

Summary
β-glucans are recognized by the innate immune system. This recognition plays important roles in host defense, and presents specific opportunities for clinical modulation of the host immune response. Neutrophils, macrophages, and dendritic cells among others express several receptors capable of recognizing β-glucan in its various forms. This review explores what is currently known about β-glucan recognition and how this recognition stimulates immune responses. Special emphasis is placed on Dectin-1, since we know the most about how this key β-glucan receptor translates recognition into intracellular signaling, stimulates cellular responses, and participates in orchestrating the adaptive immune response.
Keywords: Inflammation, Pattern recognition receptors, Fungal infections, macrophages, phagocytosis, signal transduction
Glucans and the immune system
The immunostimulatory properties of glucans have been recognized directly for decades and indirectly, as an active component in mushrooms, for centuries. As such, beneficial consequences claimed to be associated with exposure to purified glucans are diverse. These range from the preposterous (as a cure for baldness) to the scientifically sound (the anti-tumor effects of glucan are supported by modern clinical investigations). Modern molecular immunology is now providing rigorous mechanistic explanations for how humans recognize glucans and how this recognition influences the behavior of the immune system. The purpose of this review is to outline what is currently known about glucan recognition by immune cells and to highlight future directions.
“Glucan” actually refers to a diverse class of glucose polymers that includes cellulose (β-1,4-glucan). These glucans can be short or long, branched or unbranched, α or β isomers, and soluble or particulate. The literature on immune responses to glucans can be quite confusing as what is observed for one preparation of glucan is often inappropriately extrapolated to all glucans. When discussing the immune-modulator functions of glucans, we are most commonly considering β-1,3-glucan purified from fungal cell walls (Figure 1). Fungal β-glucans have varying numbers of 1,6 branches and the purified molecules may range from glucose dimers to large insoluble particles. Common sources of experimentally used β-1,3-glucans include yeast (typically S. cerevisiae), mushroom (Sclerotium glucanicum and others), bacteria (curdlan from Alcaligenes faecalis), and seaweed (laminarin from Laminaria digitata). β-glucans from all of these sources have been demonstrated variously to have pro- or anti-inflammatory effects on immune cells. The consequences of β-glucan recognition likely depend on the cell types involved and the receptor(s) engaged. Several receptors for β-glucans have been described and are discussed below. Of these, Dectin-1 has probably been characterized with the most molecular detail, and this receptor is thus the focus of much of the review. However, other receptors have been described, and their contributions to regulating immune responses should not be underestimated.
Figure 1:
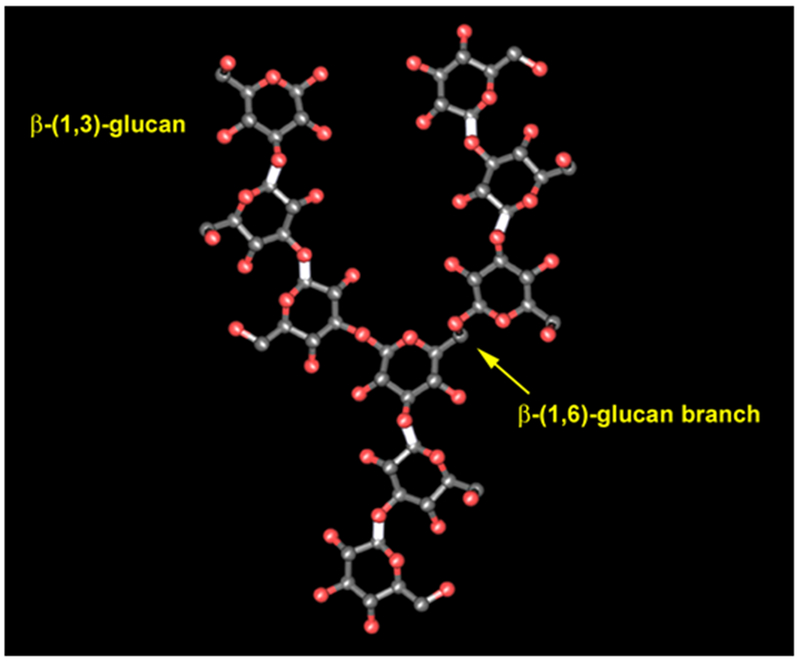
The structure of β-glucan. Immunostimulatory microbial β-glucan is most commonly β-(1,3)-glucan with various degrees of β-(1,6)-glucan branching.
β-glucan receptors: CR3 and others
Complement receptor 3 (CR3) is an integrin dimer consisting of αMβ2 (CD11b/CD18) and is expressed widely by myeloid cells including monocytes, macrophages, dendritic cells, neutrophils and NK cells (1). CR3 binds to complement component iC3b and is the main receptor responsible for phagocytosis of complement-opsonized particles. CR3 has an extremely diverse range of ligands in addition to complement including, fibrinogen, coagulation Factor X and ICAM-1 as well as a variety of fungal, parasite, and bacterial cell surfaces. The broad range of ligands described for CR3 has lead to the suggestion that, in addition to its specific ligand binding activities, CR3 may help coordinate clustering of other low affinity receptors and their attachment to the cytoskeleton (1). That said, the αM chain of CR3 has high affinity (5 × 10−8 M) β-glucan binding activity that maps to a region that is distinct from the site used to bind to iC3b (2, 3). This “lectin domain” area does not contain any known lectin-like sequences, and the mechanism of binding has not been fully defined. While the site seems to have the highest affinity for β-1,3-glucans it also binds to other carbohydrates. The receptor facilitates binding to a variety of microbes including bacteria that do not contain β-glucans, suggesting specificity for other microbial carbohydrates. Also, the lectin domain is implicated in CR3 association with other endogenous surface receptors such as uPAR, indicating that it binds to certain endogenous carbohydrates. The association of CR3 with uPAR can be blocked with soluble N-acetyl-D-glucosamine and α-D-mannoside, sugars that can also block β-glucan binding (4, 5).
That the lectin domain may facilitate interactions with other membrane components is interesting in the light of a recent study on the membrane glycosphingolipid lactosylceramide. In 1998 Zimmerman et al. reported purifying a component of human leukocytes that would bind to soluble β-1,3-glucan and identified it as lactosylceramide (6). The binding was dependent on the presence of a terminal galactose, although beyond this the mechanism of the association was unclear. The use of blocking anti-lactosylceramide antibodies and excess soluble lactosylceramide has implicated the glycosphingolipid in binding to β-glucan and to β-glucan-induced cytokine production and respiratory burst activity in human polymorphonuclear leukocytes and rat alveolar epithelial cells (7–9). More recently, Nakayama et al. have reported that the src family kinase Lyn is concentrated in lactosylceramide-enriched membrane lipid rafts and that this association is necessary for β-glucan signaling via CR3 (10). These investigators reported that antibodies to either CR3 or lactosylceramide blocked β-glucan particle binding to human neutrophils and that CR3-mediated signaling required β-glucan-induced interaction of CR3 with lactosylceramide-associated Lyn kinase. These data suggest that claims that both CR3 and lactosylceramide are receptors for β-glucan may be different views of the same process.
The idea that CR3 and lactosylceramide are somehow obligate partners in detecting β-glucan does not yet line up in two crucial ways. First, Li et al. report that in order for CR3 to trigger Syk-based signaling (typically downstream of src family kinases) β-glucan must first be internalized and processed to a ≈25 kDa fragment that is preferentially bound by CR3. If this is true, it is difficult to understand how this squares with a role for CR3 in particle binding. Second, a requirement for collaboration between CR3 and lactosyceramide in β-glucan recognition and signaling is complicated by the observation that alveolar epithelial cells are reported to utilize lactosylceramide in responding to β-glucans, although these cells reportedly do not express CR3 (8). It is possible that lactosylceramide may collaborate with other proteins, perhaps other integrins, to mediate this effect.
The biological effects of β-glucan binding to CR3 have been primarily studied in neutrophils. Here binding seems to prime the receptor for enhanced activity. Neutrophils bind to complement opsonized tumor cells, but this is not sufficient to trigger target cell killing (4). Exposure of neutrophils to soluble β-glucans primes them for CR3-mediated cytotoxicity. Li et al. recently reported that this activity requires Syk (11), a sequence that would be consistent with the requirement for lactosylceramide-associated Lyn (a Src family kinase) discussed above. In vivo studies support the efficacy of this approach to enhancing tumor killing both alone and in conjunction with anti-tumor antibody therapies (11, 12).
The contribution of CR3 to phagocytosis of β-glucan-containing particles is more controversial. We can look at this two ways. First, does β-glucan binding to CR3 prime phagocytosis of complement-opsonized particles? CR3 on the surface of phagocytes binds to complement-opsonized particles, but this is generally not sufficient to trigger phagocytosis or respiratory burst activity; a second signal such as adhesion to extracellular matrix or artificial activation by phorbol ester is additionally required (13, 14). There are, as yet, no data to indicate whether the priming effects of β-glucan noted above for tumor cell killing extend to priming phagocytosis of complement-opsonized particles, although this is likely. Second, does CR3 binding to β-glucan contribute to phagocytosis of β-glucan particles in the absence of complement. Data are conflicting as to the contribution of CR3 to binding and phagocytosis of β-glucan particles. Transfection of CHO cells with CR3 has been reported to be sufficient to confer on the cells the ability to phagocytose zymosan via a mechanism that is modestly suppressed in the presence of soluble β-glucan (15). Similarly, phagocytosis of zymosan by mouse neutrophils is modestly suppressed by CR3 antibodies or genetic deletion of CR3 (4). We and others have specifically observed that CR3 contributes little to macrophage responses to zymosan (16, 17). Together with data implicating Dectin-1 in phagocytosis of β-glucan particles (discussed below), the consensus is that CR3 contributes very little to phagocytosis of pure β-glucan particles.
An intriguing recent study has added CD5 to our list of immune receptors that recognize β-glucan. CD5 is a scavenger receptor family member that is expressed primarily on T cells and B-1 cells where it associates with antigen receptors. The intracellular tail of CD5 contains an immunoreceptor tyrosine-based inhibitory motif (ITIM). The tyrosine in this motif is constitutively phosphorylated and associated with SHP-1 in B-1 cells suggesting that CD5 plays an inhibitory role in B cell receptor signaling (18). Indeed, B cell receptor signaling is enhanced in B-1 cells from CD5-deficient mice (19). However, ligands for CD5 have not been described. The extracellular portion of CD5 consists of three repeats of a scavenger receptor cysteine-rich (SRCR) domain. SRCRs in other scavenger receptors such as the macrophage scavenger receptor MARCO have been implicated in microbial recognition. Vera et al. recently observed that the extracellular domain of human CD5 binds to β-glucan with an affinity of 3.7 × 10−9 M and that each of the three SRCRs has binding activity (20). Further, human CD5 binds to a variety of yeast cell walls and can aggregate them. Contrary to the apparent inhibitory role of CD5 in B cell signaling, transfection of HEK293 cells with CD5 facilitated zymosan-induced IL-8 secretion, suggesting an activating pro-inflammatory role for the receptor. Similarly, CD5 expression in Jurkat T cells supported zymosan-induced ERK and p38 MAP kinase activation. It is not yet clear whether mouse CD5 has similar β-glucan binding and signaling activity, nor has the exact role of CD5 in T cell and B cell responses to fungi yet been explored.
Dectin-1
Dectin-1 is a type II membrane receptor that belongs to the C-type lectin family of receptors, containing an extracellular C-terminal C-type lectin domain, a short stalk region, a single transmembrane domain, and a short 40 amino acid amino-terminal intracellular tail (21). The receptor is expressed primarily by cells of myeloid origin, including macrophages, dendritic cells and neutrophils in mice. In addition, Dectin-1 has been detected by flow cytometry on a subset of B and T lymphocytes though its function on these cells is undetermined (22). Dectin-1 signals contribute to a variety of macrophage, DC and neutrophil responses, including phagocytosis, oxidative burst, neutrophil degranulation, fungal killing, and the production of inflammatory lipid mediators, cytokines and chemokines that recruit and coordinate the activation of other immune cells. Dectin-1 knockout mouse studies with live pathogenic fungi (Candida albicans, Pneumocystis carinii, and Aspergillus fumigatus) have recently confirmed much of the previous in vitro work with zymosan, curdlan and other β-glucan-containing particles that indicated a central role for Dectin-1 in β-glucan recognition and combating fungal infection (23–25).
Originally cloned from mouse dendritic cells (21), two isoforms of Dectin-1 have been identified in mouse, a full length A isoform and a “stalkless” B isoform, with a truncated extracellular stalk region (26) (Figure 2). While much of the work regarding the recognition of fungal pathogens has focused on the recognition of β-glucans by Dectin-1 in the mouse system, the human version of Dectin-1 has also been cloned and studied in vitro. Human Dectin-1 shares 60% sequence identity and 71% sequence homology with the mouse homologue (27). However, 8 isoforms of Dectin-1 have been cloned from human tissues, resulting from alternate splicing of the mRNA (26). In spite of the large number of mRNA transcripts identified in humans, both humans and mice tend to preferentially express the B or stalkless isoform of Dectin-1. In addition, the extracellular domain in mice is decorated by 2 N-linked glycosylations, whereas in humans there is only one glycosylation, located in the stalk region which is lost in the B isoform (26) (Figure 2).
Figure 2:
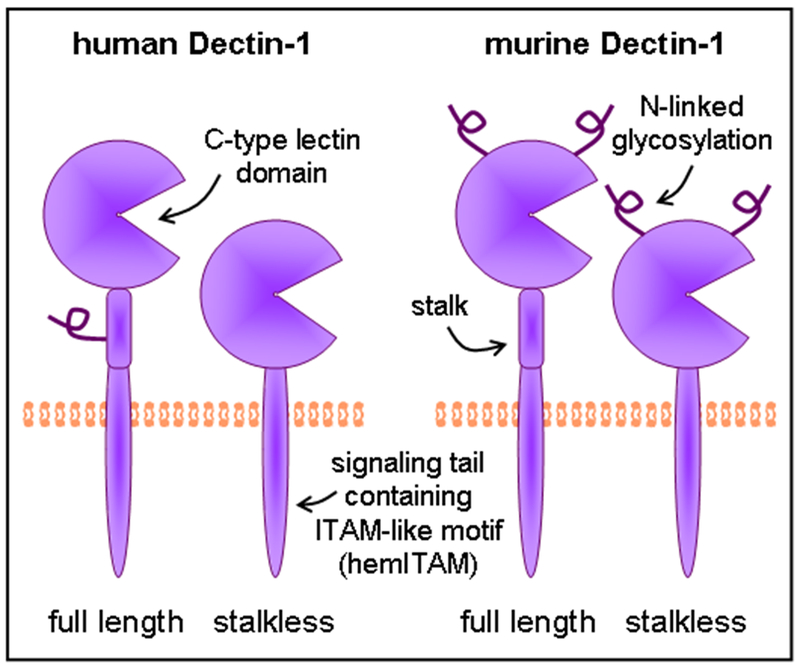
The β-glucan receptor Dectin-1. Dectin-1 consists of an extracellular C-type lectin domain for β-(1,3)-glucan detection, linked via a transmembrane domain to an intracellular signaling tail. A stalk region linking the C-type lectin domain to the transmembrane domain is present in some isoforms. The stalk region of human Dectin-1 (full length only) and the C-type lectin domain of murine Dectin-1 are glycosylated.
While not completely elucidated, indications from in vitro experiments are that both human and mouse stalkless forms of Dectin-1 demonstrate diminished binding to zymosan at low temperatures compared to the full length isoform (28). In a coccidioidomycosis model comparing the C57BL/6 and DBA/2 mice, which preferentially express the stalkless and full length forms of Dectin-1 respectively, the increased susceptibility of the C567BL/6 mice seems to be correlated with expression of the stalkless form of Dectin-1, although how this might work is not clear (29). Very little information exists regarding isoforms C-H in humans, or whether they actually generate functional proteins. Some of the additional isoforms might conceivably generate soluble forms of the receptor, although none have yet been detected, and as a type II transmembrane protein, it is unclear how secretion would occur. Work on the related C-type lectin, DC-SIGN, has lead to similar observations; putative soluble isoforms of DC-SIGN localize to the cytosol, and secretion has not been detected (30).
The β-glucan binding activity of Dectin-1 has been extensively evaluated. Depending on the source of the glucan, Adams et al. determined that Dectin-1 binds the carbohydrate polymers with affinities ranging from very low (3 × 10−3 M) to very high (2 × 10−12 M) (31). The wide range of affinities appears to be due to the differing sizes and numbers of branches in β-glucans from various sources. Structurally, the single C-type lectin domain of Dectin-1 is highly similar to other C-type lectin domains, although β-glucan binding is not dependent on calcium. All C-type lectin domains are held together by the formation of three disulfide bonds between six highly conserved essential cysteine residues. In addition to this structural feature, substitution studies by Adachi et al. showed an essential role for Try 221 and His 223 in the recognition of β-glucans by Dectin-1 (32). Recent work from our own lab has demonstrated an essential role for the Iso 222 residue in proper folding of the lectin domain of the receptor (unpublished data). The crystal structure of the lectin domain suggested that while individual Dectin-1 molecules can bind β-glucans, cooperative binding in dimers may be more efficient (33).
In addition to β-glucan recognition, studies on human Dectin-1 have also indicated that the receptor may function as a T cell costimulatory molecule (21). While the significance of the observations are unclear, HeLa cells transfected with Dectin-1 were reported to be able to enhance anti-CD3-activated expression of CD25, CD69, and CD40 by T cells as well as proliferation and production of IFN-γ (34). The soluble β-glucan laminarin does not block T cell binding, suggesting that a region independent of the β-glucan binding site is important for interaction with T cells (26). It seems to be becoming the rule rather than the exception that lectin receptors bind to endogenous as well as foreign carbohydrates, and these data suggest that this may also be true for Dectin-1. However, so far no endogenous ligands have been identified.
Fungal morphology as a determinant of Dectin-1 recognition
Fungal morphology is major determinant in the detection of fungal cell wall components by host cells. β-glucan exposure at the surface varies with fungal morphotype, and consequently different forms of the same fungus can induce dramatically different responses. For example, β-glucan in the cell wall of C. albicans is masked by a thick layer of mannoprotein which restricts its detection by Dectin-1 (Figure 3A). The ability to switch between different forms is a key virulence strategy, permitting C. albicans to evade host immune responses (35–37). Indeed, C. albicans mutants that lack the ability to switch from yeast to filamentous growth are avirulent (36). We observed that C. albicans yeast trigger Dectin-1 responses by macrophages, while filaments do not (38, 39). Consistent with this, we demonstrated using the extracellular portion of Dectin-1 (soluble Dectin-1) that β-glucan is exposed at discrete patches on the surface of C. albicans yeast (38). This staining, which was blocked by pre-treatment of yeast with a β-glucanase enzyme, corresponded to bud and birth scars, areas of altered cell wall structure resulting from separation of mother and daughter yeast cells. Hence, it appears that β-glucan, which is normally buried under the outer mannan layer of the yeast cell wall, is exposed upon separation of the budding yeast cells and becomes accessible to Dectin-1. In contrast, soluble Dectin-1 failed to detect filamentous C. albicans, including filaments isolated from mouse kidneys 24 hours after intraperitoneal infection. This form of the fungus grows but daughter cells do not separate from each other, bud and birth scars are not formed, and hence the β-glucan remains concealed beneath the surface. Thus Dectin-1 plays a more important role in the detection of and inflammatory response to C. albicans yeast than filaments.
Figure 3:
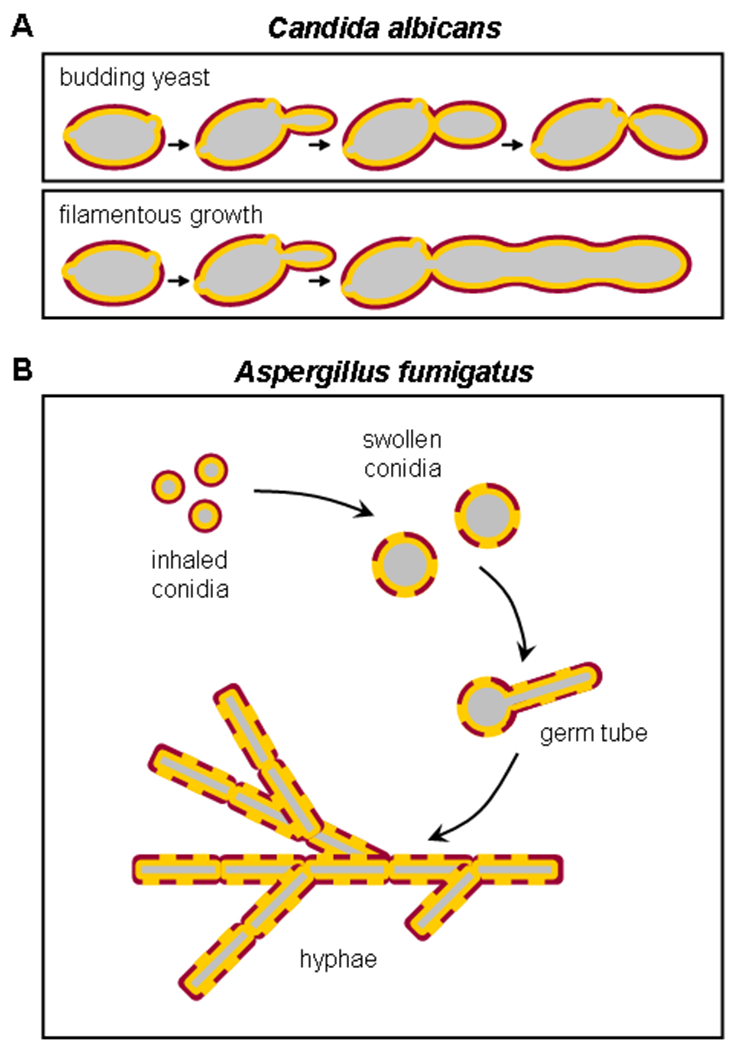
Fungal morphology can influence the ability of Dectin-1 to recognize β-glucan in cell walls. A) Candida albicans can grow in a budding yeast form, or a filamentous form. During budding growth, cell separation results in the formation of bud and birth scars in the cell wall which expose the β-glucan core to recognition by the receptor. During filamentous growth, the core β-glucan layer is obscured by the outer layer (mostly mannan). B) Aspergillus fumigatus also exists in several morphological stages, but the sequence of β-glucan exposure is quite different. Aspergillus spores have hard waxy coating that covers β-glucan, and the spores are thus not recognized by Dectin-1. When activated, the spores swell, germinate, and grow as long branched hyphae. β-glucan in the cell wall is exposed once the spores begin to swell and is readily accessible to Dectin-1 in the filament cell walls.
Wheeler et al. recently reported that C. albicans filaments extracted from the kidneys of infected mice exhibit a higher degree of β-glucan exposure 5-7 days post-infection (40), although it is not clear whether this represents deliberate structural reorganization by the fungus or is a consequence of its exposure to host factors. However, the same group also demonstrated that treatment of C. albicans in vitro, or treatment of C. albicans-infected mice, with subinhibitory doses of the anti-fungal drug caspofungin, which targets cell wall biosynthetic pathways, disrupts the fungal cell wall architecture to expose β-glucan and is hence capable of “unmasking” filaments (40, 41). Furthermore, these investigators have used an anti-β-glucan antibody to screen a genome-wide library of approximately 4,800 S. cerevisiae mutants for increased β-glucan exposure to identify genes involved in β-glucan masking. Of the 79 mutants exhibiting increased β-glucan exposure, 68 (86%) also displayed increased binding to soluble Dectin-1 (41).
Dectin-1 detection of Aspergillus fumigatus β-glucan is also dependent on morphotype (42–44) (Figure 3B). Aspergillus conidia (resting spores) are ubiquitous in the environment, inhaled frequently and rapidly phagocytosed and cleared by alveolar macrophages and recruited neutrophils of an immunocompetent host. However, the failure of immunocompromised individuals to clear germinating conidia can result in invasive pneumonia and disseminated infection (45). Following inhalation, the conidia swell and, if they are not cleared, produce germ tubes that eventually extend to form filamentous hyphae. Exposed β-glucan was detected using an anti-β-glucan antibody on the surface of swollen conidia but not on resting spores, and soluble Dectin-1 stained swollen but not resting Aspergillus conidia (42–44). Consistent with this, germinating but not resting spores induced neutrophil recruitment to the airways and cytokine and chemokine production by alveolar macrophages of infected mice. Unlike C. albicans, β-glucan was also exposed on the surface of Aspergillus germ tubes and hyphae, and therefore detected by Dectin-1. The resting conidia of an A. fumigatus mutant, pksP, which has reduced virulence and trigger an elevated oxidative burst (46, 47), have a smooth surface with high levels of exposed β-glucan (48–50). Luther et al. reported that while wild type resting conidia are poorly phagocytosed, pksP mutant resting conidia are internalized as efficiently as swollen conidia (49). Furthermore, treatment of A. fumigatus hyphae with caspofungin (or other echinocandins) increased β-glucan exposure and Dectin-1-mediated inflammatory responses (51, 52). Ultimately, experiments with Dectin-1-deficient mice demonstrate that Dectin-1 is crucial for control of airway infection with Aspergillus; Dectin-1−/− succumb to lethal airway inflammation after infection, although the infection largely remains restricted to the lung (25). This suggests that Dectin-1 is crucial for local antimicrobial host defense in the lung, but that other factors conspire to prevent dissemination of the fungus.
α-1,3-glucans have recently been shown to play a role in the pathogenicity of the respiratory fungus Histoplasma capsulatum by shielding β-glucan from detection by Dectin-1 (53). Upon inhalation, Histoplasma conidia germinate in response to mammalian body temperature to produce yeast, a process which is absolutely required for pathogenicity (54, 55). Rappleye et al. showed that upon incubation of conidia at 37ºC, which induces the switch to yeast growth, Histoplasma synthesize α-1,3-glucan that forms a layer at the exterior surface of the cell wall, concealing β-glucan beneath it (Rappleye 1366). Wild type yeasts therefore failed to bind to Dectin-1-expressing 3T3 cells. In contrast, α-1,3-glucan synthase mutant yeasts, which can’t make α-1,3-glucan, were readily detected by Dectin-1.
Collectively these studies have identified β-glucan exposure as a fungal Achilles’ heel, which is targeted effectively by a subset of anti-fungal drugs, and demonstrated that β-glucan masking is an important virulence strategy utilized by pathogenic fungi to evade host detection.
Dectin-1 signaling mechanisms
The intracellular tail of Dectin-1 contains a sequence resembling an immunoreceptor tyrosine-based activation motif (ITAM). ITAMs, which are responsible for transducing signals downstream of the T cell and B cell antigen receptors (TCR and BCR) and the Fc receptors (FcR), typically contain two tyrosines in the context of YxxL/I motifs (YxxL/Ix6-12YxxL/I), which are phosphorylated by Src family kinases to initiate signaling (56). Phosphorylation of these dual ITAM tyrosines permits recruitment of Syk family kinases (Zap70 and Syk) via binding of their dual SH2 domains to the phosphorylated tyrosines. The Dectin-1 tail contains two appropriately spaced tyrosines. However, while the membrane-proximal tyrosine is located in a YxxL motif, the membrane-distal tyrosine is found in a motif containing an additional amino acid (YHPDL in human, YHSHI in mouse). Mutation studies indicated that the membrane-proximal tyrosine is required while the membrane-distal tyrosine is dispensable for Dectin-1 signaling (57–59), and hence the Dectin-1 ITAM has been described as a “hemITAM”.
Despite its unconventional ITAM sequence, Dectin-1 signals are transduced in a similar manner to conventional ITAM signals. Dectin-1 ligation by β-glucan particles results in phosphorylation of the membrane proximal tyrosine and activation of Src and Syk family kinases in both macrophages and DC (57–59). In contrast to Src family kinases which have a single SH2 domain and can be recruited by a single phosphorylated tyrosine, Syk (or Zap70 in T cells) typically requires phosphorylation of both ITAM tyrosines for binding of its dual SH2 domains. Thus it was initially unclear how Syk could be recruited to the monophosphorylated hemITAM of Dectin-1. Two models were proposed: either one phosphorylated tyrosine is sufficient for recruiting Syk to Dectin-1, or Syk could be recruited by bridging two monophosphorylated Dectin-1 molecules. The former hypothesis seemed unlikely given the previously demonstrated requirement for both SH2 domains for Syk interaction with other receptors, and indeed mutation of each SH2 domain confirmed that both SH2 domains are required for recruitment to Dectin-1 (60). Hence it appears that Syk binds two phosphorylated tyrosines on adjacent clustered Dectin-1 receptors (Figure 4).
Figure 4:
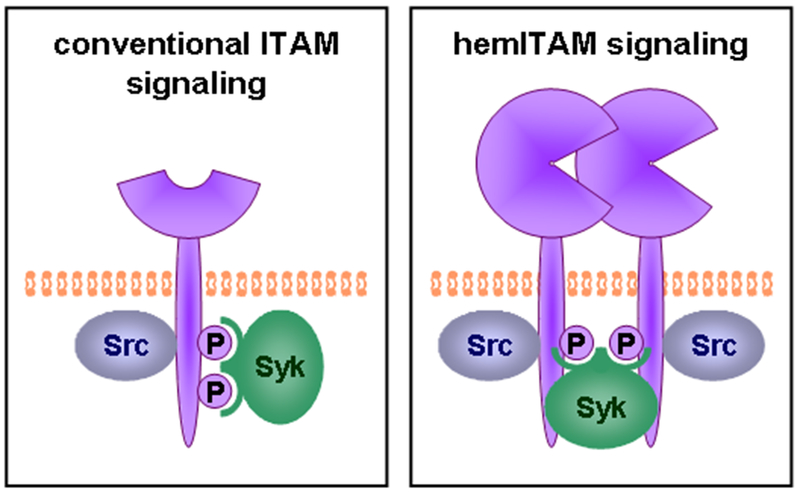
ITAM-like signaling by Dectin-1. The conventional model of ITAM signaling requires phosphorylation (by Src family kinases) of two appropriately-spaced tyrosines on a receptor tail. These phospho-tyrosines serve as binding sites for the two SH2 domains of Syk. The cytoplasmic tail of Dectin-1 requires only one phospho-tyrosine to signal via Syk. The “hemITAM” model proposes that the two SH2 domains of Syk can bind to two phospho-tyrosines on appropriately clustered Dectin-1 molecules.
Our studies indicate that an additional acidic motif upstream of the membrane-proximal tyrosine (DEDGYxxL) is important for Dectin-1 signaling (59). Another C-type lectin, CLEC-2 (CLEC1B), which is expressed by platelets and detects the snake venom toxin rhodocytin, has an identical DEDGYxxL hemITAM and signals in a similar manner (60). It is not yet clear whether this motif stabilizes the Syk interaction with Dectin-1 homodimers or performs another role in Dectin-1 signaling.
Phospholipase C (PLC) is a key mediator of ITAM signaling downstream of Src and Syk kinases. PLCγ1 in T cells and PLCγ2 in B cells are responsible for the induction of calcium fluxes and protein kinase C activation upon antigen receptor (TCR and BCR) activation. Exposure of myeloid cells to fungal particles induces calcium fluxes (61, 62), and Xu et al. recently reported that PLCγ2, and not PLCγ1, is responsible for Dectin-1-mediated calcium flux and activation of other downstream signaling pathways (63).
Another feature of ITAM signaling by the lymphocyte antigen receptors is activation of NF-κB transcription factors by the adaptor protein CARMA1 and its interacting partners Bcl10 and Malt1 (64, 65). Recent studies have indicated that CARMA1 is not involved in Dectin-1 signaling, but rather that a related adaptor, CARD9, is a key transducer of Dectin-1 signals (66–69). Gross et al. demonstrated that CARD9-deficient mice are highly susceptible to C. albicans infection (67). Cytokine production by CARD9-deficient bmM and bmDC in response to zymosan and C. albicans was impaired, and zymosan-induced NF-κB activation was reduced. bmDC from Bcl10 and Malt1 knockout mice also exhibited defective cytokine production in response to zymosan, indicating that CARD9/Bcl10/Malt1 complexes perform an analogous role downstream of Dectin-1 in myeloid cells to CARMA1/Bcl10/Malt1 complexes in lymphocyte antigen receptor signaling (see also below). The use of CARD9, and not CARMA1, by Dectin-1 appears to be attributable to cell type rather than the unusual ITAM sequence, since myeloid cells use CARD9 for cytokine production downstream of a variety of receptors that contain conventional ITAMs, including FcR, while FcR and other ITAM-containing receptors signal via CARMA1 in NK cells as well as T and B lymphocytes (68, 70, 71). Thus, there are many parallels between Dectin-1 and lymphocyte antigen receptor signaling, and despite its unusual ITAM sequence, Dectin-1 appears to transduce classical ITAM signals.
Dectin-1 activated immune functions
Phagocytosis
Internalization of fungal particles by phagocytosis permits their destruction by reactive oxygen and nitrogen species, as well as by lytic enzymes in the acidic environment of the phagolysosome. TLRs are recruited to yeast-containing phagosomes where they detect fungal components and induce production of pro-inflammatory cytokines (72), but they are not responsible for phagocytosis of fungal particles. In contrast, a role for a β-glucan receptor in the internalization of fungal particles was established by Janusz et al. in the mid-1980s, who showed that soluble β-glucans block zymosan internalization (73). Brown and Gordon and colleagues defined Dectin-1 as a phagocytic receptor for β-glucan particles by showing that expression of the receptor in nonphagocytic cells was sufficient to confer the ability to bind and phagocytose β-glucan particles (16). Subsequent studies, including Dectin-1 knockout mouse studies, have established that Dectin-1 is the primary receptor for phagocytosis of zymosan and live fungi by macrophages and DC (16, 23, 24, 38). However, Dectin-1-deficient macrophages are not incapable of fungal recognition (23, 24); indeed, other receptors, such as the mannose receptor, may also contribute to fungal binding and internalization. Although the relative contributions of different receptors likely depends on the repertoire of receptors expressed by each phagocyte subtype, as well as the fungal species and morphotype, the data strongly suggest that Dectin-1 is the most efficient phagocytic receptor for yeast with β-glucan-rich cell walls.
Dectin-1-mediated phagocytosis is dependent on ITAM phosphorylation and the activation of Src family kinases (57, 59). Mutation of the Dectin-1 ITAM tyrosine (membrane-proximal tyrosine) prevented zymosan phagocytosis (59), and Src inhibitors blocked Dectin-1-mediated phagocytosis (57), although it is as yet unclear which Src family members are responsible. Syk kinase may also participate in Dectin-1-mediated phagocytosis of β-glucan particles in a cell type-dependent manner. Evidence from inhibitor and knock-out studies indicates that Syk plays a role in Dectin-1-induced phagocytosis by DC but not macrophages (57–59). Studies using CARD9-deficient macrophages and DC indicated that CARD9 plays no role in zymosan phagocytosis by either cell type (66, 67). However, we recently showed that CARD9 is recruited to zymosan-containing phagosomes, and that the CARD domain of CARD9 is sufficient for CARD9 recruitment (66). Our data indicate that the Dectin-1 phagosome serves as a scaffold for the assembly of the downstream signaling complex.
Oxidative Burst
The phagosome is also the site of assembly of the phagocyte oxidase complex, which is responsible for the production of reactive oxygen species (ROS) upon detection and internalization of fungi. The oxidative burst is an important anti-microbial response of myeloid phagocytes. Stimulation of macrophages, DC and neutrophils with β-glucan particles induces an oxidative burst (58, 59, 74). We demonstrated by cross-linking Dectin-1 tagged with a streptavidin binding peptide using streptavidin-coated beads that Dectin-1 signals are sufficient to induce an oxidative burst in macrophages (59). We also showed that ITAM signaling, including Src and Syk kinase activation, is responsible for Dectin-1-induced ROS production.
Subsequent studies using knockdown and knockout approaches confirmed a role for Dectin-1 in the oxidative burst response to zymosan (23, 24, 39). Opsonization of zymosan with complement, which permits equivalent binding and uptake of the β-glucan particles by both wild type and Dectin-1-deficient cells, revealed a direct role for Dectin-1 in the induction of ROS that could not be attributed to the failure of the cells to detect the particles (23, 24). However, not all myeloid populations were similarly affected; neutrophil and alveolar macrophage responses to opsonized zymosan were impaired by Dectin-1 deletion, whereas the response of thioglycollate-elicited macrophages was unaltered. Hence the relative contributions of glucan, Dectin-1, complement receptors and other receptors to ROS induction are dependent on the myeloid cell type.
Regulation of transcription
Production of a range of inflammatory mediators is a classic feature of TLR responses to microbes. Detection of fungal components by TLRs, including TLR2 and TLR4, undoubtedly accounts for much of the cytokine and chemokine production triggered by fungal exposure (reviewed in (75)). To determine whether non-TLRs, including perhaps Dectin-1, also contribute to the anti-fungal transcriptional response we performed microarray analysis using macrophages from mice deficient in the TLR adaptor MyD88. Following zymosan stimulation for 2 h, more than 100 genes were induced in MyD88-deficient macrophages (39). Detection of β-glucans by Dectin-1 has now been demonstrated by us and others to drive the production of a variety of cytokines and chemokines by macrophages and DC upon fungal exposure, either by directly triggering gene induction or by collaborating with other receptors, including TLRs (17, 23, 24, 39, 58, 69, 76) (Figure 5). As discussed below, gene induction by Dectin-1 is regulated by canonical and non-canonical NF-κB pathways, MAP kinase signaling, and NFAT transcription factors (17, 39, 67, 77).
Figure 5:
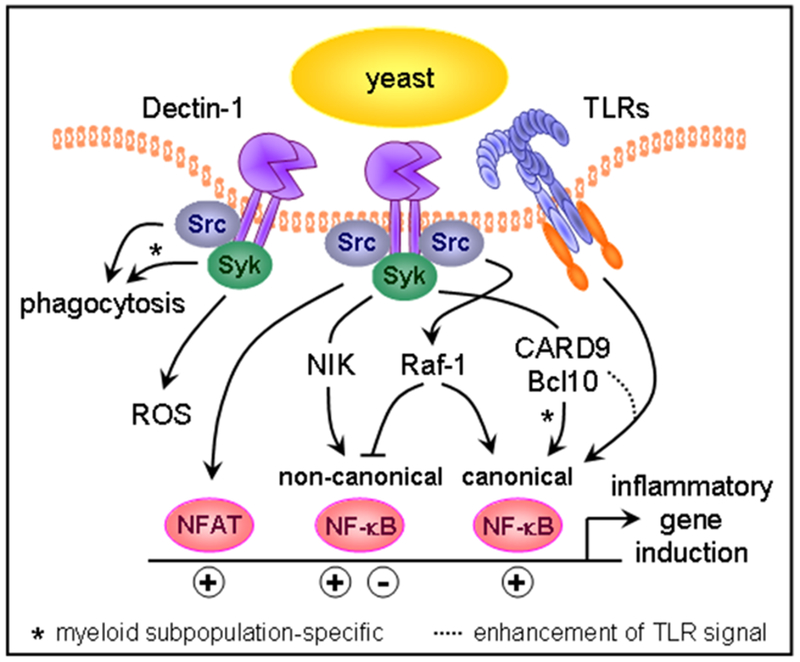
Dectin-1 signal transduction. Dectin-1 signaling via Src and Syk kinases triggers phagocytosis and respiratory burst activity. Dectin-1 signals also control the inflammatory response by activating transcription factors, including NFAT and the canonical and non-canonical NF-κB family members. The signaling adaptor CARD9 participates in signaling downstream of Dectin-1 in two ways. First, it enhances TLR-induced responses, likely through activation of MAP kinases. Second, in specific myeloid cell populations CARD9 directly promotes activation of NF-κB.
Early studies using the RAW264.7 macrophage cell line as well as primary murine macrophages derived from the bone marrow or elicited to the peritoneum using thioglycollate, indicated that, unlike TLRs, Dectin-1 signals do not activate NF-κB transcription factors or induce NF-κB-dependent cytokines such as TNF-α, MIP2 and IL-6 in the absence of TLR signaling (17, 78). Despite this, we and others showed that Dectin-1 signals could collaborate with TLR signals to enhance TLR-induced NF-κB activation and cytokine induction (17, 76). However, Gross et al. subsequently reported that Dectin-1 can activate NF-κB and drive pro-inflammatory cytokine production via the CARD9 adaptor in murine bmDC (67).
It is now evident that the ability of Dectin-1 to directly activate NF-κB and drive cytokine induction depends on the specific cell type and microenvironment (66, 79, 80). In a recent study we showed that β-glucan particles activate NF-κB in GM-CSF-derived bmDC, but not M-CSF-derived bmM. Furthermore, Dectin-1 directly induced TNF-α production by bmDC, resident peritoneal cells and alveolar macrophages, but not by bmM, thioglycollate-elicited peritoneal macrophages or Flt3L-derived DC. Thus certain cell populations are intrinsically wired for cytokine induction upon Dectin-1 ligation, while others are not. Indeed, Shah et al. reported that microglia also express Dectin-1, and that while β-glucan particles trigger Dectin-1-dependent phagocytosis, Syk activation and ROS production by these cells, they fail to induce cytokine production (80). However, we and others have shown that unresponsive macrophages can be primed for CARD9-dependent cytokine induction by environmental stimuli. bmM treated overnight with GM-CSF or IFN-γ produce TNF-α in a CARD9-dependent manner upon Dectin-1 ligation (66, 79). These data explain the apparent inconsistencies between previous studies and indicate an additional level of regulation of Dectin-1 signaling, whereby the precise response is dictated by cell programming and the influence of environmental stimuli.
The mechanisms underlying these differences remain unclear. Responsiveness does not correlate with expression of Dectin-1 or any components of the CARD9-NF-κB pathway, but rather seems to be controlled by an inhibitor of the pathway present in the unresponsive cells (66). This inhibitor does not however block all CARD9-mediated Dectin-1 signals, as evidenced by a partial reduction in β-glucan particle-induced p38 MAP kinase activation in CARD9-deficient bmM, which indicates that Dectin-1-CARD9 signals can activate p38 even when NF-κB activation is prohibited. The identity of the inhibitor is as yet unclear, but one candidate is the tetraspanin CD37, which has previously been shown to interact with Dectin-1 and regulate cytokine induction (81).
As mentioned above, Dectin-1 can also collaborate with TLRs, including TLR2, TLR4, TLR5, TLR7 and TLR9, to amplify TLR-mediated NF-κB activation and cytokine induction, even in cells where Dectin-1 signals cannot activate NF-κB directly (17, 76). Co-stimulation with TLR and Dectin-1 ligands results in enhanced and sustained I-κB degradation and NF-κB nuclear translocation (76). The precise mechanism is unclear but the collaboration is Syk- and CARD9-dependent (66, 76).
In addition to the CARD9-dependent activation of the canonical p65 and c-Rel NF-κB subunits, Gringhuis et al. recently reported that Dectin-1 signals also activate the non-canonical NF-κB subunit RelB (77). They showed that Syk-dependent Dectin-1 signals activate RelB, which acts in competition with the canonical NF-κB subunits, and that the canonical and non-canonical subunits differentially regulate gene induction to influence the repertoire of cytokines produced. Furthermore, they demonstrated that regulation of the RelB pathway, and hence NF-κB-driven transcription, by Raf-1 permits the fine-tuning of cytokine production.
Induction of cytokine production by β-glucan particles is also regulated by MAP kinases. Activation of Erk, p38 and Jnk MAP kinases is Dectin-1-, Src- and Syk-dependent and at least partially mediated by CARD9 (66, 69, 82, 83). We have also shown that Dectin-1 signals activate NFAT transcription factors in macrophages and DC exposed to zymosan and live C. albicans yeast (39). Dectin-1-NFAT signaling regulates cyclooxygenase-2 (Cox-2) and prostaglandin production by macrophages, and the induction of IL-2, IL-10, IL-12 p70 and IL-23 by DC ((39) and our unpublished data). Xu et al. subsequently reported that the NFAT isoforms NFATc1, NFATc2 and NFATc3 are induced by curdlan in bmDC (63).
Dectin-1 and the adaptive immune response
Prevailing evidence suggests that stimulation of Dectin-1 on dendritic cells preferentially directs the subsequent polarization of Th17 cells. Human and murine dendritic cells stimulated with purified β-glucan particles produce large amounts of IL-23 (69, 84). This IL-23 production requires Dectin-1, Syk, and CARD9. Glucan-induced polarization of Th17 cells has been demonstrated in vitro with dendritic cells and naïve T cells from humans and mice. However, the role of glucan-stimulated Th17 polarization in coordinating effective host defense against fungal infections is more difficult to establish.
One hint that the role of Dectin-1 in polarizing the adaptive immune response may be variable and context-dependent comes from the limited and contradicting studies on fungal infection in Dectin-1 knockout mice. Sajio et al. found little role for Dectin-1 in the control of systemic C. albicans infection, but did find a role in the control of P. carinii infection in the lungs (23). In contrast, Taylor et al. saw a significant decrease in mouse survival and increased fungal load following i.p. infection with C. albicans (24). The apparent differences may arise from the fact that the mice were independently generated on backgrounds with differing inherent T cell polarization biases; Balb/cA verses 129/C57BL/6.
These background differences have played out before. The control of fungal infections has classically been attributed to an effective innate antimicrobial immune response that is enhanced by the cytokines produced by Th1 cells, although this view is evolving. Initial studies looking at the control of systemic C. albicans infection in Balb/c mice lacking the key Th1 cytokine, IFN-γ, demonstrated no requirement for IFN-γ for protective immunity (85). However, subsequent studies in C57BL/6 mice lacking IFN-γ showed that the cytokine is essential for host defense (86–88). A variety of experimental details were suggested to explain this discrepancy including Candida dose and strain, but the genetic backgrounds of the mice were shown to be an important contributing factor (89). These studies thus implied a role for other types of adaptive immune responses in addition to Th1 in the control of fungal infections. Recent studies have now explored the contribution of Th17 or Treg cells in controlling fungal infections, albeit with varying results.
Huang et al. observed that C57BL/6 mice deficient in IL-17A were significantly more susceptible to systemic C. albicans infection (90), and Conti et al. similarly observed that IL-17R-deficient mice are highly susceptible to oropharyngeal Candida infection (91). In both cases the mice were unable to control fungal growth and had reduced neutrophil mobilization. Further, Conti et al. observed that mice lacking IL-23p19, which is required for maintenance of Th17 cells were susceptible to oropharyngeal Candida infection, while mice lacking IL-12p35, which is required for Th1 polarization, were protected (91). Further, Werner et al. have recently demonstrated that blocking IL-17 in the lungs significantly exacerbates Aspergillus infection, and that optimal IL-17 production in the lungs after infection is requires β-glucan recognition by Dectin-1 (25) . Together these studies support the importance of Th17 immune responses during fungal infection. In contrast, Zelante et al. observed that deletion of IL-23 did not impact survival of mice to intragastric Candida infection or intranasal Aspergillus infection (92). Instead, IL-23-deficient mice exhibited enhanced Th1 mediated inflammation, suggesting that IL-23 serves as a natural break on tissue damaging Th1 responses.
The divergent observations surrounding the effects of Th17 cells may be a consequence of the different routes and/or doses of infection used. At the same time, these and other studies seem to point toward the importance of a balance between Th1 and Th17 responses for effective control of fungal infections. Specifically, we might hypothesize that IL-17 is important for early steps involving the recruitment of sufficient numbers of neutrophils to deal with the pathogen, and that subsequent IFN-γ produced by Th1 cells is important for activation of these neutrophils and other phagocytes for killing fungi.
Additional regulation of the immune response to fungal pathogens is provided by regulatory T cells (Tregs) which are thought to help prevent excessive pathology due to the immune response as well contribute to the development of persistent immune memory. TLR2 activation in response to fungi or zymosan is thought to preferentially promote the generation of Tregs, and this is countered by β-glucan activation of Dectin-1 (93). Consistent with this, TLR2−/− (and TLR6−/−) mice infected with Candida show reduced Treg production, enhanced IFNγ production, and normal or enhanced resistance to infection (94, 95).
An important feature of all of the current studies is that they have at best looked at only two of these three T cell types at a time. For a more comprehensive understanding of the true balance between the regulatory and inflammatory cell subsets during fungal infection, a simultaneous analysis of the activity and contribution of Th1, Treg, and Th17 cells during the course of a fungal infection will be valuable. It may well be that differing infections engage different subsets of antigen presenting cells and that this influences the role of β-glucan recognition in defining the adaptive immune response. Work by Dillon et al. demonstrated that different murine splenic antigen presenting cells differentially produced IL-12p70 in response to β-glucan particles (96). Also, as described above, we and others have demonstrated that different antigen presenting cells utilize CARD9 signaling differently. The specific contribution of β-glucan recognition and Dectin-1 will be better elucidated by following this polarization in Dectin-1-deficient mice.
Future directions in β-glucan recognition
A better understanding of the mechanisms of β-glucan recognition will inform both our attempts to improve host defense against fungal pathogens and our attempts to utilize soluble and particulate glucans to modulate innate and adaptive immune responses. Although we may hope that we have already identified the main receptors involved in β-glucan recognition, it is likely that others remain to be described. Optimal exploitation of glucans in anti-cancer treatments, vaccines, and in priming host defenses will require a mechanistic understanding of how each receptor works. We will need a complete model for how each receptor signals in order to understand how β-glucan is similar to and different from other microbe-derived compounds being evaluated. For example, signaling studies help to inform how we may expect glucans to behave differently from TLR agonists as vaccine adjuvants. Optimal formulation will require good models for how each receptor distinguishes its ligands. For example, how does CR3 respond to soluble glucans, while Dectin-1 does not? Why are different β-glucan branching structures better ligands for each receptor? Ultimately glucans (alone and coupled to other compounds) are likely to become highly useful immune modulators.
References
- 1.Blystone SD, Brown EJ. Integrin Receptors of Phagocytes In: Gordon S, ed. Phagocytosis: The Host. Stamford, CT: JAI Press; 1999:103–47. [Google Scholar]
- 2.Thornton BP, Vetvicka V, Pitman M, Goldman RC, Ross GD. Analysis of the sugar specificity and molecular location of the beta-glucan-binding lectin site of complement receptor type 3 (CD11b/CD18). J Immunol 1996;156:1235–46. [PubMed] [Google Scholar]
- 3.Xia Y, Ross GD. Generation of recombinant fragments of CD11b expressing the functional beta-glucan-binding lectin site of CR3 (CD11b/CD18). J Immunol 1999;162:7285–93. [PubMed] [Google Scholar]
- 4.Xia Y, Vetvicka V, Yan J, Hanikyrova M, Mayadas T, Ross GD. The beta-glucan-binding lectin site of mouse CR3 (CD11b/CD18) and its function in generating a primed state of the receptor that mediates cytotoxic activation in response to iC3b-opsonized target cells. J Immunol 1999;162:2281–90. [PubMed] [Google Scholar]
- 5.Xue W, Kindzelskii AL, Todd RF, 3rd, Petty HR. Physical association of complement receptor type 3 and urokinase-type plasminogen activator receptor in neutrophil membranes. J Immunol 1994;152:4630–40. [PubMed] [Google Scholar]
- 6.Zimmerman JW, Lindermuth J, Fish PA, Palace GP, Stevenson TT, DeMong DE. A novel carbohydrate-glycosphingolipid interaction between a beta-(1–3)-glucan immunomodulator, PGG-glucan, and lactosylceramide of human leukocytes. J Biol Chem 1998;273:22014–20. [DOI] [PubMed] [Google Scholar]
- 7.Evans SE, Hahn PY, McCann F, Kottom TJ, Pavlovic ZV, Limper AH. Pneumocystis cell wall beta-glucans stimulate alveolar epithelial cell chemokine generation through nuclear factor-kappaB-dependent mechanisms. Am J Respir Cell Mol Biol 2005;32:490–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hahn PY, Evans SE, Kottom TJ, Standing JE, Pagano RE, Limper AH. Pneumocystis carinii cell wall beta-glucan induces release of macrophage inflammatory protein-2 from alveolar epithelial cells via a lactosylceramide-mediated mechanism. J Biol Chem 2003;278:2043–50. [DOI] [PubMed] [Google Scholar]
- 9.Wakshull E, et al. PGG-glucan, a soluble beta-(1,3)-glucan, enhances the oxidative burst response, microbicidal activity, and activates an NF-kappa B-like factor in human PMN: evidence for a glycosphingolipid beta-(1,3)-glucan receptor. Immunopharmacology 1999;41:89–107. [DOI] [PubMed] [Google Scholar]
- 10.Nakayama H, et al. Lyn-coupled LacCer-enriched lipid rafts are required for CD11b/CD18-mediated neutrophil phagocytosis of nonopsonized microorganisms. J Leukoc Biol 2008;83:728–41. [DOI] [PubMed] [Google Scholar]
- 11.Li B, et al. Yeast beta-glucan amplifies phagocyte killing of iC3b-opsonized tumor cells via complement receptor 3-Syk-phosphatidylinositol 3-kinase pathway. J Immunol 2006;177:1661–9. [DOI] [PubMed] [Google Scholar]
- 12.Hong F, et al. Mechanism by which orally administered beta-1,3-glucans enhance the tumoricidal activity of antitumor monoclonal antibodies in murine tumor models. J Immunol 2004;173:797–806. [DOI] [PubMed] [Google Scholar]
- 13.Allen LA, Aderem A. Molecular definition of distinct cytoskeletal structures involved in complement- and Fc receptor-mediated phagocytosis in macrophages. J Exp Med 1996;184:627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown EJ. The role of extracellular matrix proteins in the control of phagocytosis. J Leukoc Biol 1986;39:579–91. [DOI] [PubMed] [Google Scholar]
- 15.Le Cabec V, Cols C, Maridonneau-Parini I. Nonopsonic phagocytosis of zymosan and Mycobacterium kansasii by CR3 (CD11b/CD18) involves distinct molecular determinants and is or is not coupled with NADPH oxidase activation. Infect Immun 2000;68:4736–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown GD, et al. Dectin-1 is a major beta-glucan receptor on macrophages. J Exp Med 2002;196:407–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med 2003;197:1107–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sen G, Bikah G, Venkataraman C, Bondada S. Negative regulation of antigen receptor-mediated signaling by constitutive association of CD5 with the SHP-1 protein tyrosine phosphatase in B-1 B cells. Eur J Immunol 1999;29:3319–28. [DOI] [PubMed] [Google Scholar]
- 19.Bikah G, Carey J, Ciallella JR, Tarakhovsky A, Bondada S. CD5-mediated negative regulation of antigen receptor-induced growth signals in B-1 B cells. Science 1996;274:1906–9. [DOI] [PubMed] [Google Scholar]
- 20.Vera J, et al. The CD5 ectodomain interacts with conserved fungal cell wall components and protects from zymosan-induced septic shock-like syndrome. Proc Natl Acad Sci U S A 2009;106:1506–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ariizumi K, et al. Identification of a novel, dendritic cell-associated molecule, dectin-1, by subtractive cDNA cloning. J Biol Chem 2000;275:20157–67. [DOI] [PubMed] [Google Scholar]
- 22.Willment JA, et al. The human beta-glucan receptor is widely expressed and functionally equivalent to murine Dectin-1 on primary cells. Eur J Immunol 2005;35:1539–47. [DOI] [PubMed] [Google Scholar]
- 23.Saijo S, et al. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat Immunol 2007;8:39–46. [DOI] [PubMed] [Google Scholar]
- 24.Taylor PR, et al. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol 2007;8:31–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Werner J, et al. Requisite role for the Dectin-1 beta-glucan receptor in pulmonary defense against Aspergillus fumigatus. J Immunol 2009;IN PRESS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willment JA, Gordon S, Brown GD. Characterization of the human beta -glucan receptor and its alternatively spliced isoforms. J Biol Chem 2001;276:43818–23. [DOI] [PubMed] [Google Scholar]
- 27.Hermanz-Falcon P, Arce I, Roda-Navarro P, Fernandez-Ruiz E. Cloning of human DECTIN-1, a novel C-type lectin-like receptor gene expressed on dendritic cells. Immunogenetics 2001;53:288–95. [DOI] [PubMed] [Google Scholar]
- 28.Heinsbroek SE, et al. Expression of functionally different dectin-1 isoforms by murine macrophages. J Immunol 2006;176:5513–8. [DOI] [PubMed] [Google Scholar]
- 29.del Pilar Jimenez AM, et al. Susceptibility to Coccidioides species in C57BL/6 mice is associated with expression of a truncated splice variant of Dectin-1 (Clec7a). Genes Immun 2008;9:338–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinez O, Brackenridge S, El-Idrissi Mel A, Prabhakar BS. DC-SIGN, but not sDC-SIGN, can modulate IL-2 production from PMA- and anti-CD3-stimulated primary human CD4 T cells. Int Immunol 2005;17:769–78. [DOI] [PubMed] [Google Scholar]
- 31.Adams EL, et al. Differential high-affinity interaction of dectin-1 with natural or synthetic glucans is dependent upon primary structure and is influenced by polymer chain length and side-chain branching. J Pharmacol Exp Ther 2008;325:115–23. [DOI] [PubMed] [Google Scholar]
- 32.Adachi Y, et al. Characterization of beta-glucan recognition site on C-type lectin, dectin 1. Infect Immun 2004;72:4159–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown J, et al. Structure of the fungal beta-glucan-binding immune receptor dectin-1: implications for function. Protein Sci 2007;16:1042–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grunebach F, Weck MM, Reichert J, Brossart P. Molecular and functional characterization of human Dectin-1. Exp Hematol 2002;30:1309–15. [DOI] [PubMed] [Google Scholar]
- 35.Calderone RA, Fonzi WA. Virulence factors of Candida albicans. Trends Microbiol 2001;9:327–35. [DOI] [PubMed] [Google Scholar]
- 36.Lo HJ, Kohler JR, DiDomenico B, Loebenberg D, Cacciapuoti A, Fink GR. Nonfilamentous C. albicans mutants are avirulent. Cell 1997;90:939–49. [DOI] [PubMed] [Google Scholar]
- 37.Saville SP, Lazzell AL, Monteagudo C, Lopez-Ribot JL. Engineered control of cell morphology in vivo reveals distinct roles for yeast and filamentous forms of Candida albicans during infection. Eukaryot Cell 2003;2:1053–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gantner BN, Simmons RM, Underhill DM. Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. Embo J 2005;24:1277–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goodridge HS, Simmons RM, Underhill DM. Dectin-1 stimulation by Candida albicans yeast or zymosan triggers NFAT activation in macrophages and dendritic cells. J Immunol 2007;178:3107–15. [DOI] [PubMed] [Google Scholar]
- 40.Wheeler RT, Kombe D, Agarwala SD, Fink GR. Dynamic, morphotype-specific Candida albicans beta-glucan exposure during infection and drug treatment. PLoS Pathog 2008;4:e1000227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wheeler RT, Fink GR. A drug-sensitive genetic network masks fungi from the immune system. PLoS Pathog 2006;2:e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gersuk GM, Underhill DM, Zhu L, Marr KA. Dectin-1 and TLRs permit macrophages to distinguish between different Aspergillus fumigatus cellular states. J Immunol 2006;176:3717–24. [DOI] [PubMed] [Google Scholar]
- 43.Hohl TM, et al. Aspergillus fumigatus triggers inflammatory responses by stage-specific beta-glucan display. PLoS Pathog 2005;1:e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steele C, et al. The beta-glucan receptor dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathog 2005;1:e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marr KA, Patterson T, Denning D. Aspergillosis. Pathogenesis, clinical manifestations, and therapy. Infect Dis Clin North Am 2002;16:875–94, vi. [DOI] [PubMed] [Google Scholar]
- 46.Jahn B, et al. Isolation and characterization of a pigmentless-conidium mutant of Aspergillus fumigatus with altered conidial surface and reduced virulence. Infect Immun 1997;65:5110–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langfelder K, Jahn B, Gehringer H, Schmidt A, Wanner G, Brakhage AA. Identification of a polyketide synthase gene (pksP) of Aspergillus fumigatus involved in conidial pigment biosynthesis and virulence. Med Microbiol Immunol 1998;187:79–89. [DOI] [PubMed] [Google Scholar]
- 48.Jahn B, Boukhallouk F, Lotz J, Langfelder K, Wanner G, Brakhage AA. Interaction of human phagocytes with pigmentless Aspergillus conidia. Infect Immun 2000;68:3736–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luther K, Torosantucci A, Brakhage AA, Heesemann J, Ebel F. Phagocytosis of Aspergillus fumigatus conidia by murine macrophages involves recognition by the dectin-1 beta-glucan receptor and Toll-like receptor 2. Cell Microbiol 2007;9:368–81. [DOI] [PubMed] [Google Scholar]
- 50.Torosantucci A, et al. A novel glyco-conjugate vaccine against fungal pathogens. J Exp Med 2005;202:597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hohl TM, Feldmesser M, Perlin DS, Pamer EG. Caspofungin modulates inflammatory responses to Aspergillus fumigatus through stage-specific effects on fungal beta-glucan exposure. J Infect Dis 2008;198:176–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lamaris GA, et al. Caspofungin-mediated beta-glucan unmasking and enhancement of human polymorphonuclear neutrophil activity against Aspergillus and non-Aspergillus hyphae. J Infect Dis 2008;198:186–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rappleye CA, Eissenberg LG, Goldman WE. Histoplasma capsulatum alpha-(1,3)-glucan blocks innate immune recognition by the beta-glucan receptor. Proc Natl Acad Sci U S A 2007;104:1366–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Medoff G, et al. Irreversible block of the mycelial-to-yeast phase transition of Histoplasma capsulatum. Science 1986;231:476–9. [DOI] [PubMed] [Google Scholar]
- 55.Nemecek JC, Wuthrich M, Klein BS. Global control of dimorphism and virulence in fungi. Science 2006;312:583–8. [DOI] [PubMed] [Google Scholar]
- 56.Underhill DM, Goodridge HS. The many faces of ITAMs. Trends Immunol 2007;28:66–73. [DOI] [PubMed] [Google Scholar]
- 57.Herre J, et al. Dectin-1 uses novel mechanisms for yeast phagocytosis in macrophages. Blood 2004;104:4038–45. [DOI] [PubMed] [Google Scholar]
- 58.Rogers NC, et al. Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity 2005;22:507–17. [DOI] [PubMed] [Google Scholar]
- 59.Underhill DM, Rossnagle E, Lowell CA, Simmons RM. Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood 2005;106:2543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fuller GL, et al. The C-type lectin receptors CLEC-2 and Dectin-1, but not DC-SIGN, signal via a novel YXXL-dependent signaling cascade. J Biol Chem 2007;282:12397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Girotti M, Evans JH, Burke D, Leslie CC. Cytosolic phospholipase A2 translocates to forming phagosomes during phagocytosis of zymosan in macrophages. J Biol Chem 2004;279:19113–21. [DOI] [PubMed] [Google Scholar]
- 62.Qiu ZH, Gijon MA, de Carvalho MS, Spencer DM, Leslie CC. The role of calcium and phosphorylation of cytosolic phospholipase A2 in regulating arachidonic acid release in macrophages. J Biol Chem 1998;273:8203–11. [DOI] [PubMed] [Google Scholar]
- 63.Xu S, Huo J, Lee KG, Kurosaki T, Lam KP. Phospholipase Cgamma 2 is critical for dectin-1-mediated Ca2+ flux and cytokine production in dendritic cells. J Biol Chem 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gaide O, et al. CARMA1 is a critical lipid raft-associated regulator of TCR-induced NF-kappa B activation. Nat Immunol 2002;3:836–43. [DOI] [PubMed] [Google Scholar]
- 65.Wang D, et al. A requirement for CARMA1 in TCR-induced NF-kappa B activation. Nat Immunol 2002;3:830–5. [DOI] [PubMed] [Google Scholar]
- 66.Goodridge HS, et al. Differential use of CARD9 by Dectin-1 in macrophages and dendritic cells. J Immunol 2009;182:1146–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gross O, et al. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 2006;442:651–6. [DOI] [PubMed] [Google Scholar]
- 68.Hara H, et al. The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat Immunol 2007;8:619–29. [DOI] [PubMed] [Google Scholar]
- 69.LeibundGut-Landmann S, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol 2007;8:630–8. [DOI] [PubMed] [Google Scholar]
- 70.Gross O, et al. Multiple ITAM-coupled NK-cell receptors engage the Bcl10/Malt1 complex via Carma1 for NF-kappaB and MAPK activation to selectively control cytokine production. Blood 2008;112:2421–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hara H, et al. Cell type-specific regulation of ITAM-mediated NF-kappaB activation by the adaptors, CARMA1 and CARD9. J Immunol 2008;181:918–30. [DOI] [PubMed] [Google Scholar]
- 72.Underhill DM, et al. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature 1999;401:811–5. [DOI] [PubMed] [Google Scholar]
- 73.Janusz MJ, Austen KF, Czop JK. Isolation of soluble yeast beta-glucans that inhibit human monocyte phagocytosis mediated by beta-glucan receptors. J Immunol 1986;137:3270–6. [PubMed] [Google Scholar]
- 74.Kennedy AD, Willment JA, Dorward DW, Williams DL, Brown GD, DeLeo FR. Dectin-1 promotes fungicidal activity of human neutrophils. Eur J Immunol 2007;37:467–78. [DOI] [PubMed] [Google Scholar]
- 75.Netea MG, Van der Meer JW, Kullberg BJ. Role of the dual interaction of fungal pathogens with pattern recognition receptors in the activation and modulation of host defence. Clin Microbiol Infect 2006;12:404–9. [DOI] [PubMed] [Google Scholar]
- 76.Dennehy KM, et al. Syk kinase is required for collaborative cytokine production induced through Dectin-1 and Toll-like receptors. Eur J Immunol 2008;38:500–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gringhuis SI, et al. Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat Immunol 2009;10:203–13. [DOI] [PubMed] [Google Scholar]
- 78.Brown GD, Herre J, Williams DL, Willment JA, Marshall AS, Gordon S. Dectin-1 mediates the biological effects of beta-glucans. J Exp Med 2003;197:1119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rosas M, et al. The induction of inflammation by dectin-1 in vivo is dependent on myeloid cell programming and the progression of phagocytosis. J Immunol 2008;181:3549–57. [DOI] [PubMed] [Google Scholar]
- 80.Shah VB, Huang Y, Keshwara R, Ozment-Skelton T, Williams DL, Keshvara L. Beta-glucan activates microglia without inducing cytokine production in Dectin-1-dependent manner. J Immunol 2008;180:2777–85. [DOI] [PubMed] [Google Scholar]
- 81.Meyer-Wentrup F, et al. Dectin-1 interaction with tetraspanin CD37 inhibits IL-6 production. J Immunol 2007;178:154–62. [DOI] [PubMed] [Google Scholar]
- 82.Olsson S, Sundler R. The macrophage beta-glucan receptor mediates arachidonate release induced by zymosan: essential role for Src family kinases. Mol Immunol 2007;44:1509–15. [DOI] [PubMed] [Google Scholar]
- 83.Slack EC, et al. Syk-dependent ERK activation regulates IL-2 and IL-10 production by DC stimulated with zymosan. Eur J Immunol 2007;37:1600–12. [DOI] [PubMed] [Google Scholar]
- 84.Gerosa F, et al. Differential regulation of interleukin 12 and interleukin 23 production in human dendritic cells. J Exp Med 2008;205:1447–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qian Q, Cutler JE. Gamma interferon is not essential in host defense against disseminated candidiasis in mice. Infect Immun 1997;65:1748–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Balish E, Wagner RD, Vazquez-Torres A, Pierson C, Warner T. Candidiasis in interferon-gamma knockout (IFN-gamma−/−) mice. J Infect Dis 1998;178:478–87. [DOI] [PubMed] [Google Scholar]
- 87.Kaposzta R, Tree P, Marodi L, Gordon S. Characteristics of invasive candidiasis in gamma interferon- and interleukin-4-deficient mice: role of macrophages in host defense against Candida albicans. Infect Immun 1998;66:1708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lavigne LM, Schopf LR, Chung CL, Maylor R, Sypek JP. The role of recombinant murine IL-12 and IFN-gamma in the pathogenesis of a murine systemic Candida albicans infection. J Immunol 1998;160:284–92. [PubMed] [Google Scholar]
- 89.Schofield DA, Westwater C, Balish E. Divergent chemokine, cytokine and beta-defensin responses to gastric candidiasis in immunocompetent C57BL/6 and BALB/c mice. J Med Microbiol 2005;54:87–92. [DOI] [PubMed] [Google Scholar]
- 90.Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis 2004;190:624–31. [DOI] [PubMed] [Google Scholar]
- 91.Conti HR, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zelante T, et al. IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur J Immunol 2007;37:2695–706. [DOI] [PubMed] [Google Scholar]
- 93.Osorio F, et al. DC activated via dectin-1 convert Treg into IL-17 producers. Eur J Immunol 2008;38:3274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Netea MG, et al. Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J Immunol 2004;172:3712–8. [DOI] [PubMed] [Google Scholar]
- 95.Netea MG, van de Veerdonk F, Verschueren I, van der Meer JW, Kullberg BJ. Role of TLR1 and TLR6 in the host defense against disseminated candidiasis. FEMS Immunol Med Microbiol 2008;52:118–23. [DOI] [PubMed] [Google Scholar]
- 96.Dillon S, et al. Yeast zymosan, a stimulus for TLR2 and dectin-1, induces regulatory antigen-presenting cells and immunological tolerance. J Clin Invest 2006;116:916–28. [DOI] [PMC free article] [PubMed] [Google Scholar]