

Abstract
Impairments of synaptic plasticity are a hallmark of several neurological disorders, including Parkinson's disease (PD) which results from the progressive loss of dopaminergic neurons of the substantia nigra pars compacta leading to abnormal activity within the basal ganglia (BG) network and pathological motor symptoms. Indeed, disrupted plasticity at corticostriatal glutamatergic synapses, the gateway of the BG, is correlated to the onset of PD-related movement disorders and thus has been proposed to be a key neural substrate regulating information flow and motor function in BG circuits. However, a critical question is whether similar plasticity impairments could occur at other glutamatergic connections within the BG that would also affect the inhibitory influence of the network on the motor thalamus. Here, we show that long-term plasticity at subthalamo-nigral glutamatergic synapses (STN-SNr) sculpting the activity patterns of nigral neurons, the main output of the network, is also affected in experimental parkinsonism. Using whole-cell patch-clamp in acute rat brain slices, we describe a molecular pathway supporting an activity-dependent long-term depression of STN-SNr synapses through an NMDAR-and D1/5 dopamine receptor-mediated endocytosis of synaptic AMPA glutamate receptors. We also show that this plastic property is lost in an experimental rat model of PD but can be restored through the recruitment of dopamine D1/5 receptors. Altogether, our findings suggest that pathological impairments of subthalamo-nigral plasticity may enhance BG outputs and thereby contribute to PD-related motor dysfunctions.
Introduction
Parkinson's disease (PD) is caused by the loss of dopaminergic (DAergic) projections from the substantia nigra pars compacta (SNc). The resulting dramatic rise in firing of basal ganglia (BG) output neurons leads to bradykinesia, rigidity, and tremor symptoms. Most attention has focused upon the corticostriatal synapse, i.e., the main BG input, where dopaminergic denervation in experimental models of PD is correlated with impaired long-term potentiation (LTP) and long-term depression (LTD) of glutamatergic neurotransmission that can be restored by chronic administration of the dopamine precursor L-DOPA, the most effective therapeutic treatment for PD. Corticostriatal plasticity is thus envisioned as the cellular substrate regulating information flow and motor function in BG circuits (Calabresi et al., 1992a,c; Kreitzer and Malenka, 2007). However, accumulating evidence indicates that both physiological and pathological plasticity might take place at several stages of the cortico-BG–thalamocortical network as SNc DAergic projections innervate various BG nuclei.
Among those, the substantia nigra pars reticulata (SNr) is a major output of the BG network—the functional equivalent in rodents of the internal portion of the globus pallidus in primates—that conveys information to the motor thalamus and the brainstem through the action of pacemaking GABAergic neurons (Murer et al., 1997; Atherton and Bevan, 2005). SNr tonic activity is regulated by a combination of striato-nigral GABAergic inhibitory inputs and subthalamo-nigral (STN-SNr) glutamatergic excitatory inputs that favor and prevent motor execution, respectively (Alexander and Crutcher, 1990; Kravitz et al., 2010). Therefore, the abnormal hyperactivity of the subthalamic nucleus (STN) reported in PD (Bergman et al., 1994) is considered as a major cause of pathological activity in SNr neurons, and STN-SNr synapses are viewed as critical connections shaping both normal and pathological BG outputs (Murer et al., 1997; Tseng et al., 2000). In addition, dendritically released DA from SNc neurons also regulates the excitability of SNr neurons (Cheramy et al., 1981), either directly (Zhou et al., 2009) or through the modulation of striato-nigral (Radnikow and Misgeld, 1998) and STN-SNr inputs (Ibañez-Sandoval et al., 2006). Thus, dysfunctions in the DAergic modulation of SNr neuronal activity could play an important role in the emergence of PD motor symptoms.
Yet, despite the body of evidence suggesting the importance of STN-SNr synapses in the regulation of BG outputs, activity-dependent adaptations in the efficacy of these connections remain unknown. We thus investigated long-term synaptic plasticity at STN-SNr synapses and addressed whether it is affected by chronic DA depletion. We found that tetanic stimulation of STN afferents induces a persistent LTD of synaptic transmission in naive rats. This form of synaptic plasticity occurs through a postsynaptic NMDA receptor-mediated endocytosis of GluA2-containing AMPA receptors. Moreover, we show that both acute blockade of D1/5 DAergic receptors (D1/5R) and chronic DA depletion abolish nigral LTD, and that activating D1/5R restores LTD expression in DA-depleted slices. Together, these results suggest that impairments in DA-dependent adaptations of STN-SNr synapses observed in experimental parkinsonism could contribute to the pathological activity of BG outputs and thereby promote PD-related motor dysfunctions.
Materials and Methods
Animals
Male Sprague Dawley rats were housed under a 12 h light/dark cycle with food and water provided ad libitum. Every effort was made to minimize animal suffering and to use the minimum number of animals possible. Experimental procedures were conducted in accordance with the Institutional Animal Care and Use Committee of Bordeaux guidelines and the European Communities Council Directive; 24 November, 1986 (86/6091EEC).
6-Hydroxydopamine lesion
Rats (p15–p19) were anesthetized with ketamine (75 mg/kg) and xylazine (10 mg/kg) and mounted on a Kopf stereotaxic frame, as previously described (Miguelez et al., 2012). Briefly, 2 μl of 6-hydroxydopamine (6-OHDA; 4 μg/μl) was infused at a rate of 0.5 μl/min into the right medial forebrain bundle (relative to bregma; AP: −2.4 mm; ML: −1.2 mm; DV: −7.4 and −7.9 mm; 1 μl was injected in each coordinate). After each injection, the needle was left in place for an additional 2–4 min to allow the toxin to diffuse into the structure before being slowly retracted. Thirty minutes before surgery, desipramine (20 mg/kg, i.p.) was administered to avoid damage of the noradrenergic system.
Tissue preparation
Parasagittal slices containing both STN and SNr were prepared from p25–p35 Sprague Dawley rats (Fig. 1a), i.e., from 10 to 15 d after 6-OHDA injection, a lag time we have shown adequate for DA cell loss to occur and cause PD-related dysfunctions. Rats were anesthetized with isoflurane and killed by decapitation. Brains were rapidly removed and sectioned into 350-μm-thick slices in the sagittal plane with a vibratome (VT1200S, Leica Microsystems) in an ice-cold sucrose buffer solution of the following composition (in mm): 250 sucrose, 2 KCl, 7 MgCl2, 0.5 CaCl2, 1.15 NaH2PO4, 11 glucose, and 26 NaHCO3 (gassed with 95% O2/5% CO2). Slices were then left for equilibration for ∼1 h at room temperature (∼25°C) in an artificial CSF (ACSF) solution of the following composition (in mm): 126 NaCl, 3.5 KCl, 2 CaCl2, 1.3 MgCl2, 1.2 NaH2PO4, 25 NaHCO3, and 12 glucose (gassed with 95% O2/5% CO2 at 32°C).
Figure 1.
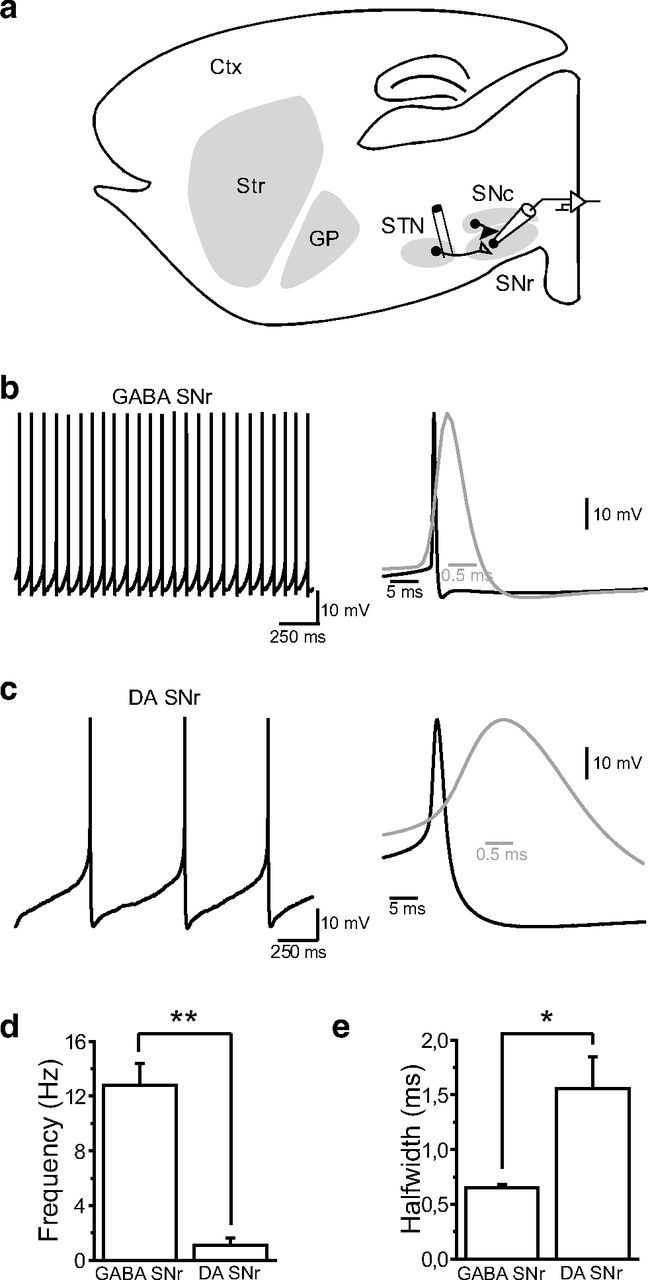
Electrophysiological properties of putative GABAergic and dopaminergic neurons in substantia nigra. a, Schematic parasagittal section of a rat brain, showing the cortex and the BG nuclei (Str, striatum; GP, globus pallidus; STN, subthalamic nucleus; SNc, substantia nigra pars compacta; SNr, substantia nigra pars reticulata). Stimulation electrodes were implanted in the posterior part of the STN and eEPSCs in SNr neurons were recorded in whole-cell voltage-clamp. b, Typical example of autonomous pacemaking of a putative GABAergic neuron from the substantia nigra pars reticulata. Right, Average action potential from the neuron in left panel. c, Typical example of autonomous pacemaking of a putative DAergic neuron from the substantia nigra pars reticulata. Right, Average action potential from the neuron in left panel. d, e, Summary histograms showing the differences in action potential firing frequency (d, GABAergic, 12.8 ± 1.56 Hz; dopaminergic, 1.12 ± 0.48 Hz; **p < 0.01) and action potential kinetics (e, GABAergic, 0.648 ± 0.031 ms; dopaminergic, 1.559 ± 0.286 ms; p < 0.05) between the two substantia nigra pars reticulata cell types.
Drugs
Unless otherwise stated, all drugs used were purchase from Abcam and prepared as concentrated stock solutions and stored at −20°C. On the day of the experiment, drugs were diluted and applied through the bath perfusion system. GABAergic synaptic transmission was blocked for all experiments. GABAA, and GABAB receptors were blocked with 10 μm 2-(3-carboxypropyl)-3-amino-6-(4 methoxyphenyl)pyridazinium bromide (SR 95531), and 5 μm (2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2 hydroxypropyl](phenylmethyl)phosphinic acid (CGP 55845), respectively. In some experiments NMDA or AMPA/kainate receptors were also blocked (in addition of GABAergic transmission) with 50 μm d-(-)-2-amino-5-phosphonopentanoic acid (d-APV), 20 μm 6,7-dinitroquinoxaline-2,3-dione (DNQX), respectively. The DAergic D1 receptor agonist (±)-6-chloro-2,3,4,5-tetrahydro-1-phenyl-1H-3-benzazepine hydrobromide (SKF81297; 2–5 μm) and antagonist (R)-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH23390; 5–10 μm) were purchased from Tocris Bioscience.
Electrophysiology
Whole-cell recordings.
Single slices were transferred to a recording chamber and perfused continuously (2 ml/min) with ACSF (see above) heated to 32°C using a temperature control system (Badcontroller V, Luigs and Neumann) and saturated with 95% O2/5% CO2. SNr neurons were visualized with infrared gradient contrast video microscopy (BX51WI, Olympus) and a 40× water-immersion objective (60×/0.80 LUMPlan FI/IR, Olympus). Whole-cell recordings were performed using 1.5 mm external diameter borosilicate pipettes (GC150F-10, Harvard Apparatus) prepared with a micropipette puller (P97, Sutter Instruments). Electrodes (4–5 MΩ) were filled with a solution containing the following (in mm): 120 potassium gluconate, 5 KCl, 10 EGTA, 10 HEPES, 1 CaCl2, 2.5 MgATP, and 0.5 Na2GTP, adjusted to pH 7.3 with KOH. For AMPA/NMDA ratio and current–voltage relationship recordings, electrodes were filled with a solution containing the following (in mm): 120 CsMeSO3, 15 CsCl, 8 NaCl, 0.2 EGTA, 10 HEPES, 2 MgATP, 0.3 Na2GTP, 10 TEA-Cl, 5 QX-314 [N-(2,6-dimethylphenylcarbamoyl-methyl) triethylammonium bromide], adjusted to pH 7.3 with CsOH. GABAergic neurons were distinguished from neighboring DAergic neurons based on their electrophysiological properties. Briefly, the spontaneous firing activity of all neurons recorded was assessed at the beginning of every experiment. This procedure enabled a clear distinction between GABAergic and dopaminergic neurons, either in terms of action potential firing frequency (GABAergic, 12.8 ± 1.56 Hz; dopaminergic, 1.12 ± 0.48 Hz; p < 0.01), action potential half-width (GABAergic, 0.648 ± 0.031 ms; dopaminergic, 1.559 ± 0.286 ms; p < 0.05), or afterhyperpolarization (GABAergic, −63 ± 0.8 mV; dopaminergic, −67.28 ± 1.95 mV; p < 0.05; Fig. 1). Glutamatergic responses were evoked in SNr neurons through monophasic electrical stimulation of the STN with a tungsten bipolar electrode (TST33A05KT Microelectrode Tungsten, World Precision Instruments) controlled by an ISO-Flex stimulator (AMPI) (Fig. 1a). Single stimuli were delivered at 0.05 Hz and the responses were observed in nigral neurons as evoked EPSCs in voltage-clamp (Vhold = −70 mV). Under these conditions and after a stable eEPSC recording had been maintained for 10 min, tetanic stimulation (four trains of 100 stimuli at 100 Hz, delivered at 20 s interval) of STN-SNr fibers was used to induce LTD, whereas the SNr neuron was recorded in current-clamp mode. All the experiments were conducted in the continuous presence of the GABAA and GABAB antagonists SR95531 (10 μm) and CGP55845 (5 μm) to block inhibitory neurotransmission. Miniature EPSCs (mEPSCs) were recorded at −70 mV in 1 μm tetrodotoxin.
Data were recorded using a Multiclamp 700A amplifier and a Digidata 1322A interface controlled by Clampex 10.1 (Molecular Devices). Signals were digitized at 20 kHz and low-pass filtered at 6 kHz, respectively. Recordings with Kgluconate-filled and CsMeSO3-filled pipettes were corrected for a junction potential of 12.6 and 7 mV, respectively. Series resistance was monitored throughout the experiment by a brief voltage step of −5 mV at the beginning of each recording. Data were discarded when the series resistance increased or decreased by >20%.
Immunohistochemistry
Tyrosine hydroxylase immunostaining.
Immunohistochemistry was performed on overnight-fixed recorded slices (4% paraformaldehyde) as previously described (Miguelez et al., 2012). After inactivation of endogenous peroxydases (3% H2O2 in PBS for 30 min), slices were blocked in 1% bovine serum albumin (BSA) in PBS containing 0.3% Triton X-100 for 30 min. Thereafter, sections were incubated in primary antibody (1:10000 monoclonal anti-TH; MAB318, Millipore Bioscience Research Reagents) overnight. After rinsing, the sections were treated with the secondary antibody (1:1000 biotinylated horse anti-mouse IgG; Vector Laboratories) for 90 min. Both immunoreagents were diluted in PBS containing 1% BSA, 0.3% Triton X-100. Finally, sections were incubated in avidin-biotin peroxidase complex (1:500; Vector Laboratories) for 60 min and immunoreactivity was revealed using diaminobenzidine tetrahydrochloride (Vector Laboratories). Sections were rinsed, mounted on gelatin-coated slides, and coverslipped in vectamount (Vector Laboratories). The entire procedure was performed at room temperature under gentle agitation.
Tyrosine-hydroxylase optical density.
Sections containing the striatum were placed on an optical bench and scanned for further processing. Mean optical density was analyzed using the Mercator image analysis system (Explora Nova). Values were corrected for background staining. TH optical density levels were expressed as a percentage of the values from the intact side. Optical density was performed in slices containing the striatum from sham and 6-OHDA-lesioned animals. Animals with unilateral 6-OHDA infusions included in the study showed 88.11 ± 1.3% (n = 15) reduction in TH-fiber density in the striatum on the side ipsilateral to the lesion.
Western blots
Single slices were transferred to a recording chamber in similar conditions as described above and processed for LTD protocol alone or in combination with SCH23390 (5 μm) or H89 (10 μm). SNr were then carefully dissected under a dissecting microscope and flash-frozen in a Snapfrost (Cryobain) 30 min after LTD induction as described for electrophysiological recordings. They were then sonicated in 1% SDS and Western blotting was performed as described previously (Santini et al., 2007) using antibodies against phospho-Ser897-GluN1 (1:1000, Millipore). Beta-actin was determined as a loading control using an antibody from Abcam (1:50000). Signal was produced by ECL Plus (GE Healthcare), detected by exposure to film (GE Healthcare Hyperfilm ECL, GE Healthcare) and quantified using software from Li-Cor.
Data analysis and statistics
The amplitudes and kinetics of eEPSCs were calculated from the baseline current preceding each individual stimulation artifact using Clampfit 10.1 (Molecular Devices). Stimulation artifacts were decreased off-line for clarity. The magnitude of LTD was calculated as the average EPSC amplitude at 30–35 min as a percentage of the average baseline (0–5 min) EPSC amplitude and reported in the text as the percentage of baseline ±SEM. Detection and analysis of mEPSCs were performed using a software developed in house. Statistical significance was evaluated using either a paired t test for intragroup comparison or a one-way ANOVA followed by Tukey's post hoc test for between-group comparison. Current amplitudes and kinetics were compared using the two-tailed unpaired t test. Given the small sample size of mEPSCs (n < 10) nonparametric Mann–Whitney U test were used for statistical comparison of this set of data. Western blot data were analyzed by one-way ANOVA, followed by Bonferroni post hoc test. Significance was considered at p < 0.05. Data are represented as group means ± SEM.
Results
Properties of intact and DA-denervated STN-SNr connection
To investigate and compare the properties of STN-SNr synapses from control and DA-depleted animals, electrical stimulations were applied to the posterior part of the STN while recording eEPSCs in SNr GABAergic neurons held at −70 mV in the presence of GABAA and GABAB receptor antagonists SR95531 (10 μm) and CGP55845 (5 μm), respectively (Fig. 1a). Briefly, the respective contributions of AMPA and NMDA receptors to the eEPSCs were evaluated by adding either the AMPA/kainate receptor antagonist DNQX (10 μm) or the NMDA receptor antagonist d-APV (50 μm) to the bath saline. Whereas DNQX reduced eEPSC amplitudes by 89.3 ± 1.4% (n = 12; p < 0.0001, two-tailed unpaired t test), d-APV yielded an amplitude reduction of 17.8 ± 9.2% (n = 12; p = 0.349) suggesting that AMPA receptors are the main contributors to evoked EPSCs at hyperpolarized holding potentials (data not shown). To determine whether the density or subunit composition of synaptic AMPA receptors might be affected by DA denervation, we further examined eEPSC amplitudes, kinetics, and current–voltage relationships in intact and DA-depleted situations. In both conditions, eEPSCs showed similar current–voltage relationships (I–V curve slope: control, 1.52 ± 0.20 pA/mV, n = 9; DA-depleted, 1.90 ± 0.31 pA/mV, n = 8; p = 0.307, two-tailed unpaired t test; Fig. 2a) and inward rectification index (control, 0.78 ± 0.12, n = 9; 6-OHDA, 0.70 ± 0.09, n = 8; p = 0.604), as well as similar amplitudes (control, 87.2 ± 2 pA, n = 35; DA-depleted, 91.1 ± 7 pA, n = 35; p = 0.55; Fig. 2bi), rise time (control, 1.22 ± 0.08 ms, n = 35; DA-depleted, 1.05 ± 0.08 ms, n = 35; p = 0.129; Fig. 2bii), and decay (control, 7.8 ± 0.5 ms, n = 35; DA-depleted, 7.1 ± 0.6 ms, n = 35; p = 0.397; Fig. 2biii), indicating that DA depletion does not affect synaptic AMPA receptor composition or properties. This conclusion was strengthened by spontaneous EPSCs recordings (sEPSCs) which showed similar amplitudes (control, 21 ± 2.2 pA, n = 10; DA-depleted, 20.9 ± 2.4 pA, n = 10; p = 0.912) and decay kinetics (control, 4.4 ± 0.4 ms, n = 10; DA-depleted, 3.5 ± 0.4 ms, n = 10; p = 0.105) in control and DA-depleted context (Fig. 2c). Comparative measurement of the ratio of AMPAR-(peak current, Vhold = −60 mV) to NMDAR-mediated EPSCs (current at 50 ms poststimulation, Vhold = +40 mV) did not reveal any significant difference (control, 3.08 ± 0.34, n = 10; DA-depleted, 3.07 ± 0.43, n = 7; p = 0.99; Fig. 2d), suggesting similar densities of synaptic NMDA receptors in intact and DA-depleted conditions. Finally, we investigated whether glutamate release from STN afferents was changed after DA denervation by applying paired stimulations over a range of frequencies (1, 2, 5, 10, 20, 50, 100 Hz). Paired-pulse recordings revealed that STN-SNr synapses from control and 6-OHDA-lesioned animals were facilitated at high stimulation frequencies and had comparable paired-pulse ratios (PPRs; 50 ms; control, 1.37 ± 0.10, n = 19; 6-OHDA, 1.38 ± 0.06, n = 22; p = 0.953; Fig. 2e). The lesion thus did not affect the probability of neurotransmitter release at STN terminals. This statement was further confirmed through the recording of mEPSCs (Fig. 2f), which did not reveal any difference in mEPSC amplitude (p = 0.138; Fig. 2fii) or frequency (p = 0.943; Fig. 2fiii) across control and DA-depleted conditions.
Figure 2.
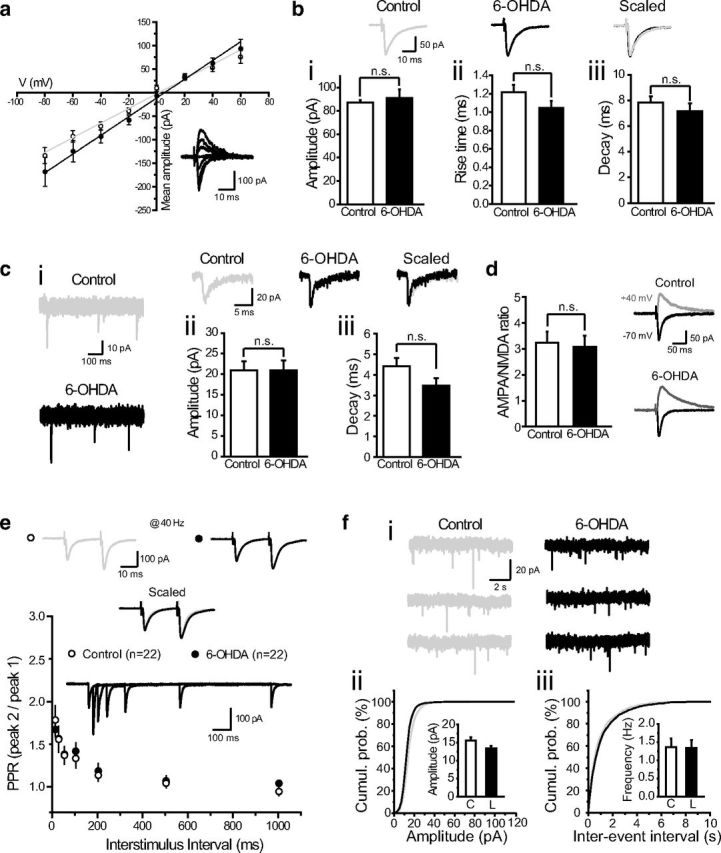
Properties of subthalamo-nigral glutamatergic synapses derived from intact and dopamine-denervated animals. a, Current–voltage relationships of AMPAR-mediated eEPSCs at STN-SNr synapses in control (open circles, n = 9) and DA-depleted animals (6-OHDA; filled circles, n = 8). Insets, Representative STN-SNr AMPAR-mediated eEPSCs recorded at membrane potentials of −80 mV to +60 mV (20 mV-step increments). b, Average amplitude (bi, peak current at −70 mV), rise time (bii), and decay (biii) of eEPSCs in control (white, n = 35) or DA-depleted (black, n = 35) conditions. Insets, Representative eEPSCs recorded at −70 mV from control (gray) and 6-OHDA-treated (black) slices. c, Spontaneous subthalamo-nigral EPSCs (ci, sEPSCs). Average amplitude (cii, peak current at −70 mV) and decay (ciii) of sEPSCs in control (white, n = 10) or DA-depleted (black, n = 10) conditions. Insets, Representative sEPSCs recorded at −70 mV from control (gray) and 6-OHDA-treated (black) slices. d, AMPA/NMDA ratio at STN-SNr synapses in control (n = 13) and DA-depleted conditions (n = 7). Insets, Representative eEPSC traces recorded at −70 mV (black) and +40 mV (gray) from control (top) and DA-depleted slices (bottom). e, Paired-pulse facilitation of STN-SNr transmission. PPRs (peak EPSC2/peak EPSC1) in control (open circles, n = 22) and DA-depleted slices (black circles, n = 22) are represented as a function of interstimulus interval. Control versus DA-depleted, p = 0.98, two-way ANOVA with Bonferroni's post hoc test. Insets, Traces from a representative individual trial, and eEPSCs from control (gray) and lesioned (black) slices recorded at a stimulation frequency of 40 Hz. f, Miniature EPSCs (fi, mEPSCs) recorded from control (gray) or lesioned (black) animals in the presence of tetrodotoxin (1 μm). Cumulative probability plots for mEPSC amplitude (fii) and interevent interval (fiii) in control (gray line, n = 7) and DA-depleted conditions (black line, n = 6). Insets, Average amplitude and frequency of mEPSCs in control (white, n = 7) or DA-depleted (black, n = 6) conditions (control vs DA-depleted, p > 0.05).
STN-SNr transmission exhibits LTD in naive animals but not in a DA-depleted context
We next studied whether activity-dependent long-term changes can occur at STN-SNr synapses. In all experiments, plasticity induction was preceded by a baseline acquisition of 10 min duration during which glutamatergic afferent inputs received paired stimulations (interstimulus interval = 50 ms) at low-frequency (0.05 Hz) to prevent any time-dependent drift in the amplitude of AMPAR-mediated eEPSCs. Plasticity protocols were then applied within 12 min after whole-cell access to avoid intracellular dialysis-related impairments of plasticity (Malinow and Tsien, 1990). Surprisingly, challenging STN-SNr synapses with tetanic stimulation (tetanus, 4 × 100 Hz − 1 s) to excitatory STN inputs while recording SNr neurons in current-clamp mode produced a robust LTD of the eEPSCs (62.6 ± 2.1% of baseline, n = 25; p < 0.0001, two-tailed paired t test; Fig. 3a). No significant difference in PPRs was observed before and after the induction of plasticity (PPR at 20 Hz before plasticity, 1.25 ± 0.03; PPR at 20 Hz after plasticity, 1.29 ± 0.06, n = 25; p = 0.359, two-tailed paired t test; Fig. 3b), suggesting no presynaptic change in the release probability of glutamate and a postsynaptic locus of expression. Interestingly, when applied to brain slices obtained from DA-depleted animals, tetanic stimulation did not produce any change in the amplitude of eEPSCs (105.1 ± 2.5% of baseline, n = 13; p = 0.09, two-tailed paired t test; Fig. 3c) nor change in PPR (PPR at 20 Hz before plasticity, 1.24 ± 0.03; PPR at 20 Hz after plasticity, 1.23 ± 0.04, n = 13; p = 0.842; Fig. 3d), indicating that this plastic property is lost in experimental parkinsonism and that the expression of tetanus-induced STN-SNr LTD might require DAergic signaling.
Figure 3.

LTD of subthalamo-nigral transmission is lost in dopamine-depleted animals. a, Tetanic stimulation-induced (tetanus) LTD of STN-SNr transmission. Normalized eEPSC amplitudes (normalization to the mean amplitude of eEPSCs recorded during baseline acquisition) are plotted against time (n = 25). a–d, Arrow indicates the time of induction (tetanus), and error bars represent SEM. Inset, Traces from a representative experiment illustrating the average eEPSC from 0 to 5 min (1) and 30 to 35 min (2). b, Evolution of the PPR during LTD induction experiments. PPRs are plotted against time. Inset, Traces from a representative experiment illustrating the average eEPSC from 0 to 5 min (1, black) and 30 to 35 min (2, gray). c, LTD induction protocol at STN-SNr synapses from DA-depleted animals (n = 13). The gray line represents the shape of control tetanus-induced LTD from Figure 2a for comparison (control vs 6-OHDA, p < 0.0001; one-way ANOVA with Tukey's post hoc test). Inset, Traces from a representative experiment illustrating the average eEPSC from 0 to 5 min (1) and 30 to 35 min (2). d, Evolution of the PPR during LTD induction experiments performed on slices from DA-depleted animals. PPRs are plotted against time. Insets, Traces from a representative experiment illustrating the average eEPSC from 0 to 5 min (1, black) and 30 to 35 min (2, gray).
NMDA-dependent endocytosis of AMPA receptors at STN-SNr synapses
We then investigated the molecular pathways involved in the onset of STN-SNr LTD. Because LTD induction at corticostriatal synapses was previously shown to require the activation of metabotropic glutamate receptors (Calabresi et al., 1992b; Kreitzer and Malenka, 2007), we examined their possible involvement in tetanus-induced LTD at STN-SNr synapses. We repeated the protocol in the presence of the large-spectrum group I/II metabotropic glutamate receptor antagonist, MCPG (250 μm). Surprisingly, MCPG did not prevent LTD induction (71.7 ± 1.4% of baseline, n = 15; p < 0.0001, two-tailed paired t test; Fig. 4a). Implication of endocannabinoid-associated signalization was also ruled out as bath application of the cannabinoid CB1 receptor antagonist AM251 comparably did not prevent LTD induction (10 μm; 59 ± 1.4% of baseline, n = 13; p = 0.035, two-tailed paired t test; Fig. 4b).
Figure 4.
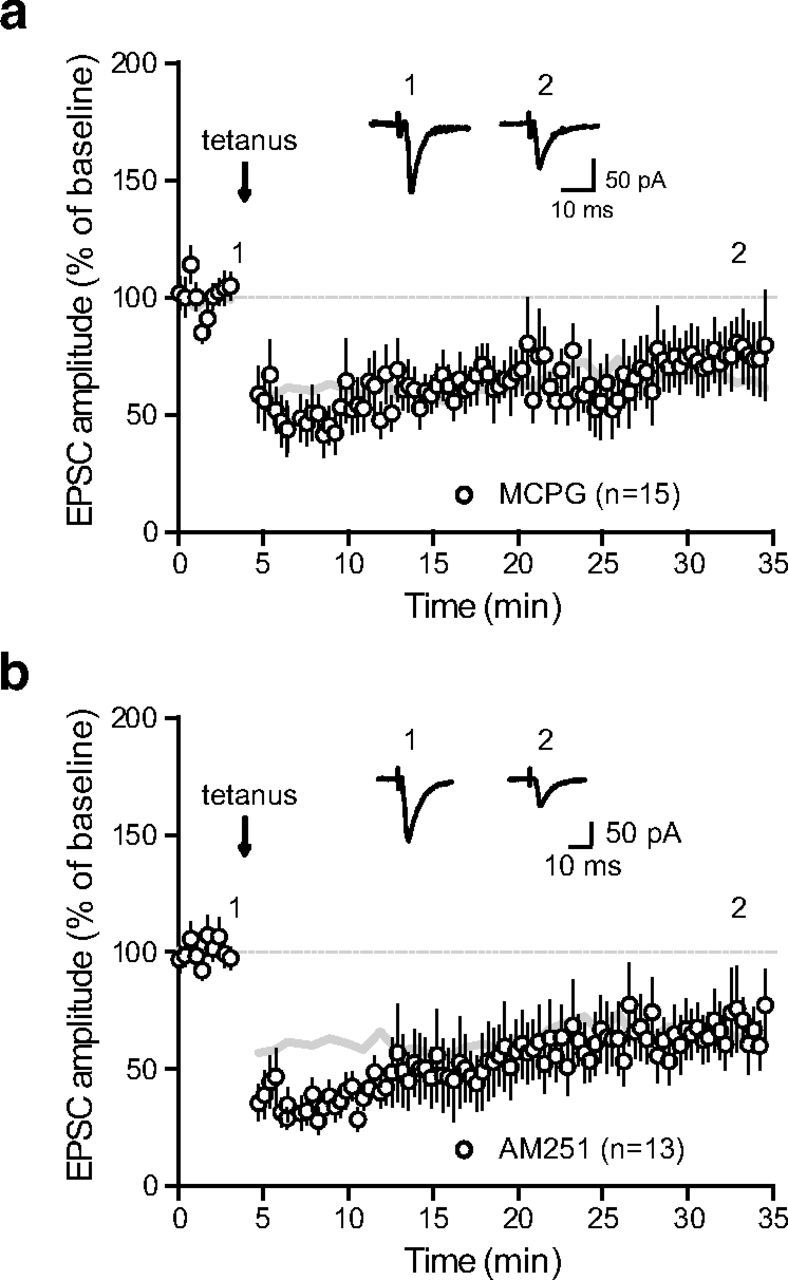
LTD is mGluR-, eCB-independent. a, LTD induction protocol in the presence of the group I/II metabotropic glutamate receptor antagonist, MCPG (250 μm; n = 15). Inset: traces from a representative experiment illustrating the average eEPSC from 0 to 5 min (1) and 30 to 35 min (2). The gray lines in panels (a) and (b) represent the shape of control tetanus-induced LTD from Figure 2a for comparison. b, LTD induction protocol in the presence of the cannabinoid CB1 receptor antagonist AM251 (10 μm; n = 13; control vs AM251, p = 0.0002, one-way ANOVA with Tukey's post hoc test). Insets, Traces from a representative experiment illustrating the average eEPSC from 0 to 5 min (1) and 30 to 35 min (2).
As unchanged PPR argued in favor of a postsynaptic mechanism, we then turned to assess the implication NMDA receptors in the induction of LTD by repeating the protocol in the presence of the NMDA receptor antagonist d-APV (50 μm). Tetanus-induced LTD was completely prevented under this condition (100.8 ± 2.8% of baseline, n = 19; p = 0.647, two-tailed paired t test; Fig. 5a). Importantly, LTD was also abolished when applying the calcium chelator BAPTA (10 μm) inside the patch pipette, indicating that chelating postsynaptic calcium entry, presumably through NMDA receptors, is sufficient to prevent plasticity induction (110.4 ± 2.9% of baseline, n = 11; p = 0.536, two-tailed paired t test; Fig. 5b). Because several studies have provided evidence that various forms of NMDA-dependent LTD involve a reduction in the number of postsynaptic AMPA receptors through enhanced clathrin-mediated endocytosis of GluA2-containing AMPAR (Lüscher et al., 1999; Bredt and Nicoll, 2003), we next examined whether tetanus-induced STN-SNr LTD involved a regulation of AMPA receptor trafficking. To do so, patch pipettes were filled with a synthetic peptide derived from the GluA2 carboxy tail, GluA23Y (YKEGYNVYG, 100 μg/ml), known to block activity-dependent, but not constitutive, endocytosis of GluA2-containing AMPA receptors without affecting basal synaptic transmission (Ahmadian et al., 2004; Yu et al., 2008). LTD was prevented by the postsynaptic application of GluA23Y (100.9 ± 2% of baseline, n = 19; p = 0.6, two-tailed paired t test) but not by the mutated inactive peptide GluA23A (AKEGANVAG, 72.6 ± 1% of baseline, n = 16; p < 0.0001, two-tailed paired t test; Fig. 5c). These results indicate that tetanus-induced LTD occurs through the activity-dependent endocytosis of GluA2-containing AMPA receptors (Fig. 5d).
Figure 5.

LTD occurs through postsynaptic, NMDA-dependent endocytosis of GluA2-containing AMPA receptors. a, LTD induction protocol in the presence of the NMDA receptor antagonist d-APV (50 μm; n = 19). Inset, Traces from a representative experiment illustrating the average eEPSC from 0 to 5 min (1) and 30 to 35 min (2). The gray lines in a and b represent the shape of control tetanus-induced LTD from Figure 2a for comparison (control vs d-APV, p < 0.0001; one-way ANOVA with Tukey's post hoc test). b, LTD induction protocol in the presence of the calcium chelator BAPTA inside the patch pipette (10 μm; n = 11). Inset, Traces from a representative experiment illustrating the average eEPSC from 0 to 5 min (1) and 30 to 35 min (2). c, LTD induction is prevented in the presence of GluA23Y peptide (filled circles, n = 19) but not GluA23A peptide (open circles, n = 16) inside the patch pipette (GluA23A vs GluA23Y, p < 0.0001; one-way ANOVA with Tukey's post hoc test). Inset, Traces from representative experiments illustrating the average eEPSC from 0 to 5 min (black) and 30 to 35 min (gray), in the presence of GluA23A (open circles) or GluA23Y (filled circles). d, Schematic representation illustrating the potential signaling pathway underlying tetanus-induced LTD. Tetanic-stimulation induced calcium entry through NMDA receptors activates phosphorylation cascades, leading to the C-terminal phosphorylation of AMPA receptors and their subsequent internalization.
Thus, it appears that nigral NMDA receptors are activated during tetanic stimulation of STN excitatory inputs to SNr, leading to a long-term downregulation of glutamatergic transmission through the activity-dependent endocytosis of AMPA receptors.
DAergic modulation of STN-SNr LTD
GABAergic neurons from the SNr receive an important dendritic DAergic innervation coming from the SNc which contributes to the regulation of their excitability, either directly (Zhou et al., 2009) or indirectly through the modulation of striato-nigral inhibitory transmission (Radnikow and Misgeld, 1998). However, DA might also modulate SNr neuronal firing by acting on STN-SNr excitatory transmission (Ibañez-Sandoval et al., 2006). We therefore investigated whether endogenous DA release might contribute to the regulation of STN-SNr synaptic plasticity, as shown for excitatory transmission in the striatum. Considering the postsynaptic location of LTD expression and the fact that SNr neurons predominantly express D1/5R (Zhou et al., 2009; Kliem et al., 2010), we studied the action of the D1/5R-selective antagonist SCH23390 on the expression of STN-SNr LTD. Plasticity expression was prevented by the addition of SCH23390 (5 μm) to the bath (99.6 ± 2.1% of baseline, n = 16; p = 0.911, two-tailed paired t test; Fig. 6a). D1/5R-associated intracellular signaling therefore contributes to the induction of LTD. To confirm this observation, we next tested potential signaling pathways downstream of D1/5R. The Gs-coupled D1/5R positively regulates the production of cAMP by adenylyl cyclase, which, in turn, targets well characterized effectors including protein kinase A (PKA; Greengard, 2001). To question whether the D1/5R-associated contribution to LTD induction depends on PKA activation, we repeated the protocol in the presence of H89 (10 μm), a membrane-permeant inhibitor of PKA. Blocking PKA activity resulted in impaired plasticity, demonstrating that LTD induction requires D1/5R-associated signaling (103.1 ± 1.6% of baseline, n = 14; p = 0.138, two-tailed paired t test; Fig. 6b). These data suggest that D1/5R activation likely modulates NMDA signaling through PKA-dependent mechanisms. This hypothesis was further supported by monitoring the phosphorylation levels of the NMDA receptor GluN1 subunit at Ser897, a residue targeted by PKA (Tingley et al., 1997). Although LTD induction triggered a significant increase in the PKA-dependent phosphorylation of GluN1 (p = 0.007, one-way ANOVA with Bonferroni's post hoc test; Fig. 6c), this rise was prevented by the application of either the D1/5R antagonist SCH23390 or the PKA inhibitor H89 (Fig. 6c). Together, these results indicate that SNr NMDAR and D1/5R synergistically contribute to the long-term downregulation of STN-SNr excitatory transmission.
Figure 6.
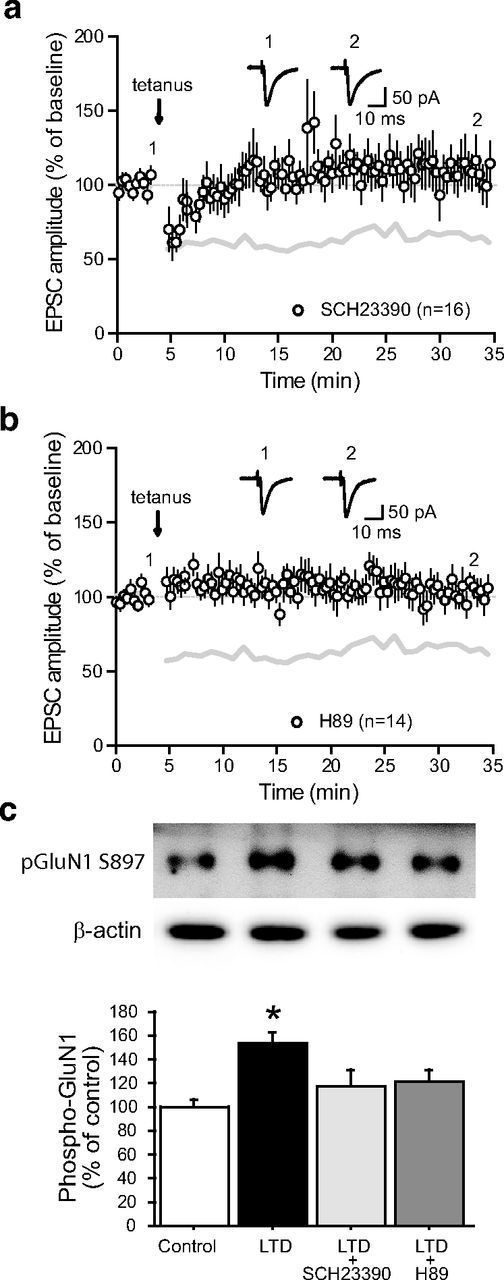
Acute dopaminergic modulation of subthalamo-nigral LTD. a, LTD induction protocol in the presence of the D1/5 receptor antagonist SCH23390 (5 μm; n = 16). Inset, Traces from a representative experiment illustrating the average eEPSC from 0 to 5 min (1) and 30 to 35 min (2). The gray lines in a and b represent the shape of control tetanus-induced LTD from Figure 2a for comparison (control vs SCH23390, p < 0.0001; one-way ANOVA with Tukey's post hoc test). b, LTD induction protocol in the presence of the PKA antagonist H89 (10 μm; n = 14; control vs H89, p < 0.0001; one-way ANOVA with Tukey's post hoc test). Inset, Traces from a representative experiment illustrating the average eEPSC from 0 to 5 min (1) and 30 to 35 min (2). c, D1/5 receptors and PKA are specifically involved in the phosphorylation of the NMDA receptor GluN1 subunit during LTD induction. The levels of phospho[Ser-897]-GluN1 were determined as described in Materials and Methods. Top, Representative autoradiograms. Bottom, Summary of data expressed as means ± SEM (n = 3). The amount of phosphorylated GluN1 is expressed as a percentage of that determined in basal condition (control). The increased phosphorylation of GluN1 triggered by LTD induction was prevented by the D1/5 receptor antagonist SCH23390 (10 μm) and the PKA inhibitor H89 (10 μm). *p = 0.007 versus respective control group; one-way ANOVA followed by Bonferroni post hoc test.
D1/5R activation restores STN-SNr LTD in experimental parkinsonism
SNr neurons from DA-depleted slices did not show tetanus-induced LTD (Fig. 3c). Because similar results were observed when perfusing the D1/5R antagonist SCH23390 in slices from control animals, one may hypothesize that impaired plasticity in slices from 6-OHDA-lesioned animals results from disrupted D1/5R-mediated signaling. We thus finally tested whether activating D1/5R would rescue plasticity expression in a DA-depleted context. Interestingly, the simple addition of the D1/5R agonist SKF81297 (2 μm) to the bath was sufficient to restore LTD expression, confirming that endogenous DA-mediated activation of D1/5R is necessary to induce LTD at STN-SNr synapses (74.3 ± 1.8% of baseline, n = 13; p < 0.0001, two-tailed paired t test; Fig. 7a). To note, similar experiments performed while bathing slices from 6-OHDA-lesioned animals with the D2 receptor agonist quinpirole (5 μm) failed to reveal any plastic change (103.5 ± 1.5% of baseline, n = 13; p > 0.05; Fig. 7b), suggesting that D1-like dopamine receptors only are involved in the dopaminergic modulation of subthalamo-nigral LTD. Altogether, our results demonstrate that repetitive activation of STN inputs produces a LTD of excitatory synaptic transmission in the SNr that requires the proper coordination of both NMDAR-mediated glutamate signaling and D1/5R-mediated DAergic modulation, a property that is lost in a chronic DA depletion experimental model of PD. More importantly, this suggests that DA loss induces plastic impairments at this connection that governs the output of the basal ganglia and could thus contribute to the onset of parkinsonian symptoms.
Figure 7.
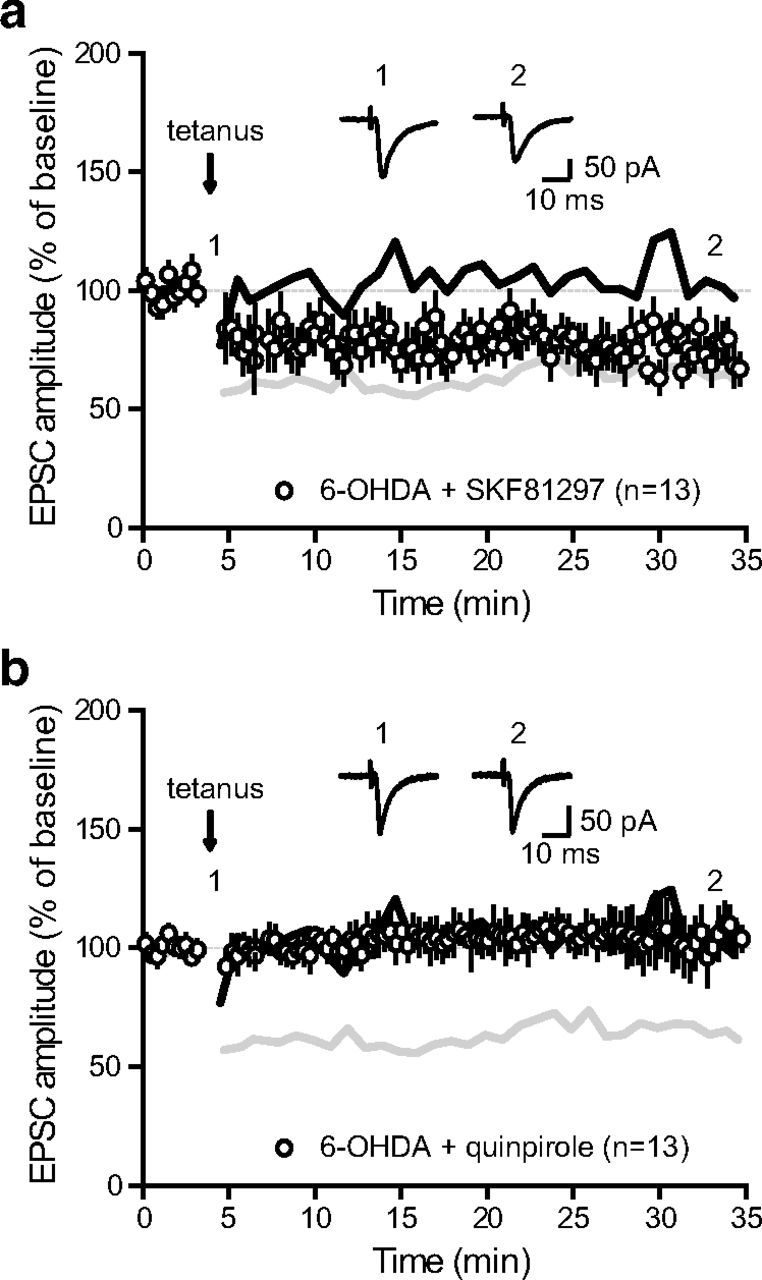
Activation of D1 receptors restores LTD induction in dopamine-depleted animals. a, LTD induction protocol at STN-SNr synapses from DA-depleted animals in the presence of the D1/5 receptor agonist SKF81297 (2 μm; n = 13). The gray and black lines in a and b represent the shapes of control LTD from Figure 2a and deficient LTD in DA-depleted animals from Figure 3c, respectively (6-OHDA + SKF81297 vs 6-OHDA, p < 0.0001; one-way ANOVA with Tukey's post hoc test). Inset, Traces from a representative experiment illustrating the average eEPSC from 0 to 5 min (1) and 30 to 35 min (2). b, LTD induction protocol at STN-SNr synapses from DA-depleted animals in the presence of the D2 receptor agonist quinpirole (5 μm; n = 13; 6-OHDA + quinpirole vs 6-OHDA, p > 0.05; one-way ANOVA with Tukey's post hoc test). Inset, Traces from a representative experiment illustrating the average eEPSC from 0 to 5 min (1) and 30 to 35 min (2).
Discussion
SNr neurons represent the main BG output and tonically inhibit motor portions of the thalamus. Thus, understanding the plastic properties of synaptic inputs regulating their activity is of particular relevance in the context of movement execution. We show here that challenging subthalamic afferents with tetanic stimulations triggers a robust LTD of subthalamo-nigral transmission through an NMDAR-mediated internalization of postsynaptic GluA2-AMPAR. Interestingly, we also demonstrate that nigral LTD requires endogenous DA release activating D1/5R-associated signaling, and that chronic DA depletion prevents this form of plasticity (Fig. 8), which has major implications regarding PD pathophysiology.
Figure 8.

Schematic representation illustrating the proposed signaling pathway by which D1/5 receptors modulate tetanus-induced LTD. Through the Gs-protein they are coupled with, D1/5 receptors enhance PKA activity which in turn modulates NMDA-dependent pathways including NMDA-dependent AMPA receptor endocytosis (a). This signaling pathway is lost in PD, preventing D1/5 receptor-mediated phosphorylation of the NMDA receptor GluN1 subunit and subsequent NMDA-dependent endocytosis of AMPAR (b).
LTD of subthalamo-nigral glutamatergic transmission
LTD of excitatory transmission can either occur through persistent presynaptic alterations in glutamate release or postsynaptic changes in glutamate receptor density. AMPAR removal-mediated LTD has been extensively described throughout the brain and is commonly triggered by the synaptic activation of either NMDAR or mGluR. Here, we report that tetanic stimulation-induced LTD of STN-SNr transmission requires the activation of NMDAR and postsynaptic calcium influxes. Several pathways can lead to the activity-dependent internalization of AMPAR, including C-terminal tyrosine phosphorylation of GluA2 subunits by Src family kinases (Ahmadian et al., 2004), calcineurin/AKAP-related pathways (Jurado et al., 2010) or PKC signaling (Seidenman et al., 2003). Interestingly, infusing a mimetic peptide competing with GluA2-containing AMPAR for C-terminal tyrosine phosphorylation (GluA23Y) prevented plasticity induction, suggesting that tyrosine phosphorylation-induced removal of AMPAR is the mechanism underlying NMDAR-dependent subthalamo-nigral LTD.
NMDA receptors are heterotetrameric proteins formed of two mandatory GluN1 subunits combined with two GluN2 (A–C or D) or GluN3 (A or B) subunits, GluN2A- and GluN2B-containing receptors being the most frequently encountered in the CNS. Although both GluN2A- and GluN2B-NMDAR are required for plasticity induction (Yashiro and Philpot, 2008), several studies suggest that NMDAR-mediated LTD preferentially involves GluN2B-NMDAR (Liu et al., 2004; Tigaret et al., 2006). Interestingly, GluN2B is the main NMDA subunit expressed in SNr neurons (Tse and Yung, 2000; Suárez et al., 2010), which could explain why plasticity induction at these synapses essentially leans toward LTD. Moreover, although the consequences of DA depletion on nigral NMDAR composition have not been as extensively evaluated as in the striatum where a shift toward GluN2A-predominant NMDAR is observed (Paillé et al., 2010), a recent study reported that 6-OHDA-treated rats exhibit an identical expression of nigral NMDAR, consistent with our data (Fig. 2d), but display higher levels of phosphorylated GluN2B suggesting that changes in NMDA-mediated signaling could occur following DAergic denervation (Quintana et al., 2012).
DAergic control of STN-SNr LTD
DA is a major regulator of SNr neuronal activity and BG outputs. At the presynaptic level, both DA D1 and D2 receptors have been shown to regulate GABA (Radnikow and Misgeld, 1998; de Jesús Aceves et al., 2011) and glutamate release (Ibañez-Sandoval et al., 2006). Additionally, a D1/5R-mediated postsynaptic modulation of SNr neuronal excitability has also been reported (Zhou et al., 2009). Furthermore, acute blockade of both DA D1R and D2R induces a switch in the pattern of SNr neurons from tonic single-spike pacemaking to rhythmic bursting in vitro, presumably through combined modulations of synaptic efficacy and changes in membrane excitability (Zhou et al., 2009; de Jesús Aceves et al., 2011).
In this study, we describe a new postsynaptic D1/5R-mediated signaling pathway controlling the strength of STN-SNr synaptic inputs in an activity-dependent manner. This effect is mediated by endogenous DA presumably released from SNc neuronal dendrites (Cheramy et al., 1981) or SNr DAergic neurons which could represent up to 20% of the SN DAergic cells (González-Hernández and Rodríguez, 2000), as it is completely abolished by acute D1/5R blockade or chronic DA deprivation. To date, the crosstalk between D1/5R- and NMDAR-mediated signaling has been shown to occur either through indirect intracellular signaling pathways (for review, see Cepeda and Levine, 2006) or via direct interactions between receptors at the plasma membrane (Lee et al., 2002). Here, we show that the DAergic control of nigral LTD involves an intracellular interplay between D1/5R and NMDAR because tetanic stimulation induces a D1/5R-mediated phosphorylation of the GluN1 NMDAR subunit at Ser897, a residue targeted by the PKA pathway (Tingley et al., 1997). The intracellular D1/5R-mediated modulation of NMDAR function usually relies on cAMP/PKA- (Cepeda et al., 1993; Snyder et al., 1998), Src kinase- (Dunah et al., 2004; Hallett et al., 2006; Yang et al., 2012), or PKC-dependent phosphorylation processes (Chen et al., 2004; Li et al., 2010). Although we have not addressed the nature of these interactions in detail, the prevention of GluN1 phosphorylation and consequent disruption of SNr LTD by the PKA inhibitor H-89 strongly support the involvement of cAMP/PKA-mediated intracellular signaling in the control exerted by D1/5R on NMDAR-dependent nigral LTD.
Synaptic plasticity in the basal ganglia
Within the BG network, the corticostriatal synapse is by far the most studied excitatory connection because the early observation that long-term plasticity at this major BG input is impaired in rodent models of PD (Calabresi et al., 1992b), leading to several pioneering experiments demonstrating the critical role of corticostriatal plasticity in motor control (for review, see Lerner and Kreitzer, 2011). Here, we found that STN-SNr synapses governing the output of the network present similarities but also differences regarding plasticity when compared with this connection. Although both synapses are depressed by tetanic stimulation, the routes leading to LTD in the striatum and the SNr diverge. Striatal LTD has a presynaptic locus of expression and involves an mGluR- and L-type calcium channel-dependent postsynaptic release of endocannabinoids mediating a retrograde downregulation of presynaptic neurotransmitter release (Ronesi et al., 2004; Kreitzer and Malenka, 2005). In contrast, blocking group I/II mGluR or CB1 receptors does not prevent nigral LTD, which rather requires an NMDA-dependent endocytosis of postsynaptic AMPAR.
Another common feature is the requirement for endogenous DA and the consequent plasticity impairments in hypodopaminergic conditions, although striatal LTD induction requires D2-mediated signaling (Calabresi et al., 1992a; Kreitzer and Malenka, 2007) when nigral LTD depends on D1/5R activation. This finding suggests that synaptic plasticity at both the input and output of the BG network are impaired under DA deprivation. Interestingly, recent investigations at corticosubthalamic connections revealed both tetanic stimulation-induced postsynaptic forms of LTP and presynaptic forms of LTD, and a low-frequency stimulation-elicited presynaptic form of LTP, all of which are sensitive to DA depletion (Shen et al., 2003; Yamawaki et al., 2012). Thus, there could be a large diversity of DA-dependent plastic mechanisms within mesencephalic glutamatergic synapses regulating BG outputs.
Functional implications
We show here that subthalamo-nigral LTD is lost in experimental parkinsonism. According to BG functional models, STN-SNr LTD could be a synaptic mechanism involved in action selection by the BG output by favoring the selection of desired motor programs (Mink, 1996). Consequently, when this physiological process is impaired in PD, action selection is not properly achieved, leading to paucity of movement and bradykinesia.
Interestingly, lesioning the STN has been shown to decrease the bursting activity of SNr neurons in hemiparkinsonian rats (Murer et al., 1997; Tseng et al., 2000, 2001) and to reduce motor symptoms in MPTP-treated monkeys and PD patients (Bergman et al., 1990; Guridi et al., 1996), suggesting a participation of STN inputs in the emergence of abnormal nigral activity and motor impairment. This is consistent with a recent study on hemiparkinsonian rats providing evidence that the entrainment of SNr neurons to pathological rhythms could be achieved either through the striato-nigral direct pathway and/or the STN-SNr pathway (Brazhnik et al., 2012). Therefore, losing the ability to downregulate STN-SNr transmission through LTD might indeed facilitate the spreading of pathological activity to the SNr (Belluscio et al., 2003; Brazhnik et al., 2012) and contribute to the abnormal activity pattern of BG outputs observed in PD.
The fact that activating D1/5 receptors restores LTD expression suggests that, in addition to reinstating a proper regulation of cortical inputs at the striatal level, some of the effects of DA replacement therapies might occur through a rescue of STN-SNr plasticity. This could be the case for L-DOPA which has been suggested to provide therapeutic benefit from both striatal and nigral sites of action (Robertson and Robertson, 1988, 1989). This hypothesis is further supported by a recent clinical study on PD patients demonstrating that L-DOPA restores synaptic plasticity of striato- and pallido-nigral inhibitory inputs to SNr neurons through the recruitment of SNr DAergic D1R (Prescott et al., 2009). Furthermore, Aceves et al. (2011) recently demonstrated that striato-nigral GABAergic synapses show tetanus-induced bidirectional plasticity, expressing either LTD or D1/5R-dependent facilitation depending on whether nigral NMDAR are simultaneously activated by subthalamo-nigral glutamatergic inputs or not. Importantly, avoiding dopamine D1/5R activation favors striato-nigral LTD, suggesting that dopamine deprivation could at the same time prevent subthalamo-nigral downregulation and striato-nigral facilitation, both of which would increase the firing activity of SNr neurons and repress movement execution. SNr neurons can thus be envisioned as coincidence detectors switching the gate between direct and indirect basal ganglia pathways, and dysfunctions in the DAergic modulation of synaptic plasticity at excitatory and inhibitory inputs to SNr neurons could contribute to the genesis of PD-related pathological BG outputs and thereby participate in the emergence of pathological motor symptoms.
These results also provide interesting information regarding deep-brain stimulation of the STN (STN DBS), the most effective neurosurgical therapy for treating PD-related movement disorders (Benazzouz et al., 1993; Limousin et al., 1995). Particularly, because STN-SNr synapses propagate either pathological or STN DBS-generated inputs to nigral neurons (Bosch et al., 2011; Brazhnik et al., 2012), the absence of synaptic plasticity at these connections in a DA-depleted context could explain the clinical observation that motor symptoms of PD reappear within seconds to minutes after switching STN DBS off (Temperli et al., 2003), suggesting that the synaptic transmission of pathological rhythms to SNr neurons is rapidly reinstated after therapeutic stimulation ceases.
Footnotes
This work was supported by Agence Nationale pour la Recherche (Grant No. ANR-08-MNPS-035), Centre National de la Recherche Scientifique, and the ERA-NET Neuron 2nd Call for transnational research projects 2009 (Grant No. 2009 NEUR 005 03) and the Swedish Research Council (13482). C.M. was supported by a postdoctoral grant from the Basque government and M.F. by the Karolinska Institutet/National Institute of Health Graduate Partnership Program. The Université Bordeaux Segalen and the Centre National de la Recherche Scientifique provided the infrastructural support. We thank Michel Goillandeau who developed the software for the detection and analysis of synaptic events.
References
- Aceves JJ, Rueda-Orozco PE, Hernandez-Martinez R, Galarraga E, Bargas J. Bidirectional plasticity in striatonigral synapses: a switch to balance direct and indirect basal ganglia pathways. Learn Mem. 2011;18:764–773. doi: 10.1101/lm.023432.111. [DOI] [PubMed] [Google Scholar]
- Ahmadian G, Ju W, Liu L, Wyszynski M, Lee SH, Dunah AW, Taghibiglou C, Wang Y, Lu J, Wong TP, Sheng M, Wang YT. Tyrosine phosphorylation of GluR2 is required for insulin-stimulated AMPA receptor endocytosis and LTD. EMBO J. 2004;23:1040–1050. doi: 10.1038/sj.emboj.7600126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander GE, Crutcher MD. Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci. 1990;13:266–271. doi: 10.1016/0166-2236(90)90107-L. [DOI] [PubMed] [Google Scholar]
- Atherton JF, Bevan MD. Ionic mechanisms underlying autonomous action potential generation in the somata and dendrites of GABAergic substantia nigra pars reticulata neurons in vitro. J Neurosci. 2005;25:8272–8281. doi: 10.1523/JNEUROSCI.1475-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belluscio MA, Kasanetz F, Riquelme LA, Murer MG. Spreading of slow cortical rhythms to the basal ganglia output nuclei in rats with nigrostriatal lesions. Eur J Neurosci. 2003;17:1046–1052. doi: 10.1046/j.1460-9568.2003.02543.x. [DOI] [PubMed] [Google Scholar]
- Benazzouz A, Gross C, Féger J, Boraud T, Bioulac B. Reversal of rigidity and improvement in motor performance by subthalamic high-frequency stimulation in MPTP-treated monkeys. Eur J Neurosci. 1993;5:382–389. doi: 10.1111/j.1460-9568.1993.tb00505.x. [DOI] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, DeLong MR. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249:1436–1438. doi: 10.1126/science.2402638. [DOI] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, Karmon B, DeLong MR. The primate subthalamic nucleus: II. Neuronal activity in the MPTP model of parkinsonism. J Neurophysiol. 1994;72:507–520. doi: 10.1152/jn.1994.72.2.507. [DOI] [PubMed] [Google Scholar]
- Bosch C, Degos B, Deniau JM, Venance L. Subthalamic nucleus high-frequency stimulation generates a concomitant synaptic excitation–inhibition in substantia nigra pars reticulata. J Physiol. 2011;589:4189–4207. doi: 10.1113/jphysiol.2011.211367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazhnik E, Cruz AV, Avila I, Wahba MI, Novikov N, Ilieva NM, McCoy AJ, Gerber C, Walters JR. State-dependent spike and local field synchronization between motor cortex and substantia nigra in hemiparkinsonian rats. J Neurosci. 2012;32:7869–7880. doi: 10.1523/JNEUROSCI.0943-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/S0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Maj R, Mercuri NB, Bernardi G. Coactivation of D1 and D2 dopamine receptors is required for long-term synaptic depression in the striatum. Neurosci Lett. 1992ab;142:95–99. doi: 10.1016/0304-3940(92)90628-K. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci. 1992b;12:4224–4233. doi: 10.1523/JNEUROSCI.12-11-04224.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Pisani A, Mercuri NB, Bernardi G. Long-term potentiation in the striatum is unmasked by removing the voltage-dependent magnesium block of NMDA receptor channels. Eur J Neurosci. 1992c;4:929–935. doi: 10.1111/j.1460-9568.1992.tb00119.x. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Levine MS. Where do you think you are going? The NMDA-D1 receptor trap. Sci STKE. 2006;333:pe20. doi: 10.1126/stke.3332006pe20. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Buchwald NA, Levine MS. Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci U S A. 1993;90:9576–9580. doi: 10.1073/pnas.90.20.9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Greengard P, Yan Z. Potentiation of NMDA receptor currents by dopamine D1 receptors in prefrontal cortex. Proc Natl Acad Sci U S A. 2004;101:2596–2600. doi: 10.1073/pnas.0308618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheramy A, Leviel V, Glowinski J. Dendritic release of dopamine in the substantia nigra. Nature. 1981;289:537–542. doi: 10.1038/289537a0. [DOI] [PubMed] [Google Scholar]
- de Jesús Aceves J, Rueda-Orozco PE, Hernández R, Plata V, Ibañez-Sandoval O, Galarraga E, Bargas J. Dopaminergic presynaptic modulation of nigral afferents: its role in the generation of recurrent bursting in substantia nigra pars reticulata neurons. Front Syst Neurosci. 2011;5:6. doi: 10.3389/fnsys.2011.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Sirianni AC, Fienberg AA, Bastia E, Schwarzschild MA, Standaert DG. Dopamine D1-dependent trafficking of striatal N-methyl-d-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol Pharmacol. 2004;65:121–129. doi: 10.1124/mol.65.1.121. [DOI] [PubMed] [Google Scholar]
- González-Hernández T, Rodríguez M. Compartmental organization and chemical profile of dopaminergic and GABAergic neurons in the substantia nigra of the rat. J Comp Neurol. 2000;421:107–135. doi: 10.1002/(SICI)1096-9861(20000522)421:1<107::AID-CNE7>3.3.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Greengard P. The neurobiology of slow synaptic transmission. Science. 2001;294:1024–1030. doi: 10.1126/science.294.5544.1024. [DOI] [PubMed] [Google Scholar]
- Guridi J, Herrero MT, Luquin MR, Guillén J, Ruberg M, Laguna J, Vila M, Javoy-Agid F, Agid Y, Hirsch E, Obeso JA. Subthalamotomy in parkinsonian monkeys: behavioural and biochemical analysis. Brain. 1996;119:1717–1727. doi: 10.1093/brain/119.5.1717. [DOI] [PubMed] [Google Scholar]
- Hallett PJ, Spoelgen R, Hyman BT, Standaert DG, Dunah AW. Dopamine D1 activation potentiates striatal NMDA receptors by tyrosine phosphorylation-dependent subunit trafficking. J Neurosci. 2006;26:4690–4700. doi: 10.1523/JNEUROSCI.0792-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibañez-Sandoval O, Hernández A, Florán B, Galarraga E, Tapia D, Valdiosera R, Erlij D, Aceves J, Bargas J. Control of the subthalamic innervation of substantia nigra pars reticulata by D1 and D2 dopamine receptors. J Neurophysiol. 2006;95:1800–1811. doi: 10.1152/jn.01074.2005. [DOI] [PubMed] [Google Scholar]
- Jurado S, Biou V, Malenka RC. A calcineurin/AKAP complex is required for NMDA receptor-dependent long-term depression. Nat Neurosci. 2010;13:1053–1055. doi: 10.1038/nn.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliem MA, Pare JF, Khan ZU, Wichmann T, Smith Y. Ultrastructural localization and function of dopamine D1-like receptors in the substantia nigra pars reticulata and the internal segment of the globus pallidus of parkinsonian monkeys. Eur J Neurosci. 2010;31:836–851. doi: 10.1111/j.1460-9568.2010.07109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K, Kreitzer AC. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010;466:622–626. doi: 10.1038/nature09159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Lee FJ, Xue S, Pei L, Vukusic B, Chéry N, Wang Y, Wang YT, Niznik HB, Yu XM, Liu F. Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell. 2002;111:219–230. doi: 10.1016/S0092-8674(02)00962-5. [DOI] [PubMed] [Google Scholar]
- Lerner TN, Kreitzer AC. Neuromodulatory control of striatal plasticity and behavior. Curr Opin Neurobiol. 2011;21:322–327. doi: 10.1016/j.conb.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YC, Liu G, Hu JL, Gao WJ, Huang YQ. Dopamine D(1) receptor-mediated enhancement of NMDA receptor trafficking requires rapid PKC-dependent synaptic insertion in the prefrontal neurons. J Neurochem. 2010;114:62–73. doi: 10.1111/j.1471-4159.2010.06720.x. [DOI] [PubMed] [Google Scholar]
- Limousin P, Pollak P, Benazzouz A, Hoffmann D, Le Bas JF, Broussolle E, Perret JE, Benabid AL. Effect of parkinsonian signs and symptoms of bilateral subthalamic nucleus stimulation. Lancet. 1995;345:91–95. doi: 10.1016/S0140-6736(95)90062-4. [DOI] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, Nicoll RA. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24:649–658. doi: 10.1016/S0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature. 1990;346:177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- Miguelez C, Morin S, Martinez A, Goillandeau M, Bezard E, Bioulac B, Baufreton J. Altered pallido-pallidal synaptic transmission leads to aberrant firing of globus pallidus neurons in a rat model of Parkinson's disease. J Physiol. 2012;590:5861–5875. doi: 10.1113/jphysiol.2012.241331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mink J. The basal ganglia: focused selection and inhibition of competing motor programs. Prog Neurobiol. 1996;50:381–425. doi: 10.1016/S0301-0082(96)00042-1. [DOI] [PubMed] [Google Scholar]
- Murer MG, Riquelme LA, Tseng KY, Pazo JH. Substantia nigra pars reticulata single unit activity in normal and 60HDA-lesioned rats: effects of intrastriatal apomorphine and subthalamic lesions. Synapse. 1997;27:278–293. doi: 10.1002/(SICI)1098-2396(199712)27:4<278::AID-SYN2>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Paillé V, Picconi B, Bagetta V, Ghiglieri V, Sgobio C, Di Filippo M, Viscomi MT, Giampà C, Fusco FR, Gardoni F, Bernardi G, Greengard P, Di Luca M, Calabresi P. Distinct levels of dopamine denervation differentially alter striatal synaptic plasticity and NMDA receptor subunit composition. J Neurosci. 2010;30:14182–14193. doi: 10.1523/JNEUROSCI.2149-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott IA, Dostrovsky JO, Moro E, Hodaie M, Lozano AM, Hutchison WD. Levodopa enhances synaptic plasticity in the substantia nigra pars reticulata of Parkinson's disease patients. Brain. 2009;132:309–318. doi: 10.1093/brain/awn322. [DOI] [PubMed] [Google Scholar]
- Quintana A, Sgambato-Faure V, Savasta M. Effects of L-DOPA and STN-HFS dyskinesiogenic treatments on NR2B regulation in basal ganglia in the rat model of Parkinson's disease. Neurobiol Dis. 2012;48:379–390. doi: 10.1016/j.nbd.2012.06.009. [DOI] [PubMed] [Google Scholar]
- Radnikow G, Misgeld U. Dopamine D1 receptors facilitate GABAA synaptic currents in the rat substantia nigra pars reticulata. J Neurosci. 1998;18:2009–2016. doi: 10.1523/JNEUROSCI.18-06-02009.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson GS, Robertson HA. Evidence that the substantia nigra is a site of action for L-DOPA. Neurosci Lett. 1988;89:204–208. doi: 10.1016/0304-3940(88)90382-5. [DOI] [PubMed] [Google Scholar]
- Robertson GS, Robertson HA. Evidence that L-dopa-induced rotational behavior is dependent on both striatal and nigral mechanisms. J Neurosci. 1989;9:3326–3331. doi: 10.1523/JNEUROSCI.09-09-03326.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi J, Gerdeman GL, Lovinger DM. Disruption of endocannabinoid release and striatal long-term depression by postsynaptic blockade of endocannabinoid membrane transport. J Neurosci. 2004;24:1673–1679. doi: 10.1523/JNEUROSCI.5214-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Valjent E, Usiello A, Carta M, Borgkvist A, Girault JA, Hervé D, Greengard P, Fisone G. Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J Neurosci. 2007;27:6995–7005. doi: 10.1523/JNEUROSCI.0852-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidenman KJ, Steinberg JP, Huganir R, Malinow R. Glutamate receptor subunit 2 serine 880 phosphorylation modulates synaptic transmission and mediates plasticity in CA1 pyramidal cells. J Neurosci. 2003;23:9220–9228. doi: 10.1523/JNEUROSCI.23-27-09220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen KZ, Zhu ZT, Munhall A, Johnson SW. Synaptic plasticity in rat subthalamic nucleus induced by high-frequency stimulation. Synapse. 2003;50:314–319. doi: 10.1002/syn.10274. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Fienberg AA, Huganir RL, Greengard P. A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci. 1998;18:10297–10303. doi: 10.1523/JNEUROSCI.18-24-10297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez F, Zhao Q, Monaghan DT, Jane DE, Jones S, Gibb AJ. Functional heterogeneity of NMDA receptors in rat substantia nigra pars compacta and reticulata neurones. Eur J Neurosci. 2010;32:359–367. doi: 10.1111/j.1460-9568.2010.07298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temperli P, Ghika J, Villemure JG, Burkhard PR, Bogousslavsky J, Vingerhoets FJ. How do parkinsonian signs return after discontinuation of subthalamic DBS? Neurology. 2003;60:78–81. doi: 10.1212/WNL.60.1.78. [DOI] [PubMed] [Google Scholar]
- Tigaret CM, Thalhammer A, Rast GF, Specht CG, Auberson YP, Stewart MG, Schoepfer R. Subunit dependencies of N-methyl-d-aspartate (NMDA) receptor-induced alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor internalization. Mol Pharmacol. 2006;69:1251–1259. doi: 10.1124/mol.105.018580. [DOI] [PubMed] [Google Scholar]
- Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-d-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem. 1997;272:5157–5166. doi: 10.1074/jbc.272.8.5157. [DOI] [PubMed] [Google Scholar]
- Tse YC, Yung KK. Cellular expression of ionotropic glutamate receptor subunits in subpopulations of neurons in the rat substantia nigra pars reticulata. Brain Res. 2000;854:57–69. doi: 10.1016/S0006-8993(99)02292-1. [DOI] [PubMed] [Google Scholar]
- Tseng KY, Riquelme LA, Belforte JE, Pazo JH, Murer MG. Substantia nigra pars reticulata units in 6-hydroxydopamine-lesioned rats: responses to striatal D2 dopamine receptor stimulation and subthalamic lesions. Eur J Neurosci. 2000;12:247–256. doi: 10.1046/j.1460-9568.2000.00910.x. [DOI] [PubMed] [Google Scholar]
- Tseng KY, Kasanetz F, Kargieman L, Pazo JH, Murer MG, Riquelme LA. Subthalamic nucleus lesions reduce low-frequency oscillatory firing of substantia nigra pars reticulata neurons in a rat model of Parkinson's disease. Brain Res. 2001;904:93–103. doi: 10.1016/S0006-8993(01)02489-1. [DOI] [PubMed] [Google Scholar]
- Yamawaki N, Magill PJ, Woodhall GL, Hall SD, Stanford IM. Frequency selectivity and dopamine-dependence of plasticity at glutamatergic synapses in the subthalamic nucleus. Neuroscience. 2012;203:1–11. doi: 10.1016/j.neuroscience.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Trepanier C, Sidhu B, Xie YF, Li H, Lei G, Salter MW, Orser BA, Nakazawa T, Yamamoto T, Jackson MF, Macdonald JF. Metaplasticity gated through differential regulation of GluN2A versus GluN2B receptors by Src family kinases. EMBO J. 2012;31:805–816. doi: 10.1038/emboj.2011.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SY, Wu DC, Liu L, Ge Y, Wang YT. Role of AMPA receptor trafficking in NMDA receptor-dependent synaptic plasticity in the rat lateral amygdala. J Neurochem. 2008;106:889–899. doi: 10.1111/j.1471-4159.2008.05461.x. [DOI] [PubMed] [Google Scholar]
- Zhou FW, Jin Y, Matta SG, Xu M, Zhou FM. An ultra-short dopamine pathway regulates basal ganglia output. J Neurosci. 2009;29:10424–10435. doi: 10.1523/JNEUROSCI.4402-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]