRIF was diminished upon DDB1 depletion by RNA interference technology, indicating that a species of CRL4 complex that participates in regulating the formation of ssDNA filaments and resection is down‐regulated (Supporting Fig. S3A). CPT‐induced phosphorylation of RPA32/NBS1 upon siDDB1 or siCUL4A treatment was also affected, resembling the phenotype of HBx expression (Supporting Fig. S3B,C). The similar HR defects caused by CRL4 ablation and HBx introduction suggest a possible mechanistic link between these factors in affecting DNA end resection.
To elucidate how HBx impairs the resection process by interfering with CUL4A‐DDB1, various mutants encompassing the H‐box motif and the HBx carboxyl domain were constructed (Supporting Fig. S3D). Ectopic expression of these HBx mutants near or within the H‐box (d3:C105‐154Δ, Hdel: C88‐100Δ and R96E) abolished the HBx–DDB1 association (Supporting Fig. S3E‐G), consistent with a previous report.12 Interestingly, upon ectopic expression in L02 cells, abolition of the H‐box impaired the antiresection potential of HBx, and the mutants displayed no profound impact on RPA32 phosphorylation or foci formation, in contrast to HBx wild type (Fig. 2D‐F). Therefore, we conclude that HBx through its H‐box targets the function of CRL4 complex to impede DNA end resection.
HBx Interferes With CRL4WDR70‐Mediated DNA End Resection
To identify the CRL4 complex targeted by HBx in respect of end resection, we examined the recently identified CRL4WDR70 complex that regulates resection in fission yeast.30 Similar to the yeast homologue, WDR70 interacts with DDB1 (Supporting Fig. S4A). WDR70 protein contains a canonical DWD motif (amino acids 405‐408 in the human homologue) as well as a conserved C‐terminal H‐box (amino acids 653‐662) (Fig. 3A,B). Both domains are indispensable in mediating the interaction of WDR70 with DDB1 (Fig. 3C).12, 30 Strikingly, the C terminus of WDR70 contains a similar feature to that characterized for the DDB2 sequence: an additional α‐helix adjunct to the H‐box that provides further binding capacity between DDB1 and DDB2. The presence of this conserved additional α‐helix is predicted to provide extra binding capabilities for WDR70 and DDB1 (Fig. 3B; Supporting Fig. S4B).
Figure 3.
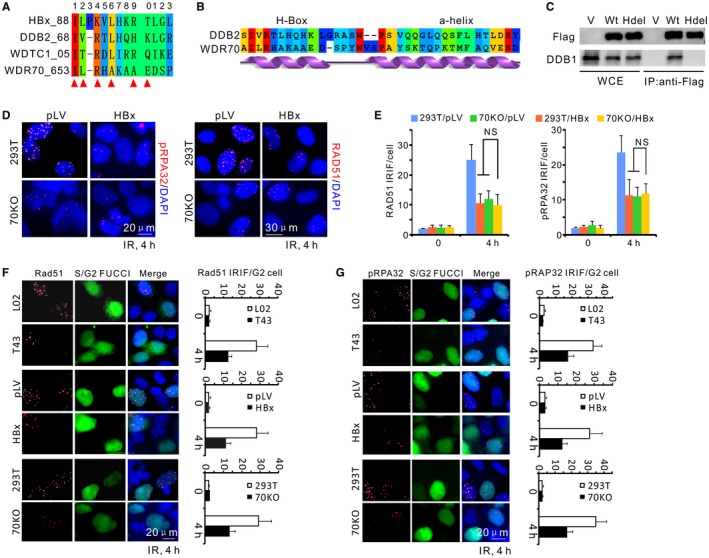
Disruption of CRL4WDR70 complex by HBx results in a resection defect. (A) Sequence alignment for the H‐boxes of cellular DCAF proteins and HBx (WDTC1, alias of DCAF9). Red triangle, binding hotspots serving as key DDB1‐contacting residues. (B) By sequence alignment and secondary structure prediction, the WDR70 C‐terminal sequence possesses a similar H‐box sequence and an adjunct α‐helical structure to that of DDB2, indicating their similar docking mode to DDB1. (C) Immunoprecipitation of endogenous DDB1 in 293T cells by Flag‐tagged WDR70 with or without H‐boxes (amino acids 653‐662). (D) Immunofluorescent staining for phosphorylated RPA32 or RAD51 IRIF in parental and WDR70 knockout 293T cells with or without HBx expression. IRIF defects in 70KO cells were not exacerbated by the presence of HBx. (E) Quantification for IRIF as in (D). n = 3 biological repeats. Error bars = SD. (F,G) Representative images and quantification for RAD51 (F) and pRPA32 (G) foci in IR‐treated cells (4 Gy). Geminin‐green fluorescent protein represents S/G2 cells (S/G2‐FUCCI). Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; FUCCI, fluorescent ubiquitylation‐based cell cycle indicator; Hdel, H‐box deletion mutant of WDR70; IP, immunoprecipitation; 70KO, WDR70 knockout; LV, lentivirus; NS, no significant difference; WCE, whole cell extracts; V, empty vector.
The structural similarities between WDR70 and DDB2 suggest that WDR70 resembles the subclass of DCAF proteins (including DDB2) whose engagement to DDB1 through their H‐box is liable to competition by the homologous sequence of HBx.28 This would be consistent with the fact that knockout of human WDR70 by clustered regularly interspaced short palindromic repeats (CRISPR) in 293T cells30 resulted in decreased NBS1/RPA32 phosphorylation and diminished RAD51 recruitment. If HBx is influencing HR mainly through an interaction with WDR70, we would predict that expressing HBx in WDR70 knockout cells would not further decrease NBS1 or RAP32 phosphorylation. Indeed, when HBx was expressed in WDR70 knockout cells, there was no further decrease in NBS1 or RPA32 phosphorylation (Supporting Fig. S4C) following IR treatment, and RPA plus RAD51 foci formation was also not decreased further (Fig. 3D,E). This strongly suggests that the antiresection activity of HBx can primarily be attributed to impairment of CRL4WDR70 function.
DSB repair is mainly mediated by two pathways, nonhomologous end joining (NHEJ) and homologous recombination. NHEJ is active throughout the cell cycle, while HR is restricted to the S and G2 phases due to the requirement for sister chromatid template.31 Therefore, a reduction of the S/G2 population could indirectly influence the observed resection efficiency without impairing HR machineries. However, fluorescence‐activated cell sorting analysis showed only a modest <20% increase of G1‐phase cells (with a corresponding decrease of S/G2 phases) in WDR70‐ablated or DDB1‐ablated cells. This could not account for the >50% reduction observed in IRIF and immunoblotting assays (Supporting Fig. S4D). Thus, the malfunction of HR observed upon loss of WDR70 is primarily caused by inhibited end processing per se and not by cell cycle perturbation. We confirmed that this was also the case in HBV‐containing cells by distinguishing G1 and S/G2 populations of T43 and HepG2.2.15 cells using the fluorescent ubiquitylation–based cell cycle indicator system.32 No apparent perturbation to the cell cycle distributions were observed (data not shown). Moreover, in WDR70 knockout, T43 and HBx‐expressing cells, the reduction of RAD51/RPA32 IRIF was only apparent in Geminin‐positive cells. Geminin staining specifically marks S/G2 cells (Fig. 3F,G). Thus, HBx interferes with the CRL4WDR70 complex and suppresses resection in S/G2.
HBx Inhibits Resection by Acting as a Dominant Negative for the Assembly of CRL4WDR70
To test our hypothesis that HBx can engage with DDB1 through its H‐box and consequently displaces WDR70 from the CRL4 scaffold, we examined the impact of HBx on CRL4WDR70 complex integrity. As predicted, the binding of WDR70 to DDB1 was effectively completed by HBx expressed either from lentivirus (Fig. 4A) or at low levels from integrated HBV genomes (T43) (Fig. 4B; Supporting Fig. S5A,B). The attenuation of the DDB1–WDR70 interaction in T43 cells could be effectively reversed by ablating HBx with siRNA (Fig. 4C). Moreover, overexpression of both DDB1 and WDR70 overrode the reduction of RPA32 phosphorylation in T43 and HBx‐expressing L02 cells (Fig. 4D). These data strongly indicate that HBx, even at relatively low levels of expression, disrupts the formation of the CRL4WDR70 complex.
Figure 4.
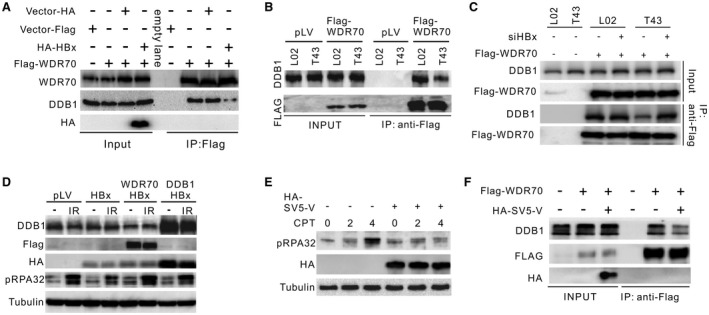
HBx influences CRL4WDR70‐dependent end resection through its H‐box. (A) Immunoprecipitation of endogenous DDB1 by Flag‐WDR70 in the presence or not of overexpressed HBx. Flag‐WDR70 plasmid was transfected into L02 cells 24 hours before infection by pLV‐HA‐HBx lentivirus. Cells were lysed for coimmunoprecipitation analysis 48 hours after infection. α‐WDR70 and α‐DDB1 were used to detected individual proteins. (B) Immunoprecipitation of endogenous DDB1 by Flag‐WDR70 in L02 and T43 cells. (C) Rescue of DDB1–WDR70 interaction in T43 cells by introducing siHBx. (D) Restoration of IR‐induced pRPA32 by forced expression of DDB1 or Flag‐WDR70 in HBx‐expressing L02 cells. Note that the protein level of HBx is elevated upon DDB1 overexpression, possibly due to a stabilizing effect of their interaction.29 (E,F) Decreased RPA32 phosphorylation (E) and interaction between endogenous Flag‐WDR70 and DDB1 (F) in L02 cells in the presence of HA‐tagged SV5‐V serving as an H‐box surrogate for HBx. Abbreviations: HA, hemagglutinin; IP, immunoprecipitation.
It is likely that the HR defect observed when HBx is expressed results from loss of the CRL4WDR70 complex because we have shown that expressing HBx in WDR70 knockout cells did not exacerbate the HR phenotype (Fig 3D,E). Nonetheless, it is possible that, because HBx forms a viral CRL4 complex and redirects the ubiquitin ligase activity to decrease the levels of specific cellular proteins,15 it could induce HR defects directly, for example, by degrading a key HR protein.11, 33 To further address this possibility, SV5‐V, a protein encoded by a paramyxovirus (simian virus 5) that shares a homologous H‐box with HBx33 (Supporting Fig. S5C), was employed.
Apart from their similar ability to bind DDB1, the amino acid sequence and functions of SV5‐V protein and HBx are unique and they cannot substitute for each other in terms of viral–host interaction. We reasoned that if HBx forms a CRL4 E3 ligase to target an HR protein, SV5‐V protein will not be able to substitute for HBx in terms of its inhibition of resection. Interestingly, we observed a clear impact of SV5‐V protein on RPA phosphorylation (Fig. 4E) and on the assembly of the of CRL4WDR70 complex, equivalent to that seen for HBx (Fig. 4F). This corroborates that viral H‐boxes, regardless of their origin, are sufficient to disrupt CRL4WDR70‐dependent resection. It also strongly argues against the possibility that HBx reconstitutes the CRL4 ubiquitin ligase activity to target a repair protein that is key for resection. We therefore conclude that HBx directly blocks the assembly of CRL4WDR70 and that this, rather than the reprogramming of CRL4 activity, is the cause of the HR defects we observed.
Reduction of CRL4WDR70‐Dependent H2B Monoubiquitylation in the Presence of HBx
DNA lesions are repaired in the context of chromatin, and the compact nucleosomal structure needs to be remodeled to allow damaged DNA to become accessible to repair factors.34, 35, 36 Given the role of yeast CRL4Wdr70 in coordinating DSB repair and histone modification,30 we investigated a variety of histone modifications under the condition of HBx expression and CRL4WDR70 depletion and in HepG2.2.15 cells. Notably, levels of H2B monoubiquitylation (K120) and multiple forms of methylated H3K4 and H3K9 were reduced when HBx was present (Fig. 5A‐C; Supporting Fig. S6A). Again, natural HBV infection in PHHs and ectopic expression of SV5‐V also strongly decreased H2B modification at K120 (Fig. 5D; Supporting Fig. S6B).
Figure 5.
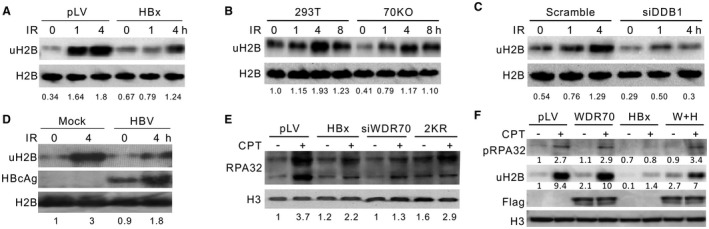
HBx inhibits damage‐induced H2B monoubiquitylation. (A‐D) Evaluation of damage‐induced H2B monoubiquitylation in HBx‐expressing L02 cells (A), WDR70‐depleted (B) or DDB1‐depleted (C) 293T cells, and PHHs with HBV replication (D). The densitometry of uH2B was determined relative to corresponding loading controls. H2B, loading control. (E) Immunoblotting for chromatin fractions of RPA32 probed with α‐RAP32 serum in L02 cells with indicated treatments. Cells were treated with 2 μM CPT for 2 hours. (F) Partial restoration of H2B monoubiquitylation levels by overexpression of pLV‐Flag‐WDR70 in HBx‐expressing L02 (E). Abbreviations: H3, histone H3 level as loading control; W+H, coexpression of Flag‐WDR70 with HBx.
H2B monoubiquitylation is required for the timely repair of DSBs, and malfunction of this pathway is implicated in tumorigenesis.37, 38 Similar to the impact of HBx expression and WDR70 depletion, the introduction of nonubiquitylatable H2B (2KR: K120R/K125R) into L02 cells prevented RPA32 chromatin association (Fig. 5E). This is consistent with H2B monoubiquitylation being required for resection. We could again link this defect directly to CRL4WDR70 because the deficiency in H2B monoubiquitylation observed in T43 and HBx‐expressing L02 cells could be complemented by WDR70 expression, congruent with the rescue of RPA phosphorylation (Fig. 5F; Supporting Fig. S6C). In addition, ablation of HBx by siRNA in T43 cells restored the level of H2B monoubiquitylation at lysine 120 (uH2B) (Supporting Fig. S6D). Furthermore, ablating the components of HULCRNF20, the histone H2B ubiquitin ligase complex,39 inhibited RPA phosphorylation as well as the foci formation of RPA32 and RAD51, which again displayed epistasis with WDR70 depletion (Supporting Fig. S6E‐G). These results consolidate the notion that the anti‐HR effect of HBx is exerted through chromatin remodeling events coregulated by HULCRNF20 and CRL4WDR70.
HBx Impedes Extensive H2B Monoubiquitylation and Long‐Range Resection
To demonstrate that the perturbation of CRL4WDR70‐dependent H2B monoubiquitylation by HBx occurs at the chromatin level, we employed ChIP assays to characterize the chromatin dynamics at intron 1 of the protein phosphatase 1 regulatory subunit 12C (PPP1R12C/p84) gene (Fig. 6A). When DSBs were artificially induced by CRISPR genomic RNA (gRNA) at this locus, the enrichment of H2B monoubiquitylation at regions surrounding an induced DSB was weakened upon WDR70 knockout or in the presence of HBV/HBx (Fig. 6B). Intriguingly, the presence of HBx or the loss of WDR70 markedly impaired the enrichment of H2B monoubiquitylation at regions distal to DNA breaks (>3.5 kb) but exerted no detectable impacts in the proximal regions tested (0.5 and 3.5 kb). In concordance with the impact of HBx on CRL4WDR70 complex integrity, this regional effect was correlated with the ability of HBx to exclude the loading of DDB1 and WDR70 from distal chromatin domains of DSBs (data not shown). Thus, HBx disables an evolutionarily conserved role of CRL4WDR70 in expanding the H2B monoubiquitylation toward DSB‐distal regions.
Figure 6.
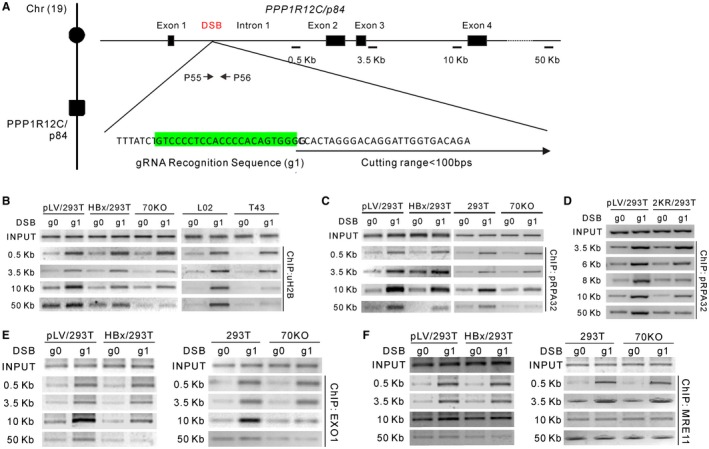
HBx impairs CRL4WDR70‐mediated extensive monoubiquitylation of H2B in the vicinity of DSBs. (A) Schematic representation for CRISPR‐gRNA‐DSB at the human PPP1R12C/p84 locus of chromosome 19. The gRNA targeting sequence in intron 1 is highlighted in green. Upon transfection, CRISPR‐RNA complexes digest DNA within a range of 100 bp downstream of the recognition site. Arrows, primer sets across cutting site; black bars, PCR amplicons for ChIP assays. The sizes of individual amplicons are 171 bp (0.5 kb), 169 bp (3.5 kb), 168 bp (10 kb), and 162 bp (50 kb). See also Supporting Fig. S7A,B. (B) Enrichment of H2B monoubiquitylation assayed by ChIP in HBx‐expressing and WDR70 knockout 293 cells or in L02 and T43 cells. Distances from PPP1R12C/p84‐specific CRISPR‐gRNA (g1)–induced DSBs are indicated at left. g0 is the control gRNA plasmid expressing no targeting sequence. (C,D) DSB loading of RPA32 assayed by ChIP in indicated cells and distance from breakpoint. (E,F) Chromatin loading of EXO1 (E) and MRE11 (F) assayed by ChIP in HBx‐expressing and WDR70 knockout 293T cells.
The progression of resection is catalyzed by the cooperation of several enzymes: C‐terminal binding protein–interacting protein and the MRE11‐RAD50‐NBS1 nuclease initiate resection to generate a short ssDNA substrate. This is subsequently extended by exonuclease 1 (EXO1) and Bloom syndrome RecQ‐like helicase (BLMRqh1)–DNA2, channeling repair into the HR pathways.40 To dissect how HBx influences the chromatin loading of resection factors through down‐regulation of H2B monoubiquitylation, patterns of chromatin recruitment for a variety of repair factors were analyzed by ChIP assays using the CRISPR‐induced DSB assay. Strikingly, we detected a reduction of RPA32 recruitment at 10 kb and 50 kb from the DSB site in the presence of HBx or following the loss of WDR70 (Fig. 6C). This suggests that the failed extension of H2B modification to the DSB‐distal prevents long‐range resection. Indeed, introduction of a nonubiquitylatable 2KR mutant of H2B into L02 cells reduced the RPA32 loading, replicating the defect caused by HBx and WDR70 depletion (Fig. 6D). Consistent with the resection defects monitored by RPA loading, the chromatin association of the long‐range resection nuclease (EXO1) displayed a dramatic decrease exclusively at sites distal to the DSB upon depletion of WDR70 or HBx expression. No alteration of DSB enrichment of the resection initiator (meiotic recombination 11 [MRE11]) was detected in the same set of experiments (Fig. 6E,F). Therefore, HBx induces CRL4WDR70 disruption, and this results in a reduction of H2B monoubiquitylation and specifically prevents the chromatin loading of EXO1 and the ensuing extensive resection.
Defective H2B Monoubiquitylation Promotes Tumorigenesis
We have shown that HBx ablates the CRL4WDR70 complex and that this results in a resection defect. The first observed event we see when CRL4WDR70 is defective is a failure of H2B monoubiquitylation. To investigate if CRL4WDR70‐dependent H2B monoubiquitylation could contribute to tumor suppression, a stable cell line of L02 integrated with a lentiviral 2KR construct was established. Two million cells were then injected subcutaneously into immune defective athymic nude mice, and tumor growth was monitored. The 2KR‐expressing L02 cells exhibited uncontrolled neoplastic proliferation when compared with control cells. Remarkably, coexpression of WDR70 and DDB1 genes suppressed this increased tumorigenesis (Fig. 7A). Consistent with H2B monoubiquitylation acting in a tumor suppressor pathway, overexpression of the deubiquitinase ubiquitin‐specific peptidase 22 (USP22, which eliminates H2B monoubiquitylation) also significantly promoted tumor growth (Fig. 7B).
Figure 7.
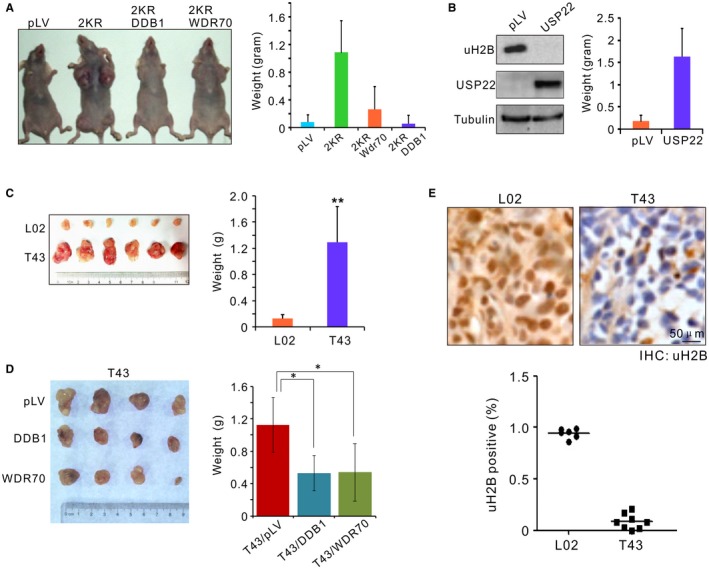
Loss of H2B monoubiquitylation promotes tumorigenesis. (A) Subcutaneous xenograft of L02 cells preinfected by indicated lentivirus. Note the suppression of tumorigenesis of 2KR by coexpression of DDB1 or WDR70. (B) Immunoblotting for H2B monoubiquitylation in L02 cells by overexpression of human USP22 (left). Cells expressing USP22 were inoculated in nude mice and measured for tumor formation (right). (C) Xenograft tumor formation assay in nude mice. Individual tumor samples were dissected from nude nice 10 days after subcutaneous injection of L02 or T43 cells (left), and average tumor weights (right) were calculated. n = 9 dissected samples. Error bars, SD. **P < 0.01. (D) As in (C), tumor formation assays for T43 cells preinfected with pLV‐vector, pLV‐DDB1, or pLV‐WDR70 lentivirus. (E) IHC images (inset) and quantification of uH2B positivity for paraffin‐embedded xenografts. Nuclei were counterstained by hematoxylin (blue). n = 10 dissected samples. Abbreviation: LV, lentivirus.
We also generated HBV‐positive xenograft models by embedding L02 and T43 cells in nude mice. Resembling the 2KR inoculums, T43 (but not L02) inoculums resulted in aggressive tumor outgrowth (Fig. 7C) that could be significantly repressed by overexpression of DDB1 or WDR70 or by in vivo knockdown with siHBx (Fig. 7D; Supporting Fig. S7A). Examination of these tumors by immunohistochemical (IHC) staining revealed that H2B monoubiquitylation was absent in T43‐derived xenografts (Fig. 7E). Taken together, these data support the idea that the CRL4WDR70‐H2B signaling pathway acts as a crucial anticancer mechanism, inhibition of which by HBx potentially promotes tumorigenesis.
Implication of HBx‐Mediated Antiresection Activity in Human HBVHCC
As HBx is expressed in a significant proportion of human HBVHCC cases, H2B monoubiquitylation, whose levels are correlated to the function of CRL4WDR70‐mediated resection, could serve as the predictive marker of HR status. We therefore examined H2B monoubiquitylation in clinically isolated liver cancer tissues. Paraffin‐embedded sections of cohorts of either HBV‐positive or HBV‐negative HCC biopsies were subjected to IHC staining with antibody specific to ubiquityl H2B (K120) (Fig. 8A). In comparison to HCC samples that were negative for serological HBV surface antigen, sections from HBVHCC patients displayed a statistically lower intensity and percentage of uH2B when visualized by staining of α‐uH2B (Fig. 8B). A small proportion of HBVHCCs displayed a modest level of H2B monoubiquitylation, possibly due to occult infection41 or an acquired second mutation such as p53 binding protein 1, which can potentially rescue the HBx‐induced HR defect. Similarly, uH2B levels in HBV‐negative non‐HCC tissues are also significantly higher than those of HBV‐positive tissues (Fig. 8B; Supporting Fig. S7B). Thus, given the mechanistic link between HBx and CRL4WDR70‐mediated H2B monoubiquitylation, a reduction of uH2B levels in the majority of HBVHCC tissues can be potentially correlated with the antiresection activity of HBx.
Figure 8.
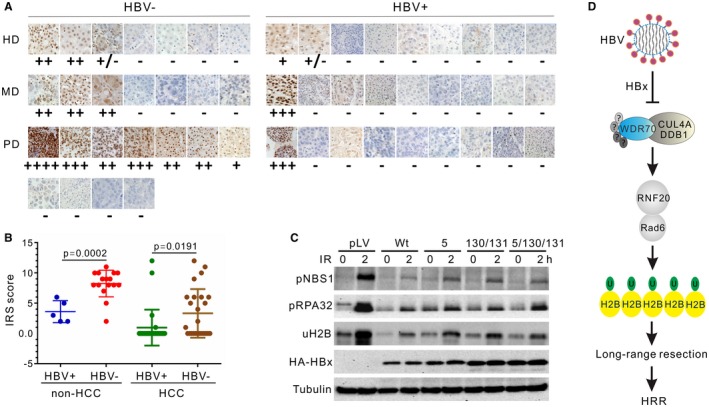
Implication of H2B monoubiquitylation and resection defects in human HBVHCC. (A) Comparison of H2B monoubiquitylation by IHC in hepatocarcinoma with or without HBV infection. HBV carriers were diagnosed according to enzyme‐linked immunosorbent assay for serological positivity of surface antigen. (B) Quantitative analysis by internal reflection spectroscopy for IHC staining of uH2B in HCC and non‐HCC samples, see also Supporting Information. P = 0.0057. (C) Impact on RPA32 phosphorylation and H2B monoubiquitylation in L02 cells by wild type and three clinically prevalent mutants of HBx at residuals 5, 130, and 131. None of the single, double, or triple combinations of mutations significantly diminishes the inhibitory effects of HBx on these cellular activities. (D) Model for the antiresection activity of HBx on HR through CRL4WDR70‐dependent chromatin modification. Mirroring the deletion of yeast Wdr70, HBx dissembles the functional CRL4WDR70 complex and results in decreased H2B monoubiquitylation, leading to an aberrant chromatin landscape. HBx impairs long‐range resection. The resultant loss of homologous recombination promotes genome instability and thus provides a driver for HBV‐associated carcinogenesis. Abbreviations: HA, hemagglutinin; HD, highly differentiated HCC; HRR, homologous recombinational repair; IRS, immunoreactive score; MD, moderately differentiated HCC; PD, poorly differentiated HCC.
Genetic variants of the HBx gene, such as missense mutations at residuals 5, 130, and 131, emerge during the course of HBV‐associated liver diseases and finally prevail in late‐stage HCC.42 These mutant forms, especially those located in the C terminus of HBx (130 and 131), could potentially disable the capability of HBx to block end resection. However, we found that these clinically prevalent HBx mutants largely preserved the suppressive effect on resection: none of these variants reduced the ability of HBx to suppress H2B monoubiquitylation or RPA32 and NBS1 phosphorylation (Fig. 8C). This result correlates with the IHC assays that demonstrated diminished H2B monoubiquitylation in the majority of HBVHCC biopsies (Fig. 8A). Thus, despite the prevalence of mutations in late stages of HBVHCC, HBx maintains the potential to influence epigenetic regulation and end resection.
Discussion
HBx is the oncoprotein of HBV and is tightly linked to the progression of HBVHCC. HBx transcripts and protein can be detected in hepatocytes from 70% of chronically infected patients and in many HBVHCC biopsies.43, 44 However, how HBV and HBx promote liver cancer is not adequately understood. Here, we report that HBV impedes an early step of homologous recombination. We demonstrate that, through the HBx protein, HBV interferes with DNA end resection and HR repair. This occurs during both phases of acute infection (PHHs) and integration in the host genome (HepaG2.2.15 and T43 cells). These data demonstrate that HBx‐mediated antiresection activity has a genuine viral impact on host cells, as opposed to an artificial phenomenon caused by random viral integration. Equally importantly, our analysis establishes that the integration of HBV fragments can cause long‐term effects on genomic stability: as long as viral replication is active or HBx is expressed (even at relatively low levels) during any stage of the disease (chronic infection, cirrhosis, and hepatocarcinoma), resection and thus HR would be restrained. This can potentially render host genomes genetically unstable over extended periods of time (Fig. 8D).
HBx disassembles specific CRL4 subcomplexes or retargets CRL4 to support the viral life cycle.12, 15 A subset of DCAFs, including WDR70, is strongly dissociated from DDB1 in the presence of HBx. Our sequence alignment of the H‐boxes of HBx and several DCAFs identified residues important for the DDB1 interaction (Fig. 3A). Interestingly, WDR70 and DDB2 share C‐terminal sequences that fold into successive α‐helices and provide additional surfaces that bind to DDB1 (Fig. 3B; Supporting Fig. S4B). Upon direct competition from HBx, this additional binding is likely to be disrupted, leading to the dissociation of WDR70 from the CRL4 scaffold.
Our data establish a functional link between HBx and CRL4‐mediated DNA end resection based on the competition between HBx and WDR70 for DDB1 binding. We identified that it is ablation of the CRL4WDR70 subcomplex that is responsible for the HR defect in HBV‐infected or HBV‐integrated cells. We can rule out the possibility that HBx causes the HR defect through hijacking and reprogramming the ubiquitin ligase activity of CRL4s toward proteins required for resection for two reasons. First, expression of HBx does not further decrease the resection defects observed in WDR70 knockout cells. Second, the antiresection activity of HBx is entirely dependent on its H‐box motif, and the introduction of the SV5 H‐box peptide is fully capable of disrupting CRL4WDR70 complex integrity and subsequent HR events. These data directly link HBV‐mediated HR interference to the structural integrity of CRL4 complexes. Therefore, the impact of HBx on DDB1 results in two major HBV‐related pathologies: by forming a viral CRL4HBx ubiquitin ligase, HBx promotes viral replication by targeting SMC5/6 for proteasome‐dependent ubiquitination.45 In this case, CRL4 ubiquitination activity is obligatory for viral replication. Independent of CRL4HBx activities, HBx also exerts an antiresection activity by blocking CRL4WDR70 complex formation, which causes genomic instability in host cells (data not shown). The complex influence of HBx on the CUL4‐DDB1‐related complexes sheds lights on future drug design: for example, small molecules could be identified as antiviral compounds which prevent the DDB1–HBx interaction and/or restore the integrity of CRL4WDR70.
In the context of HBV‐induced carcinogenesis, we show that loss of CRL4WDR70 phenocopies HBV infection and HBx expression and has the potential to drive oncogenic transformation by promoting genomic instability. Ablation of WDR70 caused an equivalent decrease of uH2B and DSB recruitment of RPA/RAD51 as is seen during HBV infection and the postintegration stage. Resembling the well‐established model of breast cancer inflicted by BRCA1 mutation,46, 47 a reduction of HR upon HBV infection predicts a threat to genomic stability and renders host cells susceptible to oncogenic transformation. We further support the prediction that HBV promotes carcinogenesis by interfering with CRL4WDR70 by showing that the ablation of H2B ubiquitylation (by expressing a 2KR mutant) dramatically promoted oncogenic proliferation and that this could be effectively suppressed by concomitant overexpression of WDR70. Again, this correlates well with the observation that the aggressive growth driven by HBV in T43 cells was accompanied by a low level of uH2B in comparison to control cells. Therefore, we conclude that the CRL4WDR70‐uH2B regulatory network is an anticancer mechanism and meets the criteria for being a direct oncogenic target of HBx.
HBx is the crucial factor for HBV replication and antigen expression. This poses the question as to whether the modulation of resection benefits viral replication. In previous work we showed that ablating DDB1 inhibited viral production and gene expression of HBV, indicating that DDB1 is required for viral production.18 Our preliminary data also suggest that loss of WDR70 facilitates viral replication (data not shown). There could be several putative mechanisms by which CRL4WDR70 and H2B monoubiquitylation influence HBV replication: aberrant DNA structures (i.e., hairpins or ssDNA) are formed during HBV replication, and antiviral mechanisms are likely to be activated in response to these potential signatures of viral infection. In such a scenario, a reduction of CRL4WDR70‐uH2B activity may be advantageous to curb unwanted processing of intermediate structures by host nuclease (such as BLM and EXO1). Alternatively, it is likely that CRL4WDR70 is involved in regulating viral gene expression through modulating the levels of H2B monoubiquitylation in promoter regions (data not shown). Therefore, from the perspective of viral replication, HBx may facilitate viral gene expression and protect viral genomes by influencing histone modifications, transcription, and host nucleases.
Taken together, our findings reveal that HBx interferes with CRL4WDR70‐mediated DNA end resection and causes HR deficiency in HBV‐infected hepatocytes. Impaired DSB repair contributes to genomic instability and drives hepatocarcinogenesis.
Supporting information
Acknowledgment
We thank Zhixiong Xiao, Dingxiang Liu, Lilin Du, Huiqiang Lou, and Qun He for critical comments on the manuscript. We also especially thank Prof. Yuquan Wei for his expert advice on tumors and pathological analysis.
This article is dedicated to the memory of our deceased coauthor, Y.T.
Supported by the National Natural Science Foundation of China (31471276, 31771580, 81702795, 81630038, and 81330016), the Ministry of Science and Technology of the People's Republic of China (2013CB911000), the Department of Science and Technology of Sichuan Province (2017FZ0034), and the Department of Science and Technology of Shanxi Province (201703D321013). N.Z. is a Howard Hughes Medical Institute Investigator.
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. El‐Serag HB. Hepatocellular carcinoma. N Engl J Med 2011;365:1118‐1127. [DOI] [PubMed] [Google Scholar]
- 2. Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer 2006;6:674‐687. [DOI] [PubMed] [Google Scholar]
- 3. Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22,707 men in Taiwan. Lancet 1981;2:1129‐1133. [DOI] [PubMed] [Google Scholar]
- 4. Brechot C, Pourcel C, Louise A, Rain B, Tiollais P. Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature 1980;286:533‐535. [DOI] [PubMed] [Google Scholar]
- 5. Seeger C, Mason WS. Molecular biology of hepatitis B virus infection. Virology 2015;479‐480:672‐686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bouchard MJ, Schneider RJ. The enigmatic X gene of hepatitis B virus. J Virol 2004;78:12725‐12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lucifora J, Arzberger S, Durantel D, Belloni L, Strubin M, Levrero M, et al. Hepatitis B virus x protein is essential to initiate and maintain virus replication after infection. J Hepatol 2011;55:996‐1003. [DOI] [PubMed] [Google Scholar]
- 8. Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991;351:317‐320. [DOI] [PubMed] [Google Scholar]
- 9. Becker SA, Lee TH, Butel JS, Slagle BL. Hepatitis B virus x protein interferes with cellular DNA repair. J Virol 1998;72:266‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Scrima A, Konickova R, Czyzewski BK, Kawasaki Y, Jeffrey PD, Groisman R, et al. Structural basis of UV DNA‐damage recognition by the DDB1–DDB2 complex. Cell 2008;135:1213‐1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N. Molecular architecture and assembly of the DDB1–CUL4A ubiquitin ligase machinery. Nature 2006;443:590‐593. [DOI] [PubMed] [Google Scholar]
- 12. Li T, Robert EI, van Breugel PC, Strubin M, Zheng N. A promiscuous alpha‐helical motif anchors viral hijackers and substrate receptors to the CUL4–DDB1 ubiquitin ligase machinery. Nat Struct Mol Biol 2010;17:105‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fischer ES, Scrima A, Bohm K, Matsumoto S, Lingaraju GM, Faty M, et al. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 2011;147:1024‐1039. [DOI] [PubMed] [Google Scholar]
- 14. He YJ, McCall CM, Hu J, Zeng Y, Xiong Y. DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4–ROC1 ubiquitin ligases. Genes Dev 2006;20:2949‐2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Decorsière A, Mueller H, van Breugel PC, Abdul F, Gerossier L, Beran RK, et al. Hepatitis B virus x protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016;531:386‐389. [DOI] [PubMed] [Google Scholar]
- 16. Yamaji S, Zhang M, Zhang J, Endo Y, Bibikova E, Goff SP, et al. Hepatocyte‐specific deletion of DDB1 induces liver regeneration and tumorigenesis. Proc Natl Acad Sci USA 2010;107:22237‐22242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee ATC, Ren J, Wong ET, Ban KHK, Lee LA, Lee CGL. The hepatitis B virus x protein sensitizes HepG2 cells to UV light‐induced DNA damage. J Biol Chem 2005;280:33525‐33535. [DOI] [PubMed] [Google Scholar]
- 18. Guo L, Wang X, Ren L, Zeng M, Wang S, Weng Y, et al. HBx affects CUL4–DDB1 function in both positive and negative manners. Biochem Biophys Res Commun 2014;450:1492‐1497. [DOI] [PubMed] [Google Scholar]
- 19. Ni Y, Urban S. Hepatitis B virus infection of HepaRG cells, HepaRG‐hNTCP cells, and primary human hepatocytes. Methods Mol Biol 2017;1540:15‐25. [DOI] [PubMed] [Google Scholar]
- 20. Schulze A, Mills K, Weiss TS, Urban S. Hepatocyte polarization is essential for the productive entry of the hepatitis B virus. Hepatology 2012;55:373‐383. [DOI] [PubMed] [Google Scholar]
- 21. Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, et al. Whole‐genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet 2012;44:760‐764. [DOI] [PubMed] [Google Scholar]
- 22. Sells M, Chen M, Acs G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci USA 1987;84:1005‐1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA–ssDNA complexes. Science 2003;300:1542‐1548. [DOI] [PubMed] [Google Scholar]
- 24. Cejka P. DNA end resection: nucleases team up with the right partners to initiate homologous recombination. J Biol Chem 2015;290:22931‐22938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 2008;77:229‐257. [DOI] [PubMed] [Google Scholar]
- 26. Serena NZ, Alexandrov LB, Wedge DC, Peter VL, Greenman CD, Keiran R, et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012;149:979‐993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Venkitaraman AR. Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science 2014;343:1470‐1475. [DOI] [PubMed] [Google Scholar]
- 28. Bontron S, Lin‐Marq N, Strubin M. Hepatitis B virus x protein associated with UV–DDB1 induces cell death in the nucleus and is functionally antagonized by UV–DDB2. J Biol Chem 2002;277:38847‐38854. [DOI] [PubMed] [Google Scholar]
- 29. Bergametti F, Sitterlin D, Transy C. Turnover of hepatitis B virus x protein is regulated by damaged DNA‐binding complex. J Virol 2002;76:6495‐6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zeng M, Ren L, Mizuno K, Nestoras K, Wang H, Tang Z, et al. CRL4(Wdr70) regulates H2B monoubiquitination and facilitates Exo1‐dependent resection. Nat Commun 2016;7:11364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matsumoto Y. Smart choice between two DNA double‐strand break repair mechanisms. [in Japanese] Igaku Butsuri 2014;34:57‐64. [PubMed] [Google Scholar]
- 32. Sakaue‐Sawano A, Ohtawa K, Hama H, Kawano M, Ogawa M, Miyawaki A. Tracing the silhouette of individual cells in S/G/M phases with fluorescence. Chem Biol 2008;15:1243‐1248. [DOI] [PubMed] [Google Scholar]
- 33. Leupin O, Bontron S, Strubin M. Hepatitis B virus x protein and simian virus 5 V protein exhibit similar UV‐DDB1 binding properties to mediate distinct activities. J Virol 2003;77:6274‐6283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Costelloe T, Louge R, Tomimatsu N, Mukherjee B, Martini E, Khadaroo B, et al. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 2012;489:581‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jackson SP, Durocher D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell 2013;49:795‐807. [DOI] [PubMed] [Google Scholar]
- 36. Chen X, Cui D, Papusha A, Zhang X, Chu C‐D, Tang J, et al. The Fun30 nucleosome remodeller promotes resection of DNA double‐strand break ends. Nature 2012;489:576‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moyal L, Lerenthal Y, Gana‐Weisz M, Mass G, So S, Wang S‐Y, et al. Requirement of ATM‐dependent monoubiquitylation of histone H2B for timely repair of DNA double‐strand breaks. Mol Cell 2011;41:529‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Blank M, Tang Y, Yamashita M, Burkett SS, Cheng SY, Zhang YE. A tumor suppressor function of Smurf2 associated with controlling chromatin landscape and genome stability through RNF20. Nat Med 2012;18:227‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nakamura K, Kato A, Kobayashi J, Yanagihara H, Sakamoto S, Oliveira DV, et al. Regulation of homologous recombination by RNF20‐dependent H2B ubiquitination. Mol Cell 2011;41:515‐528. [DOI] [PubMed] [Google Scholar]
- 40. Cannavo E, Cejka P. Sae2 promotes dsDNA endonuclease activity within Mre11‐Rad50‐Xrs2 to resect DNA breaks. Nature 2014;514:122‐125. [DOI] [PubMed] [Google Scholar]
- 41. Torbenson M, Thomas D. Occult hepatitis B. Lancet Infect Dis 2002;2:479‐486. [DOI] [PubMed] [Google Scholar]
- 42. Lee JH, Han KH, Lee JM, Park JH, Kim HS. Impact of hepatitis B virus (HBV) x gene mutations on hepatocellular carcinoma development in chronic HBV infection. Clin Vaccine Immunol 2011;18:914‐921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Paterlini P, Poussin K, Kew M, Franco D, Brechot C. Selective accumulation of the x transcript of hepatitis B virus in patients negative for hepatitis B surface antigen with hepatocellular carcinoma. Hepatology 1995;21:313‐321. [PubMed] [Google Scholar]
- 44. Hoare J, Henkler F, Dowling JJ, Errington W, Goldin RD, Fish D, et al. Subcellular localisation of the x protein in HBV infected hepatocytes. J Med Virol 2001;64:419‐426. [DOI] [PubMed] [Google Scholar]
- 45. Murphy CM, Xu Y, Li F, Nio K, Reszka‐Blanco N, Li X, et al. Hepatitis B virus x protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep 2016;16:2846‐2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C, et al. BRCA1 controls homologous recombination at Tus/Ter‐stalled mammalian replication forks. Nature 2014;510:556‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM, et al. BRCA1 tumour suppression occurs via heterochromatin‐mediated silencing. Nature 2011;477:179‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials