

Summary
Cell migration is essential during development, regeneration, homeostasis, and disease. Depending on the microenvironment, cells use different mechanisms to migrate. Yet, all modes of migration require the establishment of an intracellular front–rear polarity axis for directional movement. Although front–rear polarity can be easily identified in in vitro conditions, its assessment in vivo by live‐imaging is challenging due to tissue complexity and lack of reliable markers. Here, we describe a novel and unique double fluorescent reporter mouse line to study front–rear cell polarity in living tissues, called GNrep. This mouse line simultaneously labels Golgi complexes and nuclei allowing the assignment of a nucleus‐to‐Golgi axis to each cell, which functions as a readout for cell front–rear polarity. As a proof‐of‐principle, we validated the efficiency of the GNrep line using an endothelial‐specific Cre mouse line. We show that the GNrep labels the nucleus and the Golgi apparatus of endothelial cells with very high efficiency and high specificity. Importantly, the features of fluorescent intensity and localization for both mCherry and eGFP fluorescent intensity and localization allow automated segmentation and assignment of polarity vectors in complex tissues, making GNrep a great tool to study cell behavior in large‐scale automated analyses. Altogether, the GNrep mouse line, in combination with different Cre recombinase lines, is a novel and unique tool to study of front–rear polarity in mice, both in fixed tissues or in intravital live imaging. This new line will be instrumental to understand cell migration and polarity in development, homeostasis, and disease.
Keywords: cell migration, cell polarity, endothelial cells, fluorescent reporter
1. INTRODUCTION
Cell migration plays a pivotal role during development and maintenance of multicellular organisms. During embryogenesis, extensive single and collective movement of cells drives gastrulation, convergent extension, and organogenesis. In the adult, migration of cells is essential for wound healing, tissue regeneration, immune surveillance, and tissue homeostasis. In cancer, cell migration leads to cell dissemination and metastasis (Dogterom & Koenderink, 2019; Hamidi & Ivaska, 2018; Ladoux & Mege, 2017).
A large variety of cell types uses different modes of migration (Friedl & Wolf, 2010), yet it is well recognized that they use a set of common core components, which are conserved in evolution from protozoa to mammals. Migration of cells depends on two key coexistent and interrelated events: (a) force generation, necessary to push and retract cell membranes in relation to their environment and (b) sustained polarization, required for directional and persistent displacement. Cell polarization is characterized by asymmetrical activation and redistribution of intracellular components, proteins (aPKC/Par complex, and Cdc42), lipids (PtdIns(3,4,5)P3 and PtdIns(3,4)P2) and organelles (microtubule organization center [MTOC], and the Golgi apparatus), leading to the formation of front and rear ends in the cell (Iden & Collard, 2008; Ridley et al., 2003). The establishment of cell polarity requires an initial symmetry‐breaking event (Ridley et al., 2003; Wedlich‐Soldner & Li, 2003). This often arises from the cell's microenvironment, like growth factors and extracellular matrix chemical and mechanical properties, which regulate front–rear cell polarity through spatially controlled activation of signaling complexes (Iden & Collard, 2008; Ridley et al., 2003).
Despite being a core aspect of cell migration, most of our knowledge related to front–rear cell polarity in cell migration has come from in vitro experimentation. This is explained because front–rear cell polarity can be easily identified in in vitro conditions. However, its live‐imaging in vivo in mice is challenging due to tissue complexity and the lack of reliable markers. Live‐imaging techniques, such as two‐photon excitation, point‐scanning or light‐sheet microscopy, rely on expressing fluorescent markers and/or using dyes that highlight proteins, organelles, or cells. The combination of these imaging techniques with the power of mouse genetics allows researchers to selectively image specific cell populations with spatiotemporal resolution (Zong, Espinosa, Su, Muzumdar, & Luo, 2005). These imaging modalities have revolutionized our knowledge regarding individual and collective cell dynamics in the context of health and disease (Nobis et al., 2018). Given the multiple signaling pathways regulating cell polarity and the limitations in sensitivity and tractability of using leading edge markers in 3D tissues, we decided to generate a mouse reporter line to image front–rear cell polarity in living tissues by simultaneously tagging Golgi apparatuses and nuclei of cells, called GNrep. During cell migration, the MTOC/Golgi apparatus is positioned in front of the cell nucleus, in the same direction of migration, thus representing an internal polarity axis of cells (Etienne‐Manneville, 2013; Etienne‐Manneville & Hall, 2003; Mayor & Etienne‐Manneville, 2016). In particular, the Golgi apparatus has been previously used to visualize endothelial cell front–rear polarity in vivo in zebrafish, ex‐vivo in mouse, and in in vitro conditions (Franco et al., 2015; Gotlieb, May, Subrahmanyan, & Kalnins, 1981; Kwon et al., 2016). Individual tagging of each organelle has been achieved in the past (Abe et al., 2011), yet the combination of both had not been established yet. The GNrep mouse line expresses Golgi‐localized mCherry and nuclear‐localized eGFP upon Cre‐mediated recombination. The construct was introduced into the ubiquitous ROSA26 locus (Soriano, 1999) and it is under the control of the strong CAGGS promoter (Miyazaki et al., 1989), enabling bright fluorescent labeling of all nuclei and Golgi apparatuses. With this genetic tool and by using different Cre lines, it is possible to label and to track cell populations in different physiological conditions, and to analyze the dynamics of cell polarization and migration in vivo, using a variety of microscopy techniques.
2. RESULTS AND DISCUSSION
2.1. Generation and validation of the targeting GNrep construct
We aimed at generating a mouse reporter line to image cell polarity in vivo by fluorescently labeling nuclei and Golgi complexes. We started by cloning fluorescently labeled Golgi complex and nucleus reporter sequences (GNrep cassette) into the pUC57‐2A plasmid (Plasmid1‐Table 1), which had been previously generated in the lab. The reporter sequences consist on the coding sequence for the Golgi targeting sequence (GTS) (Hu, Li, Xie, Gu, & Peng, 2011) fused to mCherry, from the pGTS‐mCherry (Plasmid2‐Table 1), and the coding sequence for Histone 2B (H2B) fused to GFP, from pCAGGS‐G2m (Plasmid3‐Table 1). Insertion of the two reporter sequences into the pUC57‐2A plasmid led to the creation of the pUC57‐GNrep plasmid (Plasmid4‐Table 1). In this plasmid, the two reporter sequences are linked by the 2A self‐cleaving peptide (Szymczak‐Workman, Vignali, & Vignali, 2012) to allow expression of stoichiometric amounts of both fluorescent reporters (Figure 1a).
Table 1.
Plasmids used for the generation of the GNrep polarity construct
| Plasmid | Name | Origin |
|---|---|---|
| 1 | pUC57‐2A | Generated in the lab by synthesis |
| 2 | pGTS‐mCherry | https://www.addgene.org/55052/ |
| 3 | pCAGGS‐G2 m | Gift from Holger Gerhardt Lab |
| 4 | pUC57‐GNrep | Generated in the lab |
| 5 | pME‐GNrep | Generated in the lab |
| 6 | pEF1‐GNrep | Generated in the lab |
| 7 | pROSA26‐DV2 | Nyabi et al. (2009) |
| 8 | p5E‐CAGGS‐MCS | Gift from Holger Gerhardt Lab |
| 9 | p5E‐CAGGS‐LSL | Generated in the lab |
| 10 | p3E‐pA‐FRT‐PGK.Neo‐pA‐FRT | Generated in the lab |
| 11 | pROSA26‐GNrep | Generated in the lab |
| 12 | pJFRC172‐10XUAS‐loxP‐dSTOP‐loxP‐myr::GFP | https://www.addgene.org/32145/ |
| 13 | pEF1‐DEST51 | ThermoFisher (12285011) |
Figure 1.
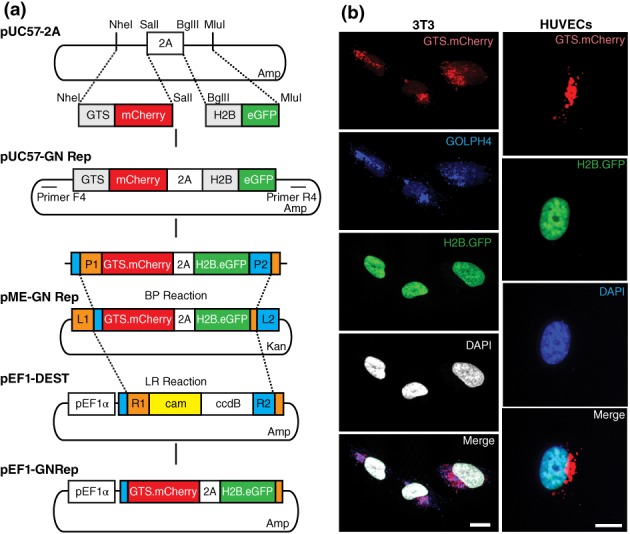
Generation and validation of the targeting GNrep construct. (a) GN reporter cassette was generated by subcloning the nucleus and the Golgi complex reporter sequences into the pUC57‐2A plasmid. This plasmid codes for the GTS‐mCherry and the H2B‐GFP sequences. The pUC57‐GNrep was used to generate a pME‐GNrep and subsequently the pEF1‐GNrep plasmid, by Gateway cloning, using the pEF1‐DEST as a destination vector. (b) NIH/3T3 fibroblasts and human umbilical vein endothelial cells transfected with pEF1‐GNrep plasmid revealed efficient expression of the GN reporter cassette. Nuclei are labeled with DAPI. Golgi apparatus is stained for GOLPH4 in NIH/3T3 cells. Scale bar, 10 μm
To validate the construct in cells in vitro, we generated a GNrep expression vector, pEF1‐GNrep. For that, the pUC57‐GNrep was used to generate a pME‐GNrep (Plasmid5‐Table 1) and subsequently the pEF1‐GNrep plasmid (Plasmid6‐Table 1), by Gateway cloning, using the pEF1‐DEST as a destination vector (Figure 1a). Transfection of both NIH/3T3 fibroblasts and human umbilical vein endothelial cells revealed efficient expression of the expression cassette with bright fluorescence levels both for nuclear GFP and Golgi apparatus‐localized mCherry (Figure 1b).
2.2. Generation of the GNrep mouse line
We next generated a plasmid suitable for homologous recombination into the Rosa26 locus. As a backbone, we used the pROSA26‐DV2 vector (Plasmid7‐Table 1) that carries Rosa26 5′‐ and 3′‐homology arms and enables rapid, modular, and efficient generation of plasmid DNA suitable for homologous recombination into the Rosa26 genomic locus of mouse embryonic stem cells (mESCs) (Nyabi et al., 2009). This plasmid allows for multisite Gateway recombination‐mediated insertion of three entry DNA sequences. We used this strategy to generate the final targeting vector (pROSA26‐GNrep vector, Plasmid11‐Table 1) containing a CAGGS synthetic promoter followed by a loxP‐dSTOP‐loxP cassette, the GNrep cassette, by a selection cassette pA‐FRT‐PGK‐Neo‐pA‐FRT (Figure 2a). The complete list of plasmids used to build this construct can be found in Table 1 (Plasmid5, 7‐10‐Table 1).
Figure 2.
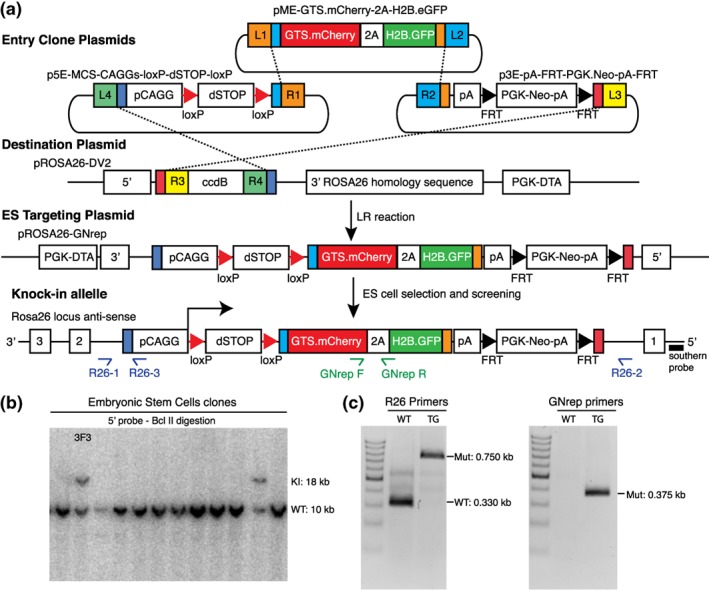
Generation of the GNrep mouse line. (a) pROSA26‐LSL‐GNrep vector was generated by multisite Gateway using a vector containing a CAGGS synthetic promoter followed by a loxP‐dSTOP‐loxP cassette, the GNrep cassette and a selection cassette FRT‐PGK‐Neo‐pA‐FRT. (b) pROSA26‐LSL‐GNrep was electroporated into mESCs and the integration by homologous recombination of the GNrep cassette was screened by Southern blot with a ROSA26 5′‐probe and BclII digestion. Clone 3F3 had the GNrep cassette (18 kb) and was the clone selected for mouse blastocyst injection and to generate knock‐in mice. (c) Genomic PCR was carried out using two sets of primers to confirm the insertion of the construct in the mouse genome. GNrep primers identify the presence or absence of the GNrep cassette (0.375 kb). R26 primers are used to distinguish between homozygous and heterozygous (wild type: 0.330 kb and homozygous: 0.750 kb)
The pROSA26‐GNrep was linearized by digestion with EcoRV and electroporated into mESCs. Homologous recombination of the GNrep cassette was screened by Southern blot with a ROSA26 5′‐probe and BclII digestion, generating a 18 kb band (Figure 2b). From the positively targeted mESCs, the clone 3F3 was selected for mouse blastocyst injection in order to produce knock‐in mice. Founders and progeny were screened by genomic PCR, using two sets of primers to confirm the insertion of the construct in the mouse genome. GNrep primers identify the presence or absence of the GNrep cassette. R26 primers allow to distinguish between homozygous and heterozygous‐targeted alleles (Figure 2c). Heterozygous and homozygous GNrep mice are viable, fertile, and produce progeny at near Mendelian rates, obtaining 17.8%–61.6%–20.5% for wild type–heterozygous–homozygous genotypes, respectively, from heterozygous × heterozygous crosses (n = 73 animals, from 10 different litters). Moreover, homozygous GNrep mice are phenotypically normal and indistinguishable from wild type littermates (data not shown), confirming that insertion of the GNrep cassette into the Rosa26 locus does not interfere with major biological processes.
2.3. Validation and characterization of the GNrep mouse line
Subsequently, we wanted to validate the specificity, sensitivity, and tractability of the GNrep line. One example of patent front–rear cell polarity occurs in endothelial cells, the cells lining the interior of blood vessels. Endothelial cells show strong front–rear and apicobasal cell polarity during vascular development in response to different cues. At the angiogenic‐sprouting front, endothelial cells are guided by and migrate toward chemokine gradients (Lavina et al., 2018). In quiescent vessels, endothelial cells establish a strong apicobasal polarity, to form a lumenized network, and a flow‐dependent front–rear polarity, necessary for vascular remodeling (Franco et al., 2015; Franco et al., 2016). Moreover, nucleus‐to‐Golgi axis was previously used to identify endothelial cell front–rear polarity in zebrafish in vivo (Franco et al., 2015; Kwon et al., 2016). Thus, we decided to validate our GNrep line using an endothelial‐specific Cre recombinase transgenic line. The Cre/loxP recombination system is a powerful tool to generate tissue and time‐specific mouse mutants. This system is also widely used to label particular cell populations or specific cell structures, and map their fate, during development or in the context of other complex physiological or pathological processes.
To control the spatial and temporal expression of the polarity reporter in endothelial cells, we crossed the GNrep line with the Cdh5(PAC)‐CreERT2 line (Sörensen, Adams, & Gossler, 2009; Figure 3a). This line is widely used within the vascular community and expresses Cre recombinase specifically in endothelial cells from both blood and lymphatic lineages. As a model system, we used the postnatal mouse retina, which is widely used in the vascular community to study vascular sprouting and vascular remodeling (Selvam, Kumar, & Fruttiger, 2018). To activate the GNrep cassette in mice, tamoxifen was injected at postnatal day 1 (P1) and P3 prior to eye collection and retina dissection at P6 or P8. Cre activity, under the regulation of the endothelial cell promoter, was sufficient to excise the LoxP‐STOP‐LoxP cassette in endothelial cells (Figure 3b) and to trigger the expression of GNrep fluorescent reporters (Figure 3c). GNrep‐recombined retinas showed strong GFP and mCherry signals in endothelial cell nuclei and Golgi apparatuses, respectively, in vessels labeled with the endothelial‐specific cell marker CD31 (Video S1). Co‐expression of GFP+ nuclei and mCherry+ Golgi occurred in all recombined cells. No expression of GFP or mCherry was detected in Cre− nor in non‐tamoxifen‐induced Cre+ animals (Figure 3c). Moreover, using the endothelial‐specific ERG transcription factor to label endothelial cell nuclei (Franco et al., 2015), we observed high level of colocalization in tamoxifen‐induced Cre+ animals (Figure 3c). mCherry fluorescence correlated with GFP‐nuclear staining and was strictly restricted to endothelial cells, as expected by the high specificity of the Cdh5(PAC)‐CreERT2 line. Golph4, a Golgi apparatus‐specific marker (Franco et al., 2015), was used to confirm the colocalization between Golgi complexes and the signal from our GNrep cassette (Figure 3d). Interestingly, endothelial cells at the vascular sprouting front showed polarization toward hypoxic avascular areas (Figure 3e).
Figure 3.
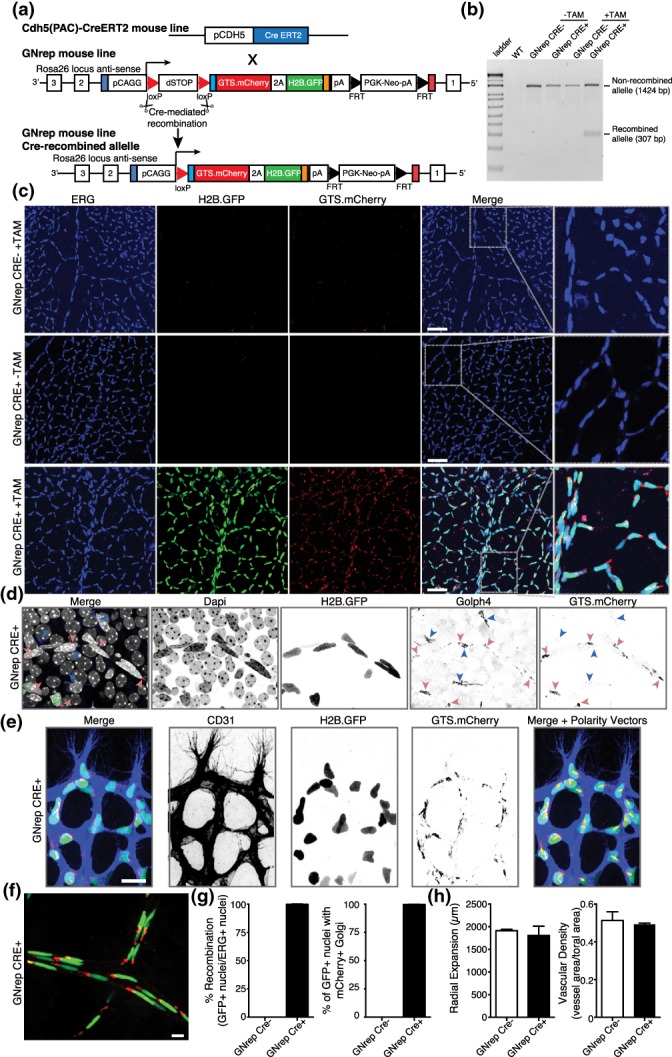
Validation and characterization of the GNrep mouse line. (a) GNrep mouse line was crossed with the Cdh5(PAC)‐CreERT2 line. Tamoxifen‐mediated activation of Cre activity leads to excision of loxP‐STOP‐loxP cassette and expression of the double fluorescent reporter cassette. (b) Analysis of the Cre‐mediated excision of the LoxP‐STOP‐LoxP cassette by genomic PCR (presence of a band at 307 bp) upon tamoxifen treatment, in designated genotypes. (c) Representative images of GFP, mCherry and anti‐ERG antibody (endothelial nuclei) fluorescent signals in P8 GNrep− or GNrep+ retinas, untreated (−TAM) or treated (+TAM) with tamoxifen. GFP and mCherry can be only detected in retinas GNrep+ treated with tamoxifen. Inserts (right panel) highlight Golgi and nuclear localization of mCherry and GFP signals. Scale bar, 50 μm. (d) High‐magnification image of GFP, mCherry, anti‐Golph4 antibody (pan‐Golgi marker), and Dapi fluorescent signals from a GNrep Cre+ mouse retina highlighting the colocalization between Golph4 and mCherry signal in endothelial cells only (red arrowheads). Non‐endothelial Golgi complexes (blue arrowheads) do not express mCherry. Scale bar, 20 μm. (e) High‐magnification image of GFP, mCherry, anti‐CD31 antibody (endothelial cell membrane marker) from a P6 GNrep Cre+ mouse retina sprouting front, highlighting the polarization patterns of endothelial cells at the angiogenic vascular front (yellow arrows). Scale bar, 20 μm. (f) Representative images of GFP and mCherry fluorescent signals in P21 GNrep Cre+ retinas, after tamoxifen treatment. Scale bar, 10 μm. (g) Quantification of recombination (%) and colocalization (%) between GFP+ nuclei and mCherry+ in P8 retinas after tamoxifen injections. (h) Quantification of vascular outgrowth (μm) and vascular density between GNrep Cre− and GNrep Cre+ mice, after tamoxifen treatment. n = 3
To demonstrate that the GNrep cassette can also be activated in more mature and quiescent endothelium, we administrated tamoxifen at P16 and P18 and collected retinas at P21. Retinal vessels showed widespread nuclei labeled with GFP and the Golgi apparatus labeled with mCherry (Figure 3f and Video S2). In addition, we observed a pan‐activation of the GNrep cassette in endothelial cells of several other tissues like brain, skin, and intestine, including in lymphatic endothelial cells (data not shown).
Next, we analyzed the efficiency of activation of the GNrep cassette. Quantification of the colocalization of the GFP+ nuclei with ERG+ nuclei revealed a recombination efficiency around 99% in endothelial cells (Figure 3g). As we observed a 100% correlation between nuclear GFP and Golgi‐located mCherry (Figure 3d), we estimate that around 99% of endothelial Golgi complexes are also labeled, similar to GFP+ nuclei. Finally, to assess if expression of the GNrep cassette in endothelial cells could induce specific phenotypes, we quantified vascular outgrowth and vessel density, in GNrep mice and their control littermates. However, there was no statistical difference between GNrep Cre+ and Cre− animals following tamoxifen treatment (Figure 3h). Thus, our results show that the GNrep cassette labels endothelial cell nuclei and Golgi complexes without affecting their basic biological functions, such as migration, sprouting, and remodeling, which ensures the formation of a properly patterned and organized vasculature.
Importantly, compartmentalization and fluorescent signal intensities for both mCherry and GFP are of sufficiently quality for automated segmentation (Figure 4a,b). Our initial and preliminary image segmentation efforts enabled a robust assignment of polarity vectors for endothelial cells in the mouse retina (Figure 4c). Integration of GNrep‐derived automated polarity segmentation with our recent open‐source image analysis software tool for the quantification of cell polarity and blood flow patterns, PolNet (Bernabeu et al., 2018), will allow to perform large‐scale automated analyses of cell behavior in three‐dimensional tissues. For instance, this tool will enable to perform pharmacological screening for modifiers of flow‐migration coupling in vascular morphogenesis. Depending on specific signal‐to‐noise ratios on each target tissue and cell type, the usefulness of this line for genetic or pharmacological screenings could naturally be extended to areas other than vascular research.
Figure 4.
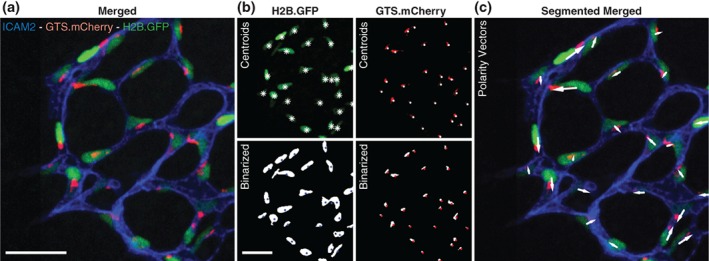
Automated segmentation and polarity assignment of GNrep endothelium. (a) Original merged image of P6 GNrep Cre+ retina (after tamoxifen injections, stained for the endothelial cell apical membrane marker ICAM‐2) used for automatic analysis. Scale bar, 100 μm. (b) Original images for GFP and mCherry signals (top) and their respective automatic segmentation (bottom). Nuclei (top: white asterisks; bottom: blue dots) and Golgi (top: white dots; bottom: red dots) centroids were determined by automated 3D analysis of segmented objects. Scale bar, 100 μm. (c) Original image merged with polarity vectors (white arrows) automatically computed from nuclei and Golgi centroids
Despite the general usefulness of the GNrep mouse to report cell front–rear polarity, there are some considerations worth to be mentioned. For instance, even if nucleus‐to‐Golgi axis represents front–rear cell polarity in most cell types, this internal reorganization is not universal. It is known that in neutrophils, T‐cells, and some cancer cell lines, the MTOC/Golgi apparatus is normally located behind the nucleus (Anderson, Wible, Hughes, Smith, & Brinkley, 1982; Dow et al., 2007; Yvon et al., 2002). This dichotomy seems to be related to the speed of migration, since slow‐moving cells appear to rely heavily on this type of internal organization, while fast‐moving cells do not (Mayor & Etienne‐Manneville, 2016; Serrador, Nieto, & Sanchez‐Madrid, 1999). Also, many cancer cells or normal cell types under certain drug‐treatments, such as brefeldin A (Fujiwara, Oda, Yokota, Takatsuki, & Ikehara, 1988), exhibit fragmented Golgi, which will restrict the ability to use the nucleus‐to‐Golgi axis as a readout for cell polarity. Thus, the usage of the GNrep mouse as a model to measure front–rear polarity requires preliminary information regarding the position and organization of the Golgi complex in the cell type of interest.
In summary, we showed that the GNrep mouse strain constitutes a novel and unique tool to study front–rear polarity of cells in fixed tissues and for live‐cell imaging. Its suitability for automated image segmentation further broadens the applicability of this line to perform high‐throughput analyses, such as pharmacological screenings to identify drugs affecting front–rear polarity in a diverse range of cell types in mice.
3. MATERIALS AND METHODS
3.1. Generation of the GNrep polarity reporter construct
We obtained a synthetized pUC57‐2A plasmid from Eurofins Genomics. This plasmid includes 5′UTR(Cdh5 Homology Arm)‐RE site(NheI/SalI)‐2A‐REsite(BglII/MluI)‐SV40pA‐FRT‐PGK‐Neo‐pA‐FRT‐3′UTR(Cdh5 Homology Arm) and was used for insertion of GTS‐mCherry and H2B.GFP, by performing the following steps: (a) pGTS‐mCherry (Plasmid2‐Table 1) was amplified by PCR using primers F1 and R1 (Table 2) and inserted into pUC57‐2A empty by restriction enzyme cloning with NheI and SalI; (b) pH2B.eGFP was amplified by PCR from pCAGGS‐G2m (Plasmid3‐Table 1) using primers F2 and R2 (Table 2) and inserted into pUC57‐2A containing GTS‐mCherry by restriction enzyme cloning with BglII and MluI; (c) The sequence of the final plasmid pUC57‐GNrep (Plasmid4‐Table 1) was then confirmed by Sanger sequencing. Thereafter, Plasmid4 was amplified by PCR using primers F4 and R4 (Table 2). This PCR product has attB sites to be used for Gateway cloning. The amplified product was used to perform a BP reaction with a pDonorT2K218 to obtain pME‐GTS.mCherry‐2A‐H2B.eGFP (Plasmid5‐Table 1), using BP Clonase II enzyme mix (11789020, Thermo Fisher Scientific, MA, USA), following manufacturer's instructions.
Table 2.
Primers used for the generation of the GNrep polarity construct
| Primer name | Sequence (5′‐3′) | Orientation |
|---|---|---|
| F1 | ATCTCGCTAGCGCCGCCAGCATGAGGCTTCGGGAG | Forward |
| R1 | GACACGTCGACCTTGTACAGCTCGTCCATGC | Reverse |
| F2 | ATCTCAGATCTGCCGCCACCATGCCAGAGCCAGCGAAGTC | Forward |
| R2 | GACACACGCGTGACTTGTACAGCTCGTCCATGC | Reverse |
| F4 | GGGGACAAGTTTGTACAAAAAAGCAGGCTTACCAGATCAGCTCCTCCACGAAG | Forward |
| R4 | GGGGACCACTTTGTACAAGAAAGCTGGGTTAGGGTGAACATACGCGTCTT | Reverse |
| F5 | GGGGACAGCTTTCTTGTACAAAGTGGAAACGCGTATGTTCACCCTATAG | Forward |
| R5 | GGGGACAACTTTGTATAATAAAGTTGTCTGTAATACGACTCACTATAGGG | Reverse |
| LSL F | GCTAGCAAGCTTCTCTGAATAGATCTATAACTTCGT | Forward |
| LSL R | GACATCGGATCCCTGCAGCATTTGTTGCCCA | Reverse |
| GNrep F | CCCGTAATGCAGAAGAAGAC | Forward |
| GNrep R | GCAGATCTTCTGCGAGGACC | Reverse |
| R26‐1 | CTCTGCTGCCTCCTGGCTTCT | Forward |
| R26‐2 | CGAGGCGGATCACAAGCAATA | Reverse |
| R26‐3 | TCAATGGGCGGGGGTCGTT | Reverse |
In parallel, Plasmid4 was also used as template to amplify the fragment pA‐FRT‐PGK.Neo‐pA‐FRT by PCR using primers F5 and R5 (Table 2). The amplicon was then used to perform a BP reaction with pDonorT2K220 to obtain p3E‐pA‐FRT‐PGK.Neo‐pA‐FRT (Plasmid10‐Table 1), using BP Clonase II enzyme mix (11789020, ThermoFisher Scientific, MA, USA), following manufacturer's instructions.
The plasmid pJFRC172‐10XUAS‐loxP‐dSTOP‐loxP‐myr::GFP (Plasmid12‐Table 1) was amplified with primers LSL F and R (Table 2) to amplify the loxP‐dSTOP‐loxP sequence, which then was inserted into the plasmid p5E‐CAGGS‐MCS (Plasmid8‐Table 1) by restriction enzyme cloning with HindIII and BamHI to obtain the plasmid p5E‐CAGGS‐LSL (Plasmid9‐Table 1).
The polarity reporter construct was then generated by a three‐way Gateway LR reaction using Plasmids 5, 9, and 10 as entry vectors with the destination vector pROSA26‐DV2 (Plasmid7‐Table 1), to obtain the final ES targeting plasmid pROSA26‐GNrep (Plasmid11‐Table 1), using LR Clonase II Plus enzyme (12538120, ThermoFisher Scientific, MA, USA) following manufacturer's instructions.
3.2. Knock‐in mice and treatments
For the generation of GNrep mice, stem cell targeting was performed by an external collaboration with Polygene (Rümlang, Switzerland). The GNrep construct was linearized with EcoRV and electroporated into embryonic mouse stem cells, following Polygene's protocols. Specific clones were selected by Southern blot using Polygene services. Confirmation of the integration of the GNrep cassette in F0 mice was performed by genotyping PCR with GNrep primers F and R and with the Rosa26 primers 1, 2, and 3 (Table 2). For the analysis of GNrep in endothelial cells, GNrep mice were crossed with Cdh5(PAC)‐CreERT2 mice (Sörensen et al., 2009), in order to ensure a stable expression of the vector in the vasculature of the mice.
Tamoxifen (Sigma, Germany) was injected intraperitoneally (20 μg/g) at different time points at least 3 days before eyes were collected.
Knock‐in mice were maintained at the Instituto de Medicina Molecular (iMM) under standard husbandry conditions and under national regulations. The GNrep mouse will be available to the research community upon acceptance of the manuscript.
3.3. Cell culture and transfection
Human umbilical vein endothelial cells (HUVECs; C2519A, Lonza, Germany) were routinely cultured following the manufacturer's guidelines, in filter‐cap T75 flasks Nunclon surface treatment (VWR International, LLC) with complete medium EGM‐2 Bulletkit (CC‐3162, Lonza, Germany) supplemented with 0.01% penicillin/streptomycin (#15140122, Gibco, MA, USA) at 37 °C and 5% CO2 to ensure a stable environment for optimal cell growth. All experiments were performed with HUVECs between passages 3 and 6.
NIH/3T3 fibroblasts were cultured following the manufacturer's guidelines, in filter‐cap T75 flasks Nunclon surface treatment (VWR International, LLC) with DMEM (41965‐039, Gibco, MA, USA) supplemented with 0.01% penicillin/streptomycin (#15140122, Gibco, MA, USA), HEPES buffer solution (15630‐056, Gibco, MA, USA), and 10% fetal bovine serum (10500‐064, Gibco, MA, USA) at 37 °C, and 5% CO2 to ensure a stable environment for optimal cell growth.
When passaging these cells for experiments, cells were washed twice in sterile PBS (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4, pH 7.4) and then incubated for 2–3 min in Trypsin/EDTA (#15400054, Gibco, MA, USA) at 37 °C, 5% CO2. When 95% of the cells detached, complete medium was added to each flask to inhibit the activity of the trypsin/EDTA and the cell suspension was transferred into a falcon tube. To maximize the number of cells collected, all the flasks were washed again with complete medium, which was added to the cell suspension collected previously. Cells were then centrifuged at 700 rpm for 5 min at RT and the pellet was re‐suspended in fresh complete medium. The cell concentration present in the suspension was determined using a Neubauer chamber (Hirschmann EM Techcolor).
To validate the construct in cells in vitro, we generated a mammalian GNrep expression vector, pEF1‐GNrep (Plasmid6‐Table 1). Plasmid6 was generated by one‐way Gateway LR reaction using Plasmid5 as entry vector with the destination vector pEF1‐DEST51 (Plasmid13‐Table 1), using LR Clonase II Plus enzyme (12538120, ThermoFisher Scientific, MA, USA) and following manufacturer's instructions.
HUVECs and NIH/3T3 fibroblasts were then transfected with this plasmid to validate the efficiency of the reporter. Cells at 60–80% of confluency were transfected with 100 ng/μL of the pEF1‐GTS.mCherry‐H2B.GFP. For each fluorodish (FD35PDL100, ThermoFisher Scientific, MA, USA) with cells, 125 μL of Opti‐Mem Reduced Serum media (31985047, ThermoFisher Scientific, MA, USA) with 7.5 μL of Lipofectamine LTX Reagent (15338500, ThermoFisher Scientific, MA, USA) was mixed with 125 μL of Opti‐Mem Reduced Serum media (31985047, ThermoFisher Scientific, MA, USA) with 5 μL of Plus Reagent (11514015, ThermoFisher Scientific, MA, USA), and plasmid (100 ng/μL), and this mixture added to the cells in droplets. Cells were incubated for 48 hr at 37 °C with 5% CO2 and then expression of the two fluorescent markers was assessed using a confocal Laser Point‐Scanning Microscope 880 (Zeiss, Germany) equipped with the Zen black software with a Plan Apochromat 63× NA 1.40 oil DIC M27 objective.
3.4. Immunofluorescence on cells
For immunofluorescence of in vitro cultured cells, they were seeded on 24‐well plates with glass coverslips, previously coated with 0.2% Gelatin in sterile water (G1393, Sigma‐Aldrich, Germany). Cells were fixed in 1% paraformaldehyde (PFA) supplemented with 0.5mM MgCl2 and 0.5mM CaCl2 in PBS for 30 min at room temperature (RT). Cells were then washed with PBS to remove the remaining PFA and the immunostaining protocol initiated. When the PBS was removed, cells were blocked and permeabilized with blocking solution containing 3% BSA in PBS‐T (PBS with 0.1% Triton X‐100) for 30 min at RT. Then cells were incubated for 2 hr at RT with anti‐GOLPH4 (ab28049, 1:400, Abcam, UK) diluted in the blocking solution and washed 3 × 15 min in PBS‐T. Afterward, cells were incubated in blocking solution containing the secondary fluorophore conjugated antibody for 1 hr at RT in the dark, followed again by three washes of 15 min in PBS‐T. Finally, cells were incubated with 1× DAPI (D1306, Thermo Fischer Scientific, MA, USA) diluted in PBS‐T for 5 min in the dark. Coverslips were then mounted on microscopy glass slides using Mowiol DABCO (Sigma‐Aldrich, Germany). Images were acquired on a confocal Laser Point‐Scanning Microscope 880 (Zeiss, Germany) equipped with the Zen black software with a Plan Apochromat 63× NA 1.40 oil DIC M27 objective.
3.5. Immunofluorescence on mouse retinas
Eyes were collected at different time points and fixed with 2% PFA in PBS for 5 hr at 4 °C, thereafter retinas were dissected in PBS. Stainings were performed according to previous published protocols (Bernabeu et al., 2018; Franco et al., 2013). Briefly, blocking/permeabilization was performed using Claudio's blocking buffer (CBB), consisting of 1% FBS (ThermoFisher Scientific, MA, USA), 3% BSA (Nzytech, Portugal), 0.5% Triton X100 (Sigma, Germany), 0.01% Sodium deoxycholate (Sigma, Germany), 0.02% Sodium azide (Sigma, Germany) in PBS, pH = 7.4 for 2 hr in a rocking platform. Primary antibodies anti‐ERG (ab92513, 1:200, Abcam, UK), anti‐CD31 (AF3628, 1:400, R&D, MN, USA), or anti‐GOLPH4 (ab28049, 1:400, Abcam, UK) were incubated in 1:1 CBB:PBS at 4 °C overnight in a rocking platform and afterward washed 3× 60 min in PBS‐T. Retinas were then incubated in 1:1 CBB:PBS solution containing the secondary fluorophore‐conjugated antibodies (Donkey anti‐Rabbit Alexa 647, A32795, 1:400; Donkey anti‐Goat Alexa 647, A21447, 1:400, ThermoFischer Scientific, MA, USA) at 4 °C overnight in the dark. Retinas were mounted on slides using Vectashield mounting medium (H‐1000, VectorLabs, CA, USA). For analysis, a tile‐scan spanning the retina was acquired on a Zeiss LSM880 (Zeiss, Germany) confocal microscope, equipped with the Zen software with a Plan‐Apochromat 40×/1.4 Oil DIC M27 objective.
3.6. Quantification measurements and statistical analysis
Tiled scans of whole retinas were analyzed with ImageJ. Radial expansion corresponds to the mean distance from the optic nerve to the edge of the sprouting blood vessels (four measurements per retina were done and averaged). Vessel density corresponds to the vascular area (skeletonized using a Voronoi diagram based method (Bernabeu et al., 2018) divided by the total area of vascularized tissue (1–2 20× objective images of petals per retina). Statistical analysis was performed using Prism 5.0 (GraphPad, CA, USA), and the MATLAB (Mathworks, MA, USA) software.
3.7. Automated segmentation
After staining for ICAM‐2 (553326, 1:200, BD Biosciences, CA, USA), retinas were mounted and examined on a Zeiss Cell Observer SD spinning disk confocal microscope (Zeiss, Germany). Image stacks were obtained with a Plan‐Apochromat 20×/0.80 Ph 2 M27 objective using the Zen software. The algorithm, developed in MATLAB, allows the binarization of original images and assignment of polarity vectors between nuclei and their Golgi. The first step of the algorithm is the definition of a vascular frame using the ICAM‐2 staining. A binary mask is obtained using a single threshold and dilated. Nuclei and Golgi channels are subsequently processed. First, a median filter is applied to the images to remove salt and pepper noise, and the vascular mask is applied to remove spurious signal from outside the vascular bed. Then, we used a triple automatic threshold to obtain binary images including objects with different levels of signal. Binary masks are filtered according to size. The three‐dimensional centroid of every object is computed, providing 3D coordinates for every nuclei and Golgi. The distances between all the nuclei and Golgi coordinates are computed and used to assign couples of nuclei and Golgi based on minimization of the assignment matrix. Finally, a maximum limit to the length of the vector between centroids is applied.
Supporting information
Movie 1 3D overview of GNrep signal in a retinal vascular capillary. 3D rotational view of a capillary vessel (CD31, blue) in a P6 mouse retina, highlighting the signals from nuclear GFP (green) and Golgi‐mCherry (red) from the Cdh5‐CreERT2::GNrep mouse
Movie 2 3D overview of GNrep signal in a P21 mouse retina. 3D rotational view of nuclear GFP (green) and Golgi‐mCherry (red) signals from a P21 mouse retina of the Cdh5‐CreERT2::GNrep mouse, highlighting the pan‐labeling of endothelial cells. Note the planar cell polarity orientation of front–rear polarities of endothelial cells
ACKNOWLEDGMENTS
The authors thank M. Davidson (Florida State University) for mCherry‐Golgi‐7 plasmid (Addgene # 55052); G. Rubin (Howard Hughes Medical Institute) for pJFRC172‐10XUAS‐loxP>‐dSTOP‐loxP>‐myr::GFP plasmid (Addgene #32145); and A.C. Vion, R. Collins and H. Gerhardt (Max‐Delbruck Center, Berlin) for providing additional plasmids and the basis of our MATLAB script for automated polarity analysis; and R. Adams (Max Planck Institute, Munster) for providing Cdh5(PAC)‐CreERT2 mice. Research was supported by European Research Council starting grant (679368); the H2020‐Twinning grant (692322); the Fundação para a Ciência e a Tecnologia grants: IF/00412/2012; EXPL‐BEX‐BCM‐2258‐2013; PRECISE‐LISBOA‐01‐0145‐FEDER‐016394; and a grant from the Fondation Leducq (17CVD03); and personal fellowships: BD/105856/2014 to P.B., and BD/128375/2017 to C.F., and LISBOA‐01‐0145‐FEDER‐007391, project cofunded by FEDER, through POR Lisboa 2020—Programa Operacional Regional de Lisboa, PORTUGAL 2020, and UID/BIM/50005/2019, a project funded by Fundação para a Ciência e a Tecnologia (FCT)/ Ministério da Ciência,Tecnologia e Ensino Superior (MCTES) through Fundos do Orçamento de Estado. A patent application has been submitted by Instituto de Medicina Molecular based on the GNrep mouse.
Barbacena P, Ouarné M, Haigh JJ, Vasconcelos FF, Pezzarossa A, Franco CA. GNrep mouse: A reporter mouse for front–rear cell polarity. genesis. 2019;57:e23299. 10.1002/dvg.23299
Funding information Fondation Leducq, Grant/Award Number: 17CVD03; Fundação para a Ciência e a Tecnologia, Grant/Award Number: IF/00412/2012; H2020 European Research Council, Grant/Award Number: 679368; H2020 Spreading Excellence and Widening Participation, Grant/Award Number: 692322
REFERENCES
- Abe, T. , Kiyonari, H. , Shioi, G. , Inoue, K.‐I. , Nakao, K. , Aizawa, S. , & Fujimori, T. (2011). Establishment of conditional reporter mouse lines at ROSA26 locus for live cell imaging. Genesis (New York, NY), 2000(49), 579–590. [DOI] [PubMed] [Google Scholar]
- Anderson, D. C. , Wible, L. J. , Hughes, B. J. , Smith, C. W. , & Brinkley, B. R. (1982). Cytoplasmic microtubules in polymorphonuclear leukocytes: Effects of chemotactic stimulation and colchicine. Cell, 31, 719–729. [DOI] [PubMed] [Google Scholar]
- Bernabeu, M. O. , Jones, M. L. , Nash, R. W. , Pezzarossa, A. , Coveney, P. V. , Gerhardt, H. , & Franco, C. A. (2018). PolNet: A tool to quantify network‐level cell polarity and blood flow in vascular remodeling. Biophysical Journal, 114, 2052–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogterom, M. , & Koenderink, G. H. (2019). Actin‐microtubule crosstalk in cell biology. Nature Reviews Molecular Cell Biology, 20, 38–54. [DOI] [PubMed] [Google Scholar]
- Dow, L. E. , Kauffman, J. S. , Caddy, J. , Zarbalis, K. , Peterson, A. S. , Jane, S. M. , … Humbert, P. O. (2007). The tumour‐suppressor scribble dictates cell polarity during directed epithelial migration: Regulation of Rho GTPase recruitment to the leading edge. Oncogene, 26, 2272–2282. [DOI] [PubMed] [Google Scholar]
- Etienne‐Manneville, S. (2013). Microtubules in cell migration. Annual Review of Cell and Developmental Biology, 29, 471–499. [DOI] [PubMed] [Google Scholar]
- Etienne‐Manneville, S. , & Hall, A. (2003). Cdc42 regulates GSK‐3beta and adenomatous polyposis coli to control cell polarity. Nature, 421, 753–756. [DOI] [PubMed] [Google Scholar]
- Franco, C. A. , Blanc, J. , Parlakian, A. , Blanco, R. , Aspalter, I. M. , Kazakova, N. , … Li, Z. (2013). SRF selectively controls tip cell invasive behavior in angiogenesis. Development, 140, 2321–2333. [DOI] [PubMed] [Google Scholar]
- Franco, C. A. , Jones, M. L. , Bernabeu, M. O. , Geudens, I. , Mathivet, T. , Rosa, A. , … Gerhardt, H. (2015). Dynamic endothelial cell rearrangements drive developmental vessel regression. PLoS Biology, 13, e1002163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco, C. A. , Jones, M. L. , Bernabeu, M. O. , Vion, A. C. , Barbacena, P. , Fan, J. , … Gerhardt, H. (2016). Non‐canonical Wnt signalling modulates the endothelial shear stress flow sensor in vascular remodelling. eLife, 5, e07727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl, P. , & Wolf, K. (2010). Plasticity of cell migration: A multiscale tuning model. The Journal of Cell Biology, 188, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara, T. , Oda, K. , Yokota, S. , Takatsuki, A. , & Ikehara, Y. (1988). Brefeldin A causes disassembly of the Golgi complex and accumulation of secretory proteins in the endoplasmic reticulum. The Journal of Biological Chemistry, 263, 18545–18552. [PubMed] [Google Scholar]
- Gotlieb, A. I. , May, L. M. , Subrahmanyan, L. , & Kalnins, V. I. (1981). Distribution of microtubule organizing centers in migrating sheets of endothelial cells. The Journal of Cell Biology, 91, 589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamidi, H. , & Ivaska, J. (2018). Every step of the way: Integrins in cancer progression and metastasis. Nature Reviews. Cancer, 18, 533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, L. , Li, L. , Xie, H. , Gu, Y. , & Peng, T. (2011). The Golgi localization of GOLPH2 (GP73/GOLM1) is determined by the transmembrane and cytoplamic sequences. PLoS One, 6, e28207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iden, S. , & Collard, J. G. (2008). Crosstalk between small GTPases and polarity proteins in cell polarization. Nature Reviews Molecular Cell Biology, 9, 846–859. [DOI] [PubMed] [Google Scholar]
- Kwon, H. B. , Wang, S. , Helker, C. S. , Rasouli, S. J. , Maischein, H. M. , Offermanns, S. , … Stainier, D. Y. (2016). In vivo modulation of endothelial polarization by Apelin receptor signalling. Nature Communications, 7, 11805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladoux, B. , & Mege, R. M. (2017). Mechanobiology of collective cell behaviours. Nature Reviews. Molecular Cell Biology, 18, 743–757. [DOI] [PubMed] [Google Scholar]
- Lavina, B. , Castro, M. , Niaudet, C. , Cruys, B. , Alvarez‐Aznar, A. , Carmeliet, P. , … Gaengel, K. (2018). Defective endothelial cell migration in the absence of Cdc42 leads to capillary‐venous malformations. Development, 145, dev161182. [DOI] [PubMed] [Google Scholar]
- Mayor, R. , & Etienne‐Manneville, S. (2016). The front and rear of collective cell migration. Nature Reviews Molecular Cell Biology, 17, 97–109. [DOI] [PubMed] [Google Scholar]
- Miyazaki, J. , Takaki, S. , Araki, K. , Tashiro, F. , Tominaga, A. , Takatsu, K. , & Yamamura, K. (1989). Expression vector system based on the chicken beta‐actin promoter directs efficient production of interleukin‐5. Gene, 79, 269–277. [DOI] [PubMed] [Google Scholar]
- Nobis, M. , Warren, S. C. , Lucas, M. C. , Murphy, K. J. , Herrmann, D. , & Timpson, P. (2018). Molecular mobility and activity in an intravital imaging setting—Implications for cancer progression and targeting. Journal of Cell Science, 131, jcs206995. [DOI] [PubMed] [Google Scholar]
- Nyabi, O. , Naessens, M. , Haigh, K. , Gembarska, A. , Goossens, S. , Maetens, M. , … Haigh, J. J. (2009). Efficient mouse transgenesis using Gateway‐compatible ROSA26 locus targeting vectors and F1 hybrid ES cells. Nucleic Acids Research, 37, e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley, A. J. , Schwartz, M. A. , Burridge, K. , Firtel, R. A. , Ginsberg, M. H. , Borisy, G. , … Horwitz, A. R. (2003). Cell migration: Integrating signals from front to back. Science (New York, NY), 302, 1704–1709. [DOI] [PubMed] [Google Scholar]
- Selvam, S. , Kumar, T. , & Fruttiger, M. (2018). Retinal vasculature development in health and disease. Progress in Retinal and Eye Research, 63, 1–19. [DOI] [PubMed] [Google Scholar]
- Serrador, J. M. , Nieto, M. , & Sanchez‐Madrid, F. (1999). Cytoskeletal rearrangement during migration and activation of T lymphocytes. Trends in Cell Biology, 9, 228–233. [DOI] [PubMed] [Google Scholar]
- Sörensen, I. , Adams, R. H. , & Gossler, A. (2009). DLL1‐mediated notch activation regulates endothelial identity in mouse fetal arteries. Blood, 113, 5680–5688. [DOI] [PubMed] [Google Scholar]
- Soriano, P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nature Genetics, 21, 70–71. [DOI] [PubMed] [Google Scholar]
- Szymczak‐Workman, A. L. , Vignali, K. M. , & Vignali, D. A. A. (2012). Design and construction of 2A peptide‐linked multicistronic vectors. Cold Spring Harbor Protocols, 2012, 199–204. [DOI] [PubMed] [Google Scholar]
- Wedlich‐Soldner, R. , & Li, R. (2003). Spontaneous cell polarization: undermining determinism. Nature Cell Biology, 5, 267–270. [DOI] [PubMed] [Google Scholar]
- Yvon, A.‐M. C. , Walker, J. W. , Danowski, B. , Fagerstrom, C. , Khodjakov, A. , & Wadsworth, P. (2002). Centrosome reorientation in wound‐edge cells is cell type specific. Molecular Biology of the Cell, 13, 1871–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong, H. , Espinosa, J. S. , Su, H. H. , Muzumdar, M. D. , & Luo, L. (2005). Mosaic analysis with double markers in mice. Cell, 121, 479–492. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie 1 3D overview of GNrep signal in a retinal vascular capillary. 3D rotational view of a capillary vessel (CD31, blue) in a P6 mouse retina, highlighting the signals from nuclear GFP (green) and Golgi‐mCherry (red) from the Cdh5‐CreERT2::GNrep mouse
Movie 2 3D overview of GNrep signal in a P21 mouse retina. 3D rotational view of nuclear GFP (green) and Golgi‐mCherry (red) signals from a P21 mouse retina of the Cdh5‐CreERT2::GNrep mouse, highlighting the pan‐labeling of endothelial cells. Note the planar cell polarity orientation of front–rear polarities of endothelial cells