

Abstract
The pharmacokinetics and safety of a single oral dose of 200‐mg plant‐derived pharmaceutical formulation of highly purified cannabidiol (CBD) in oral solution (Epidiolex in the United States; 100 mg/mL) were assessed in subjects with mild to severe hepatic impairment (n = 8 each for mild and moderate, n = 6 for severe) relative to matched subjects with normal hepatic function (n = 8). Blood samples were collected until 48 hours after dosing and evaluated by liquid chromatography and tandem mass spectrometry. Pharmacokinetic parameters (primarily maximum measured plasma concentration, area under the plasma concentration–time curve from time zero to time t, area under the concentration‐time curve from time zero to infinity, time to maximum plasma concentration, and terminal half‐life) of CBD and its major metabolites were derived using non‐compartmental analysis. CBD was rapidly absorbed in all groups independent of hepatic function (median time to maximum plasma concentration, 2‐2.8 hours). Exposure (area under the concentration–time curve from time zero to infinity) to total CBD slightly increased in subjects with mild hepatic impairment (geometric mean ratio [GMR], 1.48; 90% confidence interval [CI], 0.90‐2.41). However, there were clinically relevant increases in subjects with moderate (GMR, 2.45; 90%CI, 1.50‐4.01) and severe (GMR, 5.15; 90%CI, 2.94‐9.00) hepatic impairment, relative to subjects with normal hepatic function. Exposure to the CBD metabolites (6‐hydroxy‐CBD and 7‐hydroxy‐CBD) also increased in subjects with moderate and severe hepatic impairment, but to a lesser extent than the parent drug. The 7‐carboxy‐CBD metabolite exposure was lower in subjects with severe hepatic impairment when compared with subjects with normal liver function. These findings indicate that dose modification is necessary in patients with moderate and severe hepatic impairment, and a lower starting dose and slower titration are necessary based on benefit‐risk. CBD was well tolerated, and there were no serious adverse events reported during the trial.
Keywords: cannabidiol, cannabinoid, hepatic impairment, liver function, pharmacokinetic
The efficacy of highly purified cannabidiol (CBD) (approved as Epidiolex in the United States) in patients with Lennox‐Gastaut syndrome or Dravet syndrome has been demonstrated in randomized controlled trials with an acceptable safety profile.1, 2, 3, 4, 5 Based on recent nonclinical scientific literature, it is likely that the cumulative anticonvulsant effect is regulated via at least 3 targets: modulation of intracellular Ca2+ via G protein–coupled receptor 55, desensitization of transient receptor potential vanilloid type I channels, and inhibition of adenosine reuptake.6, 7, 8 Importantly, because there is limited or no interaction with the cannabinoid receptors, CB1 and CB2, CBD is not associated with detectable euphoric effects linked to propensity for abuse.9
CBD is highly lipophilic, subject to considerable first‐pass metabolism, and extensively metabolized by the liver, while a large proportion (33%) is excreted unchanged in the feces.10 CBD is metabolized to form monohydroxylated and monocarboxylated metabolites, predominantly 7‐carboxy‐cannabidiol (7‐COOH‐CBD) and 7‐hydroxy‐cannabidiol (7‐OH‐CBD), and a minor metabolite, 6‐hydroxy‐cannabidiol (6‐OH‐CBD).11, 12, 13 Cytochrome P450 (CYP) isoenzymes CYP2C19 and CYP3A4 are implicated in CBD biotransformation, and various other metabolic pathways are available including other CYP‐mediated pathways (1A2, 2D6, and 2C914) and direct conjugations via uridine 5′‐diphospho‐glucuronosyltransferase (UGT) enzymes such as UGT1A7, UGT1A9, and UGT2B7.15
Of the major metabolites of CBD identified in humans, 7‐OH‐CBD has been shown to be active in the nonclinical maximal electroshock seizure threshold test in mice, whereas the most abundant metabolite, 7‐COOH‐CBD, is inactive in the same model at the concentrations tested.16 Given the extensive biotransformation by hepatic first‐pass metabolism, it is possible that patients with hepatic impairment who receive CBD may be at risk of drug accumulation or, less often, failure to form an active metabolite.17, 18 There are no previously published studies investigating the pharmacokinetics (PK) of this formulation of CBD in subjects with hepatic impairment. This trial aimed to assess the effect of hepatic impairment on the systemic exposure to a single dose of CBD.
Methods
Trial Design
All relevant trial‐related documents, including the protocol, were reviewed by 3 independent ethics committees, and approval for the trial was granted in Slovakia on September 25, 2015; in the Czech Republic on July 8, 2015; and in Hungary on August 19, 2015. All subjects provided written informed consent for participation in the trial, which was performed in full conformity with the current Declaration of Helsinki,19 the International Conference on Harmonisation guidelines for Good Clinical Practice,20 and all other applicable regulations. The trial was performed between September 14, 2015, and July 4, 2016, at 3 Pharmaceutical Research Associates (PRA) sites specializing in clinical pharmacology trials (1 each in Hungary, the Czech Republic, and Slovakia). The trial was performed considering the US Food and Drug Administration and European Medicines Agency recommendations for the evaluation of PK in subjects with impaired hepatic function.17, 18
The trial consisted of a screening period (days – 28 to –2), a treatment period (hospitalization from day –1 until day 3), and a follow‐up visit (day 14 [±2 days]). Child‐Pugh assessment for subjects with hepatic impairment was performed during screening. During the in‐house treatment period, baseline assessments were performed on day –1 (after an overnight fast of at least 8 hours). On the morning of day 1, subjects received a standardized low‐protein breakfast 2 hours before administration of a single oral 200‐mg dose of a pharmaceutical formulation of highly purified CBD derived from Cannabis sativa L. plant in oral solution (100 mg/mL; Epidiolex in the United States; GW Research Ltd, Cambridge, United Kingdom). Observations were made until subjects were released from the clinical research unit, following 48‐hour postdose assessments on day 3. A follow‐up visit was performed on day 14 (±2 days). Fluid intake was prohibited during fasting only (2 hours before dosing and 4 hours after dosing).
Inclusion and Exclusion Criteria
Trial Population
The inclusion criteria specified that the trial population consisted of male and female subjects (age, 18‐75 years; body mass index [BMI], 18‐35 kg/m2) with mild, moderate, or severe hepatic impairment, as defined by Child‐Pugh score (Table 1) and subjects with normal hepatic function (matched to subjects with hepatic impairment with respect to sex, age, and BMI [extremes covered as far as possible]).
Table 1.
Child‐Pugh Classification
| Assessment | Degree of Abnormality | Score |
|---|---|---|
| Encephalopathy | NoneModerateSevere | 123 |
| Ascites | AbsentSlightModerate | 123 |
| Bilirubin (mg/dL) | <22.1‐3>3 | 123 |
| Albumin (g/dL) | >3.52.8‐3.5<2.8 | 123 |
| Prothrombin time (INR) | <1.71.7‐2.3>2.3 | 123 |
| Total score | Group | Severity |
| 5‐6 | A | Mild |
| 7‐9 | B | Moderate |
| 10‐15 | C | Severe |
INR, international normalized ratio.
Female subjects of childbearing potential were nonpregnant and nonlactating at screening. Male and female subjects agreed to use effective contraception for the duration of the trial and for 3 months and 30 days thereafter, respectively. Subjects with impaired hepatic function were deemed to have stable disease status, as judged by the investigator. At screening and baseline, platelet counts were >50 × 109/L for subjects with mild and moderate hepatic impairment and >30 × 109/L for subjects with severe hepatic impairment.
Trial Assessments
Materials
As Δ9‐tetrahydrocannabinol (THC) is present as a trace impurity in the CBD formulation under development, plasma concentrations of THC and its metabolites, 11‐hydroxy‐Δ9‐tetrahydrocannabinol (11‐OH‐THC) and 11‐nor‐9‐carboxy‐ Δ9‐tetrahydrocannabinol (11‐COOH‐THC), were also determined. Reference and internal standards for 6‐OH‐CBD, 7‐OH‐CBD, 7‐COOH‐CBD, THC, 11‐OH‐THC, and 11‐COOH‐THC bioanalysis were supplied by GW Pharma Ltd (Cambridge, United Kingdom), Cerilliant (Round Rock, Texas) or BDG Synthesis (Wellington, New Zealand).
Plasma Sample Preparation
Total (bound and unbound) CBD and metabolite samples were extracted from whole plasma samples by protein precipitation with isopropyl alcohol and acetonitrile. THC and metabolite samples were extracted by liquid‐liquid extraction. Ultracentrifugation (238,859 g max, 20 hours, 37°C) was applied to aliquots of each plasma sample prior to protein precipitation step to separate the plasma proteins and generate protein‐free samples. Although this approach was employed, the process and stability of the free fraction (ultracentrifugate) was not supported during bioanalytical method validation and therefore the data are not presented in this paper.
Bioanalysis and PK Assessment
Validated liquid chromatographic‐tandem mass spectrometric bioanalytical methods were used to quantify concentrations of CBD, THC, and their metabolites, 6‐OH‐CBD, 7‐OH‐CBD, 7‐COOH‐CBD, 11‐OH‐THC, and 11‐COOH‐THC, in human plasma. The assay ranges were 2.00 to 10 000 ng/mL for CBD, 0.250 to 250 ng/mL for 6‐OH‐CBD, 0.250 to 1250 ng/mL for 7‐OH‐CBD, and 0.250 to 20 000 ng/mL for 7‐COOH‐CBD. Calibration standards were between 0.125 to 62.5 ng/mL for THC, and 0.250 to 125 ng/mL for 11‐OH‐THC and 11‐COOH‐THC.
The precision (coefficient of variation) and accuracy (relative error/mean % different) of the high‐performance liquid chromatography method was acceptable for all analytes (≤15% [20% at the lower limit of quantification (LLOQ)]). Recovery was >90% for CBD, 6‐OH‐CBD, 7‐OH‐CBD, and 7‐COOH‐CBD and considered acceptable. The recovery of THC, 11‐OH‐THC, and 11‐COOH‐THC was considered adequate (56.3% to 124%).
At the times specified below, 6‐mL blood samples were taken from subjects via an indwelling intravenous catheter or direct venipuncture into lithium heparin vacutainers; blood samples were then centrifuged for 10 minutes at ambient temperature. The resultant plasma was stored upright in a freezer at −80°C.
Blood samples for PK analysis were taken at the following time points: before dosing, then at 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 7, 8, 9, 10, 12, 24, 36, and 48 hours after dosing.
PK parameters were determined from total (ie, bound and unbound) plasma concentrations of analytes, by noncompartmental analysis using WinNonlin version 6.3 (Pharsight Inc., Princeton, New Jersey). PK parameters evaluated included maximum measured plasma concentration (Cmax), area under the concentration‐time curve from time zero to infinity (AUC0‐∞), area under the plasma concentration–time curve from time zero to time t (AUC0‐t), terminal (elimination) half‐life (t½), time to maximum plasma concentration (tmax), oral clearance of drug from plasma, apparent volume of distribution, rate constant of the terminal phase, and percentage of estimated part for the calculation of AUC0‐∞ ([AUC0‐∞ minus AUC0‐t] / AUC0‐∞) × 100 (%AUCextra).
Safety Assessments
The safety and tolerability of CBD were evaluated by recording the incidence of adverse events (AEs) throughout the trial, and the review of clinical laboratory tests, vital signs, 12‐lead electrocardiogram (ECG), and physical examinations.
Statistical Analysis
The primary objective of the trial was to assess the PK of CBD and its major metabolites in subjects with impaired hepatic function compared with subjects with normal hepatic function. Secondary objectives were to evaluate the safety and tolerability of CBD in the same population. Descriptive statistics of subject demographics and safety outcomes were based on the safety analysis set (all subjects who received CBD).
The PK parameters of CBD, THC, and their metabolites were calculated for the PK analysis set (all subjects who received CBD, had no major protocol deviations, and had evaluable PK assessments), using Phoenix WinNonlin version 6.3. Noncompartmental methods were used to estimate PK parameters for all analytes with sufficient data above LLOQ from the concentration‐time profiles for individuals in the PK analysis set. At least 3 data points (not including Cmax) were required to calculate first‐order rate constant for elimination of drug and percent extrapolation of ≤30% was required to retain AUC0‐∞ and t½; subjects who did not satisfy this criterion were excluded from the analysis. Analysis of variance was used to compare primary PK parameters (Cmax, AUC0‐∞, AUC0‐t, t½, and tmax) between the control group (normal hepatic function) and each of the groups with hepatic impairment. PK values were log‐transformed before analysis. Covariates included sex, age, and BMI, if statistically significant. Geometric least squares means were used to calculate the ratio of primary PK parameters in each hepatic impairment group to those in the control group, together with 90% confidence intervals (CIs). A Wilcoxon rank‐sum test was used for comparison of the tmax values between the control and hepatic impairment groups; estimates of median of the differences were determined along with 90%CIs.
The relationship between log‐transformed primary PK parameters and continuous variables contributing to the Child‐Pugh score (baseline values for serum albumin and bilirubin concentrations and prothrombin time) were explored by linear regression. Secondary PK parameters and safety data were analyzed descriptively.
Sample Size
The planned sample size was 8 subjects per group, based on practical considerations and guidance from the US Food and Drug Administration and European Medicines Agency.17, 18
Results
Subject Demographics
A total of 30 subjects were enrolled into 1 of 4 subject groups (mild [n = 8], moderate [n = 8], or severe [n = 6] hepatic impairment or normal hepatic function [n = 8]). All 30 subjects completed the trial without any major protocol deviations and were included in the safety and PK analysis sets.
All subjects enrolled were white. In the mild and severe hepatic impairment groups and the normal hepatic function group, there were equal numbers of men and women. In the moderate hepatic impairment group, there were marginally more male (n = 5) than female (n = 3) subjects; other baseline characteristics (age, weight, height, and BMI) were similar across treatment groups. Mean age across the groups ranged from 52.7 to 57.5 years, and mean BMI ranged from 25.8 to 30.0 kg/m2 (Table 2).
Table 2.
Demographics and Baseline Characteristics: Safety Analysis Set
| Demographic | Mild Hepatic Impairment (N = 8) | Moderate Hepatic Impairment (N = 8) | Severe Hepatic Impairment (N = 6) | Normal Hepatic Function (N = 8) |
|---|---|---|---|---|
| Number of subjects (%) | ||||
| Sex | ||||
| Male | 4 (50.0) | 5 (62.5) | 3 (50.0) | 4 (50.0) |
| Female | 4 (50.0) | 3 (37.5) | 3 (50.0) | 4 (50.0) |
| Race | ||||
| White | 8 (100) | 8 (100) | 6 (100) | 8 (100) |
| Mean (standard deviation) | ||||
| Age (years) | 57.5 (8.1) | 55.6 (11.1) | 52.7 (6.9) | 55.0 (10.0) |
| Weight (kg) | 76.1 (23.5) | 85.2 (15.8) | 89.0 (18.7) | 89.4 (11.6) |
| Height (cm) | 170.5 (9.6) | 169.5 (11.9) | 171.8 (10.8) | 174.1 (5.9) |
| BMI (kg/m2) | 25.8 (5.7) | 29.6 (3.8) | 30.0 (4.7) | 29.4 (3.2) |
BMI, body mass index; N, number of subjects exposed.
All subjects with hepatic impairment (except 1 subject with mild hepatic impairment) took at least 1 concomitant medication during the trial. The most common classes of concomitant medications taken throughout the trial were beta‐blocking agents, diuretics, and drugs for acid‐related disorders.
Pharmacokinetics
Systemic exposure to intact CBD was more rapid in the moderate and severe hepatic impairment groups compared with the mild hepatic impairment and normal hepatic function groups (Figure 1). However, statistical analysis demonstrated that there was no effect on tmax, with a median CBD tmax of 2 to 2.8 hours independent of hepatic status (Tables 3 and 4). The same trend was seen for the CBD metabolites.
Figure 1.
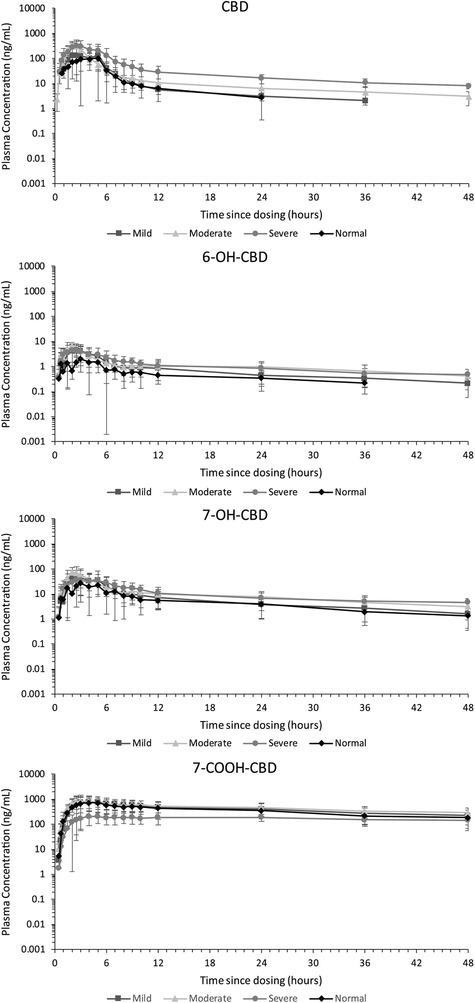
Arithmetic mean (± standard deviation) plasma concentration–time profiles for cannabidiol (CBD), 6‐OH‐CBD, 7‐OH‐CBD, and 7‐COOH‐CBD after a single oral 200 mg CBD dose, by hepatic function group (semilogarithmic); pharmacokinetic analysis set.
Table 3.
PK Parameters for Total CBD, 6‐OH‐CBD, 7‐OH‐CBD, and 7‐COOH‐CBD Following a Single Oral 200 mg CBD Dose: PK Analysis Set
| Geometric Mean (Geometric CV%)a | ||||
|---|---|---|---|---|
| Parameter | Mild Hepatic Impairment (N = 8) | Moderate Hepatic Impairment (N = 8) | Severe Hepatic Impairment (N = 6) | Normal Hepatic Function (N = 8) |
| CBD | ||||
| Cmax (ng/mL) | 233 (70.5) | 354 (42.3) | 381 (52.2) | 148 (65.0) |
| AUC0‐∞ (ng·h/mL)b | 699 (44.2) | 1163 (39.9) | 2439 (29.5)d | 474 (73.8) |
| AUC0‐t (ng·h/mL) | 648 (44.2) | 1054 (38.9) | 1855 (52.0) | 449 (73.5) |
| tmax | 2.8 (1.5‐5.0) | 2.0 (1.5‐3.0) | 2.5 (2.0‐5.0) | 2.3 (1.5‐5.0) |
| t½ (h)c | 15.7 (58.3) | 20.5 (39.2) | 22.1 (44.9)d | 8.58 (68.4) |
| CL/F (L/h) | 286 (44.2) | 172 (39.9) | 82.0 (29.5)d | 422 (73.8) |
| Vz/F (L) | 5302 (60.1) | 4668 (40.1) | 2437 (70.5)d | 4105 (37.5) |
| 6‐OH‐CBD | ||||
| Cmax (ng/mL) | 5.78 (70.7) | 7.56 (37.5) | 5.35 (50.5) | 3.19 (63.6) |
| AUC0‐∞ (ng·h/mL)b | 59.6 (14.7)e | 67.6 (16.2)f | 65.8 (39.1)e | 24.8 (71.5)e |
| AUC0‐t (ng·h/mL) | 29.4 (89.5) | 52.3 (35.1) | 51.2 (51.9) | 15.4 (116) |
| tmax | 2.5 (1.0‐5.0) | 1.5 (1.5‐3.0) | 2.0 (1.0‐5.0) | 2.5 (0.7‐6.0) |
| t½ (h)c | 17.5 (15.0)e | 20.8 (20.7)f | 20.3 (17.7)e | 13.2 (66.9)e |
| 7‐OH‐CBD | ||||
| Cmax (ng/mL) | 54.9 (121) | 76.4 (58.1) | 45.5 (45.5) | 41.8 (60.2) |
| AUC0‐∞ (ng·h/mL)b | 331 (95.7) | 612 (42.1)d | 694 (47.6)g | 301(43.8) |
| AUC0‐t (ng·h/mL) | 305 (93.0) | 525 (35.2) | 532 (46.2) | 277 (40.7) |
| tmax | 3.5 (1.5‐5.0) | 2.0 (1.5‐4.0) | 3.5 (2.1‐5.0) | 2.8 (1.5‐6.0) |
| t½ (h)c | 14.8 (18.0) | 15.6 (25.8)d | 21.7 (21.7)g | 13.3 (19.8) |
| 7‐COOH‐CBD | ||||
| Cmax (ng/mL) | 706 (113.3) | 804 (70.6) | 221 (51.1) | 823 (45.6) |
| AUC0‐∞ (ng·h/mL)b | 14075 (114)h | 28273 (6.80)h | NCi | 16239 (46.4)g |
| AUC0‐t (ng·h/mL) | 14105 (102) | 18789 (63.7) | 7226 (45.6) | 14910 (45.8) |
| tmax | 3.5 (2.5‐6.0) | 2.8 (2.4‐5.0) | 4.0 (2.5‐23.2) | 4.5 (2.1‐5.0) |
| t½ (h)c | 21.8 (18.2)h | 22.8 (3.13) | NCi | 19.8 (16.2)g |
6‐OH‐CBD, 6‐hydroxy‐cannabidiol; 7‐COOH‐CBD, 7‐carboxy‐cannabidiol; 7‐OH‐CBD, 7‐hydroxy‐cannabidiol; AUC0‐∞, area under the plasma concentration–time curve from time zero to infinity; AUC0‐t, area under the plasma concentration–time curve from time zero to time t; CBD, cannabidiol; CL/F, oral clearance of drug from plasma; Cmax, maximum measured plasma concentration; CV%, coefficient of variation; N, number of subjects exposed; NC, not calculable; t½, terminal (elimination) half‐life; tmax, time to maximum plasma concentration; PK, pharmacokinetic; Vz/F, apparent volume of distribution.
Except for tmax where median and range are presented, and t½ where arithmetic mean and CV% are presented.
Percent extrapolation ≤30% was required to retain AUC0‐∞; subjects that did not satisfy this criterion were excluded from the analysis.
Percent extrapolation ≤30% and r2 >0.80 was required to retain t½; subjects that did not satisfy these criteria were excluded from the analysis.
n = 5.
n = 4.
n = 3.
n = 6.
n = 2.
n = 0.
Table 4.
CBD, 6‐OH‐CBD, 7‐OH‐CBD, and 7‐COOH‐CBD Comparing PK Parameters Between Hepatic Impairment Groups and the Normal Hepatic Function Group: PK Analysis Set
| Ratio of Geometric Least Squares Means (90%CI) | |||||
|---|---|---|---|---|---|
| Comparison | Cmax (ng/mL) | AUC0‐∞ (ng·h/mL) | AUC0‐t (ng·h/mL) | CL/F (L/h) | tmax a (h) |
| CBD | |||||
| Mild/Normal | 1.57 (0.90 to 2.75) | 1.48 (0.90 to 2.41) | 1.44 (0.86 to 2.41) | 0.68 (0.41 to 1.11) | 0.23 (–0.55 to 1.00) |
| Moderate/Normal | 2.39 (1.37 to 4.18) | 2.45 (1.50 to 4.01) | 2.35 (1.41 to 3.92) | 0.41 (0.25 to 0.67) | –0.52 (–2.03 to 0.02) |
| Severe/Normal | 2.57 (1.41 to 4.70) | 5.15 (2.94 to 9.00) | 4.13 (2.38 to 7.18) | 0.19 (0.11 to 0.34) | 0.00 (–1.50 to 1.00) |
| 6‐OH‐CBD | |||||
| Mild/Normal | 1.81 (1.05 to 3.12) | 1.50 (0.78 to 2.88) | 1.91 (0.95 to 3.84) | … | 0.00 (–1.50 to 1.30) |
| Moderate/Normal | 2.37 (1.38 to 4.08) | 2.59 (1.40 to 4.82) | 3.40 (1.69 to 6.84) | … | –0.99 (–2.53 to 0.70) |
| Severe/Normal | 1.68 (0.93 to 3.02) | 2.16 (1.16 to 4.01) | 3.32 (1.56 to 7.08) | … | –0.47 (–2.03 to 0.98) |
| 7‐OH‐CBD | |||||
| Mild/Normal | 1.31 (0.66 to 2.59) | 1.10 (0.63 to 1.92) | 1.10 (0.64 to 1.89) | … | 0.02 (–0.98 to 1.97) |
| Moderate/Normal | 1.83 (0.92 to 3.62) | 2.12 (1.22 to 3.69) | 1.89 (1.10 to 3.26) | … | –0.76 (–2.90 to 0.03) |
| Severe/Normal | 1.09 (0.52 to 2.27) | 2.28 (1.26 to 4.14) | 1.92 (1.07 to 3.44) | … | 0.23 (–0.98 to 1.97) |
| 7‐COOH‐CBD | |||||
| Mild/Normal | 0.86 (0.44 to 1.68) | 1.02 (0.51 to 2.03) | 0.95 (0.51 to 1.77) | … | –0.02 (–1.52 to 0.98) |
| Moderate/Normal | 0.98 (0.50 to 1.91) | 1.60 (0.78 to 3.27) | 1.26 (0.67 to 2.36) | … | –1.02 (–2.05 to 0.07) |
| Severe/Normal | 0.27 (0.13 to 0.55) | 0.84 (0.33 to 2.14) | 0.48 (0.25 to 0.95) | … | 0.93 (–0.97 to 1.97) |
6‐OH‐CBD, 6‐hydroxy‐cannabidiol; 7‐COOH‐CBD, 7‐carboxy‐cannabidiol; 7‐OH‐CBD, 7‐hydroxy‐cannabidiol; AUC0‐∞, area under the plasma concentration–time curve from time zero to infinity; AUC0‐t, area under the plasma concentration–time curve from time zero to time t; CBD, cannabidiol; CI, confidence interval; CL/F, oral clearance of drug from plasma; Cmax, maximum measured plasma concentration; PK, pharmacokinetic; tmax, time to maximum plasma concentration.
Hodges‐Lehmann estimate (90%CI).
Geometric mean Cmax was 233 ng/mL for mild, 354 ng/mL for moderate, and 381 ng/mL for severe hepatic impairment versus 148 ng/mL for subjects with normal hepatic function (Table 3). Other exposure parameters (AUC0‐t and AUC0‐∞) also showed that total plasma CBD increased with severity of hepatic impairment. Respective geometric mean AUC0‐t and AUC0‐∞ were 648 and 699 ng·h/mL for subjects with mild, 1054 and 1163 ng·h/mL for subjects with moderate, and 1855 and 2439 ng·h/mL for subjects with severe hepatic impairment versus 449 and 474 ng·h/mL for subjects with normal hepatic function. Statistical analysis showed AUC0‐∞ was increased slightly in subjects with mild hepatic impairment compared with those with normal hepatic function (geometric mean ratio [GMR, 1.48; 90%CI 0.90‐2.41]), and significantly increased in subjects with moderate (GMR, 2.45; 90%CI, 1.50‐4.01) or severe (GMR, 5.15; 90%CI, 2.94‐9.00) hepatic impairment relative to subjects with normal hepatic function (Table 4). Exposure to 6‐OH‐CBD and 7‐OH‐CBD also increased in moderate and subjects with severe hepatic impairment, but to a lesser extent than the parent drug (Tables 3 and 4).
7‐COOH‐CBD was the most abundant circulating product in plasma, followed by CBD, 7‐OH‐CBD, then 6‐OH‐CBD (Figure 1). In contrast to CBD, 7‐COOH‐CBD exposure was lowest in subjects with severe hepatic impairment (Table 3).
Arithmetic mean CBD t½ increased by 1.83‐, 2.39‐ and 2.57‐fold in the mild, moderate and severe hepatic impairment groups, respectively, compared with the normal hepatic function group (Table 3).
Apparent clearance of CBD reduced in all hepatic impairment groups relative to subjects with normal hepatic function (normal hepatic function: 422.23 L/h; mild, moderate, and severe hepatic impairment: 285.93, 172.01, and 82.02 L/h, respectively [Table 3]); the statistical analysis revealed an important effect on plasma clearance in moderate and severe hepatic impairment subjects (Table 4). There was no clear trend in apparent volume of distribution relating to hepatic impairment status (range across groups, 2437‐5302 L).
Total subject variability in PK parameters was moderate to high during the trial.
Plasma concentrations of THC were below the LLOQ at all time points for most subjects in all groups. 11‐COOH‐THC was detectable in most subjects in all groups until 12 hours after dosing (data not shown).
Unbound Fraction of CBD
There was a trend to an increase in the unbound fraction of CBD by severity of hepatic impairment (4.88%, 9.42%, and 11.69% of CBD was unbound in the mild, moderate, and severe hepatic impairment groups, respectively, and 6.98% in the normal hepatic function group), and to a lesser extent the metabolites. However, data could only be considered qualitative due to bioanalytical challenges (the process and stability of the free fraction was not supported during bioanalytical method validation), and therefore only total drug (bound and unbound) PK parameters are presented in this paper. Overall, the unbound concentrations tended to reflect the effects of hepatic function that were observed for total drug in plasma.
Safety
A single oral 200‐mg dose of CBD was well tolerated across all groups. All treatment‐emergent AEs were mild in severity (see summary of AEs in Table 5). There were no serious AEs, deaths, or early withdrawals due to AEs. One (12.5%) subject each from the mild and moderate hepatic impairment groups reported ≥1 AE in the trial. Diarrhea was the only AE considered to be treatment related by the investigator and reported by 1 subject. There was no increase in AE frequency or severity with increasing degrees of hepatic impairment.
Table 5.
Adverse Events Experienced by All Subjects per Treatment Group, by MedDRA Preferred Term: Safety Analysis Set
| Mild Hepatic Impairment (N = 8) | Moderate Hepatic Impairment (N = 8) | Severe Hepatic Impairment (N = 6) | Normal Hepatic Function (N = 8) | |||||
|---|---|---|---|---|---|---|---|---|
| SOC MedDRA PT | e | n (%) | e | n (%) | e | n (%) | e | n (%) |
| Total | 4 | 1 (12.5) | 1 | 1 (12.5) | 0 | 0 | 0 | 0 |
| Gastrointestinal disorders | 3 | 1 (12.5) | 0 | 0 | 0 | 0 | 0 | 0 |
| Diarrhea | 3 | 1 (12.5) | 0 | 0 | 0 | 0 | 0 | 0 |
| Investigations | 0 | 0 | 1 | 1 (12.5) | 0 | 0 | 0 | 0 |
| Platelet count low | 0 | 0 | 1 | 1 (12.5) | 0 | 0 | 0 | 0 |
| Nervous system disorders | 1 | 1 (12.5) | 0 | 0 | 0 | 0 | 0 | 0 |
| Dizziness | 1 | 1 (12.5) | 0 | 0 | 0 | 0 | 0 | 0 |
e, number of times the adverse event occurred; MedDRA PT, Medical Dictionary for Regulatory Activities preferred term; N, number of subjects exposed; n, number of subjects that experienced the AE; SOC, system organ class.
No subjects in the normal hepatic function group had a clinically significant laboratory value during the trial. Seven (87.5%) subjects in the mild hepatic impairment group and all (100%) subjects in the moderate and severe hepatic impairment groups had clinically significant hematology and/or biochemistry values during the trial; however, in each case, these values were consistent with the subject's underlying level of hepatic impairment. Only 1 event of low platelet count (59.0 × 109/L) was reported as a mild AE. This AE affected a subject from the moderate hepatic impairment group who had a clinically significant low platelet count at baseline and was considered to be unrelated to CBD by the investigator. This was the only AE ongoing at the end of the trial. There were no clinically significant ECG or new physical examination findings. One subject in the moderate hepatic impairment group had a clinically significant vital sign finding of increased pulse rate of 102 beats per minutes; the investigator considered this change to be a result of the subject's underlying hepatic impairment status. There were no other clinically significant findings and no AEs related to vital signs.
Discussion
This trial is the first to investigate the PK of this oral CBD formulation in subjects with hepatic impairment. CBD is an oral formulation recently approved in the United States for the treatment of seizures associated with Lennox‐Gastaut and Dravet syndromes, which are severe and refractory pediatric epilepsies.1, 2, 3, 4, 5 As patients who receive CBD may have coexisting but rare hepatic morbidities that can lead to drug accumulation, or possibly failure to form an active metabolite,17, 18 the PK of CBD in the current trial population are of clinical interest to manage dosing in these rare cases. In the current trial, demographic and baseline characteristics (outside that of hepatic status) were similar across the 4 subject groups.
CBD and Metabolite PK
CBD was rapidly absorbed in all hepatic‐impaired subject groups (median tmax, 2 to 2.8 hours) with no systematic trend with hepatic insufficiency. Exposure (AUC0‐∞) to total CBD slightly increased in subjects with mild hepatic impairment (GMR, 1.48; 90%CI, 0.90‐2.41), and resulted in a clinically relevant increase in subjects with moderate (GMR, 2.45; 90%CI, 1.50‐4.01) and severe (GMR, 5.15; 90%CI, 2.94‐9.00) hepatic impairment, relative to subjects with normal hepatic function. Apparent clearance was reduced in subjects with moderate and severe hepatic impairment compared with subjects with normal hepatic function.
Exposures to 6‐OH‐CBD and 7‐OH‐CBD were low in contrast to the parent drug but, like CBD, also increased in subjects with moderate and severe hepatic impairment, although to a lesser extent than CBD. Elimination of 6‐OH‐CBD and 7‐OH‐CBD in the severe hepatic impairment group appeared to be different from that of CBD, with secondary absorption peaks present in the plasma concentration–time profiles. However, on inspection of individual subject data, the second absorption peak in the 6‐OH‐CBD and 7‐OH‐CBD profiles corresponded to a delayed tmax in 2 subjects and 1 subject, respectively.
Exposure to 7‐COOH‐CBD was much greater than the parent drug; however, in contrast to CBD, exposure to 7‐COOH‐CBD was lowest in subjects with severe hepatic impairment (compared with the other impairment groups and the normal hepatic function group), likely reflecting a reduced metabolic capacity and altered biotransformation of CBD in subjects with severe hepatic impairment.21
CBD is highly protein bound, and the major human binding protein for CBD has been reported to be mainly albumin.22 Plasma total protein binding of CBD and metabolites was high in this trial (≥88% in all groups). Data for unbound drug was considered qualitative only due to bioanalytical challenges. However, an increase in the free fraction of CBD (and to a lesser extent the metabolites) was observed in subjects with moderate and severe hepatic impairment consistent with total drug in plasma. Free drug was notably higher in severely hepatically impaired subjects compared to mild, moderate, or normal subjects, which could be expected since baseline albumin levels were consistently lower within this cohort of subjects. Although there was an increase in the free fraction of CBD, there was no trend toward an increase in the apparent volume of distribution.
The large total subject variability in this trial is common with cannabinoids, and similar levels of variability have been previously reported in other trials with cannabinoids.23, 24
Although the PK of CBD and its metabolites have been explored following a single administration in this trial, exposures have been reported following multiple bidaily administration; results show there is no significant time dependency in the PK other than accumulation approximating to 3‐fold for the parent drug CBD.25 It is anticipated this single‐dose assessment would predict the extent of change in drug clearances observed following multiple administration.
Δ9‐Tetrahydrocannabinol PK
Extracts from the plants used to formulate CBD also contain <0.1% (w/w) THC. Due to the psychoactive and potentially addictive nature of THC, plasma concentrations of THC and its metabolites were also monitored in this trial. Exposure to THC was low throughout the trial; plasma concentrations were below the LLOQ in most subjects at most time points. THC therefore has no significance for dose adjustment in special populations with hepatic impairment.
Safety
A single dose of 200 mg was well tolerated across the groups and no safety concerns were observed, with only mild AEs reported by 2 subjects during the trial. There were no severe or serious AEs, discontinuations due to AEs, or any clinically significant ECG or new physical examination findings. Clinically significant hematology, biochemistry, and/or vital sign values were reported in subjects with hepatic impairment only and were consistent with the subject's underlying level of hepatic impairment. All but 1 AE of low platelet count recovered during the trial. Although there was a clear relationship between CBD PKs and level of hepatic function, increased CBD concentrations within the moderate and severe hepatic impairment groups were not associated with an increased incidence or severity of AEs. There were no AEs in the severe hepatic impairment group.
Dose Adjustments in Patients With Moderate and Severe Hepatic Impairment
Dose reduction is recommended in patients with moderate (2‐fold) or severe (5‐fold) hepatic impairment. It may be necessary also to have slower dose titration in patients with moderate or severe hepatic impairment than in patients without hepatic impairment. No dose adjustment is required in patients with mild hepatic impairment.5
Conclusion
Dose reduction is recommended in patients with moderate or severe hepatic impairment due to increased exposure to CBD compared with subjects with normal hepatic function. Severe hepatic impairment appears to predominantly diminish the biotransformation of CBD to its major carboxylated metabolite (7‐COOH‐CBD). The 200‐mg CBD dose used in this trial was found to be safe and well tolerated in all subjects. Increased CBD, 6‐OH‐CBD, and 7‐OH‐CBD concentrations within the moderate and severe hepatic impairment groups (compared with the normal hepatic function group) were not associated with an increased incidence or severity of AEs. There were no serious or severe AEs in this trial, and no new safety concerns were identified in this single‐dose trial.
Acknowledgments
The authors would like to thank the volunteers who took part in the trial, as well as the staff that assisted with the trial at each site. The authors would also like to thank Dr. Linda White for her early review of the manuscript.
Funding
This trial was sponsored by GW Research Ltd.
Declaration of Conflicting Interests
LT, JC, GM, and BT are employees of GW Research Ltd. LT, JC, GM, and BT own shares in GW Pharmaceuticals plc. This trial was sponsored by GW Research Ltd.
Data Accessibility
The sponsor is adhering to current US and EU requirements and will not make individual deidentified participant data available; however, the protocol and statistical analysis plan will be made available upon request to corresponding author.
References
- 1. Devinsky O, Cross JH, Laux L, et al. Cannabidiol in Dravet Syndrome Study Group: trial of cannabidiol for drug‐resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376(21):2011‐2020. [DOI] [PubMed] [Google Scholar]
- 2. Devinsky O, Patel AD, Thiele EA, et al. Randomized, dose‐ranging safety trial of cannabidiol in Dravet syndrome. Neurology. 2018;90(14):e1204‐e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Devinsky O, Patel AD, Cross JH, et al. Effect of cannabidiol on drop seizures in the Lennox‐Gastaut syndrome. N Engl J Med. 2018;378(20):1888‐1897. [DOI] [PubMed] [Google Scholar]
- 4. Thiele EA, Marsh ED, French JA, et al. Cannabidiol in patients with seizures associated with Lennox‐Gastaut syndrome (GWPCARE4): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet 2018;391(10125):1085‐1096. [DOI] [PubMed] [Google Scholar]
- 5. EPIDIOLEX USPI . Highlights of prescribing information: EPIDIOLEX® (cannabidiol) oral solution. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210365lbl.pdf. Accessed September 5, 2018.
- 6. Ibeas Bih C, Chen T, Nunn AV, Bazelot M, Dallas M, Whalley BJ. Molecular targets of cannabidiol in neurological disorders. Neurotherapeutics. 2015;12(4):699‐730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Henstridge CM, Balenga NA, Kargl J, et al. Minireview: recent developments in the physiology and pathology of the lysophosphatidylinositol‐sensitive receptor GPR55. Mol Endocrinol. 2011;25(11):1835‐1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sylantyev S, Jensen TP, Ross RA, Rusakov DA. Cannabinoid‐ and lysophosphatidylinositol‐sensitive receptor GPR55 boosts neurotransmitter release at central synapses. Proc Natl Acad Sci U S A. 2013;110(13):5193‐5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McPartland JM, Duncan M, Di Marzo V, Pertwee RG. Are cannabidiol and Δ(9)‐tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br J Pharmacol. 2015;172(3):737‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wall ME, Brine DR, Perez‐Reyes M. Metabolism of cannabinoids in man In: Braude MC, Szara S, eds. The Pharmacology of Marihuana. New York, NY: Raven Press; 1976:93‐113. [Google Scholar]
- 11. Watanabe K, Fujinami M, Yamaori S, Yamamoto I. Possible involvement of Cyp3a enzymes in the metabolism of tetrahydrocannabinols by mouse brain microsomes. Forensic Toxicol. 2011;29:56‐60. [Google Scholar]
- 12. Huestis MA. Human cannabinoid pharmacokinetics. Chem Biodivers. 2007;4:1770‐1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ujvary I, Hanus L. Human metabolites of cannabidiol: a review on their formation, biological activity, and relevance in therapy. Cannabis Cannabinoid Res. 2016;1(1):90‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang R, Yamaori S, Takeda S, Yamamoto I, Watanabe K. Identification of cytochrome P450 enzymes responsible for metabolism of cannabidiol by human liver microsomes. Life Sci. 2011;89(5‐6):165‐170. [DOI] [PubMed] [Google Scholar]
- 15. Mazur A, Lichti CF, Prather PL et al. Characterization of human hepatic and extrahepatic UDP‐glucuronosyltransferase enzymes involved in the metabolism of classic cannabinoids. Drug Metab Dispos. 2009;37(7):1496‐1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Whalley B, Stott C, Gray R, Jones NA. Human metabolite of cannabidiol, 7‐hydroxy cannabidiol, but not 7‐carboxy cannabidiol, is anticonvulsant in the maximal electroshock seizure threshold (mEST) test in mouse. Abstract 1.435 American Epilepsy Society Meeting; 2017.
- 17. Food and Drug Administration. Guidance for Industry : Pharmacokinetics in patients with impaired hepatic function: study design, data analysis, and impact on dosing and labeling; 2003.
- 18. Committee for Medicinal Products for Human Use (CHMP). Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with impaired hepatic function. CPMP/EWP/2339/02, EMEA; 2005.
- 19. World Medical Association Declaration of Helsinki . Ethical principles for medical research involving human subjects Edinburgh, Scotland; October 2000, including the footnote of October 2002.
- 20. ICH Harmonised Tripartite Guideline : Guideline for good clinical practice E6(R1); June 1996. [PubMed]
- 21. Tarantino G, Di Minno MN, Capone D. Drug‐induced liver injury: is it somehow foreseeable? World J Gastroenterol. 2009;15(23):2817‐2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Elmes MW, Kaczocha M, Berger WT et al. Fatty acid‐binding proteins (FABPs) are intracellular carriers for Δ9‐tetrahydrocannabinol (THC) and cannabidiol (CBD). J Biol Chem. 2015;290(14):8711‐8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stott CG, White L, Wright S, Wilbraham D, Guy GW. A phase I study to assess the effect of food on the single dose bioavailability of the THC/CBD oromucosal spray. Eur J Clin Pharmacol. 2013;69(4):825‐834. [DOI] [PubMed] [Google Scholar]
- 24. Wilsey BL, Deutsch R, Samara E, et al. A preliminary evaluation of the relationship of cannabinoid blood concentrations with the analgesic response to vaporized cannabis. J Pain Res. 2016;9:587‐598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Taylor L, Gidal B, Blakey G, Tayo B, Morrison G. A phase I, randomized, double‐blind, placebo‐controlled, single ascending dose, multiple dose, and food effect trial of the safety, tolerability and pharmacokinetics of highly purified cannabidiol in healthy subjects. CNS Drugs. 2018;32(11):1053‐1067. [DOI] [PMC free article] [PubMed] [Google Scholar]