

Abstract
In demyelinating diseases such as multiple sclerosis, a critical problem is failure of remyelination, which is important for protecting axons against degeneration and restoring conduction deficits. However, the underlying mechanism of demyelination/remyelination remains unclear. N-acetylglucosaminyltransferase-IX (GnT-IX; also known as GnT-Vb) is a brain-specific glycosyltransferase that catalyzes the branched formation of O-mannosyl glycan structures. O-Mannosylation of α-dystroglycan is critical for its function as an extracellular matrix receptor, but the biological significance of its branched structures, which are exclusively found in the brain, is unclear. In this study, we found that GnT-IX formed branched O-mannosyl glycans on receptor protein tyrosine phosphatase β (RPTPβ) in vivo. Since RPTPβ is thought to play a regulatory role in demyelinating diseases, GnT-IX-deficient mice were subjected to cuprizone-induced demyelination. Cuprizone feeding for 8 weeks gradually promoted demyelination in wild-type mice. In GnT-IX-deficient mice, the myelin content in the corpus callosum was reduced after 4 weeks of treatment, but markedly increased at 8 weeks, suggesting enhanced remyelination under GnT-IX deficiency. Furthermore, astrocyte activation in the corpus callosum of GnT-IX-deficient mice was significantly attenuated, and an oligodendrocyte cell lineage analysis indicated that more oligodendrocyte precursor cells differentiated into mature oligodendrocytes. Together, branched O-mannosyl glycans in the corpus callosum in the brain are a necessary component of remyelination inhibition in the cuprizone-induced demyelination model, suggesting that modulation of O-mannosyl glycans is a likely candidate for therapeutic strategies.
Introduction
Demyelination occurs in many pathological conditions, but mainly in multiple sclerosis and spinal cord injury, and is accompanied by neurodegenerative disease. Cuprizone, a toxic copper chelator, has been used to generate demyelination model mice, because their demyelination and remyelination processes are easily observed. Cuprizone-induced demyelination involves a two-step mechanism for oligodendrocyte cell loss. Since oligodendrocytes are particularly vulnerable to oxidative damage and mitochondrial injury (Bradl and Lassmann, 2010), cuprizone administration initially causes metabolic stress in these cells (Torkildsen et al., 2008). The subsequent inflammatory response finally causes oligodendrocyte death (Pasquini et al., 2007; Liu et al., 2010). Withdrawal of cuprizone feeding during this process stimulates remyelination, which takes place in different steps, including proliferation of oligodendrocyte precursor cells, followed by their recruitment to demyelinated areas and differentiation into mature remyelinating oligodendrocytes. However, the mechanism for how microglial and astrocytic activation contributes to demyelination and remyelination remains unclear (Gao et al., 2000; Arnett et al., 2001; Mason et al., 2001; Pasquini et al., 2007).
Glycans, comprising sugar chains of variable length from a few to several hundred, have become increasingly recognized as important players in neural cell interactions in the developmental and adult nervous systems (Kleene and Schachner, 2004). For example, a brain-specific polysialic acid moiety on neural cell adhesion molecule negatively regulates myelin formation (Charles et al., 2000). Brain-specific glycans are synthesized by glycosyltransferases that are specifically expressed in the brain. We have focused on the brain-specific N-acetylglucosaminyltransferase-IX (GnT-IX), which was originally cloned as a homologous enzyme of GnT-V, an N-glycan-branching enzyme (Inamori et al., 2003; Kaneko et al., 2003). However, recent in vitro and cell culture studies have strongly suggested that GnT-IX is involved in β1,6-branch formation toward O-mannosyl glycans (Inamori et al., 2004; Abbott et al., 2008). An O-mannosyl glycan in α-dystroglycan plays a critical role for its proper function as an extracellular matrix receptor (Endo, 2004; Yoshida-Moriguchi et al., 2010). The limited occurrence of branched O-mannosyl glycans in the brain (Yuen et al., 1997; Stalnaker et al., 2011) can be explained by the brain-specific GnT-IX expression (Kizuka et al., 2011). Even though mutations in a glycosyltransferase for O-mannosyl glycans are associated with brain malformations (Yoshida et al., 2001; Michele et al., 2002; Inamori et al., 2012), the exact roles of O-mannosyl glycans in brain pathology are largely unknown.
In the present study, we used the Cat-315 antibody, which reacts with O-mannosyl glycans modified with the HNK-1 epitope (Matthews et al., 2002; Dino et al., 2006; Abbott et al., 2008), as a probe for GnT-IX reaction products, and found that GnT-IX modifies a brain-specific chondroitin sulfate proteoglycan, receptor protein tyrosine phosphatase β (RPTPβ; also designated Ptpz). Given the recent finding that RPTPβ plays a pivotal role in recovery from demyelinating disease (Harroch et al., 2002), we used the cuprizone-induced demyelination model, and found that GnT-IX-deficient mice, which lack branched O-mannosyl glycans, show enhanced remyelination with reduced astrogliosis. Our findings suggest that branched O-mannosyl glycans may act as a critical component for inhibition of the remyelination process.
Materials and Methods
Generation of GnT-IX-deficient mice.
A 19 kb region of the mouse GnT-IX (MGAT5B) gene containing exons 9 to 14 was isolated from the CITB Mouse BAC DNA Library (ResGen). A targeting vector was constructed by inserting a 1.6 kb pgk-neo cassette between two homologous flanking fragments of 8.3 kb KpnI-BamHI/BamHI-XbaI and 1.3 kb XbaI (artificial site generated by PCR)-HindIII, flanked with a pHSV-tk gene. The targeting vector was transfected into embryonic stem cells, and clones were selected with G418. Southern blot analysis for the detection of targeted embryonic stem cells was performed using a digoxigenin-labeled DNA probe generated by PCR with the primers 5′-TTCCCAGTCAGCTCCTC-3′ and 5′-GAGATGAATCAAGCAGG-3′. Genotyping was performed by either Southern blot analysis or PCR amplification of tail DNA. The primers for the PCR were 5′-GTGCAGAATCCGAGTCATCG-3′ (forward primer for wild-type allele), 5′-TCAGTCCCAGCTCTGGACACAGCTTCTA-3′ (reverse primer for wild-type and targeted alleles), and 5′-GCCTTCTATCGCCTTCTTGACGAGTTCTTC-3′ (forward primer for targeted allele). The targeted cell clones were then injected into C57BL/6 blastocysts and chimeric mice were generated (Wang et al., 2005).
Mice and antibodies.
GnT-V-deficient mice were described previously (Granovsky et al., 2000). All animal experiments were performed in compliance with the Institutional Guidelines for Animal Experiments of RIKEN. Anti-RPTPβ and anti-HNK-1 antibodies were generous gifts from Dr. Shogo Oka (Graduate School of Medicine, Kyoto University, Japan). The other antibodies used in this study were as follows: mouse monoclonal Cat-315 IgM (Millipore); anti-RPTPβ (BD Transduction Laboratories); anti-microtubule-associated protein 2 (MAP2) (Santa Cruz Biotechnology); anti-Olig2 (Millipore); anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Millipore Bioscience Research Reagents); anti-clathrin heavy chain (BD Transduction Laboratories); anti-5-bromo-2′-deoxyuridine (BrdU) IgG (Cell Signaling Technology); rat monoclonal anti-myelin basic protein (MBP) (Millipore); anti-glial fibrillary acidic protein (GFAP) (Invitrogen); anti-platelet-derived growth factor receptor α (PDGFRα) IgG (BD PharMingen); rabbit polyclonal anti-CD11b (Abcam); anti-GFAP (Dako Cytomation); anti-flotillin-1 (BD Transduction Laboratories); anti-Iba1 (Wako); anti-aggrecan (Millipore); and anti-PDGFRα IgG (Cell Signaling Technology).
Cuprizone administration.
Wild-type and GnT-IX-deficient mice (age and sex matched) were fed a powdered diet containing 0.2% (w/w) cuprizone, as described previously (Chen et al., 2004).
Isolation of primary neurons, astrocytes, oligodendrocytes, and oligodendrocyte precursor cells.
Cerebral neurons and astrocytes were isolated from the cerebral cortices of embryonic or neonatal C57BL/6NCrSlc mice, as previously described (Hama et al., 2001). Astrocytes were cultured in DMEM/F12 containing 10% fetal bovine serum and passaged twice before use. Oligodendrocytes were prepared from whole brains of adult mice by a Percoll gradient method, as previously described (Ma et al., 2006). Oligodendrocyte precursor cells were prepared from the cerebral cortices of neonatal mice by immunopanning using an anti-PDGFRα antibody (Cahoy et al., 2008) and cultured, as previously described (Dincman et al., 2012).
Immunofluorescence.
After fixation with methanol and blocking with 3% bovine serum albumin (BSA) in PBS, the cells were incubated with Cat-315 (1:500) and anti-GFAP (1:500), anti-RPTPβ (1:200), anti-MBP (1:500), or anti-MAP2 (1:200) antibodies. The cells were then incubated with appropriate Alexa Fluor 488- and 546-conjugated secondary antibodies (Invitrogen). To label proliferating oligodendrocyte precursor cells, the cultured cells were pulsed with 10 μm BrdU for 4 h, followed by immunostaining with the anti-BrdU antibody (1:1000). DAPI (Wako Pure Chemical Industries) was used as a counterstain for nuclei. After washing with PBS, the samples were mounted in CC/Mount (Diagnostic BioSystems) and observed using an FV1000-D laser-scanning confocal microscope (Olympus).
Immunohistochemistry.
To prepare brain sections, mice were transcardially perfused with PBS and 0.1 m phosphate-buffered 4% paraformaldehyde, and then sequentially immersed in the same fixative for 16 h and phosphate-buffered 30% sucrose for 16 h at 4°C. Klüver–Barrera staining was performed on brain sections (10 μm thickness) using Luxol fast blue solution (Muto Pure Chemical) for myelin sheath labeling and cresyl violet solution (Sigma-Aldrich) for Nissl staining. Briefly, brain sections were incubated in the Luxol fast blue solution for 24 h at 57°C, and then differentiated with 0.05% lithium carbonate solution. The sections were subsequently counterstained with 0.1% cresyl violet solution for 5 min at 37°C, and rinsed with 95% ethanol. Sagittal (10 μm thickness) or coronal (30 μm thickness) sections were immunostained based on a standard protocol using Alexa Fluor 546- or 488-conjugated secondary antibodies. For detection of intracellular proteins, the sections were permeabilized with 0.3% Triton X-100 in 1% BSA-PBS for 30 min before incubation with the primary antibodies. To detect perineuronal nets, we used biotinylated Wisteria floribunda agglutinin (WFA; 1:500 dilution) and Alexa Fluor 488-streptavidin. All stained sections were observed using the FV1000-D confocal laser-scanning microscope. Quantitative analysis was performed with the MetaMorph software (Molecular Devices).
Western and lectin blotting analyses.
Mouse brain tissues or corpus callosum parts, which were isolated from frozen cerebral hemispheres by microscopic analysis, were homogenized in T-PER Tissue Protein Extraction Reagent (Pierce) containing a protease inhibitor mixture (Roche). Brain lysates (50 μg protein) or immunoprecipitated samples were separated by SDS-PAGE using 4–20% gradient gels, and transferred to nitrocellulose membranes. After blocking with 5% nonfat milk in 20 mm Tris-buffered saline (TBS), pH 7.4, containing 0.5% Tween 20, the membranes were incubated with the Cat-315 (1:2000), anti-RPTPβ (1:1000), anti-HNK-1 (1:1000), anti-aggrecan (1:500), anti-GAPDH (1:1000), anti-clathrin heavy chain (1:1000), or anti-flotillin-1 (1:1000) antibodies. Horseradish peroxidase (HRP)-conjugated donkey anti-rabbit IgG (1:5000; GE Healthcare Life Sciences), sheep anti-mouse IgG (1:1000; GE Healthcare Life Sciences), and goat anti-mouse IgM (1:1000; Stressgen Bioreagents) were then used as the secondary antibodies. For lectin blot analysis, the membrane was blocked with 3% BSA in TBS, and incubated with HRP-conjugated PHA-L4 (1:1000). A SuperSignal chemiluminescent substrate (Thermo Fisher Scientific) or an ECL Advance Western Blotting Detection Kit (GE Healthcare) was used for detection. The signal intensities of the detected bands were quantified using a Luminoimage Analyzer LAS-1000 PLUS (Fuji Film).
Glycan analysis.
Tissue lysates (50 μg protein) were incubated with 1.6 mU of peptide N-glycosidase (PNGase) (New England BioLabs) for 2 h and/or 10 mU of chondroitinase ABC (Seikagaku) for 16 h at 37°C. For chemical deglycosylation, tissue lysates (50 μg protein) were lyophilized, dissolved in 50 μl of trifluoromethanesulfonic acid (TFMS), and incubated for 1 h on ice.
Real-time PCR analysis.
Total RNA was isolated from mouse brain tissues using TRIzol reagent (Invitrogen), and 5–10 μg of the RNA was reverse transcribed with random hexamers using a SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen) according to the manufacturer's protocol. The cDNA amplification and PCR conditions were described previously (Sugimoto et al., 2007). The probe for MBP was labeled with the fluorescent reporter dye FAM. The probe for GAPDH was labeled with VIC at its 5′ end and the quencher dye TAMRA at its 3′ end. The expression levels of the target gene were measured in duplicate and normalized by the corresponding GAPDH expression levels.
Glycosyltransferase activity assays.
GnT-IX activity was measured in brain microsomal fractions from wild-type and GnT-IX-deficient mice using GlcNAcβ1,2-Manα1-O-Ser-N-(7-nitro-2,1,3-benzoxadiazole-4-yl) (GnM-S-NBD) as an acceptor substrate according to our previously reported method (Inamori et al., 2003), with some modifications. Briefly, the enzyme reaction was performed at 37°C for 6 h in a final volume of 10 μl using a reaction mixture consisting of 50 mm HEPES buffer, pH 7.5, 5 mm EDTA, 0.5% Triton X-100, 20 mm UDP-GlcNAc, 50 μm GnM-S-NBD, 0.5 m l-glutamic acid, 1 mg/ml BSA, and 5 μl of crude enzyme extract.
Sucrose density gradient fractionation.
Detergent-resistant membrane fractions (DRMs) were isolated from the brains of wild-type and GnT-IX-deficient mice after 8 weeks of cuprizone administration, as described previously (Kitazume et al., 2009) with minor modifications. Briefly, half-brains were homogenized in five volumes of lysis buffer (25 mm MES, pH 6.5, 150 mm NaCl, 1% Triton X-100, and Complete protease inhibitor mixture), and incubated on ice for 30 min. The homogenates were centrifuged at 2000 × g for 10 min. The resulting supernatants were adjusted to a final sucrose concentration of 40%, and 2 ml aliquots were transferred to 11 ml ultracentrifuge tubes (13PA; Hitachi). A discontinuous sucrose gradient was formed by sequential layering of 30% sucrose (7 ml) and 5% sucrose (1 ml), and the tubes were ultracentrifuged at 260,000 × g for 21 h in a P40ST rotor (Hitachi). Ten 1 ml fractions were collected from the top. All fractions were treated with chondroitinase ABC, and analyzed by SDS-PAGE and Western blotting as described above.
Results
RPTPβ is a physiological substrate of GnT-IX
To generate GnT-IX-deficient mice, we designed a GnT-IX gene targeting vector in which exon 10, corresponding to part of the GnT-IX catalytic region, was replaced with a neomycin-resistance cassette to cause a frameshift downstream (Fig. 1A), and then disrupted the GnT-IX gene by homologous recombination in embryonic stem cells. The genotypes of pups from intercrosses between heterozygous mice were determined by Southern blotting (Fig. 1B, top) and PCR (Fig. 1B, bottom) analyses. Intercrosses of GnT-IX+/− mice produced ∼26% GnT-IX−/− mice, which was close to the normal Mendelian frequency. GnT-IX-deficient mice were equivalent in weight to littermate controls, healthy, fertile, and behaviorally normal. Using fluorescently labeled GlcNAcβ1,2-Manα1-O-Ser as an acceptor substrate for GnT-IX enzyme assays, we found that GnT-IX-deficient mouse brains completely lacked the enzyme activity (Fig. 1C).
Figure 1.
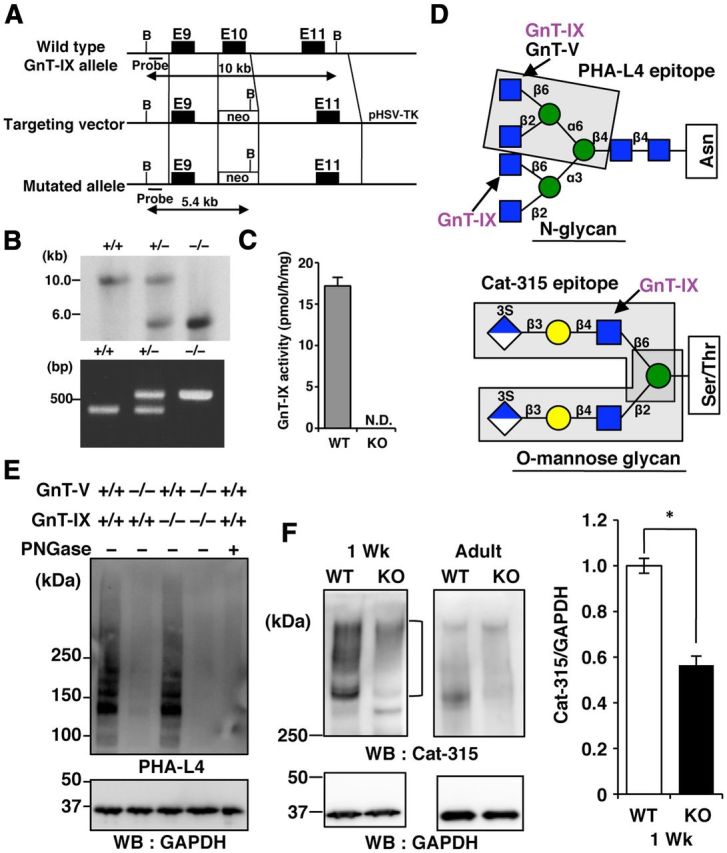
Cat-315 epitopes are reduced in the brain of GnT-IX-deficient (KO) mice. A, Structures of the wild-type (WT) GnT-IX allele, targeting construct, and targeted GnT-IX allele. The dark boxes represent exons. The open box shows the pgk-neo cassette. B, BamHI restriction site. The expected sizes of the BamHI digestion products of the gene are shown for the WT allele (10 kb) and mutated allele (5.4 kb). B, The top shows a Southern blot analysis of BamHI-digested genomic DNA derived from WT, heterozygous, and homozygous mutant mice hybridized with the probe. The bottom shows a PCR analysis of genomic DNA extracted from the tails of these mice. The wild-type allele generates a PCR product of 380 bp, while the mutant allele generates a product of 550 bp. C, The GnT-IX enzyme activities in the brain microsomal fractions prepared from WT and GnT-IX-deficient mice (KO) are presented as means ± SD (n = 3). N.D., not detected. D, The upper structure shows an N-linked glycan in which GnT-IX catalyzes the transfer of GlcNAc to both mannose arms via β1,6-linkage in vitro (Inamori et al., 2003). The enclosed residues show the PHA-L4 epitope. The lower structure shows an O-mannosyl glycan in which GnT-IX catalyzes the branched structure (Inamori et al., 2004). The enclosed residues show possible Cat-315 epitopes (Dino et al., 2006). E, Brain lysates from WT, GnT-V-deficient, GnT-IX-deficient, or double KO mice were incubated with or without PNGase, and then analyzed by PHA-L4 lectin blotting or Western blotting with an anti-GAPDH antibody. F, Brain lysates from WT and GnT-IX-deficient KO mice at 1 week of age (1 Wk) or 8–12 months of age (Adult) were analyzed by Western blotting with Cat-315 and anti-GAPDH antibodies (left). In the right graph, the bars represent the relative Cat-315 immunoreactivities shown as means ± SEM (n = 5). *p < 0.00005.
To clarify the enzymatic role of GnT-IX in vivo, we first decided to use PHA-L4 lectin, which preferentially recognizes a β1–6-GlcNAc branch on the mannose 6 arm of N-glycans, and the Cat-315 antibody, which is a probe for O-mannosyl glycans (Fig. 1D). The Cat-315 antibody was originally described to detect brain-specific chondroitin sulfate proteoglycans (Lander et al., 1998; Matthews et al., 2002), and has since been shown to react with O-mannosyl glycans with the HNK-1 epitope (Abbott et al., 2006, 2008; Dino et al., 2006). Deficiency of GnT-V, a homolog of GnT-IX, almost completely abolished the PHA-L4 epitope in the brain (Fig. 1E), as previously reported (Granovsky et al., 2000). On the other hand, GnT-IX deficiency had a negligible effect (Fig. 1E). These findings confirm that most PHA-L4 epitopes in the brain are derived from GnT-V products, rather than GnT-IX products.
It is known that RPTPβ is the only major molecule carrying the Cat-315 epitope at an early developmental stage (around postnatal day 14), and that the emergence of aggrecan, another Cat-315-carrying molecule, is observed at a later stage (Dino et al., 2006). Therefore, we used 1- or 2-week-old and adult brains from both wild-type and GnT-IX-deficient mice for biochemical analyses. The Cat-315 immunoreactivity observed at >300 kDa in the wild-type mouse brain at 1 week of age, which was considered to be RPTPβ, was significantly reduced in the brain of GnT-IX-deficient mice (Fig. 1F). Even though the Cat-315 epitope level was downregulated in the adult mouse brain, we were still able to observe that GnT-IX-deficient mice had a lower level of this epitope. Together, these findings suggest that GnT-IX is involved in branched O-mannosyl glycan formation in vivo, and that the remaining Cat-315 epitopes in GnT-IX-deficient mice are presumably HNK-1 epitopes connected to β1,2-linked O-mannosyl glycans (Stalnaker et al., 2011).
Next, we characterized the Cat-315 epitopes in wild-type and GnT-IX-deficient mouse brains. Western blot analyses using brain lysates from wild-type and GnT-IX-deficient mice showed that the HNK-1-positive signals with diffuse bands migrated at >300 kDa, similar to the Cat-315-positive signals, and that these signals were reduced in GnT-IX-deficient mice (Fig. 2A), confirming previous reports that Cat-315 detects the HNK-1 epitope (Dino et al., 2006; Abbott et al., 2008). After treatment with chondroitinase ABC or PNGase, the broad Cat-315-positive band was converted to a clearer and more intense discrete band (Fig. 2B), indicating that the Cat-315 epitope is carried by a chondroitin sulfate proteoglycan possessing N-glycans, as previously reported (Matthews et al., 2002; Dino et al., 2006). Furthermore, the finding that TFMS treatment, which removes both N- and O-glycans, abolished the Cat-315-positive signals (Fig. 2B), supports the previous suggestion that the Cat-315 epitope is an O-mannosyl glycan with the HNK-1 epitope (Dino et al., 2006).
Figure 2.
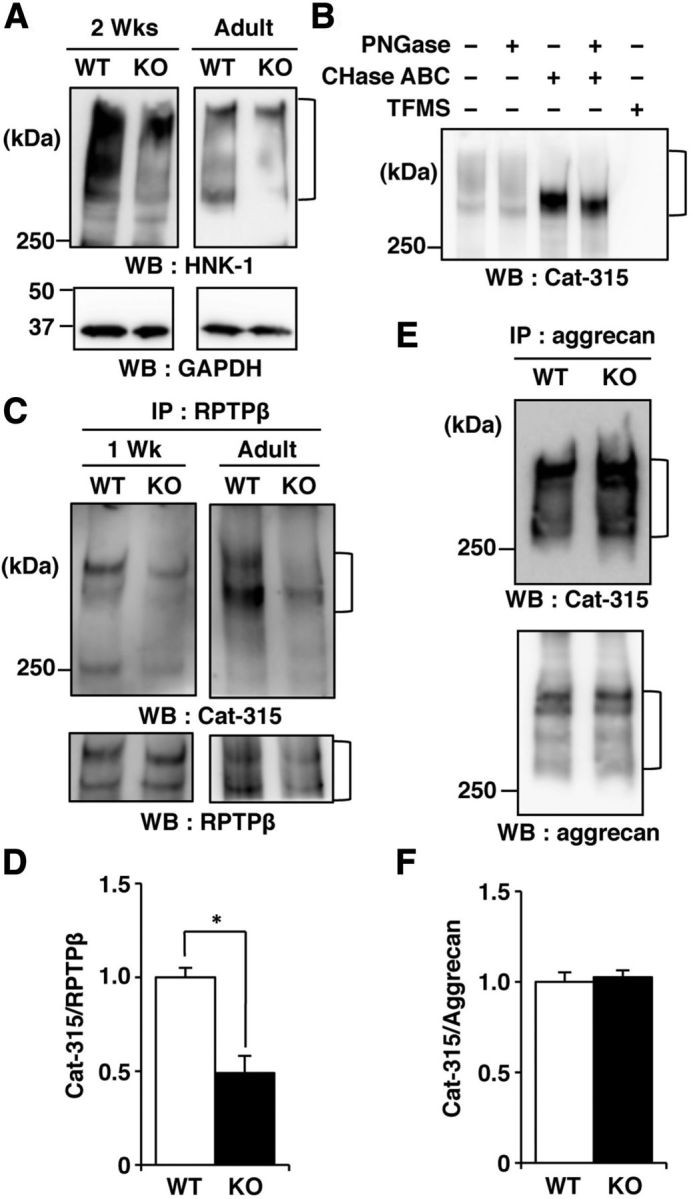
The Cat-315 epitope level on RPTPβ is reduced in GnT-IX-deficient mice. A, Brain lysates from wild-type (WT) and GnT-IX-deficient (KO) mice at 2 weeks of age (2 Wks) or 8–12 months of age (Adult) were analyzed by Western blotting with anti-HNK-1 and anti-GAPDH antibodies. B, Brain lysates from WT mice at 2 weeks of age were incubated in the presence or absence of PNGase and/or chondroitinase ABC (CHase ABC), or treated with TFMS, and then analyzed by Western blotting with the Cat-315 antibody. C, RPTPβ immunoprecipitated from brain microsomal fractions of WT and GnT-IX-deficient mice were analyzed by Western blotting with Cat-315 and anti-RPTPβ antibodies. D, Graph representing the relative signal intensities of Cat-315/RPTPβ in the immunoprecipitates with the anti-RPTPβ antibody. The data shown are means ± SEM (n = 5). *p < 0.005. E, Immunoprecipitated aggrecan from brain lysates was analyzed as described for RPTPβ (C). F, Graph representing the relative signal intensities of Cat-315/aggrecan in the immunoprecipitates with the anti-aggrecan antibody. The data shown are means ± SEM (n = 5).
Even though Western blot analyses with the Cat-315 antibody using brains from both mouse genotypes at an early developmental stage suggested that RPTPβ probably carried the Cat-315 epitope, we next tried to identify the protein on which the Cat-315 epitope was reduced by GnT-IX deficiency. We found that immunoprecipitated RPTPβ from the brains of both 1-week-old and adult GnT-IX-deficient mice had significantly lower levels of the Cat-315 epitope than that from the brains of wild-type mice (Fig. 2C,D). Meanwhile, immunoprecipitated aggrecan from the adult brains of wild-type and GnT-IX-deficient mice had similar levels of the Cat-315 epitope (Fig. 2E,F). Together, we concluded that RPTPβ, but not aggrecan, is a physiological substrate for GnT-IX, and is most likely to possess branched O-mannosyl glycans with the HNK-1 epitope.
GnT-IX-deficient mice show mild demyelination after cuprizone administration
A recent report showing a critical role of RPTPβ in the demyelination process (Harroch et al., 2002) prompted us to apply the cuprizone-induced demyelination model to GnT-IX-deficient mice. To compare the effects of cuprizone feeding, we first performed Klüver–Barrera staining to evaluate the demyelination after 8 weeks of cuprizone feeding. Our observations of robust demyelination in wild-type mice and a compact but retained myelin sheath morphology in GnT-IX-deficient mice (Fig. 3A,B) raised the possibility that GnT-IX-deficient mice exhibit enhanced remyelination, since remyelination results in a thinner and shorter myelin sheath than would be expected for a given diameter of axon (Blakemore, 1974; Ludwin and Maitland, 1984). We then evaluated whether oligodendrocytes from wild-type and GnT-IX-deficient mice suffered equally from the initial metabolic stress induced by cuprizone feeding. After 1 week of cuprizone feeding, we found that MBP mRNA was equivalently reduced in the brains of both mouse strains (Fig. 3C), indicating that cuprizone exerted equal metabolic toxicity toward the oligodendrocytes in both types of mice. Furthermore, immunohistochemical analysis of brain sections after 4 and 8 weeks of cuprizone feeding with an anti-MBP antibody showed that the reduction in myelin continued in wild-type mice (Fig. 3D,E). On the other hand, the GnT-IX-deficient mouse brains exhibited a lower level of myelin at 4 weeks, but a significantly higher level of myelin at the later stage of 8 weeks (Fig. 3D,E). These findings indicate that subsequent remyelination, rather than initial oligodendrocyte changes, is enhanced in GnT-IX-deficient mice, leading to a milder demyelination phenotype.
Figure 3.
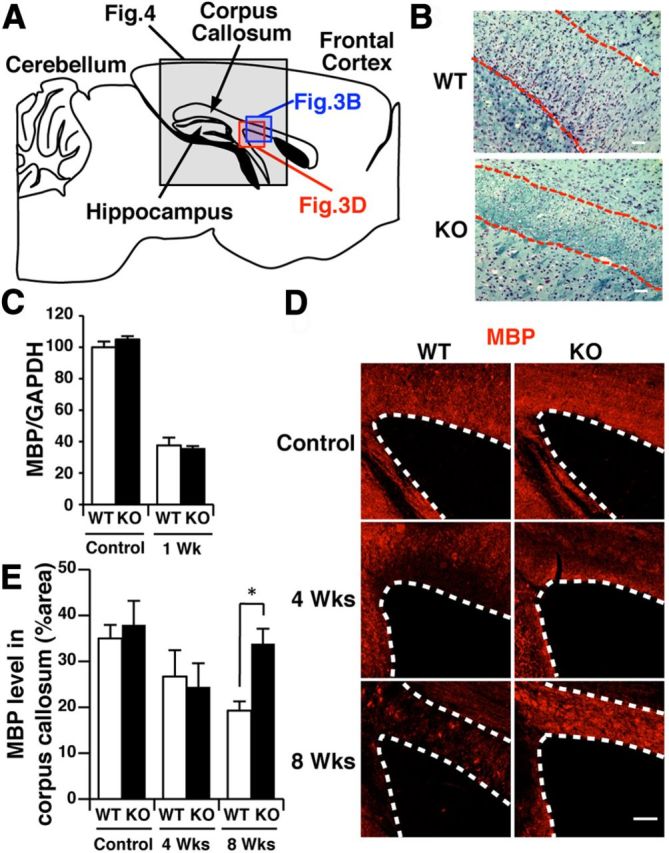
GnT-IX-deficient mice are relatively resistant to cuprizone-induced demyelination. A, Schematic diagram of a sagittal section of the corpus callosum in the mouse brain. The enclosed region was mainly observed in the immunohistochemical analyses shown in B and D and Figure 4. B, Klüver–Barrera staining was performed for brain sagittal sections from wild-type (WT) and GnT-IX-deficient (KO) mice after 8 weeks of cuprizone treatment. The dashed lines indicate the borders of the corpus callosum. Scale bars, 50 μm. C, The expression levels of MBP mRNA in the brains of WT and GnT-IX-deficient mice were measured and normalized by the corresponding GAPDH expression levels. The data shown are means ± SEM (n = 4). D, Brain sections from WT and GnT-IX-deficient mice during demyelination were stained with an anti-MBP antibody. The dashed lines indicate the borders of the corpus callosum. Scale bar, 100 μm. E, The relative MBP-positive areas in the corpus callosum in WT and GnT-IX-deficient mice are shown as means ± SEM (n = 6 for controls and n = 9 for 4 and 8 weeks). *p < 0.05.
Induction of the Cat-315 epitope in the corpus callosum by cuprizone feeding
We anticipated that cuprizone administration would affect the Cat-315 epitope level on RPTPβ in a GnT-IX-dependent manner. Immunohistochemical analysis with the Cat-315 antibody of brain sections from untreated wild-type and GnT-IX-deficient adult mice showed comparable distribution patterns of the Cat-315 epitope (Fig. 4A). Most of the Cat-315 epitopes were colocalized with WFA lectin, a marker for perineuronal nets, in lattice-like condensations of the extracellular matrix surrounding neurons, and were therefore considered to be derived from aggrecan (McRae et al., 2007). In contrast, cuprizone administration caused marked increases in both RPTPβ and the Cat-315 epitope in the corpus callosum of wild-type mice by 8 weeks, but not 4 weeks, of cuprizone administration, and these signals were mostly colocalized (Fig. 4B). Meanwhile, this induction of both RPTPβ and the Cat-315 epitope was attenuated in GnT-IX-deficient mice (Fig. 4B). These findings indicate that cuprizone treatment induces expression of RPTPβ with branched O-mannosyl glycans in the corpus callosum.
Figure 4.
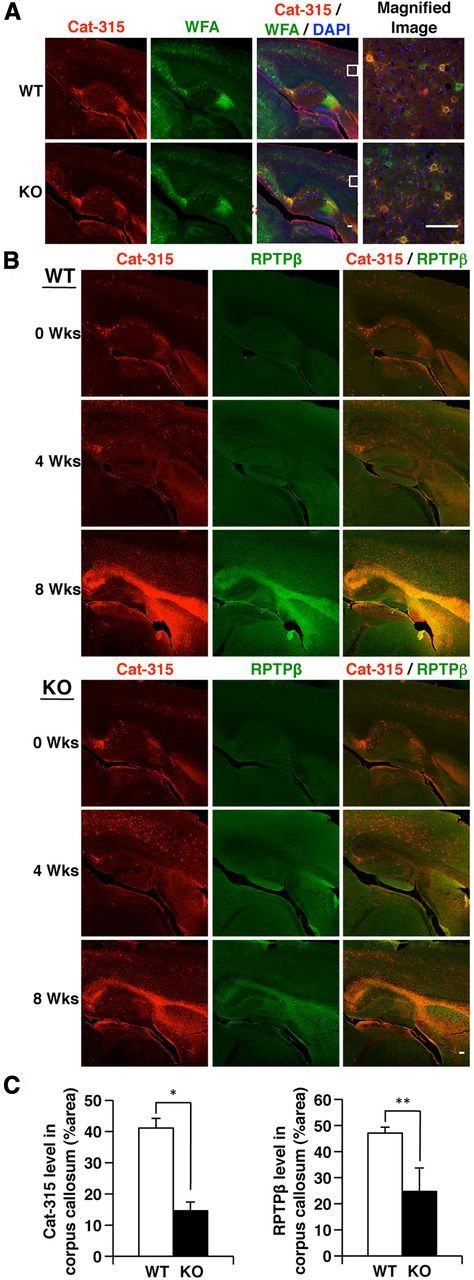
The Cat-315 epitope level in the corpus callosum is significantly increased in wild-type (WT) mice and moderately increased in GnT-IX-deficient mice with cuprizone-induced demyelination. A, Brain sagittal sections from WT and GnT-IX-deficient (KO) mice were stained with the Cat-315 antibody (red) and WFA lectin (green) to detect perineuronal nets. Nuclei were counterstained with DAPI (blue). The enclosed areas are magnified on the right. Scale bars, 100 μm. B, After 4 and 8 weeks of cuprizone treatment, brain sections containing the corpus callosum from WT and GnT-IX-deficient mice were stained with Cat-315 (red) and anti-RPTPβ (green) antibodies. Scale bar, 100 μm. C, Graphs representing the relative signal intensities of Cat-315 (left) and anti-RPTPβ (right) in the corpus callosum from WT and GnT-IX-deficient mice. The data shown are means ± SEM (n = 6).
Brain inflammation is modulated by GnT-IX deficiency
Based on our findings that the initial toxic effects of cuprizone on oligodendrocytes were similar between wild-type and GnT-IX-deficient mice, while Cat-315 immunoreactivity was decreased in the corpus callosum of GnT-IX-deficient mice at the later stage of cuprizone treatment, we next tried to evaluate the role of GnT-IX in cuprizone-induced neuroinflammation. Initially, activated astrocytes in the corpus callosum were identified by GFAP staining. In untreated wild-type and GnT-IX-deficient mice, limited populations of the cells had GFAP-positive and Cat-315-positive signals (Fig. 5A). After cuprizone administration, prominent astrogliosis was observed in the corpus callosum of wild-type mice, and nearly all of the Cat-315-positive cells were identified as GFAP-positive astrocytes (Fig. 5A). Notably, such astrogliosis was markedly decreased in GnT-IX-deficient mice (Fig. 5C). Next, the microglial populations in the corpus callosum were assessed by Iba1 staining (Fig. 5B,D), which reflects microglial invasion. Untreated wild-type and GnT-IX-deficient mice exhibited few Iba1-positive cells. After 8 weeks of cuprizone administration, we found that wild-type and GnT-IX-deficient mice showed comparable levels of increased microglial infiltration, and nearly all of the Iba1-positive cells were Cat-315-negative. These findings imply that GnT-IX deficiency modulates cuprizone-induced brain inflammation by decreasing astrogliosis around the corpus callosum and that the Cat-315 epitope is expressed in activated astrocytes, but not in infiltrated microglia.
Figure 5.
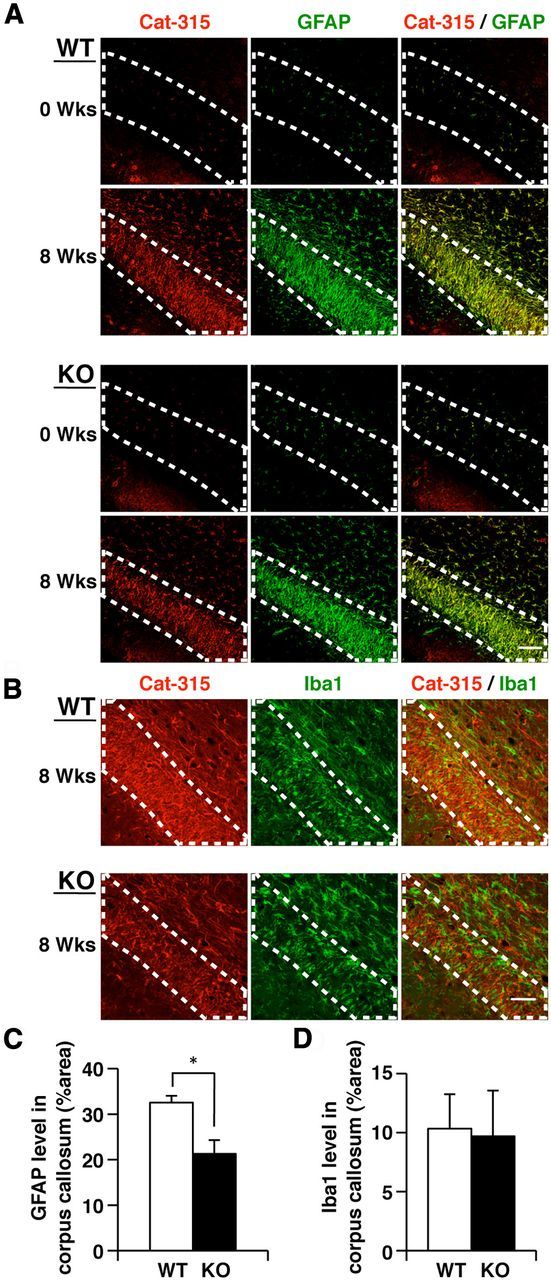
Inflammatory reactions are altered in the corpus callosum in cuprizone-fed GnT-IX-deficient mice. A, Astrogliosis is decreased in GnT-IX-deficient mice. After 8 weeks of cuprizone treatment, brain sections from wild-type (WT) and GnT-IX-deficient (KO) mice were stained with Cat-315 (red) and anti-GFAP (green) antibodies. Nuclei were counterstained with DAPI (blue). Scale bar, 50 μm. B, Microglia recruitment to the corpus callosum is increased in GnT-IX-deficient mice. Brain sections were stained with Cat-315 (red) and anti-Iba1 (green) antibodies to detect microglia. The enclosed areas indicate the borders of the corpus callosum. Nuclei were counterstained with DAPI (blue). Scale bar, 50 μm. C, Astrogliosis in the corpus callosum was quantified by measuring the relative GFAP-positive areas. The data shown are means ± SEM (n = 9). *p < 0.05. D, Quantification of Iba1-positive cells in the corpus callosum. The data shown are means ± SEM (n = 6).
Both RPTPβ and the Cat-315 epitope are expressed in astrocytes
To extend our knowledge of which cell types exhibit increased expression of RPTPβ and the Cat-315 epitope in the corpus callosum after cuprizone administration, we performed immunohistochemical analyses using the anti-RPTPβ or Cat-315 antibodies plus several kinds of neural cell-specific markers. We found that most of the activated astrocytes expressed RPTPβ (Shintani et al., 1998), but some of the RPTPβ-positive cells were GFAP negative (Fig. 6A), indicating that increased RPTPβ expression after cuprizone feeding is not limited to astrocytes, in accord with a previous report concerning the critical role of oligodendrocyte RPTPβ in recovery from demyelinating disease (Harroch et al., 2002). On the other hand, the Cat-315-positive signals were solely found in activated astrocytes, and did not merge with the signals of oligodendrocyte markers (Fig. 6A). These results indicate that cuprizone treatment caused increased RPTPβ expression in several types of neural cells, whereas the increase in branched O-mannose glycan expression was limited to activated astrocytes. We also prepared primary astrocytes, neurons, and oligodendrocytes from mice for immunostaining analyses, and found that both astrocytes and neurons, but not oligodendrocytes, carried the Cat-315 epitope (Fig. 6B). The Cat-315 epitope found in neurons is most likely attached to aggrecan (Matthews et al., 2002), and therefore does not carry branched O-mannose glycans (Fig. 2E,F). Meanwhile, it is expected that the Cat-315 epitope in astrocytes would be attached to RPTPβ. The finding that oligodendrocytes did not carry a significant level of the Cat-315 epitope is consistent with our observation that the sensitivity of oligodendrocytes to cuprizone-induced metabolic stress at the initial stage was not modified by GnT-IX expression.
Figure 6.
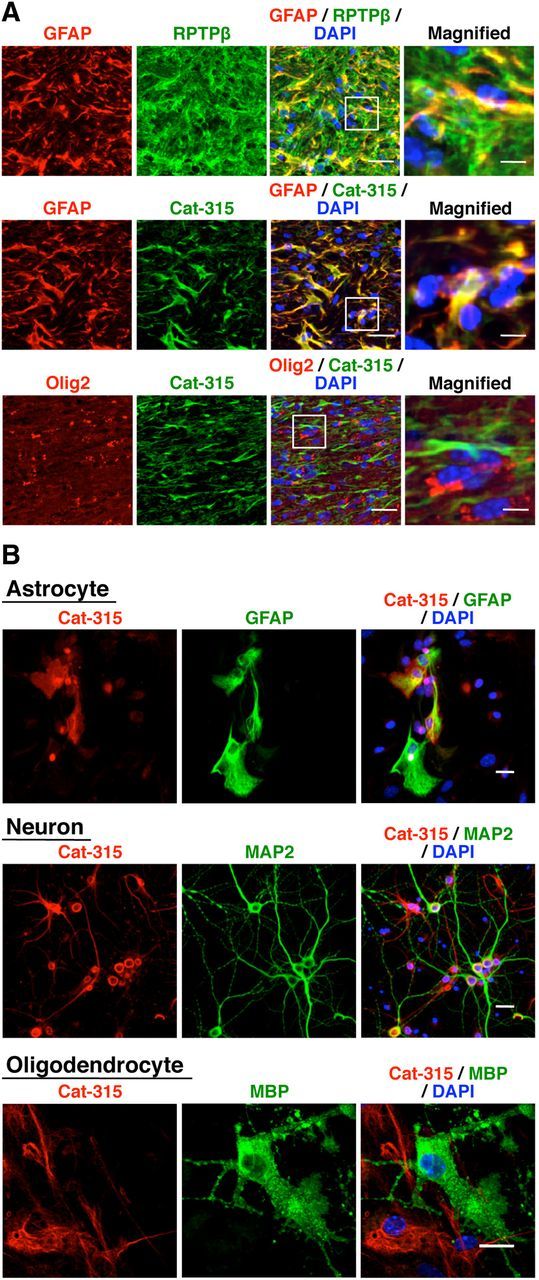
Both the Cat-315 and RPTPβ epitopes are increased in activated astrocytes in the corpus callosum by cuprizone feeding. A, After 8 weeks of cuprizone treatment, brain sections from wild-type mice were stained with Cat-315 (green), anti-RPTPβ (green), anti-GFAP (red), and anti-Olig2 (red) antibodies. Scale bars, 20 μm. The enclosed areas are magnified on the right (Scale bars, 5 μm). B, A series of cell cultures that mainly contained astrocytes, neurons, or oligodendrocytes were prepared from wild-type mice, fixed, and stained with Cat-315 (red) and anti-GFAP, anti-MAP2, and anti-MBP (green) antibodies, respectively. Nuclei were counterstained with DAPI (blue). Scale bars, 20 μm.
GnT-IX deficiency causes impairment of astrocyte activation
Since we found that cuprizone treatment specifically induced the Cat-315 epitope in astrocytes in the corpus callosum, we next prepared astrocytes from wild-type and GnT-IX-deficient mouse brains and compared the activation statuses in these cells. Unexpectedly, we found that GnT-IX-deficient astrocytes exhibited attenuated GFAP immunoreactivity compared with wild-type astrocytes in the absence of activation stimuli (Fig. 7A,B). We found that addition of insulin-like growth factor-1 (IGF-1) and epidermal growth factor (EGF) to wild-type astrocytes successfully led to astrocyte activation, as judged by enhanced GFAP immunoreactivity (Fig. 7C) (Suzuki et al., 2001). In this condition, both Cat-315- and RPTPβ-positive signals were significantly increased in wild-type cells (Fig. 7D,E). However, in GnT-IX-deficient astrocytes, these growth factors poorly increased the GFAP immunoreactivity as well as those of RPTPβ and Cat-315. These findings indicate that GnT-IX deficiency is somehow involved in the impaired astrocyte activation.
Figure 7.
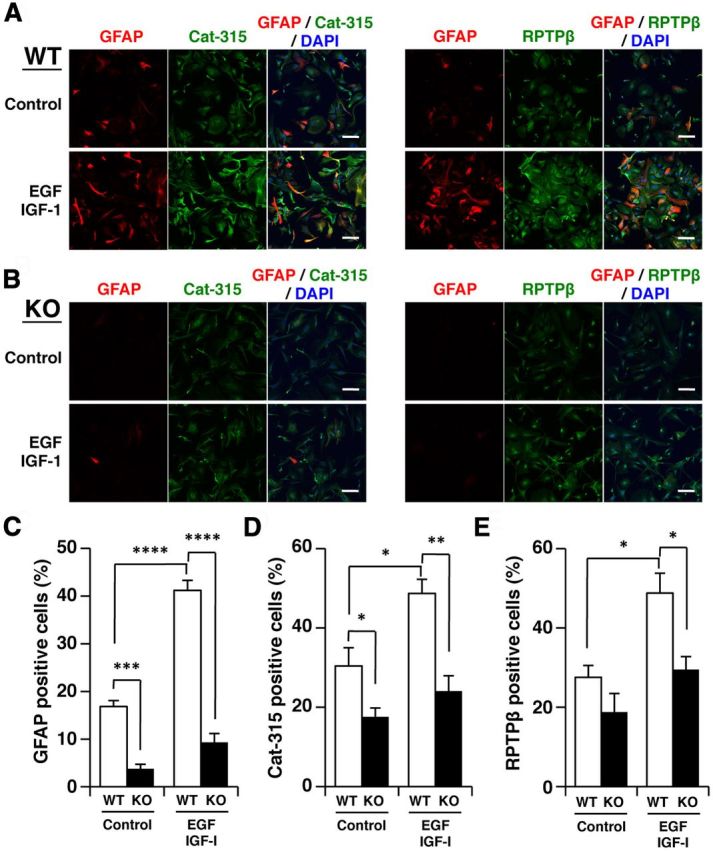
GnT-IX deficiency is involved in the impaired astrocyte activation. A, B, Primary astrocytes from wild-type (WT) (A) and GnT-IX-deficient (KO) (B) mice were cultured with or without 100 ng/ml of IGF-1 and EGF for 72 h, fixed, and stained with anti-GFAP (red) and Cat-315 (green) or anti-RPTPβ (green) antibodies. Nuclei were counterstained with DAPI (blue). Scale bars, 100 μm. C–E, The graphs show quantification of GFAP-positive (C), Cat-315-positive (D), and RPTPβ-positive cells (E). The data shown are means ± SEM (n = 6, 3, and 3), respectively. *p < 0.05; **p < 0.005, ***p < 10−5; ****p < 10−6.
Astrocyte RPTPβ has a different localization in GnT-IX-deficient mice
Western blot analyses of the corpus callosum isolated from cuprizone-fed wild-type and GnT-IX-deficient mouse brains showed similar increases in RPTPβ in both types of mice (Fig. 8A). Given the recent information that particular glycosylation profiles are critical for the localization of membrane glycoproteins to their functional places (Lau et al., 2007), we hypothesized that the lack of branched O-mannosyl glycans affects the subcellular RPTPβ localization, such as lipid raft targeting, and reduces the accessibility of the anti-RPTPβ antibody for immunohistochemical staining, as shown in Figure 4B. Indeed, a lipid raft fraction prepared from the brain of GnT-IX-deficient mice contained a significantly higher level of RPTPβ (Fig. 8B,C), indicating that the lack of branched O-mannosyl glycans by GnT-IX deficiency enhanced the lipid raft targeting of RPTPβ.
Figure 8.

Aberrant RPTPβ localization is observed in the GnT-IX-deficient mouse brain. A, Corpus callosum parts were collected from the brains of wild-type (WT) and GnT-IX-deficient (KO) mice and analyzed by Western blotting with anti-RPTPβ and anti-GAPDH antibodies (left). In the right graph, the relative RPTPβ immunoreactivities to those of GAPDH are shown as means ± SEM (n = 3). B, Brains from WT and GnT-IX-deficient mice were solubilized, and DRM fractions were isolated. After chondroitinase ABC treatment, each fraction was subjected to SDS-PAGE and immunostaining with anti-RPTPβ, anti-clathrin, and anti-flotillin-1 antibodies. Floating fractions containing DRMs are indicated by “DRM.” C, The percentages of the total RPTPβ level are shown as means ± SEM (n = 4). *p < 0.01.
Oligodendrocyte differentiation is enhanced in GnT-IX-deficient mice
Our findings raised the possibility that the reactivity of astrocytes to cuprizone treatment is somehow modulated by GnT-IX. Even though activated microglia and astrocytes perform several overlapping roles during brain inflammation (Dong and Benveniste, 2001), a recent report showed that reactive astrocytes have inhibitory activity for oligodendrocyte differentiation of oligodendrocyte precursor cells (Wang et al., 2011). The attenuated astrogliosis in GnT-IX-deficient mice would therefore allow more oligodendrocyte precursor cells to differentiate into oligodendrocytes, leading to an enhanced remyelination process. Thus, we analyzed the oligodendrocyte cell lineage in both types of mice. PDGFRα, a marker for oligodendrocyte precursor cells, was evaluated in both types of brains after 8 weeks of cuprizone feeding. As expected, GnT-IX-deficient mice had somewhat lower numbers of PDGFRα-positive cells within the demyelinated corpus callosum than wild-type mice (Fig. 9A,B). On the other hand, GnT-IX-deficient mice had significantly higher numbers of Olig2-positive cells representing both mature oligodendrocytes and oligodendrocyte precursor cells (Fig. 9A,C). Furthermore, oligodendrocyte precursor cells prepared from GnT-IX-deficient mice had an equivalent number of BrdU-positive proliferating cells to those from wild-type mice (Fig. 9D), thereby excluding the possibility of an inhibitory role of GnT-IX for the proliferation of oligodendrocyte precursor cells by themselves. These findings suggest that GnT-IX products in astrocytes are somehow involved in the impaired differentiation of oligodendrocyte precursor cells into oligodendrocytes, leading to enhancement of demyelination. Nevertheless, there is a possibility that oligodendrocyte proliferation is also modulated by GnT-IX products. The findings further indicate that branched O-mannosyl glycans are a necessary component for inhibition of remyelination.
Figure 9.
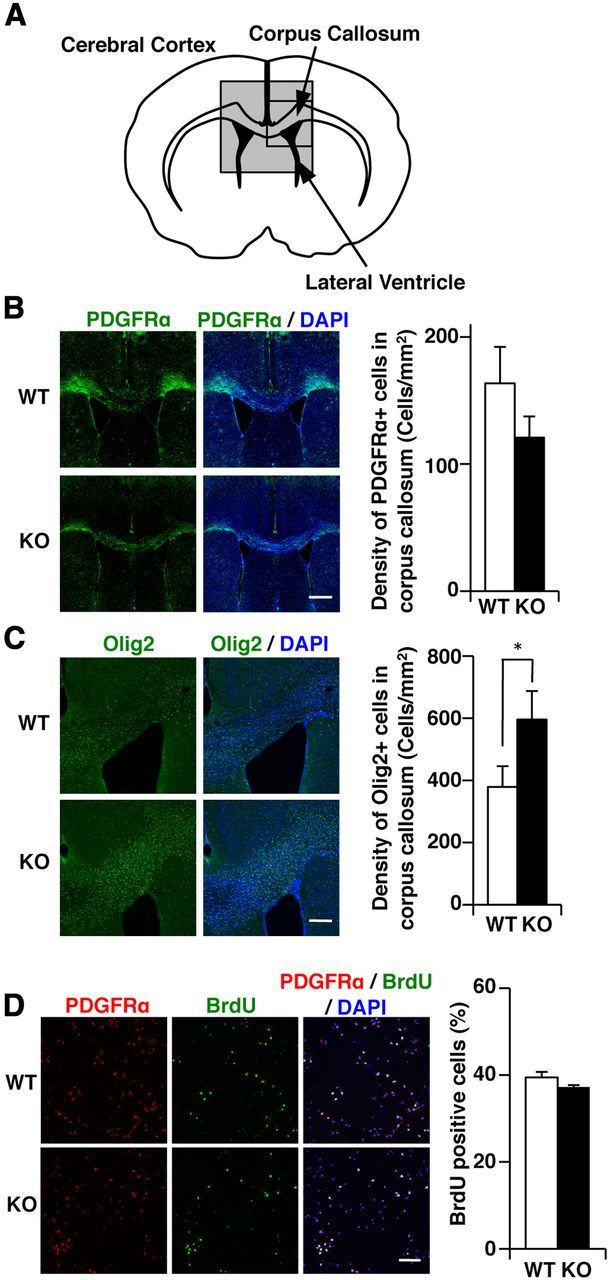
Lower accumulation of oligodendrocyte precursor cells in GnT-IX-deficient mice. A, Schematic diagram of a coronal section of the corpus callosum in the mouse brain. The enclosed regions were observed by immunohistochemical analyses. B, C, After 8 weeks of cuprizone treatment, brain sections from wild-type (WT) and GnT-IX-deficient (KO) mice were stained with anti-PDGFRα (B) and anti-Olig2 (C) antibodies. Nuclei were counterstained with DAPI (blue). Scale bars, 500 μm. The right graphs show quantification of the PDGFRα-positive and Olig2-positive cells in the corpus callosum. The data shown are means ± SEM (n = 6). *p < 0.05. D, Oligodendrocyte precursor cells were purified from WT and GnT-IX-deficient (KO) neonatal mice, cultured for 3 d, and then labeled with BrdU to identify proliferating cells. Scale bar, 100 μm. The right graph shows the percentages of BrdU-positive cells among the PDGFRα-positive cells. The data shown are means ± SEM (n = 3).
Discussion
We found that GnT-IX-deficient mice fed cuprizone showed ameliorated demyelination through an enhanced remyelination process. First, the sensitivity of oligodendrocytes to cuprizone at the initial stage was comparable in both types of mice. Second, immunohistochemical analyses revealed similar ranges of demyelination in wild-type and GnT-IX-deficient mice at the early stage, i.e., 4 weeks of cuprizone feeding. However, GnT-IX-deficient mice subsequently showed a remarkable recovery of the myelin content, even with another 4 weeks of cuprizone feeding. Third, even though microglial infiltration into the corpus callosum was comparable in the two types of mice, astrogliosis, which was recently reported to involve the secretion of an inhibitory factor for differentiation of oligodendrocytes from their precursor cells (Wang et al., 2011), was significantly reduced in GnT-IX-deficient mice. We also found that stimulating growth factors activated wild-type astrocytes, while GnT-IX-deficient astrocytes showed impaired responses to such activating stimuli. However, it is currently difficult to exclude the possibility that astrocytes with branched O-mannose glycans secrete an inhibitory factor independently of astrocyte activation. Fourth, even though the numbers of proliferating oligodendrocyte precursor cells were similar between the two types of mice, the oligodendrocyte lineage analysis revealed increased numbers of mature oligodendrocytes plus oligodendrocyte precursor cells (Olig2-positive cells), increased numbers of oligodendrocytes (MBP-positive cells), and decreased numbers of PDGFRα-positive oligodendrocyte precursor cells in GnT-IX-deficient mice. The compact myelin sheath observed in GnT-IX-deficient mice after 8 weeks of cuprizone feeding also supported enhanced remyelination (Fig. 3B). It is known that the correlation between the axon diameter and the myelin sheath thickness and length, which is observed during developmental myelination, is less apparent in remyelination, resulting in the generation of a thinner and shorter myelin sheath during remyelination compared with that during developmental myelination (Franklin and ffrench-Constant, 2008).
By using the Cat-315 antibody, which recognizes O-mannosyl glycans with the HNK-1 epitope, we found that RPTPβ is a carrier protein of branched O-mannosyl glycans that are produced by GnT-IX. Our immunohistochemical staining of the brains of cuprizone-fed mice revealed that astrocytic RPTPβ specifically possesses the Cat-315 epitope. A recent report described that RPTPβ-deficient mice show impaired recovery from demyelination (Harroch et al., 2002). Furthermore, another member of the receptor protein tyrosine phosphatase family, RPTPα, has a lipid raft-targeted form that is a Fyn activator (Vacaresse et al., 2008). Considering these observations, lipid raft-targeted RPTPβ is most likely to be involved in the remyelination process. Indeed, in our lipid raft separation experiment, more RPTPβ was distributed to the lipid raft fraction in the GnT-IX-deficient mouse brain, suggesting that the branching of the O-mannosyl glycans on RPTPβ tends to shift the RPTPβ distribution to the non-lipid raft fraction. A schematic drawing for the model of the regulatory role of branched O-mannosyl glycans on astrocytic RPTPβ is shown in Figure 10. Furthermore, several reports, including ours, have shown that particular glycosylation patterns regulate the functionality of receptors by modifying their intracellular localizations by virtue of glycan recognition molecules (Ohtsubo et al., 2005; Lau et al., 2007; Kitazume et al., 2010). In GnT-IX-deficient mice, neither astrogliosis nor an increased Cat-315 epitope level was prominent compared with wild-type mice. Together, we speculate that the branching of O-mannosyl glycans on astrocytic RPTPβ by GnT-IX keeps RPTPβ away from the functional lipid raft fraction, somehow leading to enhanced astrogliosis and demyelination. Since we failed to detect the Cat-315 epitope in oligodendrocytes, we did not focus on oligodendrocyte RPTPβ, which presumably plays a role in an O-mannosyl glycan-independent manner (Harroch et al., 2002). A recent study found an unexpected level of plasticity of myelin in adult CNS (Emery, 2010). Although the underlying molecular mechanisms for how branched O-mannosyl glycans reduce the lipid raft distribution of RPTPβ and lead to astrocyte activation remain to be elucidated in future studies, our study clarifies the role of GnT-IX in vivo for the first time, and suggests that regulation of O-mannosyl glycans in the brain would be a novel therapeutic target for multiple sclerosis.
Figure 10.
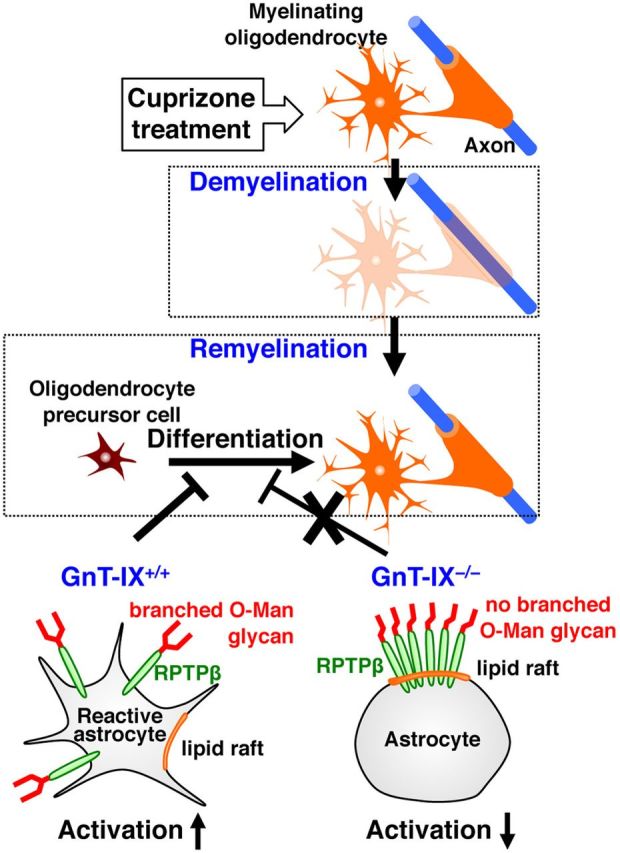
Model of the regulatory role of branched O-mannosyl glycans on astrocytic RPTPβ. Cuprizone administration causes reversible demyelination. Reactive astrocytes are known to inhibit oligodendrocyte differentiation from oligodendrocyte precursor cells, leading to impairment of remyelination (Wang et al., 2011). GnT-IX-dependent branching of the O-mannosyl glycans on RPTPβ (left, wild-type mice) shifts the RPTPβ distribution to a non-lipid raft fraction. In the absence of branched O-mannosyl glycans, astrocytic RPTPβ is targeted to a functional lipid raft fraction (right, GnT-IX-deficient; (KO), astrocyte activation is suppressed, and remyelination is enhanced.
Footnotes
This work was supported in part by grants from the Systems Glycobiology Research Project and the Strategic Programs for R&D of RIKEN (N.T.), Grants-in-Aid for Scientific Research (A) (N.T.) and (C) (S.K.), and a grant from the Takeda Science Foundation (S.K.). We thank Dr. James W. Dennis (Mount Sinai Hospital) for providing the GnT-V-deficient mice, Dr. Shogo Oka (Kyoto University) for the anti-HNK-1 and anti-RPTPβ antibodies, and the RIKEN BSI-Olympus Collaboration Center for technical support.
The authors declare no competing interests.
References
- Abbott KL, Matthews RT, Pierce M. Receptor tyrosine phosphatase (RPTP) activity and signaling are attenuated by glycosylation and subsequent cell surface galectin-1 binding. J Biol Chem. 2008;283:33026–33035. doi: 10.1074/jbc.M803646200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott KL, Troupe K, Lee I, Pierce M. Integrin-dependent neuroblastoma cell adhesion and migration on laminin is regulated by expression levels of two enzymes in the O-mannosyl-linked glycosylation pathway, PomGnT1 and GnT-Vb. Exp Cell Res. 2006;312:2837–2850. doi: 10.1016/j.yexcr.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001;4:1116–1122. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- Blakemore WF. Pattern of remyelination in the CNS. Nature. 1974;249:577–578. doi: 10.1038/249577a0. [DOI] [PubMed] [Google Scholar]
- Bradl M, Lassmann H. Oligodendrocytes: biology and pathology. Acta Neuropathol. 2010;119:37–53. doi: 10.1007/s00401-009-0601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles P, Hernandez MP, Stankoff B, Aigrot MS, Colin C, Rougon G, Zalc B, Lubetzki C. Negative regulation of central nervous system myelination by polysialylated-neural cell adhesion molecule. Proc Natl Acad Sci U S A. 2000;97:7585–7590. doi: 10.1073/pnas.100076197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MK, Baidoo K, Verina T, Guilarte TR. Peripheral benzodiazepine receptor imaging in CNS demyelination: functional implications of anatomical and cellular localization. Brain. 2004;127:1379–1392. doi: 10.1093/brain/awh161. [DOI] [PubMed] [Google Scholar]
- Dincman TA, Beare JE, Ohri SS, Whittemore SR. Isolation of cortical mouse oligodendrocyte precursor cells. J Neurosci Methods. 2012;209:219–226. doi: 10.1016/j.jneumeth.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dino MR, Harroch S, Hockfield S, Matthews RT. Monoclonal antibody Cat-315 detects a glycoform of receptor protein tyrosine phosphatase beta/phosphacan early in CNS development that localizes to extrasynaptic sites prior to synapse formation. Neuroscience. 2006;142:1055–1069. doi: 10.1016/j.neuroscience.2006.07.054. [DOI] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Emery B. Regulation of oligodendrocyte differentiation and myelination. Science. 2010;330:779–782. doi: 10.1126/science.1190927. [DOI] [PubMed] [Google Scholar]
- Endo T. Structure, function and pathology of O-mannosyl glycans. Glycoconj J. 2004;21:3–7. doi: 10.1023/B:GLYC.0000043740.26062.2c. [DOI] [PubMed] [Google Scholar]
- Franklin RJ, ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. 2008;9:839–855. doi: 10.1038/nrn2480. [DOI] [PubMed] [Google Scholar]
- Gao X, Gillig TA, Ye P, D'Ercole AJ, Matsushima GK, Popko B. Interferon-gamma protects against cuprizone-induced demyelination. Mol Cell Neurosci. 2000;16:338–349. doi: 10.1006/mcne.2000.0883. [DOI] [PubMed] [Google Scholar]
- Granovsky M, Fata J, Pawling J, Muller WJ, Khokha R, Dennis JW. Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat Med. 2000;6:306–312. doi: 10.1038/73163. [DOI] [PubMed] [Google Scholar]
- Hama E, Shirotani K, Masumoto H, Sekine-Aizawa Y, Aizawa H, Saido TC. Clearance of extracellular and cell-associated amyloid beta peptide through viral expression of neprilysin in primary neurons. J Biochem. 2001;130:721–726. doi: 10.1093/oxfordjournals.jbchem.a003040. [DOI] [PubMed] [Google Scholar]
- Harroch S, Furtado GC, Brueck W, Rosenbluth J, Lafaille J, Chao M, Buxbaum JD, Schlessinger J. A critical role for the protein tyrosine phosphatase receptor type Z in functional recovery from demyelinating lesions. Nat Genet. 2002;32:411–414. doi: 10.1038/ng1004. [DOI] [PubMed] [Google Scholar]
- Inamori K, Endo T, Ide Y, Fujii S, Gu J, Honke K, Taniguchi N. Molecular cloning and characterization of human GnT-IX, a novel beta1,6-N-acetylglucosaminyltransferase that is specifically expressed in the brain. J Biol Chem. 2003;278:43102–43109. doi: 10.1074/jbc.M308255200. [DOI] [PubMed] [Google Scholar]
- Inamori K, Endo T, Gu J, Matsuo I, Ito Y, Fujii S, Iwasaki H, Narimatsu H, Miyoshi E, Honke K, Taniguchi N. N-Acetylglucosaminyltransferase IX acts on the GlcNAc beta 1,2-Man alpha 1-Ser/Thr moiety, forming a 2,6-branched structure in brain O-mannosyl glycan. J Biol Chem. 2004;279:2337–2340. doi: 10.1074/jbc.C300480200. [DOI] [PubMed] [Google Scholar]
- Inamori K, Yoshida-Moriguchi T, Hara Y, Anderson ME, Yu L, Campbell KP. Dystroglycan function requires xylosyl- and glucuronyl transferase activities of LARGE. Science. 2012;335:93–96. doi: 10.1126/science.1214115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Alvarez-Manilla G, Kamar M, Lee I, Lee JK, Troupe K, Zhang WJ, Osawa M, Pierce M. A novel beta(1,6)-N-acetylglucosaminyltransferase V (GnT-VB) (1) FEBS Lett. 2003;554:515–519. doi: 10.1016/S0014-5793(03)01234-1. [DOI] [PubMed] [Google Scholar]
- Kitazume S, Oka R, Ogawa K, Futakawa S, Hagiwara Y, Takikawa H, Kato M, Kasahara A, Miyoshi E, Taniguchi N, Hashimoto Y. Molecular insights into beta-galactoside alpha2,6-sialyltransferase secretion in vivo. Glycobiology. 2009;19:479–487. doi: 10.1093/glycob/cwp003. [DOI] [PubMed] [Google Scholar]
- Kitazume S, Imamaki R, Ogawa K, Komi Y, Futakawa S, Kojima S, Hashimoto Y, Marth JD, Paulson JC, Taniguchi N. Alpha2,6-sialic acid on platelet endothelial cell adhesion molecule (PECAM) regulates its homophilic interactions and downstream antiapoptotic signaling. J Biol Chem. 2010;285:6515–6521. doi: 10.1074/jbc.M109.073106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizuka Y, Kitazume S, Yoshida M, Taniguchi N. Brain-specific expression of N-acetylglucosaminyltransferase IX (GnT-IX) is regulated by epigenetic histone modifications. J Biol Chem. 2011;286:31875–31884. doi: 10.1074/jbc.M111.251173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleene R, Schachner M. Glycans and neural cell interactions. Nat Rev Neurosci. 2004;5:195–208. doi: 10.1038/nrn1349. [DOI] [PubMed] [Google Scholar]
- Lander C, Zhang H, Hockfield S. Neurons produce a neuronal cell surface-associated chondroitin sulfate proteoglycan. J Neurosci. 1998;18:174–183. doi: 10.1523/JNEUROSCI.18-01-00174.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau KS, Partridge EA, Grigorian A, Silvescu CI, Reinhold VN, Demetriou M, Dennis JW. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell. 2007;129:123–134. doi: 10.1016/j.cell.2007.01.049. [DOI] [PubMed] [Google Scholar]
- Liu L, Belkadi A, Darnall L, Hu T, Drescher C, Cotleur AC, Padovani-Claudio D, He T, Choi K, Lane TE, Miller RH, Ransohoff RM. CXCR2-positive neutrophils are essential for cuprizone-induced demyelination: relevance to multiple sclerosis. Nat Neurosci. 2010;13:319–326. doi: 10.1038/nn.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwin SK, Maitland M. Long-term remyelination fails to reconstitute normal thickness of central myelin sheaths. J Neurol Sci. 1984;64:193–198. doi: 10.1016/0022-510X(84)90037-6. [DOI] [PubMed] [Google Scholar]
- Ma J, Matsumoto M, Tanaka KF, Takebayashi H, Ikenaka K. An animal model for late onset chronic demyelination disease caused by failed terminal differentiation of oligodendrocytes. Neuron Glia Biol. 2006;2:81–91. doi: 10.1017/S1740925X06000056. [DOI] [PubMed] [Google Scholar]
- Mason JL, Suzuki K, Chaplin DD, Matsushima GK. Interleukin-1beta promotes repair of the CNS. J Neurosci. 2001;21:7046–7052. doi: 10.1523/JNEUROSCI.21-18-07046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews RT, Kelly GM, Zerillo CA, Gray G, Tiemeyer M, Hockfield S. Aggrecan glycoforms contribute to the molecular heterogeneity of perineuronal nets. J Neurosci. 2002;22:7536–7547. doi: 10.1523/JNEUROSCI.22-17-07536.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae PA, Rocco MM, Kelly G, Brumberg JC, Matthews RT. Sensory deprivation alters aggrecan and perineuronal net expression in the mouse barrel cortex. J Neurosci. 2007;27:5405–5413. doi: 10.1523/JNEUROSCI.5425-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS, Dollar J, Nishino I, Kelley RI, Somer H, Straub V, Mathews KD, Moore SA, Campbell KP. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–422. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- Ohtsubo K, Takamatsu S, Minowa MT, Yoshida A, Takeuchi M, Marth JD. Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell. 2005;123:1307–1321. doi: 10.1016/j.cell.2005.09.041. [DOI] [PubMed] [Google Scholar]
- Pasquini LA, Calatayud CA, Bertone Uña AL, Millet V, Pasquini JM, Soto EF. The neurotoxic effect of cuprizone on oligodendrocytes depends on the presence of pro-inflammatory cytokines secreted by microglia. Neurochem Res. 2007;32:279–292. doi: 10.1007/s11064-006-9165-0. [DOI] [PubMed] [Google Scholar]
- Shintani T, Watanabe E, Maeda N, Noda M. Neurons as well as astrocytes express proteoglycan-type protein tyrosine phosphatase zeta/RPTPbeta: analysis of mice in which the PTPzeta/RPTPbeta gene was replaced with the LacZ gene. Neurosci Lett. 1998;247:135–138. doi: 10.1016/S0304-3940(98)00295-X. [DOI] [PubMed] [Google Scholar]
- Stalnaker SH, Aoki K, Lim JM, Porterfield M, Liu M, Satz JS, Buskirk S, Xiong Y, Zhang P, Campbell KP, Hu H, Live D, Tiemeyer M, Wells L. Glycomic analyses of mouse models of congenital muscular dystrophy. J Biol Chem. 2011;286:21180–21190. doi: 10.1074/jbc.M110.203281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto I, Futakawa S, Oka R, Ogawa K, Marth JD, Miyoshi E, Taniguchi N, Hashimoto Y, Kitazume S. ST6Gal I cleavage by BACE1 enhances the sialylation of soluble glycoproteins: a novel regulatory mechanism for alpha 2,6-sialylation. J Biol Chem. 2007;282:34896–34903. doi: 10.1074/jbc.M704766200. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Ikegaya Y, Matsuura S, Kanai Y, Endou H, Matsuki N. Transient upregulation of the glial glutamate transporter GLAST in response to fibroblast growth factor, insulin-like growth factor and epidermal growth factor in cultured astrocytes. J Cell Sci. 2001;114:3717–3725. doi: 10.1242/jcs.114.20.3717. [DOI] [PubMed] [Google Scholar]
- Torkildsen O, Brunborg LA, Myhr KM, B ø L. The cuprizone model for demyelination. Acta Neurol Scand Suppl. 2008;188:72–76. doi: 10.1111/j.1600-0404.2008.01036.x. [DOI] [PubMed] [Google Scholar]
- Vacaresse N, Møller B, Danielsen EM, Okada M, Sap J. Activation of c-Src and Fyn kinases by protein-tyrosine phosphatase RPTPalpha is substrate-specific and compatible with lipid raft localization. J Biol Chem. 2008;283:35815–35824. doi: 10.1074/jbc.M807964200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Inoue S, Gu J, Miyoshi E, Noda K, Li W, Mizuno-Horikawa Y, Nakano M, Asahi M, Takahashi M, Uozumi N, Ihara S, Lee SH, Ikeda Y, Yamaguchi Y, Aze Y, Tomiyama Y, Fujii J, Suzuki K, Kondo A, et al. Dysregulation of TGF-beta1 receptor activation leads to abnormal lung development and emphysema-like phenotype in core fucose-deficient mice. Proc Natl Acad Sci U S A. 2005;102:15791–15796. doi: 10.1073/pnas.0507375102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cheng X, He Q, Zheng Y, Kim DH, Whittemore SR, Cao QL. Astrocytes from the contused spinal cord inhibit oligodendrocyte differentiation of adult oligodendrocyte precursor cells by increasing the expression of bone morphogenetic proteins. J Neurosci. 2011;31:6053–6058. doi: 10.1523/JNEUROSCI.5524-09.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, Mizuno M, Inazu T, Mitsuhashi H, Takahashi S, Takeuchi M, Herrmann R, Straub V, Talim B, Voit T, Topaloglu H, Toda T, Endo T. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell. 2001;1:717–724. doi: 10.1016/S1534-5807(01)00070-3. [DOI] [PubMed] [Google Scholar]
- Yoshida-Moriguchi T, Yu L, Stalnaker SH, Davis S, Kunz S, Madson M, Oldstone MB, Schachter H, Wells L, Campbell KP. O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science. 2010;327:88–92. doi: 10.1126/science.1180512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen CT, Chai W, Loveless RW, Lawson AM, Margolis RU, Feizi T. Brain contains HNK-1-immunoreactive O-glycans of the sulfoglucuronyl lactosamine series that terminate in 2-linked or 2,6-linked hexose (mannose) J Biol Chem. 1997;272:8924–8931. doi: 10.1074/jbc.272.14.8924. [DOI] [PubMed] [Google Scholar]