

Abstract
Nav1.8 is a tetrodotoxin-resistant voltage-gated sodium channel selectively expressed in primary sensory neurons. Peripheral inflammation and nerve injury induce Nav1.8 accumulation in peripheral nerves. However, the mechanisms and related significance of channel accumulation in nerves remains unclear. Here we report that KIF5B promotes the forward transport of Nav1.8 to the plasma membrane and axons in dorsal root ganglion (DRG) neurons of the rat. In peripheral inflammation induced through the intraplantar injection of complete Freund's adjuvant, increased KIF5 and Nav1.8 accumulation were observed in the sciatic nerve. The knock-down of KIF5B, a highly expressed member of the KIF5 family in DRGs, reduced the current density of Nav1.8 in both cultured DRG neurons and ND7–23 cells. Overexpression of KIF5B in ND7–23 cells increased the current density and surface expression of Nav1.8, which were abolished through brefeldin A treatment, whereas the increases were lost in KIF5B mutants defective in ATP hydrolysis or cargo binding. Overexpression of KIF5B also decreased the proteasome-associated degradation of Nav1.8. In addition, coimmunoprecipitation experiments showed interactions between the N terminus of Nav1.8 and the 511–620 aa sequence in the stalk domain of KIF5B. Furthermore, KIF5B increased Nav1.8 accumulation, Nav1.8 current, and neuronal excitability detected in the axons of cultured DRG neurons, which were completely abolished by the disruption of interactions between KIF5B and the N terminus of Nav1.8. Therefore, our results reveal that KIF5B is required for the forward transport and axonal function of Nav1.8, suggesting a mechanism for axonal accumulation of Nav1.8 in inflammatory pain.
Introduction
Voltage-gated sodium channels (Nav, VGSCs) are critical for the generation of action potentials in excitable cells (Waxman et al., 1999). Particularly, subtypes of VGSCs predominantly localized in small and medium-sized dorsal root ganglion (DRG) neurons, such as tetrodotoxin-resistant (TTX-R) Nav1.8, have been associated with pain transmission (Lai et al., 2004). Nav1.8 acts as a major contributor to the generation of action potentials in small- and medium-sized DRG neurons (Renganathan et al., 2001). Results obtained from both knockout and antisense studies support a pivotal role for Nav1.8 in the development of inflammatory and neuropathic pain (Akopian et al., 1999; Gold et al., 2003; Lai et al., 2004). Intraplantar injection of complete Freund's adjuvant (CFA) induces an increase in Nav1.8 in the rat digital nerve (Coggeshall et al., 2004). Nav1.8 is accumulated at sites of nerve injury in human patients with chronic neuropathic pain and chronic local hyperalgesia (Coward et al., 2000; Yiangou et al., 2000; Kretschmer et al., 2002; Black et al., 2008). Changes in the trafficking and redistribution of Nav1.8 might be responsible for abnormal firing and the generation of ectopic activity in afferent nerves in chronic pain models (Devor, 2006); however, little is known about the molecular mechanisms of Nav1.8 accumulation in peripheral nerves in pathological pain. Previous research suggests that selective axonal transport and the local upregulation of Nav1.8 mRNA in the sciatic nerve might contribute to the hyperexcitability of peripheral nerves in some neuropathic pain states (Thakor et al., 2009).
Motor protein kinesin-1 (also known as conventional kinesin or KIF5) is composed of two identical heavy chains and two identical light chains and mediates the plus end–directed, microtubule-dependent transport of cargo proteins (Hirokawa et al., 2010). The mammalian genome contains three kinesin-1 heavy chain genes, namely KIF5A, KIF5B, and KIF5C. Of these, KIF5A and KIF5C are expressed in neurons only, whereas KIF5B is expressed ubiquitously (Xia et al., 1998). Kinesin-1 motor proteins are critical for the transport of ion channels across long distances in the axons of neurons (Hirokawa et al., 2010). This protein is also associated with the localization and transport of voltage-gated potassium channels, AMPA receptor GluR2, and GABAA receptors (Setou et al., 2002; Rivera et al., 2007; Twelvetrees et al., 2010). The involvement of kinesins in the transport of sodium channels has not been established.
In the present study, KIF5 and Nav1.8 increased in the sciatic nerve in CFA-induced peripheral inflammation. Further, KIF5B interacted with Nav1.8, promoting channel transport and increasing channel function in both the cell bodies and axons of cultured DRG neurons. This finding provides a molecular mechanism for the forward transport of sodium channels through the motor protein kinesin and reveals the function of Nav1.8 accumulation in axons.
Materials and Methods
Animal model and tissue preparation.
All interventions and animal care were performed in accordance with the policies of the Society for Neuroscience on the use of animals in neuroscience research. All experiments were approved by the Committee for the Use of Laboratory Animals and Common Facility, Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences. All efforts were made to minimize the number of animals used and their discomfort after peripheral inflammation. Forty-eight adult Sprague Dawley male rats (∼200–250 g, Shanghai Center of Experimental Animals, Chinese Academy of Sciences, Shanghai, China) were anesthetized and paw inflammation was induced through the injection of CFA (Sigma) into the plantar subcutaneous space of the hindpaw (200 μl/paw). The rats were maintained for 1, 2, 4, and 7 d after injection. The sciatic nerve (1 cm long) and L4 and L5 DRGs were dissected from the treated rats and 12 normal rats. Half of the tissues were homogenized and lysed in lysis buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Triton, 0.5% sodium deoxycholate, 0.1% SDS, 10 mm NaF, 1 mm EDTA, 0.1 mm PMSF, 1 mg/ml pepstatin A, and 1 mg/ml leupeptin) for immunoblotting. Total RNA of the remaining tissues was extracted using TRIzol reagent (Life Technologies) for real-time PCR. Total RNA from the cerebral cortex, hippocampus, and DRGs from three adult rats were extracted for semiquantitative RT-PCR.
Plasmid construction.
The construction of plasmids, including rat Nav1.8-GFP, Nav1.8-Myc, and the intracellular loops of Nav1.8, has been described previously (Zhang et al., 2008). Various truncations of the Nav1.8 N terminus were PCR amplified and added to the beginning of a modified transferrin receptor (TfR) plasmid as described previously (Li et al., 2010). Human KIF5B-GFP and GCaMP3 were gifts from Drs. Shumin Duan and Shiqing Cai (Institute of Neuroscience, Chinese Academy of Sciences). KIF5B was subcloned into pMyc and KIF5BG234C-GFP was generated through PCR from KIF5B-GFP using the KOD-Plus mutagenesis kit (Toyobo). The various truncations of KIF5B-GFP were constructed using PCR-amplified fragments inserted into the C terminus of pEGFP-N3. Both KIF5B and Ctrl shRNA plasmids were generated through the direct synthesis and insertion of the corresponding sequences into the pSUPER vector containing GFP or a modified pRNAT-H1.1/neo vector containing mCherry (obtained from Dr. Chen Gu; Xu et al., 2010). Therefore, neurons transfected with the shRNA plasmids expressed GFP or mCherry as an indicator for transfection. The probe against rat KIF5B was ACAGCAGATCCAGAGTCACAGAGAA. To construct rat KIF5A, PCR-amplified fragments were inserted into the C terminus of pEGFP-N3. The following primers were used: 5′-ATGGAATTCATGGCGGAGACCAATAAC-3′ and 5′-ATGGATCCGCTGGCTGCCGTCTCTTG-3′.
Semiquantitative and real-time RT-PCR.
The mRNA (1 μg) of different tissues was reverse transcribed to cDNA. For semiquantitative RT-PCR, the PCR products were analyzed on 1.5% agarose gels. Real-time PCR with SYBR Green detection was performed using an ABI PRISM 7500 sequence detection system (Applied Biosystems). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a control. The following primers were used: 5′-TTTTCCCGAGTGTATCAGCTAC-3′ and 5′-TTCTGCCGTCTCTTGGTGGAG-3′ for KIF5A; 5′-GGGAATAAGACTCTACGGAAC-3′ and 5′-GGCAGCATCTGTAAAACTACC-3′ for KIF5B; 5′-GCCAACGAGCAACTGACC-3′ and 5′-CAGCCTCGTCAGGTGCTC-3′ for KIF5C; and 5′-GGCAAGTTCAACGGCACAG-3′ and 5′-CGCCAGTAGACTCCACGAC-3′ for GAPDH.
Cell culture and transfection.
The HEK293T cells (American Type Culture Collection, Manassas, VA) were maintained in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Biochrom). The ND7–23 cells (rat DRG/mouse neuroblastoma hybrid; European Collection of Cell Cultures, Salisbury, United Kingdom) were maintained in DMEM (without glutamine; Invitrogen) supplemented with 2 mm glutamine (Invitrogen) and 10% fetal bovine serum. The HEK293T cells were transiently transfected with plasmids using the calcium phosphate method and the ND7–23 cells were transfected with plasmids using Lipofectamine 2000 (Invitrogen). Two days after transfection, the cells were used for different assays. The HEK293 cells (American Type Culture Collection) that stably expressed Nav1.8-GFP were cultured in MEM (Invitrogen) supplemented with 10% fetal bovine serum and 1 mg/ml G418 (Amresco) as described previously (Li et al., 2010).
Neurons were dissociated from the DRGs of 40 male Sprague Dawley rats (body weight 100–120 g). Briefly, freshly dissected DRGs were digested in DMEM containing 1 mg/ml collagenase type 1A, 0.4 mg/ml trypsin type I, and 0.1 mg/ml DNase I (all from Sigma) at 37°C for 35 min. The dissociated DRG neurons were transiently transfected with plasmids through electroporation with Nucleofector II (Amaxa) using Rat Neuron Nucleofector Solution and program O-003, and the cells were subsequently plated on poly-d-lysine precoated coverslips or microfluidic chambers (Chen et al., 2012) in DMEM containing 10% fetal bovine serum (Invitrogen). After 4 h, the medium was replaced with DMEM/F12 (1:1; Invitrogen) containing 1% N2 (Invitrogen) and the neurons were analyzed within 72 h.
Coimmunoprecipitation.
The HEK293T cells were lysed in ice-cold RIPA buffer (150 mm NaCl, 30 mm HEPES, 10 mm NaF, 1% Triton, 0.1 mm PMSF, 1 mg/ml pepstatin A, and 1 mg/ml leupeptin), and DRGs were extracted from six normal rats and homogenized in lysis buffer (35 mm PIPES, pH 7.2, 5 mm MgSO4, 1 mm EGTA, 0.5 mm EDTA, 1 mm DTT, 0.2% NP-40, 0.1 mm PMSF, 1 mg/ml pepstatin A, and 1 mg/ml leupeptin). The samples were incubated with a specific antibody overnight at 4°C, followed by incubation with protein G-Sepharose beads (GE Healthcare) for 4 h at 4°C. The immunoprecipitates were efficiently washed with lysis buffer and analyzed through immunoblotting.
Cell surface biotinylation.
Cell surface biotinylation was performed according to previously described protocols (Zhang et al., 2008). Briefly, HEK293T cells and cultured DRG neurons were incubated with Sulfo-NHS-biotin (Pierce) in cold Ca2+/Mg2+ PBS for 45 min at 4°C, with 10 mm glycine added subsequently for 20 min to terminate the reaction. The cells were rinsed with PBS, harvested in RIPA buffer, and lysed for 1 h at 4°C. The biotin-labeled proteins were precipitated overnight with Immunopure Immobilized Neutravidin Beads (Pierce), washed with RIPA buffer, and incubated at 50°C for 20 min in SDS-PAGE loading buffer. The cell surface fraction and whole-cell lysates were analyzed through immunoblotting.
Immunoblotting.
The samples were separated by SDS-PAGE, transferred, probed with specific antibodies, and visualized with enhanced chemiluminescence (GE Healthcare). The primary antibodies included mouse antibodies against GFP (1:1000, 0.40 μg/ml; Roche), Myc (1:2000, 0.45 μg/ml; Developmental Studies Hybridoma Bank, Iowa City, IA), actin (1:10,0000, 0.01 μg/ml; Millipore), α-tubulin (1:20,000, 0.01 μg/ml; Sigma), GAPDH (1:10,000, 0.01 μg/ml; Abcam), TfR (1:1000, 0.50 μg/ml; Invitrogen), ubiquitin (1:2000, 0.10 μg/ml; Santa Cruz Biotechnology), KIF5 (1:1000, 1.00 μg/ml; Millipore), and Nav1.7 (1:500, 1.00 μg/ml; Millipore) and rabbit antibodies against GFP (1:1000, 1.00 μg/ml; Invitrogen), calnexin (1:10,000, 0.50 μg/ml; Sigma), KIF5B (1:500, 0.20 μg/ml; Abcam), Nav1.6 (1:2000, 0.80 μg/ml; Millipore), Nav1.8 (1:2000, 0.40 μg/ml; Alomone Labs) and Nav1.9 (1:500, 1.60 μg/ml; Alomone Labs). The immunoreactive bands were quantified using Image Pro Plus software (Media Cybernetics). For tissue samples and cell lysates, 15 and 30 μg of total protein per lane were loaded, respectively. The specificity for KIF5, Nav1.6, Nav1.8, and Nav1.9 was verified through the preabsorption of antibodies with 10−6 M the corresponding immunogenic peptides. Due to the lack of Nav1.7 antigen (a purified protein containing a region of Nav1.7 fused with glutathione S-transferase), the specificity of Nav1.7 was verified through the transfection of the Nav1.7 plasmid in HEK293 cells.
Immunocytochemistry.
The ND7–23 cells treated with brefeldin A (BFA) and the cultured neurons in the microfluidic chamber were fixed with 4% paraformaldehyde at 4°C for 15 min. The cells were stained with mouse antibodies against GM130 (1:500, 0.5 μg/ml; BD Biosciences) or β-tubulin 3 (1:1000, 0.1 μg/ml; Millipore) or rabbit antibodies against Nav1.8 (1:1000, 0.80 μg/ml), followed by FITC-conjugated donkey anti-mouse (1:100; Jackson ImmunoResearch) or Cy3-conjugated donkey anti-rabbit (1:500; Jackson ImmunoResearch) secondary antibodies. The cells were labeled with DAPI (1 μg/ml; Invitrogen) or not, mounted in a mixture of glycerol/PBS (3:1) containing 0.1% paraphenylenediamine, and examined under a Leica SP5 laser scanning confocal microscope.
Immunohistochemistry.
For all groups, 12-μm-thick sections from the fixed sciatic nerve and L4 and L5 DRGs were cut in series using a cryostat and mounted on gelatin-coated slides. The sections were processed for double immunofluorescence staining using mouse antibodies against KIF5 (1:1000) and rabbit antibodies against Nav1.8 (1:1000) overnight at 4°C. After extensive rinsing with PBS, the sections were incubated with FITC-conjugated donkey anti-mouse (1:100) and Cy3-conjugated donkey anti-rabbit (1:500) secondary antibodies for 45 min at 37°C. The sections were mounted and examined under a Leica SP5 confocal microscope. The specificity for Nav1.8 and KIF5 was verified through the preabsorption of antibodies with 10−6 M the corresponding immunogenic peptides in DRGs.
Drug treatment.
BFA (2 μg/ml; Tocris Bioscience Cookson) was added to the culture medium of HEK293T or ND7–23 cells for 5 h. The HEK293 cells were incubated with 100 μg/ml cycloheximide (CHX; Sigma) in culture medium for 0.5, 1, 2, 4, 6, 8, and 12 h. Treatments with 10 μm MG132 (Sigma) were performed for 6 h. In all experiments, the control cells were treated with the vehicle used for drug preparation.
Electrophysiological recording.
Conventional whole-cell patch-clamp recording was conducted at room temperature using an EPC9 amplifier (HEKA Elektronik) as described previously (Liu et al., 2010). Briefly, ND7–23 cells expressing GFP or dissociated DRG neurons with a diameter of <30 μm were recorded. To isolate TTX-R Na+ currents, 1 μm TTX (Sigma) was included to inhibit TTX-sensitive Na+ currents in a bath solution containing the following (in mm): 120 NaCl, 5 CsCl, 1 MgCl2, 1 CaCl2, 0.1 CdCl2, 20 TEA-Cl, 10 HEPES, and 10 d-glucose, pH 7.4 (all from Sigma). The pipette was filled with intracellular solution containing the following (in mm): 140 CsF, 1 EGTA, 10 NaCl and 10 HEPES, pH 7.4 (all from Sigma), with a resistance of 3–4 MΩ. Whole-cell Na+ currents were filtered at 5 kHz and acquired at 50 kHz using PULSE 8.30 software (HEKA Elektronik). Series resistance errors and capacitance transients were compensated for >85% using the amplifier circuitry and linear leakage currents were digitally subtracted using the online “P/4” procedure. The Nav1.8 currents were isolated using the following protocol: a holding potential at −70 mV, followed by a series of 100 ms pulses from −40 to +40 mV. To generate activation curves, the cells were held at −70 mV and stepped to potentials of −70 to +60 mV in 10 mV increments for 100 ms. To generate fast-inactivation curves, the cells were stepped to inactivation potentials of −100 to +20 mV for 500 ms, followed by a 20 ms step to 10 mV. The current–voltage relationships (I–V curves) of peak transient and averaged persistent (60–90 ms) currents were plotted. The data were analyzed using IGOR PRO 4.01 (WaveMetrics) and PULSEFIT (HEKA Elektronik).
For axonal recording, cultured DRG neurons on polylysine-coated glass coverslips were placed in a submerged-style chamber mounted under an infrared-differential interference contrast microscope (BX-51 WI; Olympus), and continuously perfused with oxygenated extracellular solution at 36°C. The GFP-expressing axons derived from small-diameter (16–30 μm) DRG neurons with a distance of >80 μm (127.5 ± 23.2 μm) away from the soma were identified under GFP fluorescence. For voltage-clamp recording to isolate the TTX-R Na+ currents, we added 1 μm TTX and 200 μm CdCl2 to a normal artificial cerebral fluid (ACSF) solution containing the following (in mm): 126 NaCl, 2.5 KCl, 2 MgSO4, 2 CaCl2, 26 NaHCO3, 1.25 NaH2PO4 and 25 dextrose (315 mOsm), pH 7.4. The pipette was filled with CsCl-based intracellular solution containing the following (in mm): 140 CsCl, 2 MgCl2, 2 Na2ATP, 10 HEPES and 10 EGTA, 287 mOsm, pH 7.2 with CsOH, with a resistance of 7–10 MΩ. The cells were held at −70 mV and stepped to potentials from −70 to +60 mV in 10 mV increments for 30 ms. Holding potential of −70 mV was used to minimize the residual current of persistent TTX-R Nav1.9 currents. The ACSF extracellular solution was used for current–clamp recording to determine action potential. The intracellular solution contained the following (in mm): 140 K-gluconate, 3 KCl, 2 MgCl2, 10 HEPES, 0.2 EGTA, and 2 Na2ATP. To inactivate other sodium channels, except for Nav1.8, the axons of the recording sites were held at a baseline membrane potential of −40 mV using sustained current injections (Vasylyev and Waxman, 2012). The action potentials primarily mediated through Nav1.8 were evoked using a series of 200 ms depolarizing current injections from 10 to 150 pA (step: 10 pA) or from 100 to 800 pA (step: 50 pA) if the action potentials were not generated. The data were low-pass filtered at 10 kHz and digitized at 50 kHz using a MultiClamp 700B amplifier and Digidata 1440 A/D converter (Molecular Devices). The data were processed and analyzed using pClamp 10.2 (Molecular Devices) and MATLAB (MathWorks). The maximum rising slope of the action potential was determined from the peak amplitude of the first derivative of action potential (dV/dt).
To exclude the possibility that local axonal current was contaminated by somatic current, we performed simultaneous patch-clamp recordings at soma and axons. Once dual patch-clamp recording was achieved, the soma or axons were stimulated by current injection to determine whether the two recordings were from the same neuron. Voltage commands were delivered to the axons at a distance of >80 μm from the soma and the currents were recorded at soma and axon. The axonal Na+ current was evoked immediately after the onset of voltage steps, exhibiting greater peak amplitude occurring earlier than the corresponding somatic current. In addition to the immediate current, a second current was observed in half of the recordings and this late current corresponded well with the large somatic Na+ current, indicating the contribution of somatic channels (see Fig. 9A). Because the early current component was mainly attributable to the activation of local axonal channels, we focused on the properties of this current.
Figure 9.
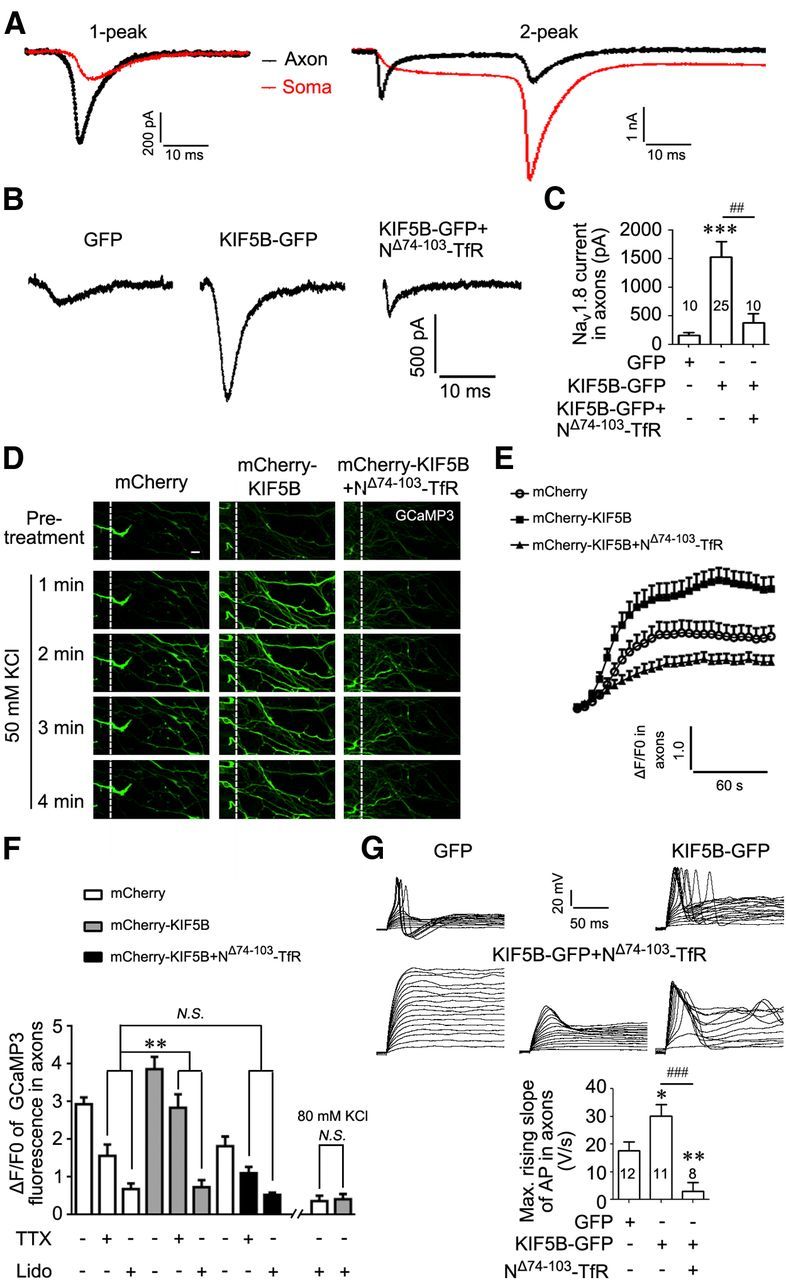
KIF5B promotes the function of Nav1.8 in axons of DRG neurons. A, Two examples of currents from dual patch-clamp recordings at the soma and axon. Black lines, currents detected at axon; red lines, currents detected at soma. The left example with the axon recorded 120 μm from the soma showing that the immediate current peak (1-peak) after the voltage step at the axon occurred earlier and exhibited greater amplitude than that at the soma (axial current arrived at the soma). The right example with the axon recorded 240 μm from the soma showing that the second peak (2-peak) was attributable to the activation of somatic channels. B, C, Representative Nav1.8 current traces recorded in the GFP-expressing axons of the small DRG neurons expressing different proteins as indicated. Summary bar chart shows the mean peak current of three groups. ***p < 0.001 versus control and ##p < 0.01 versus indicated, Mann–Whitney test. D, DRG neurons cotransfected GCaMP3 with the indicated plasmids were cultured in microfluidic chambers. Fluorescence images show the KCl-induced response of GCaMP3 in axons in the axon compartment. Scale bar, 10 μm. E, Mean ΔF/F0 kinetic curves in axons from the three groups. ΔF = F − F0, where F indicates fluorescence intensity of the axon at time t and F0 indicates fluorescence intensity of the axon before treatment. F, Bar charts showing maximum ΔF/F0 in axons from the three groups with or without 1 μm TTX or 500 μm lidocaine. **p < 0.01 versus indicated, unpaired t test. N.S., nonsignificant. G, Representative voltage traces recorded at axons (> 80 μm from soma) from a baseline membrane potential of −40 mV. The injected currents to initiate the first action potential (AP) were 120 and 50 pA for the representative neurons expressing GFP and KIF5B-GFP, respectively. For axons expressing NΔ74–103-TfR-Myc, APs were not generated in seven of eight axons (two as the left and five as the center), suggesting a low density of functional Nav1.8 channels. Only one axon generated APs and the injected current to initiate the first AP was 130 pA (right). Summary bar chart shows the maximum rising slopes of APs. Numbers within columns indicate cell number used in each experiment (C, G). *p < 0.05 versus control, unpaired t test; **p < 0.01 versus control and ###p < 0.001 versus indicated, Mann–Whitney test.
Calcium imaging.
To display calcium influx, we adopted a genetically encoded calcium indicator, GCaMP3, which was more stable and sensitive compared with GCaMP2 (Tian et al., 2009). The small-diameter (<30 μm) DRG neurons expressing GCaMP3 cultured in a microfluidic compartment chamber were imaged with a PerkinElmer UltraView Vox system using a 10× lens on a temperature-controlled workstation (37°C) with an inverted microscope. The axons were stimulated with 50 or 80 mm KCl through treatment with a combination of 1 μm TTX or 500 μm lidocaine. A 6-min video was acquired during treatment. The change in fluorescence intensity was analyzed using ImageJ 1.4 software. Data were acquired from at least three independent experiments.
Statistical analysis.
Data are presented as means ± SEM. For the biochemical experiments, data were quantified from at least three independent experiments and analyzed with Student's paired t test. Comparison of channel degradation rate was performed using two-way ANOVA. For others, normally distributed data were analyzed using Student's paired or unpaired t tests for two-group comparison using Prism 5 software (GraphPad) and non-normally distributed data were analyzed using nonparametric statistical tests (Mann–Whitney test). Differences were considered significant at a level of p < 0.05.
Results
KIF5 is increased with Nav1.8 accumulation in the sciatic nerve during peripheral inflammation
Previous studies have shown that Nav1.8 accumulates in the sciatic nerve under pathological conditions (Novakovic et al., 1998; Yiangou et al., 2000; Gold et al., 2003; Keh et al., 2008). We explored molecules to induce Nav1.8 accumulation in peripheral nerves. Motor protein kinesin-1 has been reported to involve the forward transport of ion channels (Hirokawa et al., 2010) and we detected the protein level of KIF5 in the sciatic nerve during peripheral inflammation. Representative immunoblots and quantitative data showed that the protein level of KIF5 was significantly increased in the sciatic nerve after peripheral inflammation induced through intraplantar CFA injection into the rat hindpaw (Fig. 1A), but not in DRGs (Fig. 1B). Simultaneously, the Nav1.8 level was increased in the sciatic nerve after peripheral inflammation (Fig. 1C), consistent with results obtained from other studies (Coward et al., 2000). According to previous research, all three kinesin-1 isoforms were expressed in neurons, but the expression of these proteins varied among different cell types (Kanai et al., 2000). We analyzed the expression profile of kinesin-1 isoforms using semiquantitative RT-PCR and observed that KIF5A, KIF5B, and KIF5C were distributed throughout the central and peripheral nervous systems and KIF5A and KIF5B were more abundant in DRGs (Fig. 1D). In addition, no alterations in KIF5A-C mRNA expression were detected in DRGs after the intraplantar CFA injection (Fig. 1E). The colocalization of KIF5 and Nav1.8 was further verified in DRG neurons and the sciatic nerve because KIF5 was expressed in all DRG neurons (Fig. 1F). Four days after inflammation, immunostaining showed an increase in KIF5 expression and Nav1.8 accumulation in the sciatic nerve (Fig. 1G). Therefore, kinesin-1 might be associated with the accumulation of Nav1.8 in the sciatic nerve under CFA-induced inflammatory pain.
Figure 1.
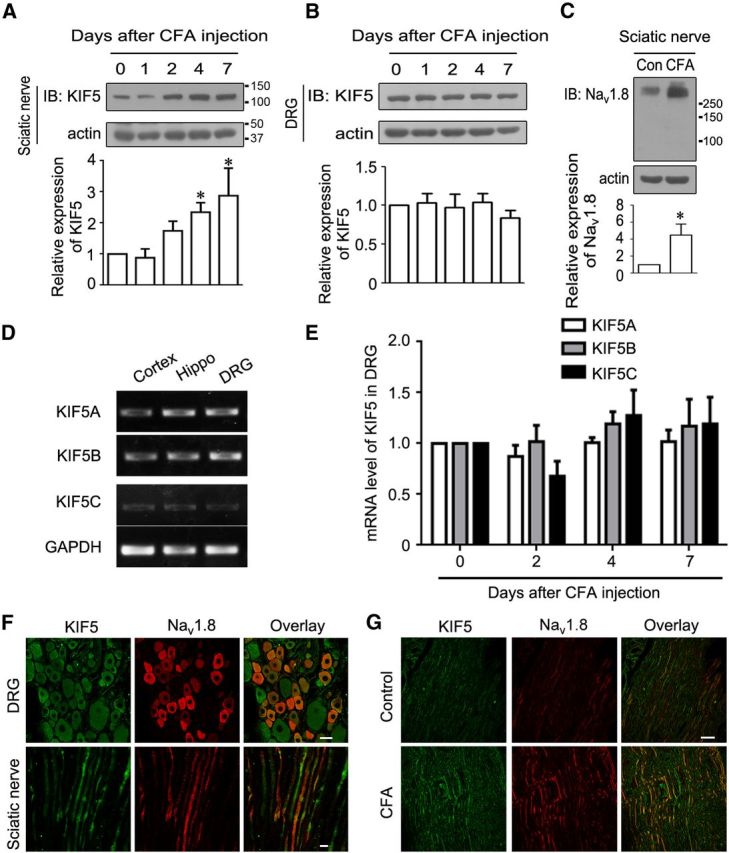
KIF5 is increased with Nav1.8 accumulation in the sciatic nerve with peripheral inflammation. A, B, Immunoblots showing the time course of the upregulated expression of KIF5 in the sciatic nerve, but no change in L4 and L5 DRGs, after intraplantar CFA injection. C, Immunoblots showing the increase in Nav1.8 protein levels in the sciatic nerve 4 d after intraplantar CFA injection. Actin served as an internal control for protein loading (A–C). D, Tissue distribution of three KIF5s using semiquantitative RT-PCR. KIF5A and KIF5B were more abundant in DRGs. GAPDH served as an internal control. E, Real-time RT-PCR showing no changes in the mRNA level of KIF5A-C in DRGs after intraplantar CFA injection. F, Immunohistochemistry showing the colocalization of Nav1.8 with KIF5 in DRG neurons and the sciatic nerve. Scale bars: top, 25 μm; bottom, 5 μm. G, Immunohistochemistry showing the increase in KIF5 and Nav1.8 in the sciatic nerve 4 d after intraplantar CFA injection. Scale bar, 100 μm. All quantitative data are plotted as normalized values versus control. *p < 0.05 versus control, paired t test. Molecule weight markers on the first blot of molecules are provided; Nav1.8, ∼260 kDa; KIF5, ∼120 kDa; and actin, ∼42 kDa.
KIF5B interacts with Nav1.8 and promotes its surface expression
The relationship between kinesin-1 and Nav1.8 was identified. Coimmunoprecipitation revealed that KIF5 interacted with Nav1.8 in DRGs (Fig. 2A). Interestingly, KIF5 also interacted with Nav1.9, but not Nav1.6 or Nav1.7 (Fig. 2A). Considering the low expression of KIF5C in DRGs, only the roles of KIF5A and KIF5B were further evaluated. An interaction between KIF5B and Nav1.8 was observed in HEK293T cells coexpressing GFP-tagged Nav1.8 at the C terminus (Nav1.8-GFP) and Myc-tagged KIF5B at the C terminus (KIF5B-Myc) or Myc-tagged Nav1.8 at the C terminus (Nav1.8-Myc) and GFP-tagged KIF5B at the C terminus (KIF5B-GFP; Fig. 2B,C), whereas a much weaker interaction between KIF5A and Nav1.8 was detected in HEK293T cells expressing GFP-tagged KIF5A at the C terminus (KIF5A-GFP) and Nav1.8-Myc (Fig. 2C). These results suggest that KIF5B interacts more strongly than KIF5A with Nav1.8.
Figure 2.

KIF5B interacts with Nav1.8 and promotes its surface expression. A, Nav1.8 interacting with KIF5 in DRGs. The DRG lysates were immunoprecipitated with IgG or KIF5-specific antibodies and analyzed using the indicated antibodies. B, C, Nav1.8 interacting with KIF5B in HEK293T cells. Nav1.8-GFP or Nav1.8-Myc was cotransfected with KIF5B-Myc, KIF5B-GFP, or KIF5A-GFP. The proteins were immunoprecipitated with GFP or Myc-specific antibodies and subjected to immunoblotting with the indicated antibodies. D, E, Surface biotinylation analysis of Nav1.8. The HEK293T cells were cotransfected Nav1.8-Myc with KIF5B-GFP, KIF5A-GFP, KIF5BG234C-GFP, or KIF5B1–330-GFP and subjected to cell surface biotinylation/immunoblotting. F, Surface biotinylation analysis of endogenous Nav1.8. DRG neurons were transfected with GFP or KIF5B-GFP and subjected to cell surface biotinylation/immunoblotting. TfR served as an internal control for protein loading (D–F). The images are representative of at least three independent experiments (A–F). All quantitative data are plotted as normalized values versus control. *p < 0.05 and **p < 0.01 versus control, paired t test. Molecular weight markers on the first blot of molecules are provided; Nav1.6, ∼230 kDa; Nav1.7, ∼230 kDa; Nav1.9, ∼250 kDa; TfR, ∼90 kDa; KIF5B-GFP, ∼135 kDa; KIF5A-GFP, ∼142 kDa; KIF5BG234C-GFP, ∼135 kDa; and KIF5B1–330-GFP, ∼60 kDa.
We investigated whether KIF5B and KIF5A influenced the surface expression of Nav1.8. For biochemical experiments, HEK293T cells with higher transfection efficiency that expressed endogenous KIF5B but not endogenous Nav1.8 were used. In HEK293T cells expressing Nav1.8-Myc, the overexpression of KIF5B-GFP increased the surface expression of Nav1.8 ∼4-fold, whereas the overexpression of KIF5A-GFP only slightly elevated the surface expression of Nav1.8 (Fig. 2D). The two KIF5B mutants, KIF5BG234C-GFP (deficient in ATP hydrolysis) and KIF5B1−330-GFP (lacking the cargo-binding domain), did not enhance the surface expression of Nav1.8 (Fig. 2E). Importantly, KIF5B overexpression also caused a remarkable increase in endogenous Nav1.8 expression on the surface of cultured DRG neurons (Fig. 2F). Therefore, KIF5B plays a major role in promoting the cell surface expression of Nav1.8 among the three kinesin-1 isoforms.
KIF5B is required for maintaining the Nav1.8 current
To examine the hypothesis that KIF5B was required for Nav1.8 function, cultured DRG neurons and ND7–23 cells derived from rat DRG neurons were used for electrophysiological recording. ND7–23 cells do not express endogenous Nav1.8 but do express KIF5B. In a previous study, we showed that the expressed Nav1.8 currents were much larger in ND7–23 cells than those in HEK293T cells and resembled the endogenously dissected Nav1.8 currents (Zhang et al., 2008). Small DRG neurons (diameter <30 μm) expressing both endogenous Nav1.8 and KIF5B were also used for electrophysiological recording. We applied shRNA technology to knock down the expression of KIF5B in ND7–23 cells and cultured DRG neurons. The specificity and efficiency of shRNA was assessed. In ND7–23 cells, the mRNA level of KIF5B, but not KIF5A or KIF5C, was reduced through KIF5B shRNA (Fig. 3A). The expression of endogenous KIF5B protein in ND7–23 and KIF5B-Myc in HEK293T cells was also significantly decreased (Fig. 3A). Subsequently, we examined the effect of knocking down endogenous KIF5B on the current density of Nav1.8. In ND7–23 cells coexpressing RFP-tagged Nav1.8 at the N terminus (RFP-Nav1.8) and KIF5B shRNA, the representative Nav1.8 currents and quantitative data on Nav1.8 current density showed a dramatic decrease compared with that of cells coexpressing RFP-Nav1.8 and Ctrl shRNA (Ctrl shRNA: 40.30 ± 3.90 pA/pF, n = 22, vs KIF5B shRNA: 24.59 ± 2.91 pA/pF, n = 22; Fig. 3B). In the presence of 1 μm TTX, which blocks TTX-sensitive sodium channels, we further dissected TTX-R Nav1.8 from TTX-R Nav1.9 currents in small DRG neurons by holding potential at −70 mV. The TTX-R currents showed a large transient peak with a small persistent component (Fig. 3C), consistent with earlier reports (Hudmon et al., 2008; Shields et al., 2012). The I–V curves of both transient peak and averaged persistent currents revealed depolarized voltage dependence (Fig. 3C), consistent with the properties of Nav1.8. Importantly, in small DRG neurons expressing KIF5B shRNA, the representative Nav1.8 currents and quantitative data on Nav1.8 current density also showed a significant decrease compared with neurons expressing Ctrl shRNA (Ctrl shRNA: 117.10 ± 14.57 pA/pF, n = 22, vs KIF5B shRNA: 77.87 ± 11.29 pA/pF, n = 21; Fig. 3D). These results suggest that endogenous KIF5B is required for maintaining Nav1.8 function.
Figure 3.

KIF5B is required for maintaining Nav1.8 current. A, Left, KIF5B mRNA levels reduced after transfection of KIF5B shRNA in ND7–23 cells. Right, Immunoblots of lysates from HEK293T cells cotransfected KIF5B-Myc with control (Ctrl) shRNA or KIF5B shRNA and from ND7–23 cells transfected with Ctrl shRNA or KIF5B shRNA. Actin served as an internal control for protein loading. The images are representative of at least three independent experiments. B, Representative Nav1.8 current traces recorded in ND7–23 cells cotransfected RFP-Nav1.8 with Ctrl or KIF5B shRNA. Summary bar chart shows the mean peak current density of two groups. C, Representative Nav1.8 current traces recorded in a dissociated rat DRG neuron. I–V curves of Nav1.8 transient peak and averaged persistent currents are provided (filled squares: transient peak current, n = 7; filled circles: persistent peak current, n = 7). D, Representative Nav1.8 current traces recorded in dissociated rat DRG neurons transfected with Ctrl or KIF5B shRNA. Summary bar chart shows the mean peak current density of two groups. E, Representative Nav1.8 current traces recorded in ND7–23 cells cotransfected RFP-Nav1.8 with KIF5B-GFP, KIF5BG234C-GFP, or KIF5B1–330-GFP. Summary bar chart shows the mean peak current density of four groups. F, Steady-state activation, fast-inactivation curve, and the I–V curve (filled squares: GFP, activation, n = 5; inactivation, n = 6; I–V curve, n = 6; open squares: KIF5B-GFP, activation, n = 9; inactivation, n = 7; I–V curve, n = 7). G, Representative Nav1.8 current traces recorded in dissociated rat DRG neurons after transfection with GFP or KIF5B-GFP. Summary bar chart shows the mean peak current density. Numbers within columns indicate cell number used in each experiment (B, D, E, G). *p < 0.05, **p < 0.01, ***p < 0.001 versus control, Mann–Whitney test. Molecular weight of KIF5B, ∼120 kDa.
KIF5B increases the Nav1.8 current
We examined the effects of KIF5B on the function of Nav1.8. In ND7–23 cells coexpressing RFP-Nav1.8 and KIF5B-GFP, the representative Nav1.8 currents and quantitative data on Nav1.8 current density showed a significant increase compared with cells expressing only RFP-Nav1.8 (control: 34.87 ± 4.15 pA/pF, n = 32, vs KIF5B-GFP: 58.52 ± 6.14 pA/pF, n = 33; Fig. 3E). However, neither KIF5BG234C (39.41 ± 6.68 pA/pF, n = 24) nor KIF5B1–330 (31.92 ± 10.28 pA/pF, n = 19) altered the current density of this channel (Fig. 3E), indicating that motor activity and cargo binding of KIF5B were required for its functional regulation of Nav1.8. In small DRG neurons expressing KIF5B-GFP, the representative Nav1.8 currents and quantitative data on Nav1.8 current density also showed a remarkable increase compared with control neurons (control: 98.20 ± 12.53 pA/pF, n = 26, vs KIF5B-GFP: 230.91 ± 39.82 pA/pF, n = 26; Fig. 3G). Therefore, KIF5B increases the Nav1.8 current.
We further analyzed whether KIF5B also affected the electrophysiological properties of Nav1.8. In ND7–23 cells coexpressing RFP-Nav1.8 and KIF5B-GFP, the voltage-dependent activation of Nav1.8 (V1/2= −4.44 ± 0.20 mV, slope k = 4.96 ± 0.17, n = 9) was not significantly altered compared with cells expressing only RFP-Nav1.8 (V1/2= −6.28 ± 0.48 mV, slope k = 5.62 ± 0.42, n = 6; Fig. 3F). The steady-state inactivation in ND7–23 cells coexpressing RFP-Nav1.8 and KIF5B-GFP (V1/2= −50.76 ± 0.34 mV, slope k = 11.37 ± 0.30, n = 8) was similar to that in cells expressing only RFP-Nav1.8 (V1/2= −49.09 ± 0.35 mV, slope k = 12.38 ± 0.31, n = 5; Fig. 3F). The I–V curves showed that the depolarized voltage dependence properties of Nav1.8 currents were not altered by overexpression of KIF5B (Fig. 3F). These results exclude the possibility that KIF5B-induced Nav1.8 currents result from the modulation of the electrophysiological properties of this channel.
KIF5B promotes the forward transport of Nav1.8 and prevents channel degradation
We used BFA to further examine whether the KIF5B-promoted surface expression of Nav1.8 came from the increased forward transport of this channel. BFA blocks the secretory pathway upstream of the Golgi (Dinter and Berger, 1998) and thus deprives forward transport of membrane proteins to the cell surface. Immunocytochemistry showed that treatment with 2 μg/ml BFA for 5 h dispersed the Golgi in ND7–23 cells (Fig. 4A). In ND7–23 cells coexpressing RFP-Nav1.8 and KIF5B-GFP, the average current density of Nav1.8 was reduced from 47.34 ± 7.48 pA/pF to 20.50 ± 4.94 pA/pF after BFA treatment (Fig. 4B). Accordingly, in HEK293T cells coexpressing Nav1.8-Myc and KIF5B-GFP, the KIF5B-induced surface expression of Nav1.8 was almost completely blocked after BFA treatment (Fig. 4C). These results indicate that KIF5B is involved in the promotion of Nav1.8 forward transport, thereby increasing the function of this channel.
Figure 4.

KIF5B promotes the forward transport of Nav1.8. A, ND7–23 cells labeled with antibodies against the Golgi marker GM130. Scale bar, 10 μm. B, Representative Nav1.8 current traces recorded in ND7–23 cells transfected with RFP-Nav1.8 or RFP-Nav1.8 and KIF5B-GFP under control conditions or after 2 μg/ml BFA treatment for 5 h. Summary bar chart shows the mean peak current density. Numbers within columns indicate cell number used in each experiment. **p < 0.01 versus control and ###p < 0.001 versus indicated, Mann–Whitney test. C, HEK293T cells cotransfected with Nav1.8-Myc or Nav1.8-Myc and KIF5B-GFP incubated in DMEM containing vehicle or 2 μg/ml BFA for 5 h and subjected to cell-surface biotinylation/immunoblotting. TfR served as an internal control for protein loading. The images are representative of four independent experiments. All quantitative data are plotted as normalized values versus control. *p < 0.05 versus control and #p < 0.05 versus indicated, paired t test.
In a previous study, we suggested a mechanism for Nav1.8 degradation through an ubiquitin-proteasome system (Li et al., 2010). Therefore, the influence of the KIF5B on the stability of Nav1.8 was examined. Degradation was assayed in the presence of CHX, which inhibits protein synthesis. In HEK293 cells stably expressing Nav1.8-GFP, treatment with CHX reduced the protein expression of this channel within 1–2 h and Nav1.8 was mostly degraded after 8–12 h (Fig. 5A). The overexpression of KIF5B reduced the degradation of Nav1.8-GFP in the presence of CHX (Fig. 5A), indicating that KIF5B protected this channel from degradation. Furthermore, the role of ubiquitination in this process was determined. After 6 h of treatment with the proteasome inhibitor MG132 (10 μm), the ubiquitination of Nav1.8-GFP increased, whereas the overexpression of KIF5B-Myc decreased the level of Nav1.8 ubiquitination in HEK293 cells (Fig. 5B). In addition, we detected the interaction between Nav1.8 and ER chaperon protein calnexin, which has been implicated in the degradation of Nav1.8 (Li et al., 2010). In HEK293 cells, KIF5B overexpression decreased the interaction between calnexin and Nav1.8 (Fig. 5C), suggesting that the amount of channel available for degradation was reduced. Therefore, KIF5B promotes the forward transport of Nav1.8, thereby protecting this channel from degradation.
Figure 5.

KIF5B prevents the degradation of Nav1.8. A, HEK293 cells stably expressing Nav1.8-GFP were transiently transfected with KIF5B-Myc, treated with 100 μg/ml CHX for the indicated times, and subjected to immunoblotting. Actin served as an internal control for protein loading. Quantitative data were plotted as a normalized value versus control. ***p < 0.001, two-way ANOVA. B, Increase in Nav1.8-GFP ubiquitination blocked after coexpression with KIF5B. The HEK293 cells stably expressing Nav1.8-GFP were transfected with KIF5B-Myc or not and treated with 10 μm MG132 for 6 h. Proteins were immunoprecipitated with GFP-specific antibodies and subjected to immunoblotting with the indicated antibodies. C, In HEK293 cells stably expressing Nav1.8-GFP transfected with KIF5B-Myc or not, the proteins were immunoprecipitated with a GFP-specific antibody and the cell lysates were subjected to immunoblotting with the indicated antibodies. The images are representative of at least three independent experiments. All quantitative data are plotted as normalized values versus control. *p < 0.05 versus cells expressing Nav1.8-GFP; #p < 0.05 versus indicated, paired t test. Molecular weight of calnexin, ∼90 kDa.
Determination of the interacting domains between Nav1.8 and KIF5B
Given that Nav1.8 was associated with KIF5B, the domains mediating this interaction were identified. As a transmembrane protein, Nav1.8 contains five cytoplasmic fragments: N, 1L, 2L, 3L, and C. Considering the different orientations associated with transmembrane segments, we constructed two chimeric plasmids to screen the domains involved in the interaction of Nav1.8 with KIF5B: type II membrane protein TfR for the N terminus (N-TfR) and type I membrane protein CD8α for the others (such as Myc-CD8α-1L). Coimmunoprecipitation experiments in HEK293T cells transfected with KIF5B and various chimeric plasmids showed that the N terminus of Nav1.8 interacted with KIF5B (Fig. 6A). To avoid interference with the function of p11 in Nav1.8 transport, we deleted the 74–103 sequence in the N terminus of Nav1.8, which interacts with p11 (Poon et al., 2004). We observed that the expression of NΔ74–103-TfR efficiently attenuated the interaction of Nav1.8 with KIF5B in HEK293T cells (Fig. 6B). Accordingly, NΔ74–103-TfR expression suppressed the KIF5B-induced cell surface expression of Nav1.8 in both HEK293T cells and DRG neurons (Fig. 6C,D). In small DRG neurons expressing NΔ74–103-TfR-GFP, the representative Nav1.8 currents and quantitative data on Nav1.8 current density showed a remarkable decrease compared with control neurons expressing GFP (control: 119.20 ± 8.31 pA/pF, n = 17, vs NΔ74–103-TfR-GFP: 51.42 ± 6.99 pA/pF, n = 20; Fig. 6E). Therefore, the N terminus of Nav1.8 mediates the effects of KIF5B on this channel.
Figure 6.

The Nav1.8 N terminus is critical for KIF5B interaction. A, KIF5B-GFP cotransfected with N-TfR-Myc, Myc-CD8α-1L, Myc-CD8α-2L, Myc-CD8α-3L, Myc-CD8α-C, or Nav1.8-Myc in HEK293T cells. Proteins immunoprecipitated with Myc-specific antibodies and cell lysates were subjected to immunoblotting with the indicated antibodies. B, KIF5B-GFP was cotransfected with Nav1.8-Myc or Nav1.8-Myc and TfR-Myc or Nav1.8-Myc and NΔ74–103-TfR-Myc in HEK293T cells. Proteins immunoprecipitated with Nav1.8-specific antibodies and cell lysates were subjected to immunoblotting with the indicated antibodies. C, D, KIF5B-GFP was cotransfected with Nav1.8-Myc or Nav1.8-Myc and NΔ74–103-TfR-Myc in HEK293T cells (C). DRG neurons were transfected with vector or NΔ74–103-TfR-Myc (D). Proteins were subjected to cell-surface biotinylation/immunoblotting. TfR served as an internal control for protein loading. Quantitative data in C and D were plotted as normalized values versus control. The images are representative of at least three independent experiments (A–D). *p < 0.05 and **p < 0.01 versus control, paired t test. E, Representative Nav1.8 current traces recorded in dissociated rat DRG neurons transfected with GFP or NΔ74–103-TfR-GFP. Summary bar chart shows the mean peak current density of two groups. Numbers within columns indicate the cell number used in each experiment. ***p < 0.001 versus control, unpaired t test. Molecular weight markers on the first blot of molecules are provided; TfR-Myc, ∼90 kDa; N-TfR-Myc, ∼104 kDa; NΔ74–103-TfR-Myc, ∼100 kDa; Myc-CD8α-1L, ∼60 kDa; Myc-CD8α-2L, ∼70 kDa; Myc-CD8α-3L, ∼30 kDa; and Myc-CD8α-C, ∼50 kDa.
We attempted to identify the domains in KIF5B responsible for its interaction with Nav1.8. Based on earlier research (Schnapp, 2003), KIF5B was divided into motor, stalk, and tail domains. Coimmunoprecipitation experiments in HEK293T cells coexpressing Nav1.8-Myc with various GFP-tagged KIF5B truncations showed that the 511–620 aa sequence in the stalk domain of KIF5B was responsible for its interaction with Nav1.8 (Fig. 7A,B). Therefore, the N terminus of Nav1.8 interacts with the 511–620 sequence in the stalk domain of KIF5B.
Figure 7.

The 511–620 aa sequence of KIF5B interacts with Nav1.8. Nav1.8-Myc was cotransfected with various plasmids of KIF5B in HEK293T cells (A, B). The proteins were immunoprecipitated with GFP-specific antibodies and the cell lysates were subjected to immunoblotting with the indicated antibodies. The images are representative of at least three independent experiments. MW markers on the first blot of molecules are provided. Molecular weights: KIF5B1–510-GFP, ∼80 kDa; KIF5B1–620-GFP, ∼93 kDa; KIF5B1–822-GFP, ∼115 kDa; and KIF5BΔ511–620-GFP, ∼120 kDa.
KIF5B increases Nav1.8 accumulation in the axons of DRG neurons
Because KIF5B promoted the forward transport of Nav1.8, we investigated whether the axonal transport of Nav1.8 was also affected. We adopted a microfluidic chamber to culture DRG neurons and detected Nav1.8 accumulation in axons. This microfluidic chamber was used in a recent study (Chen et al., 2012) and modified from previous research (Park et al., 2006). The DRG neurons transfected with different plasmids were plated in the cell-body compartment. After 3 d, the neurons stained with DAPI and β-tubulin 3 showed that only axons extended through the microchannels to the axon compartment (Fig. 8A). Immunostaining showed that Nav1.8 was distributed along the axons in neurons expressing KIF5B-GFP (Fig. 8A). Samples were collected from the cell body and axon compartment. In the axons of DRG neurons coexpressing Nav1.8-GFP and KIF5B-Myc, Nav1.8 was accumulated ∼3-fold compared with that in axons expressing only Nav1.8-GFP (Fig. 8B). This effect was lost in cells expressing KIF5BG234C-Myc (Fig. 8B). Moreover, the coexpression of NΔ74–103-TfR-Myc inhibited Nav1.8 accumulation in axons (Fig. 8C). Therefore, KIF5B promotes Nav1.8 accumulation in the axons of DRG neurons.
Figure 8.
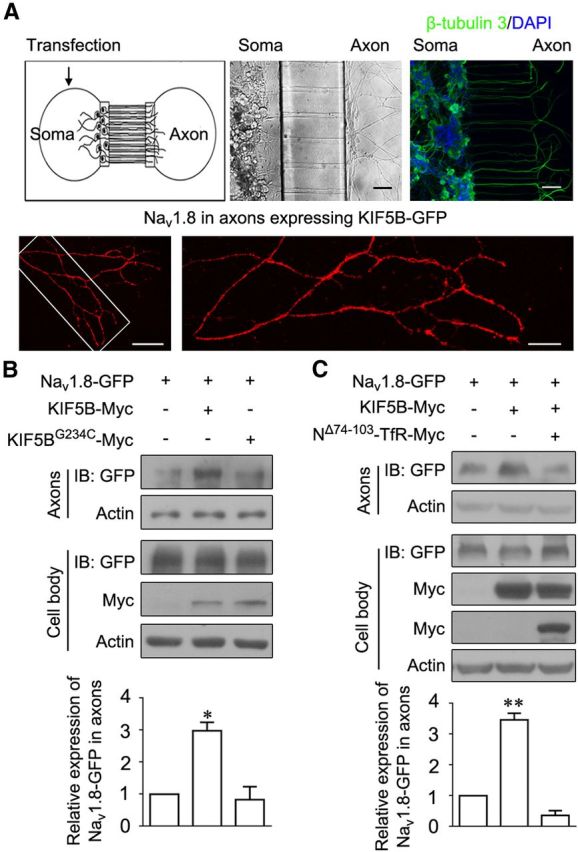
KIF5B promotes the Nav1.8 accumulation in axons of DRG neurons. A, Top left, Diagram of the microfluidic chamber. Top center, Differential interference contrast image of DRG neurons cultured for 3 d showing that the axons cross the microchannels into the axon compartment, whereas the cell bodies remain in the cell body compartment. Scale bar, 100 μm. Top right, Only axons cross the microchannels. Blue, DAPI; green, β-tubulin 3. Scale bar, 50 μm. Bottom, Immunostaining showing Nav1.8 distributed along the axons in neurons expressing KIF5B-GFP. Scale bars: bottom left, 50 μm; bottom right, 20 μm. B, Nav1.8 accumulated in the axons after cotransfection with KIF5B-Myc. Top, DRG neurons were cotransfected Nav1.8-GFP with KIF5B-Myc or KIF5BG234C-Myc deficit in ATP hydrolysis. After culturing for 3–4 d, the cell body and axons were lysed separately for immunoblotting. Actin served as an internal control for protein loading. Bottom, Quantitative analysis of the above data. C, Axonal Nav1.8 accumulation attenuated through the expression of NΔ74–103-TfR-Myc. Top, DRG neurons were cotransfected Nav1.8-GFP with KIF5B-Myc or KIF5B-Myc and NΔ74–103-TfR-Myc. After culturing for 3–4 d, the cell body and axons were lysed separately for immunoblotting. Actin served as an internal control for protein loading. Bottom, Quantitative analysis of the above data. Quantitative data were plotted as normalized values versus control (B, C). The images are representative of at least three independent experiments (B, C). *p < 0.05 and **p < 0.01 versus control, paired t test.
KIF5B increases Nav1.8 function in the axons of DRG neurons
To study the effects of KIF5B on the function of Nav1.8 in axons, we performed an electrophysiological assay in the axons of DRG neurons. Based on previous research (Vasylyev and Waxman, 2012), we performed whole-cell patch-clamp recording to measure the Nav1.8 current in the presence of 1 μm TTX. The GFP-expressing axons at a distance of >80 μm from the cell body of small DRG neurons were selected (see Materials and Methods). From a dual patch-clamp recording, the local axonal current (evoked immediately after the onset of voltage steps) was easily distinguished from that induced by the activation of somatic channels (Fig. 9A). The overexpression of KIF5B-GFP largely increased the Nav1.8 current detected in axons (control: 158.21 ± 47.00 pA, n = 10 axons, versus KIF5B-GFP: 1525.56 ± 266.18 pA, n = 25 axons; Fig. 9B,C). The coexpression of NΔ74–103-TfR completely blocked the KIF5B-promoted Nav1.8 current detected in axons (372.21 ± 159.04 pA, n = 10 axons; Fig. 9B,C). Therefore, KIF5B increases the Nav1.8 current in the axons of DRG neurons.
Next, an assay based on calcium influx was adopted to study axonal excitability. Studies have shown that the real-time imaging of calcium influx in the cell bodies of sensory neurons reliably reflects action potential firing patterns (Usoskin et al., 2010). In the present study, the fluorescence intensity in axons expressing GCaMP3 was largely increased with 50 mm KCl stimulation in the axon compartment (Fig. 9D–F), suggesting axon depolarization. In the presence of 1 μm TTX, the KCl-induced increase in GCaMP3 fluorescence intensity was reduced significantly (Fig. 9F), representing the contribution of TTX-sensitive VGSCs to axonal excitability. After pretreatment with 500 μm lidocaine, which blocks total VGSCs, the KCl-induced increase in GCaMP3 fluorescence intensity was decreased further (Fig. 9F). The contribution of TTX-R VGSCs was detected after subtracting the KCl-induced increase in GCaMP3 fluorescence intensity in the presence of lidocaine from that in the presence of TTX. The quantitative data showed that the overexpression of KIF5B significantly increased the KCl-induced calcium influx contributed by TTX-R VGSCs (Control: 1.11 ± 0.26, n = 112 axons, vs KIF5B: 2.27 ± 0.30, n = 107 axons; Fig. 9F). To evaluate the role of voltage-gated calcium channels, experiments with 80 mm KCl were further performed to generate thorough depolarization. The quantitative data showed that in the presence of lidocaine, there were no significant differences between the control group and KIF5B-overexpression group in the increase of GCaMP3 fluorescence intensity induced through treatment with 80 mm KCl (Fig. 9F). Therefore, it is likely that voltage-gated calcium channels were not involved in KIF5B-promoted axonal excitability. Importantly, the coexpression of NΔ74–103-TfR (0.73 ± 0.12, n = 93 axons) completely disrupted the KIF5B-promoted calcium influx contributed by TTX-R VGSCs (Fig. 9F). These results suggest that TTX-R VGSCs contribute to KIF5B-promoted axonal excitability.
Patch-clamp electrodes were used to obtain whole-cell current-clamp recordings of the action potentials evoked from the axons of cultured DRG neurons (Vasylyev and Waxman, 2012). The membrane potential was depolarized to −40 mV to inactivate other VGSCs, except Nav1.8, in small DRG neurons (Cummins and Waxman, 1997; Cummins et al., 2007). The maximum rising slope of the action potential, which represents channel density, showed a significant increase in axons expressing KIF5B-GFP compared with control axons (control: 17.74 ± 3.03 V/s, n = 12 axons, vs KIF5B-GFP: 30.17 ± 4.15 V/s, n = 11 axons; Fig. 9G). Moreover, the coexpression of NΔ74–103-TfR almost abolished the firing of action potential detected in axons (7/8 failure) and dramatically reduced the maximum rising slope of the generated action potential (3.05 ± 3.05 V/s, n = 8 axons; Fig. 9G). Therefore, the KIF5B-promoted accumulation of Nav1.8 in axons indeed increases neuronal excitability detected in axons.
Discussion
In the present study, ubiquitous kinesin-1 isoform KIF5B was required for the forward transport of a peripheral VGSC, Nav1.8, to the plasma membrane and axons in DRG neurons. KIF5B increased the surface expression and current density of Nav1.8, and decreased the degradation rate and ubiquitination level of this channel. Moreover, the KIF5B-promoted axonal transport of Nav1.8 led to an enhancement in Nav1.8 current and neuronal excitability detected in axons. These findings indicate the important role of motor protein KIF5B in regulating the forward trafficking and axonal transport of Nav1.8, which contributes to hypersensitivity in inflammatory pain development.
KIF5B motor protein for the forward transport of Nav1.8
Ion channels are critical for neuronal activity and depend on the number of functional channels on the cell surface. Therefore, the molecular mechanisms for the regulation of ion channel trafficking are important for channel function. As channels supporting action potential conduction in DRG neurons (Renganathan et al., 2001), several reports have shown a role for interacting molecules (such as p11 and sodium channel β subunit) and extracellular factors (such as prostaglandin E2) in promoting the surface expression of Nav1.8 (Okuse et al., 2002; Rush et al., 2005; Choi et al., 2006; Zhang et al., 2008; Thakor et al., 2009; Li et al., 2010; Liu et al., 2010). These regulations are critical to ensure physiological surface expression and to protect Nav1.8 from degradation through the ubiquitin-proteasome system. Here, we revealed an essential role for KIF5B in the forward transport of Nav1.8.
Kinesins and dyneins are motor proteins that travel along microtubule tracks to deliver cargo to their final cellular destinations. In general, kinesins move from the center of the cell toward the periphery (or the plus end of microtubules). KIF5 is the first identified and the most abundant motor protein. Both the heavy chain of KIF5 and the associated light chain contribute directly to cargo binding (Hirokawa et al., 2010). Previous studies have revealed that KIF5 is critical for the forward transport of ion channels in neurons (Setou et al., 2002; Rivera et al., 2007; Zadeh et al., 2009; Twelvetrees et al., 2010; Xu et al., 2010). The results of the present study show that ubiquitously and predominantly expressed KIF5B interacts with Nav1.8. The overexpression of this motor protein resulted in a significant increase in the cell surface expression of functional Nav1.8. This effect was caused by the augmentation in the number of channels delivered to the plasma membrane, but not through alterations in the channel properties. Moreover, suppressing endogenous KIF5B levels through RNA interference in both ND7–23 cells and DRG neurons significantly reduced the function of Nav1.8, suggesting a physiological role of KIF5B. Because treatment with the Golgi-disturbing agent BFA attenuated the KIF5B-promoted effects, the increase in surface expression of functional Nav1.8 reflected the enhanced forward delivery but not the reduced endocytosis of this channel; correspondingly, the amount of this channel for ubiquitination and degradation was reduced dramatically. The results obtained in this study suggest that KIF5B enhances the forward delivery of channel proteins to the plasma membrane and strengthens the function of Nav1.8.
Association of kinesin with Nav1.8
Previous studies have shown that KIF5 transports cargo including mitochondria and various vesicles by binding to distinct adaptor proteins through heavy or light chains (Hirokawa et al., 2010). Channels are membrane proteins synthesized in the rough endoplasmic reticulum, which are transported in vesicles and inserted into plasma membrane. However, it is not clear how these channels are loaded onto KIF5. Two possibilities exist: the motor proteins either bind to the vesicles carrying these channels or these proteins interact directly with the cytoplasmic sequences of these channels. A recent study has shown that a conserved domain in the tail of KIF5 interacts directly with the N terminus of Kv3.1 (Xu et al., 2010). KIF5B has also been reported to interact directly or indirectly with Kv1.5 and is essential for its forward trafficking (Zadeh et al., 2009). In the present study, the 511–620 aa sequence in the stalk domain of KIF5B played a major role in its interaction with Nav1.8. Due to the limitation of coimmunoprecipitation experiments, we cannot exclude the possibility that other scaffold proteins might mediate the association of KIF5B with Nav1.8.
Previous studies have identified nine other pore-forming sodium channel α-subunits in mammals (Catterall et al., 2005); of these, Nav1.6, Nav1.7, and Nav1.9, which are expressed in DRG neurons, are also critical for the sensation and transmission of pain (Dib-Hajj et al., 2010; Xie et al., 2013). Our results show that KIF5 interacted with both Nav1.8 and Nav1.9, but not with Nav1.6 or Nav1.7 in DRGs, suggesting specificity of the interaction between the channels and KIF5. Although the N terminus of Nav1.8 was primarily associated with KIF5B, the homology of this sequence was only 48% between Nav1.8 and Nav1.9. The spatial structure of this sequence or other intracellular loops of Nav1.9 might be responsible for its association with KIF5.
Role of KIF5B in Nav1.8 accumulation in peripheral nerves in pathological conditions
Previous studies have shown that Nav1.8 accumulates in peripheral nerves under pathological conditions. The translocation of Nav1.8 to myelinated and unmyelinated axons in the sciatic nerve was increased after the injection of CFA into the rat hindpaw (Coggeshall et al., 2004). A marked increase in Nav1.8 has been detected along the sciatic nerve in animal models of neuropathic pain induced through peripheral nerve injury (Gold et al., 2003). The intensity of Nav1.8 immunolabeling was enhanced in the sciatic nerve at the site of injury in chronic constriction injury models or nerve injury patients (Novakovic et al., 1998; Coward et al., 2000; Yiangou et al., 2000; Kretschmer et al., 2002; Black et al., 2008). In the present study, axonal electrophysiology showed that the increased Nav1.8 current and excitability occurred with channel accumulation in the axons of cultured DRG neurons. These results provide evidence that the accumulation of Nav1.8 in the nerve was involved in the development of peripheral hypersensitivity in inflammatory pain.
Nav1.8 mRNA can be transported peripherally to the sciatic nerve and translated locally, contributing to axonal hyperexcitability (Thakor et al., 2009). Mechanisms such as the increased transport of Nav1.8 protein to axons might also play important roles in the peripheral accumulation of this channel. However, the molecules that promote Nav1.8 transport to axons under pathological conditions remain unknown. In the present study, the protein level of KIF5B was increased in the sciatic nerve after the induction of peripheral inflammation through the intraplantar injection of CFA. In cultured DRG neurons, the overexpression of KIF5B increased Nav1.8 transport to the axons and enhanced the Nav1.8 current and neuronal excitability detected in axons. Disruption of the association between Nav1.8 and KIF5B completely abolished KIF5B-promoted channel accumulation and Nav1.8 current and excitability detected in axons. These results suggest that KIF5B plays a role in the accumulation of Nav1.8 in the sciatic nerve in the inflammatory animal model through the intraplantar injection of CFA. The importance of KIF5B in modulating inflammatory pain and the mechanisms regulating axonal transport of KIF5B need further investigation.
Footnotes
This work was supported by grants from the National Natural Science Foundation of China (30930044), the National Basic Research Program of China (2010CB912001), and the “Strategic Priority Research Program (B)” of the Chinese Academy of Sciences (XDB01020300). We thank Dr. Xiaoyang Cheng for instruction on sodium current analysis.
The authors declare no competing financial interests.
References
- Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH, Wood JN. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999;2:541–548. doi: 10.1038/9195. [DOI] [PubMed] [Google Scholar]
- Black JA, Nikolajsen L, Kroner K, Jensen TS, Waxman SG. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann Neurol. 2008;64:644–653. doi: 10.1002/ana.21527. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- Chen XQ, Wang B, Wu C, Pan J, Yuan B, Su YY, Jiang XY, Zhang X, Bao L. Endosome-mediated retrograde axonal transport of P2X3 receptor signals in primary sensory neurons. Cell Res. 2012;22:677–696. doi: 10.1038/cr.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JS, Hudmon A, Waxman SG, Dib-Hajj SD. Calmodulin regulates current density and frequency-dependent inhibition of sodium channel Nav1.8 in DRG neurons. J Neurophysiol. 2006;96:97–108. doi: 10.1152/jn.00854.2005. [DOI] [PubMed] [Google Scholar]
- Coggeshall RE, Tate S, Carlton SM. Differential expression of tetrodotoxin-resistant sodium channels Nav1.8 and Nav1.9 in normal and inflamed rats. Neurosci Lett. 2004;355:45–48. doi: 10.1016/j.neulet.2003.10.023. [DOI] [PubMed] [Google Scholar]
- Coward K, Plumpton C, Facer P, Birch R, Carlstedt T, Tate S, Bountra C, Anand P. Immunolocalization of SNS/PN3 and NaN/SNS2 sodium channels in human pain states. Pain. 2000;85:41–50. doi: 10.1016/S0304-3959(99)00251-1. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Waxman SG. Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J Neurosci. 1997;17:3503–3514. doi: 10.1523/JNEUROSCI.17-10-03503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Sheets PL, Waxman SG. The roles of sodium channels in nociception: Implications for mechanisms of pain. Pain. 2007;131:243–257. doi: 10.1016/j.pain.2007.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor M. Sodium channels and mechanisms of neuropathic pain. J Pain. 2006;7:S3–S12. doi: 10.1016/j.jpain.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu Rev Neurosci. 2010;33:325–347. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- Dinter A, Berger EG. Golgi-disturbing agents. Histochem Cell Biol. 1998;109:571–590. doi: 10.1007/s004180050256. [DOI] [PubMed] [Google Scholar]
- Gold MS, Weinreich D, Kim CS, Wang R, Treanor J, Porreca F, Lai J. Redistribution of Nav1.8 in uninjured axons enables neuropathic pain. J Neurosci. 2003;23:158–166. doi: 10.1523/JNEUROSCI.23-01-00158.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68:610–638. doi: 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Choi JS, Tyrrell L, Black JA, Rush AM, Waxman SG, Dib-Hajj SD. Phosphorylation of sodium channel Nav1.8 by p38 mitogen-activated protein kinase increases current density in dorsal root ganglion neurons. J Neurosci. 2008;28:3190–3201. doi: 10.1523/JNEUROSCI.4403-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y, Okada Y, Tanaka Y, Harada A, Terada S, Hirokawa N. KIF5C, a novel neuronal kinesin enriched in motor neurons. J Neurosci. 2000;20:6374–6384. doi: 10.1523/JNEUROSCI.20-17-06374.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keh SM, Facer P, Simpson KD, Sandhu G, Saleh HA, Anand P. Increased nerve fiber expression of sensory sodium channels Nav1.7, Nav1.8, and Nav1.7 in rhinitis. Laryngoscope. 2008;118:573–579. doi: 10.1097/MLG.0b013e3181625d5a. [DOI] [PubMed] [Google Scholar]
- Kretschmer T, Happel LT, England JD, Nguyen DH, Tiel RL, Beuerman RW, Kline DG. Accumulation of PN1 and PN3 sodium channels in painful human neuroma-evidence from immunocytochemistry. Acta Neurochirurgica. 2002;144:803–810. doi: 10.1007/s00701-002-0970-1. discussion 810. [DOI] [PubMed] [Google Scholar]
- Lai J, Porreca F, Hunter JC, Gold MS. Voltage-gated sodium channels and hyperalgesia. Annu Rev Pharmacol Toxicol. 2004;44:371–397. doi: 10.1146/annurev.pharmtox.44.101802.121627. [DOI] [PubMed] [Google Scholar]
- Li Q, Su YY, Wang H, Li L, Wang Q, Bao L. Transmembrane segments prevent surface expression of sodium channel Nav1.8 and promote calnexin-dependent channel degradation. J Biol Chem. 2010;285:32977–32987. doi: 10.1074/jbc.M110.143024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li Q, Su Y, Bao L. Prostaglandin E2 promotes Nav1.8 trafficking via its intracellular RRR motif through the protein kinase A pathway. Traffic (Copenhagen) 2010;11:405–417. doi: 10.1111/j.1600-0854.2009.01027.x. [DOI] [PubMed] [Google Scholar]
- Novakovic SD, Tzoumaka E, McGivern JG, Haraguchi M, Sangameswaran L, Gogas KR, Eglen RM, Hunter JC. Distribution of the tetrodotoxin-resistant sodium channel PN3 in rat sensory neurons in normal and neuropathic conditions. J Neurosci. 1998;18:2174–2187. doi: 10.1523/JNEUROSCI.18-06-02174.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuse K, Malik-Hall M, Baker MD, Poon WY, Kong H, Chao MV, Wood JN. Annexin II light chain regulates sensory neuron-specific sodium channel expression. Nature. 2002;417:653–656. doi: 10.1038/nature00781. [DOI] [PubMed] [Google Scholar]
- Park JW, Vahidi B, Taylor AM, Rhee SW, Jeon NL. Microfluidic culture platform for neuroscience research. Nat Protoc. 2006;1:2128–2136. doi: 10.1038/nprot.2006.316. [DOI] [PubMed] [Google Scholar]
- Poon WY, Malik-Hall M, Wood JN, Okuse K. Identification of binding domains in the sodium channel Nav1.8 intracellular N-terminal region and annexin II light chain p11. FEBS Lett. 2004;558:114–118. doi: 10.1016/S0014-5793(03)01512-6. [DOI] [PubMed] [Google Scholar]
- Renganathan M, Cummins TR, Waxman SG. Contribution of Nav1.8 sodium channels to action potential electrogenesis in DRG neurons. J Neurophysiol. 2001;86:629–640. doi: 10.1152/jn.2001.86.2.629. [DOI] [PubMed] [Google Scholar]
- Rivera J, Chu PJ, Lewis TL, Jr, Arnold DB. The role of Kif5B in axonal localization of Kv1 K+ channels. Eur J Neurosci. 2007;25:136–146. doi: 10.1111/j.1460-9568.2006.05277.x. [DOI] [PubMed] [Google Scholar]
- Rush AM, Craner MJ, Kageyama T, Dib-Hajj SD, Waxman SG, Ranscht B. Contactin regulates the current density and axonal expression of tetrodotoxin-resistant but not tetrodotoxin-sensitive sodium channels in DRG neurons. Eur J Neurosci. 2005;22:39–49. doi: 10.1111/j.1460-9568.2005.04186.x. [DOI] [PubMed] [Google Scholar]
- Schnapp BJ. Trafficking of signaling modules by kinesin motors. J Cell Sci. 2003;116:2125–2135. doi: 10.1242/jcs.00488. [DOI] [PubMed] [Google Scholar]
- Setou M, Seog DH, Tanaka Y, Kanai Y, Takei Y, Kawagishi M, Hirokawa N. Glutamate-receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature. 2002;417:83–87. doi: 10.1038/nature743. [DOI] [PubMed] [Google Scholar]
- Shields SD, Ahn HS, Yang Y, Han C, Seal RP, Wood JN, Waxman SG, Dib-Hajj SD. Nav1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain. 2012;153:2017–2030. doi: 10.1016/j.pain.2012.04.022. [DOI] [PubMed] [Google Scholar]
- Thakor DK, Lin A, Matsuka Y, Meyer EM, Ruangsri S, Nishimura I, Spigelman I. Increased peripheral nerve excitability and local NaV1.8 mRNA up-regulation in painful neuropathy. Mol Pain. 2009;5:14. doi: 10.1186/1744-8069-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, Bargmann CI, Jayaraman V, Svoboda K, Looger LL. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods. 2009;6:875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twelvetrees AE, Yuen EY, Arancibia-Carcamo IL, MacAskill AF, Rostaing P, Lumb MJ, Humbert S, Triller A, Saudou F, Yan Z, Kittler JT. Delivery of GABAARs to synapses is mediated by HAP1-KIF5 and disrupted by mutant huntingtin. Neuron. 2010;65:53–65. doi: 10.1016/j.neuron.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usoskin D, Zilberter M, Linnarsson S, Hjerling-Leffler J, Uhlén P, Harkany T, Ernfors P. En masse in vitro functional profiling of the axonal mechanosensitivity of sensory neurons. Proc Natl Acad Sci U S A. 2010;107:16336–16341. doi: 10.1073/pnas.0914705107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasylyev DV, Waxman SG. Membrane properties and electrogenesis in the distal axons of small dorsal root ganglion neurons in vitro. J Neurophysiol. 2012;108:729–740. doi: 10.1152/jn.00091.2012. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Cummins TR, Dib-Hajj S, Fjell J, Black JA. Sodium channels, excitability of primary sensory neurons, and the molecular basis of pain. Muscle Nerve. 1999;22:1177–1187. doi: 10.1002/(SICI)1097-4598(199909)22:9<1177::AID-MUS3>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Xia Ch, Rahman A, Yang Z, Goldstein LS. Chromosomal localization reveals three kinesin heavy chain genes in mouse. Genomics. 1998;52:209–213. doi: 10.1006/geno.1998.5427. [DOI] [PubMed] [Google Scholar]
- Xie W, Strong JA, Ye L, Mao JX, Zhang JM. Knockdown of sodium channel Nav1.6 blocks mechanical pain and abnormal bursting activity of afferent neurons in inflamed sensory ganglia. Pain. 2013;154:1170–1180. doi: 10.1016/j.pain.2013.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Gu Y, Barry J, Gu C. Kinesin I transports tetramerized Kv3 channels through the axon initial segment via direct binding. J Neurosci. 2010;30:15987–16001. doi: 10.1523/JNEUROSCI.3565-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiangou Y, Birch R, Sangameswaran L, Eglen R, Anand P. SNS/PN3 and SNS2/NaN sodium channel-like immunoreactivity in human adult and neonate injured sensory nerves. FEBS Lett. 2000;467:249–252. doi: 10.1016/S0014-5793(00)01166-2. [DOI] [PubMed] [Google Scholar]
- Zadeh AD, Cheng Y, Xu H, Wong N, Wang Z, Goonasekara C, Steele DF, Fedida D. Kif5b is an essential forward trafficking motor for the Kv1.5 cardiac potassium channel. J Physiol. 2009;587:4565–4574. doi: 10.1113/jphysiol.2009.178442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZN, Li Q, Liu C, Wang HB, Wang Q, Bao L. The voltage-gated Na+ channel Nav1.8 contains an ER-retention/retrieval signal antagonized by the β3 subunit. J Cell Sci. 2008;121:3243–3252. doi: 10.1242/jcs.026856. [DOI] [PubMed] [Google Scholar]