

Abstract
The neocortex in our brain stores long-term memories by changing the strength of connections between neurons. To date, the rules and mechanisms that govern activity-induced synaptic changes at human cortical synapses are poorly understood and have not been studied directly at a cellular level. Here, we made whole-cell recordings of human pyramidal neurons in slices of brain tissue resected during neurosurgery to investigate spike timing-dependent synaptic plasticity in the adult human neocortex. We find that human cortical synapses can undergo bidirectional modifications in strength throughout adulthood. Both long-term potentiation and long-term depression of synapses was dependent on postsynaptic NMDA receptors. Interestingly, we find that human cortical synapses can associate presynaptic and postsynaptic events in a wide temporal window, and that rules for synaptic plasticity in human neocortex are reversed compared with what is generally found in the rodent brain. We show this is caused by dendritic L-type voltage-gated Ca2+ channels that are prominently activated during action potential firing. Activation of these channels determines whether human synapses strengthen or weaken. These findings provide a synaptic basis for the timing rules observed in human sensory and motor plasticity in vivo, and offer insights into the physiological role of L-type voltage-gated Ca2+ channels in the human brain.
Introduction
One of the leading questions in neuroscience is how memories are formed and stored in the human brain. Learning and memory critically depend on long-term modifications in the strength of synaptic connections (Bliss and Collingridge, 1993; Sjöström et al., 2008). Changes in synaptic strength in response to timed action potential (AP) firing of connected neurons, called spike timing-dependent plasticity (STDP), resemble typical features of associative learning and are thought to underlie several forms of memory (Hebb, 1949; Kampa et al., 2007; Letzkus et al., 2007; Caporale and Dan, 2008). Depending on the millisecond timing and temporal order of presynaptic and postsynaptic spiking, synaptic strength can either increase [long-term potentiation (LTP)] or decrease [long-term depression (LTD); Levy and Steward, 1983; Markram et al., 1997; Bi and Poo, 1998]. Despite the important role attributed to synaptic plasticity in normal brain function, it is still unknown to what extent STDP plays a part in the human brain, and if so, what mechanisms govern the timing windows and whether it persists at human synapses into adulthood.
There is accumulating evidence that coincident millisecond timing of activity may indeed govern synaptic changes in humans as well (Cooke and Bliss, 2006; Müller-Dahlhaus et al., 2010). A timing-dependent form of plasticity of motor-evoked potentials (MEPs) can be induced in human subjects by pairing peripheral nerve stimulation (analogous to presynaptic stimulation) with a transcranial magnetic stimulation (TMS; analogous to postsynaptic stimulation) of the motor cortex (Stefan et al., 2000, 2002; Wolters et al., 2003; De Beaumont et al., 2012; Lu et al., 2012). Such paired associative stimulation (PAS) can induce both LTP-like increases and LTD-like decreases in MEP amplitude, with the sign of change depending on the relative timing of associated stimuli (Wolters et al., 2003, 2005; Thabit et al., 2010; De Beaumont et al., 2012; Lu et al., 2012; Conde et al., 2013; Koch et al., 2013). These changes have been suggested to be of cortical origin and related to STDP at synapses in upper cortical layers (Stefan et al., 2000; Wolters et al., 2003, 2005; Thabit et al., 2010; Conde et al., 2013; Koch et al., 2013). However, how TMS-induced changes in MEP amplitude reflect plasticity processes at the level of human synapses remains unclear.
To directly test what mechanisms govern the timing rules at neocortical synapses, and to understand the synaptic basis for plasticity-like changes in corticospinal excitability observed in vivo, we investigated STDP induced by single presynaptic and postsynaptic action potentials in pyramidal neurons of the adult human medial temporal cortex (MTC). We find that human neocortical synapses can bidirectionally change strength in response to timed presynaptic and postsynaptic activity, at least up to 65 years of age. Postsynaptic L-type voltage-gated Ca2+ channels (VGCCs) play a central role in human STDP, deciding between synaptic strengthening or weakening of the synapse and thus shaping the rules for synaptic plasticity at adult human neocortical synapses.
Materials and Methods
Human and rat brain slice preparation.
All procedures on human tissue were performed with the approval of the Medical Ethical Committee of the VU University Medical Centre, and in accordance with Dutch license procedures and the declaration of Helsinki. Human brain slices were cut from anterior medial temporal cortex tissue that had to be removed for the surgical treatment of deeper brain structures, with written informed consent of the patients before surgery, as described in Testa-Silva et al. (2010). Anesthesia was induced with fentanyl (1–3 μg/kg, i.v.) and a bolus dose of propofol (2–10 mg/kg), and was maintained with remyfentanyl (250 μg/kg/min) and propofol (4–12 mg/kg). We obtained neocortical tissue from 31 patients (18 females, 13 males; age range, 18–65 years) treated for medial temporal lobe epilepsy (24 cases), or for the removal of a hippocampal tumor (4 cases), cavernoma (2 cases), or cele (1 case). In all patients, the resected neocortical tissue was located well outside the epileptic focus or tumor, was neither gliotic nor necrotic, displayed no structural abnormalities in preoperative MRI investigations, and was judged healthy neocortical tissue by the resident pathologist of the VU University Medical Centre. After resection, the cortical tissue was placed within 30 s in ice-cold artificial CSF (aCSF) slicing solution, which contained the following (in mm): 110 choline chloride, 26 NaHCO3, 10 d-glucose, 11.6 sodium ascorbate, 7 MgCl2, 3.1 sodium pyruvate, 2.5 KCl, 1.25 NaH2PO4, and 0.5 CaCl2. The tissue was then transported to the neurophysiology laboratory, which is located within 200 m of the operating room. Transition time between resection of tissue and slice preparation was <10 min. Cortical slices (350 μm thick) were prepared in ice-cold slicing solution, and transferred to holding chambers in which they were stored for 30 min at 34°C and subsequently for at least 1 h at room temperature before recording in aCSF containing the following (in mm): 125 NaCl, 3 KCl, 1.25 NaH2PO4, 2 MgSO4, 2 CaCl2, 26 NaHCO3, and 10 glucose. All aCSF solutions were continuously bubbled with carbogen gas (95% O2, 5% CO2), and had an osmolality of 300 mOsm. Rat brain slices were prepared from male and female Wistar rats (NL; 10–15 weeks old; Harlan) in accordance with Dutch license procedures and with permission from the VU University Ethical Committee for Animal Experimentation. After decapitation, the brain was rapidly removed and dissected in ice-cold aCSF, and coronal slices of temporal association cortex were cut using procedures described for human slice preparation above.
Electrophysiology in acute human and rat neocortical slices.
Slices were placed in the recording chamber submerged in aCSF (32–35°C), and whole-cell patch-clamp recordings were made from pyramidal cells using standard borosilicate glass pipettes (3.5–5.5 MΩ resistance) filled with intracellular solution containing the following (in mm): 110 K-gluconate; 10 KCl; 10 HEPES; 10 K2Phosphocreatine; 4 ATP-Mg; 0.4 GTP, biocytin 5 mg/ml, pH adjusted with KOH to 7.3 (290–300 mOsm). Pyramidal neurons in human and rat slices were readily identified using differential interference contrast microscopy by the prominent apical dendrite and pyramid-shaped cell bodies. Cells were filled with biocytin (4–5 mg/ml) and then processed for post hoc cell identification. In humans, pyramidal neurons were recorded throughout layers 2–6. In rats, layer 5 pyramidal neurons were targeted. Slices of human cortical tissue were of good quality and showed no sign of spontaneous epileptiform activity. Recordings could be made without difficulty from apparently viable, healthy neurons for up to 24 h after slicing, but recordings included in the analysis were all made within 15 h of slice preparation. Recordings were made using Multiclamp 700A/B amplifiers (Molecular Devices), sampling at 10 kHz and low-pass filtering at 3 kHz. Recordings were digitized with an Axon Digidata 1440A and acquired using pClamp software (Axon).
Spike timing-dependent plasticity.
EPSPs were evoked every 7 s (0.14 Hz) using bipolar stimulating electrodes in glass pipettes filled with aCSF positioned ∼100–150 μm along the cell's somatodendritic axis (Fig. 1A). Duration (50 μs) and amplitude (40–80 μA) of extracellular stimulation were controlled by Isoflex stimulators (A.M.P.I.). After obtaining a stable baseline of 30–60 EPSPs, presynaptic extracellular stimulations were paired to a single postsynaptic action potential (50 times, 0.14 Hz), evoked by whole-cell current injection (1–2 nA, 1–2 ms). The timing of EPSPs and action potentials was controlled by a Master-8 stimulator (A.M.P.I.). Pairing intervals were measured from the onset of the evoked EPSP to the onset of the postsynaptic AP. Negative intervals up to −130 ms and positive intervals up to +60 ms were used to explore the temporal window for STDP. On a few occasions, extracellular stimulation evoked network activity that could sustain for tens of milliseconds, which in appearance was very similar to the “complex events” reported before in human neocortex and could be triggered by the firing of a single pyramidal neuron (Molnár et al., 2008). These events disappeared when the stimulus duration was reduced to 30 μs and only single synaptic events remained.
Figure 1.
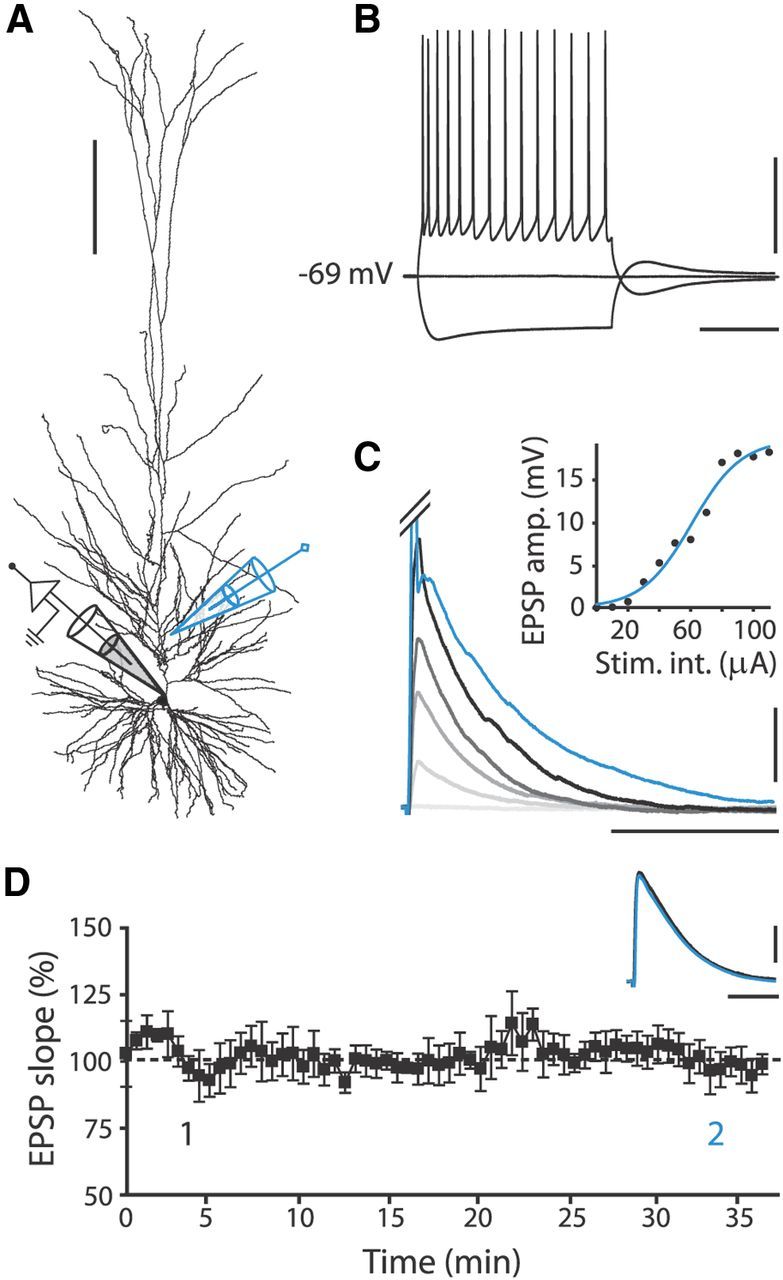
Recording pyramidal neurons in slices of human medial temporal cortex. A, Reconstruction of biocytin-labeled human pyramidal neuron, showing relative positions of recording (black) and stimulating (blue) electrode. Calibration: 200 μm. B, Typical voltage response to hyperpolarizing (−200 pA) and depolarizing (+250 pA) current steps (500 ms) of human pyramidal neuron. Calibration: 40 mV, 200 ms. C, Human EPSPs evoked by extracellular stimulation at different intensities starting at 10 μA, with 20 μA increments. Blue trace represents clipped AP evoked at 120 μA stimulation intensity (Stim. int.). Inset, Corresponding input/output curve. Calibration: 5 mV, 60 ms. D, EPSP slope over time normalized to mean EPSP slope in first 10 min of recording; no pairing to postsynaptic APs was applied. Black squares, 7 EPSP mean ± SEM. Inset traces, Example EPSPs recorded within first 5 min (1, black) and after 30 min (2, blue). Calibration: 2 mV, 40 ms.
The slope of the initial 2 ms of the EPSP was analyzed to ensure that the data reflected only the monosynaptic component of the EPSP. In general, EPSP slope had stabilized to plateau levels at 25–30 min after pairing. Change in synaptic strength was therefore measured as the percentage change in EPSP slope 25–30 min after pairing compared with baseline. When recordings lasted <20 min after pairing, the whole postpairing period was compared with baseline. Cell input resistance was monitored by applying a −100 pA, 200–500 ms hyperpolarizing pulse at the end of each sweep. Criteria for the inclusion of recordings in the STDP dataset were as follows: (1) a stable baseline EPSP slope; (2) <30% change in input resistance; (3) no more than 5 mV depolarization from original baseline resting membrane potential over the course of the experiment; and (4) baseline resting membrane potential was less than −60 mV.
Pharmacology.
All experiments were performed in the absence of blockers of GABAergic synaptic transmission. To block L-type VGCCs in STDP experiments, nifedipine (10 μm; Sigma-Aldrich) was bath applied 3–4 min before and during the pairing procedure, and washed out after pairing. To block postsynaptic NMDRs, the use-dependent NMDA receptor blocker MK801 (1 mm; Sigma-Aldrich) was added to the pipette medium (Bender et al., 2006; Rodríguez-Moreno et al., 2010).
Two-photon Ca2+ imaging.
In imaging experiments, the fluorescent dyes Alexa Fluor 594 (80 μm; Invitrogen) and Fluo-4 (100 μm; Invitrogen) were added to the intracellular solution to visualize morphology and measure [Ca2+]i changes respectively within the dendrites. After whole-cell break-in, dyes were allowed to diffuse into the dendritic tree for at least 20 min before line scans were made. To induce backpropagating APs (bAPs), single APs, or burst APs were triggered by a somatic current injection (1–2 nA), lasting 3 ms (single AP) or 15 ms (AP burst, 80–150 Hz). bAP-induced Ca2+ influx was assessed by the fluorescence change in the Fluo-4 signal relative to the corresponding constant Alexa Fluor 594 signal (Meredith et al., 2007). Fluorescence was measured using a LEICA RS2 two-photon laser scanning microscope with a 40× [0.8 numerical aperture (NA)] or 63× (0.9 NA) water-immersion objective and a Ti:Sapphire laser tuned to 830 nm excitation at a bidirectional scanning frequency of 8 kHz. Line scans (500 ms duration, 8 bit signal) synchronized with AP stimulation were made at the dendritic region of interest (ROI). To assess the effect of the L-type VGCC blocker, nifedipine (10 μm) was bath applied, and identical stimulus protocols and line scans at the ROI were repeated. At the end of the experiment, a z-stack of the dendritic ROI was made, based on the Alexa Fluor 594 signal, to calculate the distance of backpropagation from the soma. Three to six line scans were made per the stimulus protocol per ROI and were averaged for analysis. Off-line analysis was performed using custom Matlab scripts (R2008a, MathWorks). Amplitude, rise, decay, and area of fluorescence signal were extracted from the fluorescence trace by fitting a double-exponential function:
 |
Data analysis.
STDP recordings were analyzed using custom-written Matlab scripts (MathWorks). AP waveforms were analyzed in Clampfit 10.2. Statistical tests indicated in text were performed in IBM SPSS Statistics 20.0 or Sigmaplot 9.0. To assess whether EPSP–AP pairing induced a significant increase (LTP) or decrease (LTD) in synapse strength for individual STDP experiments, paired-samples t tests were used to compare baseline EPSP slope to EPSP slope 25–30 min after pairing, with Bonferroni-corrected p value to account for the familywise error rate. To extract the trend from STDP data (Fig. 2E), we used locally weighted polynomial regression (Cleveland and Devlin, 1988), which provides an unbiased estimation of the trend, referred to as a loess curve, based on local smoothing with tricube weighting and polynomial regression. Different polynomial degrees (λ = 1–3, α = 0.2) were used to determine the switch in sign, which ranged from +6.6 to +7.2 ms. 2D smoothed running averages (sampling proportion, 0.05–0.15) accounted for unequally spaced data by using the nearest-neighbor method and indicated a switch in sign between +7.3 and +8.6 ms. Figures were made in Sigmaplot 9.0 or ImageJ 1.47 and were processed using Adobe Illustrator CS2. Summary quantifications in the main text, figures, and tables are presented as means ± SEM throughout the article, and p < 0.05 was taken as the level of significance for statistical comparisons.
Figure 2.

Human cortical synapses show inverse timing rules for STDP. A, Slope data from STDP experiment with +5 ms pairing interval. Top left, EPSP–AP pairing protocol used for STDP induction. Calibration: 40 mV, 40 ms. Top middle, EPSP waveform before (black) and 25–30 min after pairing (blue). Calibration: 3 mV, 50 ms. Top right, Rising phase of EPSPs before (black) and after (blue) pairing. Calibration: 5 ms. Middle, EPSP slope over the course of an experiment normalized to baseline. Gray circles, 1 EPSP; black circles, mean of 7 EPSPs. Shaded area indicates plasticity induction period, where EPSPs where repeatedly paired to APs (50×). Bottom panels, Input resistance (Ri) and resting membrane potential (Vm) over the course of the experiment. Solid line indicates mean baseline value. B, The same as in A, for an experiment with −40 ms pairing interval. Calibration: same as in A, except for the vertical scale bar in top middle and right traces (5 mV), and horizontal scale bar of the right trace (3 ms). C, The same as in A, for an experiment with +10 ms pairing interval. Calibration same as in A. D, STDP window of adult human temporal cortex synapses. Change in synaptic strength (percentage change in EPSP slope compared with baseline) displayed versus timing of spike relative to EPSP (Δt in milliseconds). ○, EPSP slope change in individual experiments. Blue bars, Mean change in synaptic strength ± SEM for 30-ms-wide bins; red curves, single exponential fit to STDP data points with pairing intervals ≤5 and ≥10 ms. Inset traces, Examples of EPSP–AP pairings at positive and negative intervals. E, Magnification of STDP window around switch in sign from LTP to LTD. Blue bars, Mean change in synaptic strength ±SEM for 5-ms-wide bins. Note no net change in synaptic strength occurs between +5 and +10 ms, and that both running average (green) and loess curve (red, dashed) switch in sign within this range of pairing intervals. F, STDP window of adult rat temporal association cortex synapses. Data are presented as in D. Red curves, Single exponential fit to STDP data points with pairing intervals <0 and >0 ms. Inset trace, Example of EPSP–AP pairing at negative intervals in rat neuron. Calibration: 40 mV, 30 ms.
Results
To investigate synaptic plasticity in human neocortex, we made whole-cell recordings from pyramidal neurons in acute slices of healthy MTC of adult humans (31 subjects; age range, 18–65 years; see Materials and Methods). Pyramidal neurons were readily identified by their prominent apical dendrite and pyramid-shaped cell bodies, which were confirmed post hoc by inspection of neuronal morphology visualized by biocytin labeling (Fig. 1A). Recorded neurons had stable membrane potentials of −68.4 ± 0.4 mV and had regular firing properties (Fig. 1B; Table 1). Although the majority of patients were treated for epilepsy (24 of 31 patients), we found no signs of epileptiform activity in these slices; in general, spontaneous excitatory inputs to pyramidal neurons and interneurons were of small amplitude (pyramidal neurons: 14.1 ± 0.2 pA, n = 5; interneurons: 12.4 ± 1.1 pA, n = 3) and occurred at low frequencies (pyramidal neurons: 4.2 ± 0.1 Hz, n = 5; interneurons: 5.5 ± 0.3 Hz, n = 3).
Table 1.
Electrophysiological properties of human and rat temporal cortex pyramidal neurons
| Human |
Rat Total (47 cells) | |||
|---|---|---|---|---|
| Total (71 cells) | MTS epilepsy patients (50 cells) | Tumor or other patients (21 cells) | ||
| Resting membrane potential (mV) | −68.4 ± 0.4 | −68.5 ± 0.5 | −68.1 ± 0.8 | −68.3 ± 0.7 |
| Change in Vm (mV)a* | 0.9 ± 0.3 | 1.3 ± 0.3 | 0.0 ± 0.5 | −0.2 ± 0.5 |
| Input resistance (MΩ)b | 86.1 ± 8.1 | 79.6 ± 6.3 | 102.4 ± 23.6 | 63.8 ± 4.5 |
| Change in Ri (%)a,b | −2.9 ± 1.5 | −2.8 ± 1.9 | −3.0 ± 2.0 | 1.8 ± 3.4 |
| EPSP amplitude (mV) | 7.9 ± 0.3 | 7.9 ± 0.3 | 8.0 ± 0.6 | 5.7 ± 0.4 |
| EPSP slope (mV/ms) | 2.6 ± 0.1 | 2.4 ± 0.1 | 2.9 ± 0.2 | 1.9 ± 0.1 |
| EPSP decay time constant (ms) | 34.2 ± 1.7 | 36.1 ± 2.1 | 30.3 ± 2.7 | 24.7 ± 2.5 |
| ADP peak amplitude (mV) | 16.6 ± 0.8 | 16.7 ± 0.9 | 16.5 ± 1.7 | 19.0 ± 1.4 |
| ADP duration (ms) | 62.2 ± 3.1 | 64 ± 3.8 | 58.1 ± 5.0 | 35.1 ± 4.7 |
| ADP area (mV/ms) | 513.7 ± 64.8 | 517.1 ± 83.7 | 505.1 ± 92.3 | 309.9 ± 43.2 |
Data from neurons used for control STDP recordings are presented as the mean ± SEM. Vm, resting membrane potential; Ri, input resistance. Statistical testing for differences in electrophysiological properties of human neurons from two patient groups was performed using independent-samples t test.
a20–25 min after pairing compared to baseline.
bFor data not normally distributed or showing inhomogeneous variance between groups, an independent-samples Mann-Whitney U test was used.
*Indicates significant difference between two patient groups.
EPSPs were evoked by extracellular stimulation 100–150 μm away from the soma along the apical dendrite (Fig. 1A). To have a substantial dynamic range to observe both increases and decreases in synaptic strength, EPSPs were evoked at half-maximal stimulation intensity, which resulted in EPSPs of 7.9 ± 0.3 mV (n = 71; Fig. 1C; Table 1). The amplitude and slope of EPSPs evoked at 0.14 Hz remained stable for at least 50 min of recording time (2.5 mV/ms baseline slope vs 2.4 mV/ms slope 25–35 min after baseline, paired-samples t test: p > 0.05, n = 12; Fig. 1D). Other electrophysiological properties of human neocortical pyramidal neurons, such as input resistance and resting membrane potential equally changed little over the course of experiments (Table 1), and thus, the human brain slice preparation provided a good background for plasticity experiments.
Spike timing-dependent plasticity of adult human neocortical synapses
To induce STDP, EPSPs were repeatedly paired with a single postsynaptic action potential, after which EPSPs were monitored for 30–40 min (Fig. 2A–C). To characterize the temporal requirements for the induction of LTP and LTD at human cortical synapses, we varied the order and time interval between presynaptic (EPSPs) and postsynaptic (APs) activation in separate experiments. Similar to rodent synapses, we found that when presynaptic stimulation preceded the postsynaptic AP by up to 5 ms (a “pre-before-post” paradigm, hereafter referred to as pairing at “positive intervals”), robust LTP could be elicited (Fig. 2A,D). In juvenile rodent cortical synapses, LTD is induced when postsynaptic neurons fire before the presynaptic stimulation (“post-before-pre” paradigm, hereafter referred to as pairing at “negative intervals”) and the temporal window for depression at proximal synapses typically stretches from 0 to approximately −20 ms (Levy and Steward, 1983; Bell et al., 1997; Magee and Johnston, 1997; Markram et al., 1997; Bi and Poo, 1998; Wittenberg and Wang, 2006; Caporale and Dan, 2008). Interestingly, we found that in contrast to juvenile rodent synapses, induction protocols with negative timing intervals induced LTP at human neocortical synapses (Fig. 2B,D). The amount of LTP obtained at negative intervals was similar for neurons recorded in superficial and deep layers [126.0 ± 5.6% (superficial layers 2–3) vs 127.5 ± 7.8% (deep layers 4–6) for intervals from 0 to −40 ms; and 112.6 ± 7.2% (superficial) vs 119.2 ± 13.5% (deep) for intervals from −40 to −80 ms]. The window for LTP stretched far into the negative intervals, with cases of significant LTP observed even when APs preceded EPSPs by up to 100 ms during pairing (Fig. 2D).
To address the question of whether these synapses can also decrease in strength, we tested a wider range of positive intervals. Pairing intervals beyond 8 ms induced significant LTD in the majority of cells (total: n = 20, LTP: n = 1, LTD: n = 13, no significant change: n = 6; Fig. 2C,D). The switch in sign from LTP to LTD occurred at pairing intervals between +5 and +10 ms. Within this range of timing intervals, EPSP slope did not change compared with baseline (98.4 ± 13.8%; paired-samples t test: p > 0.05, n = 5; Fig. 2E). A running average and a loess curve obtained from locally weighted polynomial regression of STDP data switched in sign at +7.3 and +6.6 ms, respectively (Fig. 2E). These data show that human neocortical synapses can bidirectionally change strength in response to timed presynaptic and postsynaptic activity, and that human cortical synapses follow a reversed STDP rule compared with what is usually found in juvenile rodent cortical synapses (Bi and Poo, 1998; Froemke and Dan, 2002; Tzounopoulos et al., 2004).
Since the majority of STDP recordings were made in tissue obtained from patients with medial temporal lobe epilepsy, we questioned whether STDP results may be influenced by their disease history. We therefore compared STDP data recorded from 13 medial temporal lobe epilepsy patients to that from patients treated for a hippocampal tumor (three patients), cele (one patient), or cavernoma (one patient). We found no apparent differences in STDP magnitude and sign between patient groups; the amount of LTP and LTD obtained when pairing at intervals ranging from −30 to +5 ms and +8 to +30 ms, respectively, were both not significantly different (independent samples t tests: p > 0.05 for LTP and LTD intervals; Fig. 3A). These results suggest that the observed inverse window for STDP is not restricted to epilepsy and may apply to human neocortical synapses in general.
Figure 3.

Bidirectional STDP is independent of patient disease history or age. A, Summary of STDP data of patients treated for medial temporal lobe epilepsy (white, 13 patients) and of patients treated for tumors or other brain abnormalities (black, 5 patients). Bars represent the average EPSP slope change for pairing intervals within windows of −30 to +5 ms and +8 to +30 ms. No significant differences in STDP results were found between the two patient groups in both ranges of timing intervals (independent-samples t test). B, Efficiency of LTP when pairing at intervals ranging from −80 to +5 ms, for patients younger and older than 35 years of age, the median age of our study sample. LTP efficiency was defined as the percentage of cells where a significant increase in EPSP slope was induced after pairing (paired-samples t tests were used to compare EPSP slope during baseline to that 25–30 min after pairing, with corrected p value; see Materials and Methods). C, The same as in B, for the efficiency of LTD at pairing intervals >8 ms.
The ability of synapses to change strength decreases with age in laboratory animals, such that in older animals, synapses tend to require stronger induction protocols for plasticity to occur (Meredith et al., 2003). Studies in human subjects have also shown a considerable reduction in the amount of PAS-induced plasticity in older individuals (Sawaki et al., 2003; Müller-Dahlhaus et al., 2008). The tissue used in this study was collected from patients that ranged in age from 18 to 65 years. Both in young and older patients, LTP was induced in ∼70% of cells in pairing intervals from −80 to +5 ms (Fig. 3B). The proportion of cells undergoing LTP was not significantly different between age groups (<35 years of age: 73.7% LTP, n = 19; >35 years of age: 68.4% LTP, n = 19; χ2 test of independence: χ2(1) = 0.13, p > 0.05), suggesting no age-dependent decline in the efficacy of LTP at human synapses. In pairing intervals over 8 ms, LTD was induced in ∼65% of cells in both age groups (n = 20; Fig. 3C). The proportion of cells undergoing LTD was not different between groups (<35 years of age: 61.5% LTD, n = 13; >35 years of age: 71.4% LTD, n = 7; Fig. 3C). These results suggest no age-dependent decline in the efficacy of STDP at human synapses.
Spike timing-dependent plasticity in adult rat medial temporal cortex
The STDP window we report here for human cortical synapses is quite at odds with what is generally reported for rodent synapses. However, most rodent studies on STDP have been conducted in juvenile animals at ∼2–4 weeks of age in brain areas other than temporal cortex. To test whether STDP rules observed in adult human synapses are similar to STDP rules in adult rodent synapses, we recorded from layer 5 pyramidal neurons of adult rat temporal association cortex (10–15 weeks old). Most passive and active electrical properties of human and rat pyramidal neurons were similar, but human EPSPs did on average have larger amplitudes, faster rise times, and slower decay times than rat EPSPs (Table 1). Testing the temporal requirements for STDP in rats, we found that, similar to humans, adult rat synapses showed robust LTP when pairing at negative intervals (Fig. 2F). In contrast to human cells, however, very few adult rat synapses showed LTD at positive intervals. LTD occurred in only 4 of 32 neurons tested, whereas LTP occurred in 24 of 32 neurons at these intervals. This is in line with previous reports from other brain areas in rats, where the capacity of rodent cortical synapses to undergo LTD, in response to either spike timing-dependent plasticity paradigms (Banerjee et al., 2009) or low-frequency stimulation (Dudek and Bear, 1993; Bear and Abraham, 1996), has been shown to decline and disappear with age.
STDP at adult human synapses requires NMDA receptors and L-type voltage-gated Ca2+ channels
Neocortical synaptic plasticity depends on intracellular Ca2+ signals to trigger the intracellular machinery involved in changing synaptic strength (Couey et al., 2007; Meredith et al., 2007). Two important sources of Ca2+ in this respect are NMDA receptors (NMDARs) and VGCCs. To test whether NMDARs are involved in STDP in adult human synapses, we added the use-dependent NMDAR blocker MK801 (1 mm) to the intracellular solution of the recording pipette (Bender et al., 2006; Rodríguez-Moreno et al., 2010). Intracellular MK801 (iMK801) did not affect the amplitude or rise time of human EPSPs, but reduced EPSP decay time (Fig. 4A). Pairing EPSPs to APs at intervals between −15 and +5 ms induced clear LTP in control neurons (121.0 ± 7.0%; paired-samples t test: p = 0.019, n = 10; Fig. 4B,C), but not in neighboring iMK801-loaded neurons (94.3 ± 4.2%; paired-samples t test: p > 0.05, n = 5; Fig. 4B,C), indicating that LTP at human synapses is dependent on postsynaptic NMDA receptors.
Figure 4.

STDP in human synapses requires postsynaptic NMDA receptors and L-type VGCCs. A, iMK801 reduces human EPSP decay time. Black and gray traces, EPSPs recorded within first 5 min and >10 min after whole-cell break-in, respectively. Calibration: 2 mV, 20 ms. Inset, Effect of iMK801 on EPSP decay time in different experiments. Consistent with use-dependent blockade of NMDARs, EPSP decay time is significantly reduced after 10 min of EPSP stimulation (paired-samples t test: p = 0.002, n = 10). B, Mean normalized slope of human control STDP recordings (▴) and recordings where postsynaptic NMDARs were blocked by iMK801 (□), or where nifedipine was present during pairing (▾), for −15 to +5 ms intervals. C, Summary histograms of human pharmacology data. Bars represent the average EPSP slope change induced using pairing intervals from −15 to +5 ms for control recordings, for recordings with iMK801 and for recordings with bath-applied nifedipine. Differences between experimental groups were tested by one-way ANOVA (F(2,19) = 9.036, p = 0.002). Asterisks: significant with Tukey's HSD post hoc test (Ctrl vs iMK: p = 0.031, Ctrl vs Nif.: p = 0.003). D, The same as in C, for +8 to +15 ms intervals. Differences between groups were tested by one-way ANOVA (F(2,21) = 4.552, p = 0.024). Asterisks: significant with Tukey's HSD post hoc test (Ctrl vs iMK: p = 0.029). E, Nifedipine did not affect the slope of human EPSPs that were not paired to APs, or where APs were evoked >400 ms after the EPSP (paired-samples t test: p > 0.05, n = 9). Black trace, Baseline EPSP; red trace, 25–30 min after baseline. Inset, EPSP slope during baseline versus slope 25–30 after baseline for different experiments. Calibration: 3 mV, 20 ms. F, The same as in B, for +8 to +15 ms intervals. G, Summary histogram of rat pharmacology data for pairing intervals from −15 to 0 ms. Differences between groups were tested by one-way ANOVA (F(2,16) = 6.750, p = 0.009). Asterisks: significant with Tukey's HSD post hoc test (Ctrl vs iMK: p = 0.026, Ctrl vs Nif.: p = 0.013). H, As G, for 0 to +15 ms intervals. Differences between groups were tested by one-way ANOVA (F(2,15) = 9.124, p = 0.003). Asterisks: significant with Tukey's HSD post hoc test (Ctrl vs iMK: p = 0.003, Ctrl vs Nif.: p = 0.021). Nif., Nifedipine; Ctrl, control.
Recent reports have shown that LTD at a number of cortical synapses in the rodent brain is dependent on presynaptic NMDARs (Sjöström et al., 2003; Rodríguez-Moreno and Paulsen, 2008; Banerjee et al., 2009; Rodríguez-Moreno et al., 2010; Verhoog and Mansvelder, 2011). To test whether this holds true in adult human synapses as well, we tested whether LTD would persist when postsynaptic NMDARs were blocked by iMK801. Pairing presynaptic and postsynaptic activity at intervals ranging from +8 to +15 ms, which induced LTD in interleaved control experiments (79.1 ± 6.8%; paired-samples t test: p = 0.028, n = 9; Fig. 4D,F), failed to produce a significant decrease in EPSP slope in neurons loaded with iMK801 (101.3 ± 3.1%; paired-samples t test, p > 0.05, n = 8; Fig. 4D,F). This shows that postsynaptic NMDA receptors gate both potentiation and depression at human synapses.
In rodent cortical synapses, activation of L-type VGCCs is required for STDP (Bi and Poo, 1998; Rubin et al., 2005; Nevian and Sakmann, 2006; Meredith et al., 2007; Remy and Spruston, 2007). To test the involvement of L-type VGCCs in human cortical STDP, we blocked these channels with bath-applied nifedipine (10 μm). Nifedipine did not affect the amplitude or slope of EPSPs (Fig. 4E). Interestingly, pairing at LTP intervals between −15 and +5 ms while L-type VGCCs were blocked abolished LTP and resulted in LTD in all cells tested (83.6 ± 3.5%; paired-samples t test: p = 0.045, n = 5; Fig. 4B,C). These findings suggest that L-type VGCCs can act as a molecular switch for the polarity of synaptic plasticity at human neocortical synapses.
To test whether L-type VGCCs are also necessary for LTD at human synapses, we blocked L-type VGCCs and paired activity at intervals between +8 and +15 ms. Despite L-type VGCCs being unavailable during pairing, significant LTD was induced (80.5 ± 8.6%; paired-samples t test: p = 0.028, n = 5; Fig. 4D,F). Thus, L-type VGCCs are not involved in LTD of human neocortical synapses.
STDP in adult rats also involved NMDARs and L-type VGCCs; LTP at negative intervals (in a range of −15 to 0 ms) no longer occurred in neurons loaded with iMK-801 or in the presence of nifedipine (control: 167.5 ± 18.2%, paired-samples t test: p < 0.001, n = 6; iMK801: 114 ± 10.0%, paired-samples t test: p > 0.05, n = 6; nifedipine: 104.6 ± 5.3%, paired-samples t test: p > 0.05, n = 5; Fig. 4G). At positive intervals (in a range of 0 to +15 ms), LTP equally relied on postsynaptic NMDA receptors and L-type VGCCs (control: 165.2 ± 18.9%, paired-samples t test, p < 0.001, n = 5; iMK801: 101 ± 3.5%, paired-samples t test: p > 0.05, n = 7; nifedipine: 109.8 ± 13.9%, paired-samples t test: p > 0.05, n = 4; Fig. 4H). In contrast to human synapses, blocking L-type VGCCs did not uncover LTD at adult rat synapses. These data show that STDP in both adult human and adult rat synapses require postsynaptic NMDAR activity. L-type VGCC activation seems to be a common mechanism enabling adult human and rat synapses to potentiate in response to post-before-pre activation. Particular to human synapses, however, is the conversion of LTP into LTD when these channels are blocked.
To test the contribution of L-type VGCCs to dendritic bAP, we used two-photon Ca2+ imaging in human apical dendrites (Fig. 5). Pyramidal neurons were loaded with Fluo-4 (100 μm) and Alexa Fluor 594 (80 μm), and line scans of apical dendrites were made 100–150 μm from the soma (Fig. 5A). Single APs and bursts of APs reliably evoked Fluo-4 fluorescence transients (Fig. 5A–C). To probe the contribution of L-type VGCCs to AP-evoked dendritic Ca2+ influx, we next bath applied nifedipine (10 μm), which induced a significant decrease in amplitude of single AP-induced Ca2+ influx [amplitude: 2.9 ± 0.7%ΔG/R (aCSF) vs 2.1 ± 0.4%ΔG/R (nifedipine); paired-samples t test (log-transformed data): p = 0.035; n = 7; Fig. 5B,D]. The reduction in amplitude was similar to that found in rodent dendrites (Sabatini and Svoboda, 2000; Meredith et al., 2007). Burst-induced Ca2+ influx decreased both in amplitude and area upon blockade of L-type VGCCs [amplitude: 6.2 ± 1.4%ΔG/R (aCSF) vs 3.6 ± 0.7%ΔG/R (nifedipine); area: 1.27 ± 0.29%ΔG/R*ms (aCSF) vs 0.77 ± 0.15%ΔG/R*ms (nifedipine); paired-samples t test (log-transformed data): p < 0.001 and p = 0.007 for amplitude and area, respectively; n = 9; Figure 5C,D]. These findings indicate that bAPs generated at the soma reliably activate L-type VGCCs in the dendrites of human neurons.
Figure 5.

L-type VGCCs contribute to dendritic Ca2+ influx evoked by single APs and bursts. A, Top, 2D z-stack of patched human neuron visualized by two-photon excitation of morphology dye Alexa Fluor 594. Calibration: 20 μm. Bottom: Pseudo-colored 500 ms line scan of apical dendrite at ROI boxed in yellow above. Line scan is the average of five trials where a single AP was evoked; ▴ marks the time the AP was triggered. Calibration: 100 ms. B, Ca2+ transient traces evoked by single APs in neuron shown in A in aCSF conditions (left, corresponds to line scan in A), and in the presence of nifedipine (right). Light-colored traces are the means of four to six trials; dark superimposed traces represent 30 ms running average. ▴ marks the time the AP was triggered. Calibration same as in C. C, The same as in B, for burst of APs in neuron shown in A. Calibration: 10% ΔG/R, 200 ms. D, Ca2+ transient amplitude and area in the presence of nifedipine as percentage of that in aCSF conditions, for single APs and bursts of APs (80–150 Hz). Nifedipine significantly reduced the amplitude of Ca2+ transients triggered by single APs and bursts of APs (paired-samples t test, log-transformed values: p = 0.035 and p < 0.001 for single APs and bursts, respectively). Nifedipine also significantly reduced the area of Ca2+ transients triggered by bursts of APs (Ca2+ transient area measured over the first 300 ms after AP initiation; paired-samples t test, log-transformed values: p = 0.007). E, Overlay of ADP waveforms following single APs concurrently recorded in soma during imaging experiment in aCSF conditions (black) and in the presence of nifedipine (red). Inset shows the portion of AP–ADP waveform that has been magnified. Note reduction in both amplitude and area (shaded) of the ADP in the presence of nifedipine. Calibration: 10 mV, 20 ms.
AP afterdepolarizations shape the LTP timing window through L-type VGCC activation
Action potentials in human neocortical neurons showed prominent afterdepolarizing potentials (ADPs) that lasted 62.2 ± 3.1 ms (Figs. 6, 7A,B; Table 1). The ADP is considered a somatic readout of dendritic calcium spike initiation and possibly reflects active dendritic action potential propagation (Larkum et al., 1999; Larkum et al., 2001), which is crucial for LTP induction (Magee and Johnston, 1997; Kampa et al., 2006; Couey et al., 2007; Fuenzalida et al., 2010). Since L-type VGCCs are recruited during action potential backpropagation (Fig. 5), blocking these channels may affect the ADP waveform. Indeed, during most imaging experiments shown in Figure 5, application of nifedipine led to a reduction in the size of the ADP of the somatic APs triggered to elicit action potential backpropagation (Fig. 5E). Since these results may have been influenced by the Ca2+ buffering capacities of the intracellularly present Ca2+ indicator Fluo-4, we quantified the effect of nifedipine on the ADP of human neurons in experiments with standard intracellular (Fig. 6). ADP amplitude and area markedly decreased upon L-type VGCC blockade to, respectively, 88% and 79% of their original values in aCSF conditions (paired-samples t test, p = 0.021 and p = 0.040, for ADP amplitude and area, respectively, n = 9), suggesting activation of these receptors by single somatic action potentials (Fig. 6A). In contrast, blocking postsynaptic NMDARs with iMK801 had no effect on ADP waveform (Fig. 6B).
Figure 6.

L-type Ca2+ channel blockade reduces the ADP. A, Effect of bath-applied nifedipine on ADP area. Black trace is recorded in aCSF conditions, red trace in presence of nifedipine. Calibration: 20 mV, 20 ms. Inset, ADP area in aCSF conditions and in the presence of nifedipine for different experiments. Nifedipine significantly decreases ADP area (paired-samples t test: p = 0.040, n = 9). B, Effect of iMK801 on ADP area. Black trace recorded within first 5 min after whole-cell break-in, gray trace recorded >10 min after break-in. Calibration same as in A. Inset, Effect of iMK801 on ADP area for different experiments. iMK801 does not affect ADP area (paired-samples t test: p > 0.05, n = 7).
Figure 7.

LTP at negative timing intervals depends on the ADP following the AP. A, AP and ADP waveform recorded from three different human neurons at similar membrane potentials (−69 to −71 mV). Inset, Cumulative fraction of ADP areas. Calibration: 40 mV, 10 ms. B, Schematic showing quantification methods for ADP amplitude, ADP amplitude at EPSP onset, ADP duration, and ADP area, on a magnification of the area boxed in the inset AP. Black trace is AP recorded without EPSP, blue trace is an AP plus EPSP recorded from the same cell. Calibration: 10 mV, 20 ms. C, ADP hyperpolarization protocol. Top, Example traces of EPSP–AP pairing for human neuron in control conditions (blue trace) and where the ADP was eliminated by hyperpolarizing current injection (gray trace). Bottom, Corresponding current injections. Calibration: 25 mV (top vertical), 2 nA (bottom vertical) and 25 ms. D, Mean normalized EPSP slope in time for recordings from human neurons where ADP was eliminated by hyperpolarizing current injection (empty squares) and control recordings from same patients (filled triangles). Pairing intervals used ranged from −10 to −30 ms. E, Summary quantifications of ADP-hyperpolarization experiments. Eliminating the ADP by hyperpolarization significantly blocked LTP in human and in rat neurons (independent samples t test: p = 0.042 and p = 0.020, respectively). F, EPSP slope change in individual STDP experiments paired at negative intervals versus ADP amplitude at EPSP onset. Regression line is shown in blue. G, Summarized STDP results for neurons with ADP areas above (blue squares) and below (black squares) the median ADP area, in pairing interval bins of 40 ms. At pairing intervals between −40 and −80 ms, neurons with large ADPs potentiated significantly more than neurons with smaller ADPs (independent-samples t test, p = 0.01). Inset, Example traces of AP–ADP waveforms recorded from neuron with large (blue) and from neuron small (black) ADP area. H, ADP area plotted against somatic depth (measured as distance from pia) and corresponding layers of recorded human neurons. Horizontal blue bars show mean ± SEM ADP area per layer; filled circles, data points corresponding to reconstructed neurons to the right. Asterisks indicate where the dendrite is cut at slice surface. L, layer; W.M., white matter.
Can L-type VGCC activation during the ADP explain the wide temporal window for LTP at negative pairing intervals in human cortical pyramidal neurons? When pairing at negative intervals, EPSPs coincide with the downward slope of the ADP (Fig. 7A,B), which may support LTP induction at negative timing intervals (Testa-Silva et al., 2010). We therefore investigated whether the long-lasting depolarization of the ADP is important for the LTP window at negative intervals. First, we manipulated the levels of the ADP during pairing by injecting a 100-ms-long negative current (−600 to −1000 pA) 2 ms after evoking the postsynaptic spike during STDP induction (Bender et al., 2006; Fig. 7C). Induction of LTP was completely blocked in all human neurons where the ADP was eliminated (interleaved control experiments: 123.9 ± 7.9%, n = 9; hyperpolarization: 100.8 ± 6.3%, n = 9, independent samples t test: p = 0.042; Fig. 7D,E). Rat APs were similarly followed by ADPs, albeit of smaller area and duration (area: 309.9 ± 43.2 mV/ms, duration: 35.1 ± 4.7 ms; Fig. 2F, inset; Table 1). Eliminating the ADP by hyperpolarization during pairing also completely blocked LTP in rat neurons (control: 151.3 ± 20.7%, n = 7; hyperpolarization: 101.4 ± 8.0%, n = 9, independent samples t test: p = 0.020; Fig. 7E).
Next, we correlated the change in EPSP slope to the amplitude of the ADP at the time the EPSP was triggered during plasticity induction (Fig. 7B,F). ADP amplitude at EPSP onset showed a weak but significant correlation with EPSP slope change (humans: R2 = 0.17, p < 0.01, n = 42; Fig. 7F; rats: R2 = 0.19, p = 0.03, n = 25). The effect of the ADP was also evident when comparing EPSP slope changes between neurons with ADP areas above and below the median ADP area of human neurons, which was ∼400 mV/ms. Comparing three bins of pairing intervals (0 to −40 ms, −40 to −80 ms, and less than −80 ms; Fig. 7G), we found that neurons with ADP areas >400 mV/ms potentiated significantly more than neurons with ADP areas <400 mV/ms at intervals ranging from −40 to −80 ms (independent-samples t test, p = 0.01; n = 6 for both groups). Importantly, ADP area was not related to other electrophysiological or morphological characteristics measured; ADP areas were not significantly different between cortical layers (one-way ANOVA: p > 0.05; Fig. 7H), nor did ADP area correlate with apical, basal, or total dendritic length (data not shown). ADP area also showed no relationship with other physiological parameters, including input resistance and membrane time constant. Although the somatic ADP waveform showed a clear voltage dependence within neurons; across neurons, we found no significant correlation between ADP area and somatic resting membrane potential (r = −0.21, p > 0.05, n = 42). Together, these findings show that L-type VGCC activation during the large, long-lasting ADP following the action potential endows adult cortical synapses with the ability to associate presynaptic and postsynaptic activity at negative timing intervals over long stretches of time, resulting in a wide temporal window for STDP at these synapses.
Discussion
In this study, we show that adult human neocortical synapses can bidirectionally change strength in response to spike timing at least up to 65 years of age, and uncovered mechanisms underlying human synaptic plasticity rules. The temporal window for associative changes in strength in human synapses is wide (±100 ms) and shows LTP at negative pairing intervals and LTD at positive intervals, with a switch from LTP to LTD occurring between +5 and +10 ms. The depolarization following the action potential, the ADP, is necessary for LTP at negative timing intervals. L-type VGCCs are activated during the ADP and are required for LTP induction, but not for LTD induction. Adult rodent MTC synapses show similar features of STDP, but do not show LTD at positive timing intervals. The ADP following the action potential and L-type VGCC activation are common mechanisms enabling adult human and rat synapses to associate presynaptic and postsynaptic events in an exceptionally wide temporal window.
Inverse, anti-Hebbian STDP rules have been reported for synapses between layer 4 spiny stellate neurons in rat barrel cortex, which appear to have a symmetric depression window (Egger et al., 1999). In the cerebellum-like structure of electric fish, pairing at positive intervals induces LTD, and at negative intervals induces LTP (Bell et al., 1997). In our previous investigations of STDP at adult human hippocampal synapses, LTP was also observed at negative timing intervals; EPSPs following a postsynaptic burst of APs by up to 80 ms potentiated, while LTD was only observed if EPSPs followed the burst by >80 ms (Testa-Silva et al., 2010). Interestingly, in vivo studies in humans subjects observed STDP-like changes in corticospinal excitability following anti-Hebbian plasticity rules similar to those reported in the present study (Thabit et al., 2010; Conde et al., 2013; Koch et al., 2013). Moreover, the timing window for association of stimuli was wide, with significant changes in MEP amplitude occurring when TMS was delivered from 50 ms before to 100 ms after voluntary movement (Thabit et al., 2010). The extent to which changes in MEP amplitude reflect synaptic plasticity processes remains unclear, but the reversed and wide STDP window for human temporal cortex reported here is strikingly similar to that of paired association of TMS and voluntary movement (Thabit et al., 2010; Conde et al., 2013; Koch et al., 2013).
The shape and form of STDP windows are dictated by the intracellular machinery in postsynaptic dendrites and spines and presynaptic terminals (Caporale and Dan, 2008; Sjöström et al., 2008). Variations in these mechanisms over different cell types and brain areas, perhaps combined with other factors such as the actions of neuromodulators and/or the levels of GABAergic inhibition, can lead to a variety of synaptic learning rules (Meredith et al., 2003; Babadi and Abbott, 2010; Fino and Venance, 2010). Reports on the involvement of inhibition in shaping synaptic plasticity rules are ubiquitous, but it has perhaps been most dramatically demonstrated in corticostriatal STDP. There, a reversed STDP window was reported with LTP following post-before-pre induction protocols, and LTD following pre-before-post protocols, which could be completely reverted to the classic STDP window when GABAergic inhibition was blocked during induction (Fino et al., 2010). In vivo studies in humans have shown how inhibition can also shape human associative plasticity (De Beaumont et al., 2012; Elahi et al., 2012; Conde et al., 2013); how increased GABAB-mediated inhibition in concussed athletes leads to a suppression of LTP- and LTD-like plasticity (De Beaumont et al., 2012); and how interhemispheric inhibition of the sensorimotor network during PAS results in anti-Hebbian STDP-like plasticity rules (Conde et al., 2013). A role for inhibition in forming the reversed STDP window observed in human synapses in the present study cannot be ruled out, as GABAergic transmission was not blocked in our experiments. Inhibition may act to reduce postsynaptic depolarization and thereby reduce the Ca2+ signals that would otherwise trigger potentiation (Couey et al., 2007). However, it is not straightforward to understand how a reduction of a synaptically induced depolarization by inhibition would favor potentiation over depression of synapses when pairing at negative intervals, as we observed. Recently, in rodent striatum, depolarizing actions of GABA in distal dendrites have been suggested to mediate the switch from LTD to LTP in corticostriatal synapses paired at negative intervals (Paille et al., 2013). Such mechanisms may contribute to shaping the human cortical STDP window, but other factors will have to be considered as well (Sjöström et al., 2008).
An alternative cause for the reversed STDP observed in humans may be the location of recruited synapses on the dendritic tree of the recorded neuron (Froemke et al., 2005; Letzkus et al., 2006; Sjöström and Häusser, 2006; Sjöström et al., 2008). Within one neuron, proximal synapses can display the classic Hebbian learning rule, while distal synapses display inverse, anti-Hebbian plasticity rules (Letzkus et al., 2006). Dendritic location of synapses was proposed as an explanation for anti-Hebbian corticocortical STDP-like plasticity observed in vivo (Koch et al., 2013). In our study, synaptic inputs were evoked by extracellular stimulation 100–150 μm from the soma, which would hardly qualify as distal inputs (Letzkus et al., 2006). So, unless the stimulated fibers actually impinge on the dendrite at more distal locations than our stimulating electrode, or else that synaptic plasticity rules in human neurons have quite a different distance dependency than in rodents, it seems unlikely that this synapse location-dependent form of anti-Hebbian STDP is dominating our experiments.
In the present study on human neocortical synapses and in our previous study on human hippocampal synapses (Testa-Silva et al., 2010), we observed that EPSPs coincide with the falling flank of the ADP when pairing at negative intervals. The synaptic potentiation that follows such pairing protocols in human neocortex and hippocampus may result from activating synapses concurrently with the dendritic calcium dynamics induced by the backpropagating action potential. In rodents, apical dendrites and spines express several types of VGCCs, including L-type VGCCs, which are activated during dendritic action potential propagation (Sabatini and Svoboda, 2000; Meredith et al., 2007). In human dentate gyrus cells, AP firing was shown to recruit VGCCs; increases in ADP area when bursting above critical frequency were blocked by the N-type VGCC antagonist NPY (Hamilton et al., 2010). In our experiments in human neocortex, blocking L-type VGCCs with nifedipine caused a prominent reduction of the ADP in most neurons, indicating a substantial recruitment of L-type VGCCs by single APs. Individual differences in ADP area may therefore in part reflect different degrees of L-type VGCC activation. Pairing at negative intervals in these conditions no longer induced LTP, but resulted in LTD instead. In rodents, L-type VGCCs are also involved in STDP, but have a markedly different role than in adult humans (Meredith et al., 2007; Remy and Spruston, 2007; Sjöström et al., 2008). In juvenile mouse prefrontal cortex, blocking L-type VGCCs prevented LTP induction (Meredith et al., 2007), but it did not unmask LTD, which is in line with our findings in adult rat MTC. In hippocampal and somatosensory cortical pyramidal neurons, blockade of L-type calcium channels strongly reduced the extent of spike timing-dependent LTD (Bi and Poo, 1998; Nevian and Sakmann, 2006). In human subjects, systemic application of an L-type VGCC antagonist altered the sign of metaplasticity of evoked peripheral endplate potentials and corticospinal excitability evoked by TMS stimulation (Wankerl et al., 2010). The authors suggested that postsynaptic L-type VGCCs might act at cortical synapses. Here, we directly show that at the synaptic level, L-type VGCCs decide between spike timing-dependent potentiation and depression, and set the timing window for activity-dependent plasticity in adult human cortical synapses.
Footnotes
This research was supported by grants to H.D.M. from the Netherlands Organization for Scientific Research (NWO; 917.76.360; www.nwo.nl), Neuroscience Campus Amsterdam, and VU University board (StgVU-ERC), ERC StG “BrainSignals,” the Dutch Fund for Economic Structure Reinforcement (FES, 0908 “NeuroBasic PharmaPhenomics project”), and the European Union 7th Framework Programme (HEALTH-F2-2009-242167 “SynSys”); grants to R.M.M. from NWO (917.10.372) and Fondation Jérôme LeJeune; and grants to C.P.J.d.K. from the Dutch Brain Foundation (Hersenstichting Nederland, www.hersenstichting.nl, HSN 2010(1)-09). We thank all patients for giving their consent for use of their brain tissue in this study. Also, we thank Hans Lodder and Brendan Lodder for excellent technical assistance; Professor Dr. Ole Paulsen for inspiring discussions; Dr. Annemieke Rozemuller for inspecting resected human tissue for signs of pathology; and Dr. Philip de Witt Hamer for assistance with human brain tissue handling.
The authors declare no competing financial interests.
References
- Babadi B, Abbott LF. Intrinsic stability of temporally shifted spike-timing dependent plasticity. PLoS Comput Biol. 2010;6:e1000961. doi: 10.1371/journal.pcbi.1000961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Meredith RM, Rodríguez-Moreno A, Mierau SB, Auberson YP, Paulsen O. Double dissociation of spike timing-dependent potentiation and depression by subunit-preferring NMDA receptor antagonists in mouse barrel cortex. Cereb Cortex. 2009;19:2959–2969. doi: 10.1093/cercor/bhp067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Abraham WC. Long-term depression in hippocampus. Annu Rev Neurosci. 1996;19:437–462. doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- Bell CC, Han VZ, Sugawara Y, Grant K. Synaptic plasticity in a cerebellum-like structure depends on temporal order. Nature. 1997;387:278–281. doi: 10.1038/387278a0. [DOI] [PubMed] [Google Scholar]
- Bender VA, Bender KJ, Brasier DJ, Feldman DE. Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J Neurosci. 2006;26:4166–4177. doi: 10.1523/JNEUROSCI.0176-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi GQ, Poo MM. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Caporale N, Dan Y. Spike timing-dependent plasticity: a Hebbian learning rule. Annu Rev Neurosci. 2008;31:25–46. doi: 10.1146/annurev.neuro.31.060407.125639. [DOI] [PubMed] [Google Scholar]
- Cleveland WS, Devlin SJ. Locally-weighted regression: an approach to regression analysis by local fitting. J Am Stat Assoc. 1988;83:596–610. doi: 10.1080/01621459.1988.10478639. [DOI] [Google Scholar]
- Conde V, Vollmann H, Taubert M, Sehm B, Cohen LG, Villringer A, Ragert P. Reversed timing-dependent associative plasticity in the human brain through interhemispheric interactions. J Neurophysiol. 2013;109:2260–2271. doi: 10.1152/jn.01004.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke SF, Bliss TV. Plasticity in the human central nervous system. Brain. 2006;129:1659–1673. doi: 10.1093/brain/awl082. [DOI] [PubMed] [Google Scholar]
- Couey JJ, Meredith RM, Spijker S, Poorthuis RB, Smit AB, Brussaard AB, Mansvelder HD. Distributed network actions by nicotine increase the threshold for spike-timing-dependent plasticity in prefrontal cortex. Neuron. 2007;54:73–87. doi: 10.1016/j.neuron.2007.03.006. [DOI] [PubMed] [Google Scholar]
- De Beaumont L, Tremblay S, Poirier J, Lassonde M, Théoret H. Altered bidirectional plasticity and reduced implicit motor learning in concussed athletes. Cereb Cortex. 2012;22:112–121. doi: 10.1093/cercor/bhr096. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. J Neurosci. 1993;13:2910–2918. doi: 10.1523/JNEUROSCI.13-07-02910.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger V, Feldmeyer D, Sakmann B. Coincidence detection and changes of synaptic efficacy in spiny stellate neurons in rat barrel cortex. Nat Neurosci. 1999;2:1098–1105. doi: 10.1038/16026. [DOI] [PubMed] [Google Scholar]
- Elahi B, Gunraj C, Chen R. Short-interval intracortical inhibition blocks long-term potentiation induced by paired associative stimulation. J Neurophysiol. 2012;107:1935–1941. doi: 10.1152/jn.00202.2011. [DOI] [PubMed] [Google Scholar]
- Fino E, Venance L. Spike-timing dependent plasticity in the striatum. Front Synaptic Neurosci. 2010;2:6. doi: 10.3389/fnsyn.2010.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Paille V, Cui Y, Morera-Herreras T, Deniau JM, Venance L. Distinct coincidence detectors govern the corticostriatal spike timing-dependent plasticity. J Physiol. 2010;588:3045–3062. doi: 10.1113/jphysiol.2010.188466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froemke RC, Dan Y. Spike-timing-dependent synaptic modification induced by natural spike trains. Nature. 2002;416:433–438. doi: 10.1038/416433a. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Poo MM, Dan Y. Spike-timing-dependent synaptic plasticity depends on dendritic location. Nature. 2005;434:221–225. doi: 10.1038/nature03366. [DOI] [PubMed] [Google Scholar]
- Fuenzalida M, Fernández de Sevilla D, Couve A, Buño W. Role of AMPA and NMDA receptors and back-propagating action potentials in spike timing-dependent plasticity. J Neurophysiol. 2010;103:47–54. doi: 10.1152/jn.00416.2009. [DOI] [PubMed] [Google Scholar]
- Hamilton TJ, Wheatley BM, Sinclair DB, Bachmann M, Larkum ME, Colmers WF. Dopamine modulates synaptic plasticity in dendrites of rat and human dentate granule cells. Proc Natl Acad Sci U S A. 2010;107:18185–18190. doi: 10.1073/pnas.1011558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebb D. The organization of behavior. New York: John Wiley; 1949. [Google Scholar]
- Kampa BM, Letzkus JJ, Stuart GJ. Requirement of dendritic calcium spikes for induction of spike-timing-dependent synaptic plasticity. J Physiol. 2006;574:283–290. doi: 10.1113/jphysiol.2006.111062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampa BM, Letzkus JJ, Stuart GJ. Dendritic mechanisms controlling spike-timing-dependent synaptic plasticity. Trends Neurosci. 2007;30:456–463. doi: 10.1016/j.tins.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Koch G, Ponzo V, Di Lorenzo F, Caltagirone C, Veniero D. Hebbian and anti-hebbian spike-timing-dependent plasticity of human cortico-cortical connections. J Neurosci. 2013;33:9725–9733. doi: 10.1523/JNEUROSCI.4988-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Kaiser KM, Sakmann B. Calcium electrogenesis in distal apical dendrites of layer 5 pyramidal cells at a critical frequency of back-propagating action potentials. Proc Natl Acad Sci U S A. 1999;96:14600–14604. doi: 10.1073/pnas.96.25.14600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ, Sakmann B. Dendritic mechanisms underlying the coupling of the dendritic with the axonal action potential initiation zone of adult rat layer 5 pyramidal neurons. J Physiol. 2001;533:447–466. doi: 10.1111/j.1469-7793.2001.0447a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzkus JJ, Kampa BM, Stuart GJ. Learning rules for spike timing-dependent plasticity depend on dendritic synapse location. J Neurosci. 2006;26:10420–10429. doi: 10.1523/JNEUROSCI.2650-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzkus JJ, Kampa BM, Stuart GJ. Does spike timing-dependent synaptic plasticity underlie memory formation? Clin Exp Pharmacol Physiol. 2007;34:1070–1076. doi: 10.1111/j.1440-1681.2007.04724.x. [DOI] [PubMed] [Google Scholar]
- Levy WB, Steward O. Temporal contiguity requirements for long-term associative potentiation/depression in the hippocampus. Neuroscience. 1983;8:791–797. doi: 10.1016/0306-4522(83)90010-6. [DOI] [PubMed] [Google Scholar]
- Lu MK, Tsai CH, Ziemann U. Cerebellum to motor cortex paired associative stimulation induces bidirectional STDP-like plasticity in human motor cortex. Front Hum Neurosci. 2012;6:260. doi: 10.3389/fnhum.2012.00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Meredith RM, Floyer-Lea AM, Paulsen O. Maturation of long-term potentiation induction rules in rodent hippocampus: role of GABAergic inhibition. J Neurosci. 2003;23:11142–11146. doi: 10.1523/JNEUROSCI.23-35-11142.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith RM, Holmgren CD, Weidum M, Burnashev N, Mansvelder HD. Increased threshold for spike-timing-dependent plasticity is caused by unreliable calcium signaling in mice lacking fragile X gene FMR1. Neuron. 2007;54:627–638. doi: 10.1016/j.neuron.2007.04.028. [DOI] [PubMed] [Google Scholar]
- Molnár G, Oláh S, Komlósi G, Füle M, Szabadics J, Varga C, Barzó P, Tamás G. Complex events initiated by individual spikes in the human cerebral cortex. PLoS Biol. 2008;6:e222. doi: 10.1371/journal.pbio.0060222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-Dahlhaus F, Ziemann U, Classen J. Plasticity resembling spike-timing dependent synaptic plasticity: the evidence in human cortex. Front Synaptic Neurosci. 2010;2:34. doi: 10.3389/fnsyn.2010.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-Dahlhaus JF, Orekhov Y, Liu Y, Ziemann U. Interindividual variability and age-dependency of motor cortical plasticity induced by paired associative stimulation. Exp Brain Res. 2008;187:467–475. doi: 10.1007/s00221-008-1319-7. [DOI] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J Neurosci. 2006;26:11001–11013. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paille V, Fino E, Du K, Morera-Herreras T, Perez S, Kotaleski JH, Venance L. GABAergic circuits control spike-timing-dependent plasticity. J Neurosci. 2013;33:9353–9363. doi: 10.1523/JNEUROSCI.5796-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy S, Spruston N. Dendritic spikes induce single-burst long-term potentiation. Proc Natl Acad Sci U S A. 2007;104:17192–17197. doi: 10.1073/pnas.0707919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, Paulsen O. Spike timing-dependent long-term depression requires presynaptic NMDA receptors. Nat Neurosci. 2008;11:744–745. doi: 10.1038/nn.2125. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, Banerjee A, Paulsen O. Presynaptic NMDA receptors and spike timing-dependent depression at cortical synapses. Front Synaptic Neurosci. 2010;2:18. doi: 10.3389/fnsyn.2010.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin JE, Gerkin RC, Bi GQ, Chow CC. Calcium time course as a signal for spike-timing-dependent plasticity. J Neurophysiol. 2005;93:2600–2613. doi: 10.1152/jn.00803.2004. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Svoboda K. Analysis of calcium channels in single spines using optical fluctuation analysis. Nature. 2000;408:589–593. doi: 10.1038/35046076. [DOI] [PubMed] [Google Scholar]
- Sawaki L, Yaseen Z, Kopylev L, Cohen LG. Age-dependent changes in the ability to encode a novel elementary motor memory. Ann Neurol. 2003;53:521–524. doi: 10.1002/ana.10529. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Häusser M. A cooperative switch determines the sign of synaptic plasticity in distal dendrites of neocortical pyramidal neurons. Neuron. 2006;51:227–238. doi: 10.1016/j.neuron.2006.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/S0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Rancz EA, Roth A, Häusser M. Dendritic excitability and synaptic plasticity. Physiol Rev. 2008;88:769–840. doi: 10.1152/physrev.00016.2007. [DOI] [PubMed] [Google Scholar]
- Stefan K, Kunesch E, Cohen LG, Benecke R, Classen J. Induction of plasticity in the human motor cortex by paired associative stimulation. Brain. 2000;123:572–584. doi: 10.1093/brain/123.3.572. [DOI] [PubMed] [Google Scholar]
- Stefan K, Kunesch E, Benecke R, Cohen LG, Classen J. Mechanisms of enhancement of human motor cortex excitability induced by interventional paired associative stimulation. J Physiol. 2002;543:699–708. doi: 10.1113/jphysiol.2002.023317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa-Silva G, Verhoog MB, Goriounova NA, Loebel A, Hjorth J, Baayen JC, de Kock CP, Mansvelder HD. Human synapses show a wide temporal window for spike-timing-dependent plasticity. Front Synaptic Neurosci. 2010;2:12. doi: 10.3389/fnsyn.2010.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thabit MN, Ueki Y, Koganemaru S, Fawi G, Fukuyama H, Mima T. Movement-related cortical stimulation can induce human motor plasticity. J Neurosci. 2010;30:11529–11536. doi: 10.1523/JNEUROSCI.1829-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzounopoulos T, Kim Y, Oertel D, Trussell LO. Cell-specific, spike timing-dependent plasticities in the dorsal cochlear nucleus. Nat Neurosci. 2004;7:719–725. doi: 10.1038/nn1272. [DOI] [PubMed] [Google Scholar]
- Verhoog MB, Mansvelder HD. Presynaptic ionotropic receptors controlling and modulating the rules for spike timing-dependent plasticity. Neural Plast. 2011;2011:870763. doi: 10.1155/2011/870763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wankerl K, Weise D, Gentner R, Rumpf JJ, Classen J. L-type voltage-gated Ca2+ channels: a single molecular switch for long-term potentiation/long-term depression-like plasticity and activity-dependent metaplasticity in humans. J Neurosci. 2010;30:6197–6204. doi: 10.1523/JNEUROSCI.4673-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittenberg GM, Wang SS. Malleability of spike-timing-dependent plasticity at the CA3-CA1 synapse. J Neurosci. 2006;26:6610–6617. doi: 10.1523/JNEUROSCI.5388-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolters A, Sandbrink F, Schlottmann A, Kunesch E, Stefan K, Cohen LG, Benecke R, Classen J. A temporally asymmetric Hebbian rule governing plasticity in the human motor cortex. J Neurophysiol. 2003;89:2339–2345. doi: 10.1152/jn.00900.2002. [DOI] [PubMed] [Google Scholar]
- Wolters A, Schmidt A, Schramm A, Zeller D, Naumann M, Kunesch E, Benecke R, Reiners K, Classen J. Timing-dependent plasticity in human primary somatosensory cortex. J Physiol. 2005;565:1039–1052. doi: 10.1113/jphysiol.2005.084954. [DOI] [PMC free article] [PubMed] [Google Scholar]