

Deep brain stimulation (DBS) of the basal ganglia is an effective treatment option for intractable symptoms of neurological and psychiatric disorders. For example, movement deficits in Parkinson's disease and dystonia are improved by DBS of the internal segment of the globus pallidus (GPi), a major output structure of the basal ganglia. Despite its increasing prevalence, however, there remains substantial disagreement over the therapeutic mechanisms of DBS (Miocinovic et al., 2013). Because there is potential for adverse side effects depending on DBS parameters and location, further elucidating the mechanism of DBS of the basal ganglia may improve treatment. In a recent report published in The Journal of Neuroscience, Chiken and Nambu (2013) address this issue in the globus pallidus of normal monkeys. It is known that the GPi is inhibited by afferent GABAergic projections from the striatum and the external segment of the globus pallidus (GPe), and it is activated by a glutamatergic projection from the subthalamic nucleus. Chiken and Nambu (2013) suggest that DBS of the GPi blocks cortical and basal ganglia signals by increasing GABA release from afferent fibers to the GPi, thereby suppressing GPi activity.
Chiken and Nambu (2013) inserted an electrode bundle containing two drug delivery tubes, a recording electrode, a stimulating electrode, and an attached tungsten ground wire (to allow bipolar stimulation) into the GPi or GPe. In addition, bipolar stimulating electrodes were chronically implanted in the primary and supplementary motor areas in the cortex. This multifaceted approach allowed the authors to comprehensively investigate the method of action of DBS, including the influence of glutamatergic and GABAergic pallidal receptors.
The authors first recorded single-neuron activity in the GPi, and found a suppression of spiking during either single pulses or trains of electrical stimulation. In contrast, GPe stimulation produced mixed responses, generally consisting of decreased neuronal firing rate during stimulation and increased firing rate shortly thereafter (Chiken and Nambu, 2013, their Figs. 3, 6). They hypothesized that stimulation activates afferent fibers to the GPi or GPe, increasing both GABA and glutamate release (Fig. 1A, i). Supporting this, Chiken and Nambu (2013) found that in the GPi, suppression of neuronal activity during stimulation was prevented by local drug microinjection of gabazine (GABAA receptor antagonist). In contrast, after GABAA block, GPi stimulation caused a local increase in neuronal activity, which was in turn blocked by AMPA/kainite and NMDA ionotropic glutamate receptor antagonists. Similarly in the GPe, drug microinjection of GABA receptor antagonists blocked neuronal inhibition, and ionotropic glutamate receptor antagonists blocked neuronal excitation. This suggests a competition of both glutamatergic (excitatory) and GABAergic (inhibitory) signals during DBS. Interestingly, although glutamatergic excitation was present in both GPi and GPe, GABAergic inhibition dominated glutamatergic excitation in the GPi. Chiken and Nambu (2013) found that this GPi suppression blocked cortical motor commands, mimicked by electrical microstimulation of the primary or supplementary motor areas, from being transmitted through the basal ganglia. These results suggest that DBS activates afferent fibers to influence pallidal activity (Fig. 1A, i), which presumably may disrupt pathologically altered cortical signals in different disorders.
Figure 1.
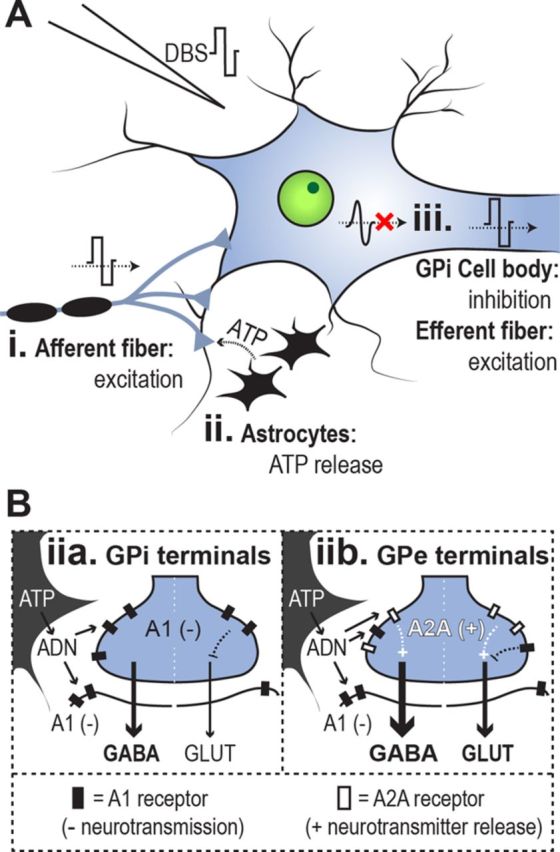
Hypothesized effects of DBS in the pallidum. Only elements discussed in the article are included. A, In the pallidum, DBS activates afferent GABAergic and glutamatergic fibers (i) and astrocytes (ii), and suppresses endogenous GPi neuronal activity (iii) while activating GPi afferents. B, DBS stimulates ATP release from astrocytes; ATP is converted to adenosine (ADN) by ecto-ATPase. Adenosine activates A1 receptors (filled rectangles) and A2A receptors (empty rectangles), which inhibit neurotransmission and facilitate neurotransmitter release, respectively. A2A receptors are preferentially located in the GPe, however. A1 and A2A receptor localization may contribute to differential GPi and GPe stimulation effects.
However, it is important to note that the decrease in GPi activity found by Chiken and Nambu (2013) in healthy monkeys may not entirely reflect the DBS mechanism underlying ameliorative effects in Parkinson's disease (PD). Chiken and Nambu (2013) argue that in PD, increased GPi GABAA receptor expression would amplify, not counteract, their effect. Nevertheless, results have been reported using a PD monkey model that may not support this conclusion. First, the efficacy of GPi GABA transporters (critical for GABA transmission) is altered in a PD model (Galvan et al., 2010), which may implicate different GPi-DBS effects in PD. Second, GPi electrical stimulation in a PD model resulted in multiphase responses of GPi neurons (McCairn and Turner, 2009), as opposed to the inhibition (only) in healthy monkeys reported by Chiken and Nambu (2013). Therefore, factors in addition to GABAA overexpression may be relevant to the DBS effect in PD, and may explain these inconsistent results between GPi stimulation of healthy monkeys (Chiken and Nambu, 2013) and PD monkeys (McCairn and Turner, 2009; Galvan et al., 2010). Further research should use the techniques described by Chiken and Nambu (2013) to address the same questions in a PD model.
In addition to the activation of afferent fibers to the GPi, we propose that DBS activation of non-neuronal glial tissues, specifically astrocytes, may be relevant to the inhibition of GPi activity. Astrocytes are directly depolarized by DBS (Vedam-Mai et al., 2012) and glia-neuronal signaling can modulate both GABA and glutamate release in the GPi (Cunha, 2005), making this a plausible contributor. In the basal ganglia, stimulation facilitates release of ATP (and its catabolic byproduct adenosine) from nearby astrocytes (Fig. 1A, ii; for review, see Vedam-Mai et al., 2012), and adenosine acts upon A1 receptors in the GPi to affect neuronal activity (Fig. 1B). A1 receptors are localized both postsynaptically and presynaptically, and their activation reduces neuronal activity through several parallel mechanisms (including presynaptic inhibition of glutamate release; Cunha, 2005). Therefore, astrocytic activation may contribute to the dominance of inhibitory over excitatory neuronal signals during GPi stimulation. Furthermore, glial activation may help explain different excitatory and inhibitory effects of electrical stimulation in GPi and GPe (Fig. 1B). Because both the GPe and GPi receive GABAergic and glutamatergic inputs, Chiken and Nambu (2013) hypothesized that differences in the balance of GABAergic and glutamatergic inputs to these structures may underlie different effects of DBS. Complimentary evidence also suggests that differences in glia–neuronal signaling between the GPe and GPi lead to different modulations of glutamate and GABA release (Cunha, 2005; Morelli et al., 2009; Luquin et al., 2012). For example, the gliotransmitter adenosine activates A2A receptors, which are expressed at higher levels in the GPe and striatum than in the GPi (Cunha, 2005; Morelli et al., 2009; Luquin et al., 2012). Activation of A2A receptors in GABAergic and glutamatergic nerve terminals within the GPe can locally increase both GABA and glutamate release (Cunha, 2005; Morelli et al., 2009). In contrast, A1 adensosine receptors are expressed widely in GPe as well as GPi. Therefore, it is plausible that the higher concentration of A2A receptors in the GPe compared with the GPi, combined with a similar expression of A1 receptors between structures, explains the different effects of DBS (Chiken and Nambu, 2013). Specifically, A2A activation may increase both GABA and glutamate release in the GPe, whereas activation of A1 receptors suppresses neurotransmission in the GPi (Fig. 1B).
The suppression of GPi activity with GPi-DBS may contribute to DBS efficacy in PD or dystonia by suppressing pathological signaling in the basal ganglia output (Miocinovic et al., 2013). However, strong evidence also indicates that GPi activity is pathologically attenuated in dystonia, in both transgenic mice and humans (Nambu et al., 2011). Hence, a factor in addition to the suppression of GPi activity must be required for GPi-DBS clinical effects to explain this conflict. One potentially critical difference between the pathological suppression of GPi activity in dystonia and the DBS-mediated suppression of GPi activity to treat dystonia symptoms is whether the axons of GPi output fibers are also excited by DBS (in addition to the inhibition of GPi cell body spiking). While Chiken and Nambu (2013) have demonstrated that GPi stimulation inhibits GPi neuronal spiking, evidence also supports an increase in neurotransmitter release in GPi efferent terminals, likely due to the transmission of the artificial (nonphysiological) DBS pulse down the axon (Anderson et al., 2003). In particular, GPi stimulation has been shown to reduce activity in thalamic neurons, presumably due to activation of pallido-thalamic GABAergic efferent fibers (Anderson et al., 2003); single-pulse GPi stimulation also evokes a single spike at a regular latency in GPi single neurons, in addition to the otherwise suppressed endogenous neuronal activity (Chiken and Nambu, 2013). Therefore GPi DBS may exert clinical effects by simultaneously inhibiting endogenous activity in cell bodies while directly activating efferent fiber output (Fig. 1A, iii).
The report by Chiken and Nambu (2013) is a thorough and well implemented study, with ambitious methods and straightforward results. The authors suggest that DBS of the globus pallidus affects neuronal activity by activating neurotransmitter release from afferent fibers. Furthermore, we hypothesize that additional, complimentary processes may underlie the mechanism of action of DBS, specifically the activation of glia and GPi efferent fibers, to produce clinical effects. Further research can expand on a few unanswered questions using Chiken and Nambu's (2013) methods. For instance, the experimental design adopted in this study can be extended to examine our hypothesis regarding the influence of adenosine (using drug microinjection). It is also important to examine the impact of DBS on endogenous motor commands during behavioral tasks, instead of those mimicked by cortical electrical stimulation. This will complement further studies using electrical stimulation in awake monkeys, and in studies examining DBS in patients with neurological and psychiatric disorders.
Footnotes
Editor's Note: These short, critical reviews of recent papers in the Journal, written exclusively by graduate students or postdoctoral fellows, are intended to summarize the important findings of the paper and provide additional insight and commentary. For more information on the format and purpose of the Journal Club, please see http://www.jneurosci.org/misc/ifa_features.shtml.
We thank Drs. Takuro Ikeda, Josh Wang, and Brian White for their assistance.
References
- Anderson ME, Postupna N, Ruffo M. Effects of high-frequency stimulation in the internal globus pallidus on the activity of thalamic neurons in the awake monkey. J Neurophysiol. 2003;89:1150–1160. doi: 10.1152/jn.00475.2002. [DOI] [PubMed] [Google Scholar]
- Chiken S, Nambu A. High-frequency pallidal stimulation disrupts information flow through the pallidum by GABAergic inhibition. J Neurosci. 2013;33:2268–2280. doi: 10.1523/JNEUROSCI.4144-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha R. Neuroprotection by adenosine in the brain: from A1 receptor activation to A2A receptor blockade. Purinergic Signaling. 2005;1:111–134. doi: 10.1007/s11302-005-0649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan A, Hu X, Smith Y, Wichmann T. Localization and function of GABA transporters in the globus pallidus of parkinsonian monkeys. Exp Neurol. 2010;223:505–515. doi: 10.1016/j.expneurol.2010.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luquin N, Sierra S, Rico AJ, Gómez-Bautista V, Roda E, Conte-Perales L, Franco R, McCormick P, Labandeira-García JL, Lanciego JL. Unmasking adenosine 2A receptors (A2ARs) in monkey basal ganglia ouput neurons using cholera toxin subunit B (CTB) Neurobiol Dis. 2012;47:347–357. doi: 10.1016/j.nbd.2012.05.006. [DOI] [PubMed] [Google Scholar]
- McCairn KW, Turner RS. Deep brain stimulation of the globus pallidus internus in the Parkinsonian primate: local entrainment and suppression of low-frequency oscillations. J Neurophysiol. 2009;101:1941–1960. doi: 10.1152/jn.91092.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miocinovic S, Somayajula S, Chitnis S, Vitek JL. History, applications, and mechanisms of deep brain stimulation. JAMA Neurol. 2013;70:163–171. doi: 10.1001/2013.jamaneurol.45. [DOI] [PubMed] [Google Scholar]
- Morelli M, Carta AR, Jenner P. Adenosine A2A receptors and Parkinson's disease. Handb Exp Pharmacol. 2009;193:589–615. doi: 10.1007/978-3-540-89615-9_18. [DOI] [PubMed] [Google Scholar]
- Nambu A, Chiken S, Shashidharan P, Nishibayashi H, Ogura M, Kakishita K, Tanaka S, Tachibana Y, Kita H, Itakura T. Reduced pallidal output causes dystonia. Front Syst Neurosci. 2011;5:89. doi: 10.3389/fnsys.2011.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedam-Mai V, van Battum EY, Kamphuis W, Feenstra MG, Denys D, Reynolds BA, Okun MS, Hol EM. Deep brain stimulation and the role of astrocytes. Mol Psychiatry. 2012;17:124–131. doi: 10.1038/mp.2011.61. [DOI] [PubMed] [Google Scholar]