

Abstract
Large dense core vesicle (LDCV) exocytosis in chromaffin cells follows a well characterized process consisting of docking, priming, and fusion. Total internal reflection fluorescence microscopy (TIRFM) studies suggest that some LDCVs, although being able to dock, are resistant to calcium-triggered release. This phenomenon termed dead-end docking has not been investigated until now. We characterized dead-end vesicles using a combination of membrane capacitance measurement and visualization of LDCVs with TIRFM. Stimulation of bovine chromaffin cells for 5 min with 6 μm free intracellular Ca2+ induced strong secretion and a large reduction of the LDCV density at the plasma membrane. Approximately 15% of the LDCVs were visible at the plasma membrane throughout experiments, indicating they were permanently docked dead-end vesicles. Overexpression of Munc18-2 or SNAP-25 reduced the fraction of dead-end vesicles. Conversely, expressing open-syntaxin increased the fraction of dead-end vesicles. These results indicate the existence of the unproductive target soluble N-ethylmaleimide-sensitive factor attachment protein receptor acceptor complex composed of 2:1 syntaxin–SNAP-25 in vivo. More importantly, they define a novel function for this acceptor complex in mediating dead-end docking.
Introduction
The adrenal chromaffin cell is one of the leading model systems to study regulated exocytosis of large dense core vesicles (LDCVs). Before fusing with the plasma membrane (PM), LDCVs dock to the PM and then become primed. The docking, i.e., tethering, process is induced by the interaction of synaptotagmin with the target soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor (t-SNARE) acceptor complex, consisting of SNAP-25 and syntaxin (de Wit et al., 2009). The formation of this t-SNARE acceptor complex is assisted by Munc18, a chaperone for syntaxin (Gulyás-Kovács et al., 2007; de Wit et al., 2009), by stabilizing functional syntaxin–SNAP-25 1:1 complexes and decreasing the formation of nonfunctional 2:1 complexes (Zilly et al., 2006; Weninger et al., 2008). Additionally, access of the LDCVs to the PM is facilitated by Munc18 which induces the depolymerization of the cortical actin network (Toonen et al., 2006). Priming occurs as the v-SNARE, e.g., synaptobrevin2, associates with the t-SNARE acceptor complex (Walter et al., 2010). This step is modulated by several factors, such as the Ca2+-dependent activator protein of secretion (CAPS), protein kinase C (PKC), or tomosyn (Smith et al., 1998; Yizhar et al., 2004; Speidel et al., 2005; Liu et al., 2008). Interestingly, Verhage and Sørensen (2008) introduced the idea that some docked LDCVs cannot fuse with the PM, designating this process dead-end docking.
Repetitive stimulation of chromaffin cells with flash photolysis of caged Ca2+ was previously shown to entirely deplete the releasable pool of vesicles, as assessed by membrane capacitance recording (Ashery et al., 2000). This result was interpreted as a depletion of all docked vesicles. Studies using total internal reflection fluorescence microscopy (TIRFM) to visualize LDCVs close to the PM, reported that LDCVs residing at the PM of chromaffin cells or PC12 cells were never entirely depleted, even when using long-lasting stimulation (Steyer et al., 1997; Lang et al., 2000; Johns et al., 2001). In fact, a maximum LDCV depletion at the PM of only 41% was obtained using Ba2+ stimulation for 20 min (Johns et al., 2001). Thus, it is possible that LDCVs remaining at the PM after the stimulus represent dead-end vesicles.
Using a combination of TIRFM and membrane capacitance measurements, we now demonstrate the existence of dead-end vesicles in bovine chromaffin cells. Perfusing the cells with an intracellular solution containing high free [Ca2+] reveals that ∼15% of all LDCVs at the PM were unreleasable dead-end vesicles. Additionally, overexpression of Munc18-2 strongly reduced the fraction of unreleasable vesicles residing at the membrane. Because Munc18 regulates the formation of the t-SNARE acceptor complex, we tested whether the unproductive 2:1 syntaxin:SNAP-25 acceptor complex was responsible for dead-end docking. Raising the syntaxin:SNAP-25 ratio to promote the 2:1 acceptor complex formation increased the size of the dead-end vesicle pool. Conversely, reducing the syntaxin:SNAP-25 ratio, to hinder the 2:1 acceptor complex formation, decreased the number of dead-end vesicles. These data support a role of the 2:1 acceptor complex in dead-end docking of LDCVs.
Materials and Methods
Cell preparation and transfection.
Chromaffin cells were isolated from bovine adrenal glands of either sex and cultivated as described by Nofal et al. (2007). Unless mentioned otherwise, cells were used after 3–5 d in vitro (DIV). Vesicles were stained by overexpressing NPY fused to mCherry. The NPY construct, which was verified by DNA sequencing, was cloned into a pMAX vector (kindly provided by Prof. Hoht, Institut für Biophysik Universität des Saarlandes, Homburg, Germany) and overexpressed by electroporation (Neon Transfection System, Invitrogen).
Plasmid pmTFP-C1 was generated by replacing GFP within pEGFP-C1 (Clontech) by monomeric Teal Fluorescent Protein (mTFP; Allele Biotech). hMunc18-2 was amplified by PCR from cDNA clone IRAUp969E1225D (ImaGenes) to add 5′ EcoRI and 3′ BamHI restriction sites and inserted into plasmid pmTFP-C1. Nine missing nucleotides (basepairs 247–255) were inserted using a PCR-based strategy to obtain the full-length hMunc18-2 cDNA transcript-I (ENST00000221283). Munc18-2-mTFP plasmid was coelectroporated in cells with NPY-mCherry resulting in 6.8-fold increase in the number of Munc18-2 mRNA copy compared with the combined wild-type Munc18-1 and -2 level (Table 1). Cells were transfected with syntaxin, open-syntaxin, or SNAP-25 using the Semliki Forest virus transfection system 6–18 h before measurement (Ashery et al., 1999). Viral particles pSFV1-syntaxin-IRES-eGFP and pSFV1-syntaxinL165A/E166A-IRES-GFP were made according to Liu et al. (2010). The plasmid of pSFV1-SNAP-25-PV-IRES-GFP was generated by subcloning SNAP-25 via the BamHI and BssHII sites from pSFV1-GFP-SNAP-25 into pSFV1-synaptobrevin2-IRES-eGFP (Sørensen et al., 2002). Control cells expressed pSFV1-eGFP. The Semliki Forest virus transfection system increased the number of mRNA copies of the transfected gene by >5000-fold (Table 1). This is in line with previous studies in which the concentration of the overexpressed gene was raised by >10-fold compared with the wild-type level (Ashery et al., 2000; Wei et al., 2000; Yizhar et al., 2004). The Semliki Forest virus transfection system reduced the number of mRNA copies of other genes (Table 1). It is unlikely that this reduction produced a marked decrease in concentration of SNARE proteins because the infection time was not longer than 18 h (Sanders et al., 1998). All cells were cotransfected with NPY-mCherry via electroporation to label the vesicles.
Table 1.
Overexpression of Munc18-2, open-syntaxin, and SNAP-25 do not affect the transcription level of unrelated proteins but overexpression of Munc18-2 increased the transcription of syntaxin
| Overexpressed protein | Tested mRNA level normalized to the endogenous concentration |
|||||
|---|---|---|---|---|---|---|
| Munc18 | Syntaxin | SNAP-25 | Rab3 | Synaptotagmin | Complexin | |
| Munc18-2 | 6.8 | 23.3 | 0.5 | 1.5 | 0.9 | 1.48 |
| Open-syntaxin | 2.5 | >5000 | 0.2 | 1.0 | 1.0 | 0.6 |
| SNAP-25 | 0.8 | 1.1 | >5000 | 0.8 | 0.9 | 0.6 |
The results are expressed as fold increase over endogenous expression level. We detected mRNA of Munc18-1 and -2 but they were pooled together since both isoforms share most features.
qRT-PCR.
Because the transfection efficiency varied from <10 to 50% between constructs and cell preparation, we isolated a pure population of overexpressing cells using the BD FACSAria III cell sorter (BD Biosciences), before performing quantitative real-time PCR (qRT-PCR). The sorting was performed after 2 DIV, 12 h, or 6 h after transfection with Munc18-2, SNAP-25, or open-syntaxin, respectively, using the coexpressed fluorescent protein as marker. The cells were then immediately lysed in TRIzol (Life Technologies GmbH) following the manufacturer's protocol. The mRNA expression level for endogenous (bovine), overexpressed, and housekeeping (UBC) genes were analyzed by qRT-PCR using forward and reverse primers designed with NCBI primer blast (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) shown in Table 2. PCRs were performed in 96-well plates in 25 μl reaction volume with the QuantiTect SYBR Green PCR master mix (Qiagen). The reaction conditions were 15 min heat activation at 95°C to activate HotStarTaq DNA polymerase, followed by 40 cycles of denaturation (15 s at 95°C), annealing (30 s at 60°C), and amplification (30 s at 72°C). We checked the specificity of the primers to their transcripts with melting curve analysis. Reactions were performed in duplicates in three independent experiments using MaxPro-Mx300P Real TimePCR System (Stratagene). We analyzed the data with the MaxPro 3000P software (Stratagene). The threshold cycle (Ct) value for the gene of interest was normalized to Ct value obtained for the housekeeping gene.
Table 2.
Forward and reverse primers used for the qRT-PCR reaction
| Gene | Genebank reference no. | Primers | 5′–3′ | Apmlicon size |
|---|---|---|---|---|
| SNAP-25 (bovine) | NM_001076246.1 | Fw | GGCCGATGAGTCTCTGGAAA | 128 |
| Rev | TCCCTTCCTCAATGCGTTCC | |||
| SNAP-25 (mouse) | NM_011428.3 | Fw | GGATGAGCAAGGCGAACAAC | 180 |
| Rev | TCCTGATTATTGCCCCAGGC | |||
| Munc18-1 (bovine) | NM_174619.2 | Fw | GCTCATAGGATCCACGCACA | 110 |
| Rev | TGGAGGGGCGGCTTATTTTT | |||
| Munc18-2 (bovine) | NM_001046208.2 | Fw | CTACGGAATGGTGTGAGCGA | 189 |
| Rev | CCAGCGAGACAGCTGATAGG | |||
| Munc18-2 (human) | NM_006949.2 | Fw | ATCAACAAACGGCGGGAACC | 96 |
| Rev | GTCTTTGATCAGGGCCTTCTCC | |||
| Syt 1 (bovine) | NM_174192.3 | Fw | AGCTGGTGGCCCTGGAAA | 177 |
| Rev | CTGGTTTTCCTGGAGCGGG | |||
| Stx 1 (bovine) | NM_001083798.1 | Fw | GAGTTCTTCGAGCAGGTGGA | 180 |
| Rev | TTGGAGCGGACTTTGTTTGC | |||
| Stx 1 (rat) | NM_053788.2 | Fw | AGCGATGACGACGATGATGT | 178 |
| Rev | CCTTGGTCTTCTCATCGGGG | |||
| Rab3a (bovine) | NM_174446.3 | Fw | CCACAGCATCAGTGCAGAAC | 160 |
| Rev | GAGTCGTCTGCATAGCGGAA | |||
| Cplx 2 (bovine) | NM_174283.2 | Fw | CATCGCCCCCAAGCCAG | 123 |
| Rev | TTGGTGGCCCCTCCAAGG | |||
| UBC (bovine) | NM_001206307.1 | Fw | GGACCGGGAGTTCAGTCTTC | 100 |
| Rev | TGATGGTTTTACCAGTGAGGGT |
Syt, synaptotagmin; Stx, syntaxin ; Cplx, complexin.
Solutions.
The bath solution contained the following (in mm): 147 NaCl, 2.4 KCl, 10 HEPES, 1.2 MgCl2, 2.5 CaCl2, and 10 glucose, pH 7.5, 305 mOsm. The internal solution compositions are given in Table 3. The [Ca2+]i of the solutions containing high free Ca2+ were calculated using an IGOR (WaveMetrics) macro Freecon. In experiments in which we stimulated the cells with flash photolysis of caged Ca2+ or depolarization, [Ca2+]i was measured with the mixture of two ratiometric indicator dyes, Fura-2 [Kd (Ca2+): 0.22 μm] and Fura-FF [Kd (Ca2+): 5.50 μm] (Invitrogen). The dyes were excited with light alternating between 350 and 380 nm with a monochromator-based system (TILL Photonics) and the emission was detected by a photodiode (Rapp OptoElectronics). An in vivo calibration curve was used to convert the ratio R of the fluorescent signals at both wavelengths into [Ca2+]i. The resting [Ca2+]i before flash 1–4 was (in nM) 737 ± 50, 757 ± 83, 807 ± 39, and 831 ± 76, respectively (n = 21). Before depolarization 1–4, the [Ca2+]i was (in nM) 718 ± 39, 798 ± 56, 862 ± 150, and 859 ± 110, respectively (n = 21).
Table 3.
Composition of intracellular solutions (in mm)
| Flash and depolarization | Solution containing high [Ca2+]i |
|||
|---|---|---|---|---|
| 1 μm | 6 μm | 15 μm | ||
| CaCl2 | 4.5 | 5 | 5 | 5 |
| Ca2+ buffer | 5 (NP-EGTA) | 5.9 (EGTA) | 9 (HEDTA) | 20 (DPTA) |
| Fura-2 | 0.1 | — | — | — |
| Fura-FF | 0.1 | — | — | — |
| Mg-ATP | 2 | 2 | 2 | 2 |
| Na2-GTP | 0.3 | 0.3 | 0.3 | 0.3 |
| Cs-glutamate | 98 | 120 | 110 | 90 |
| HEPES | 40 | 10 | 10 | 10 |
| pH | 7.3 | 7.3 | 7.2 | 7.4 |
NP-EGTA, 5 nitrophenyl-EGTA.
TIRFM setup.
The experiments were done at two setups with different characteristics. Most experiments were done with a setup based on an inverted Zeiss Axiovert 200 described by Pasche et al. (2012). Final pixel size was 160 nm. Acquisition rate was 10 Hz with an exposure time of 75 ms. For the flash photolysis of caged Ca2+ short flashes of ultraviolet light from a Xenon arc flash lamp (Rapp OptoElectronics) were applied to the cell. The other setup used for all experiments involving cells loaded with FFN511 or overexpressing Munc18-2 was as described by Becherer et al. (2007) with some modifications. The inverted IX 70 microscope (Olympus) was additionally equipped with an Ar-laser (Spectraphysics) emitting at 450 nm among others, and an Acousto Optical Tunable Filter (Visitron System). Images were acquired with a QuantEM 512SC camera (Photometrics). The acquisition rate was 10 Hz and the exposure time was 100 ms.
Membrane capacitance recording and image recording protocol.
All whole-cell recordings were performed with 3–5 MΩ pipettes and an EPC-9 patch-clamp amplifier controlled by Pulse software (Heka Elektronik). Capacitance measurements were performed using the Lindau–Neher technique implemented as the “sine+dc” mode of the “software lock-in” extension of Pulse software. A 1 kHz, 70 mV peak-to-peak sinusoid command potential stimulus was applied at a DC holding potential of −70 mV. Cells stimulated by depolarization or flash photolysis of caged Ca2+ were recorded after a loading period of 2 min without exciting the fluorescently labeled LDCVs to avoid bleaching of the fluorophore. We measured the [Ca2+]i every 2.5 s. Only cells showing a constant [Ca2+]i for >30 s before the end of the loading procedure were used for experiments. The loading period was followed by an imaging period (2 min at 10 Hz) including stimulation by either a train of 10 depolarizations (from a holding potential of −70–0 mV, each lasting 100 ms at 5 Hz) or a short UV flash (duration 1 ms, 110 V). Cells were allowed to recover for 2 min between stimuli. All experiments were performed at room temperature.
When using 1, 6, or 15 μm free [Ca2+]i the fluorescently labeled LDCVs were imaged at 10 Hz immediately after reaching whole-cell configuration. Imaging was continued until there was no more secretion, i.e., no rise of the membrane capacitance which was recorded in parallel. To control the speed of loading the cell with the pipette solution we added a mixture of Fura-2 and Fura-FF to the pipette solution. The final [Ca2+]i was already reached after few seconds.
FFN511 staining.
Cells were loaded with 5 μm fluorescent false neurotransmitter (FFN511, kindly provided by Prof. Kazmaier, Institut für Organische Chemie, Universität des Saarlandes, Saarbrücken, Germany) for 10–15 min at 37°C. New vesicles were detected in the red (NPY-mCherry) and green (FFN511) channels, whereas the old LDCVs were only visible in the green channel. Secretion of the FFN511 was difficult to detect as the density of loaded LDCVs is very high and the fluorescence intensity of the probe relatively weak. Thus identifying individual vesicles became challenging.
Immunolabeling.
Immunolabeling was performed on chromaffin cells fixed 18 h postinfection with Semliki Forest virus coding for SNAP-25 or GFP, and 6.5 h in case of open-syntaxin, respectively. For Munc18-2, cells were used 3 DIV post-transfection. Cells were fixed with 4% paraformaldehyde in PBS followed by a 50 mm glycine block. For SIM imaging, the cells were then incubated with the cis-Golgi network marker GM130 antibody (BD Transduction Laboratories) at 1:100 dilution. The secondary antibody used was Alexa 647 goat anti-mouse (Invitrogen). For TIRFM imaging, the cells were incubated with a monoclonal syntaxin 1 antibody (HPC/1 mouse ascites, from Prof. Jahn, Max-Planck-Institut für biophysikalische Chemie, Göttingen, Germany) at 1:500 dilution and colabeled with polyclonal SNAP-25 antibody (GenScript, anti-rabbit) at 1:500 dilution. The secondary antibodies used were Alexa Fluor 568 goat anti-mouse and Alexa Fluor 647 goat anti-rabbit for syntaxin 1 and SNAP-25, respectively.
SIM.
Structured illumination microscopy (SIM), which has XY and Z resolutions of 100 and 250 nm respectively, was performed as described by Pattu et al. (2011). One caveat of this technique is its rather long acquisition time for a single plane (3.5 s), that precludes the imaging of moving organelles. Thus SIM is usually used on fixed cells. However, FFN511 does not remain in fixed cells since it leaks out of LDCVs as soon as the H− ATPase is inactive. Therefore, to visualize FFN511 in living cells, we abolished all mobility in the cells by treatment with phalloidin (1 μm for 12 h at 37°C) and taxol (1 μm for 15 min at 37°C) inhibiting the depolymerization of actin and tubulin, respectively (see Fig. 2). In these experiments, the excitation light was 561 and 450 nm to visualize the NPY-mCherry and FFN511, respectively. To analyze the spatial distribution of LDCVs we measured the integrated fluorescence in evenly spaced concentric rings placed on the red and green channel of a single midsection plane of the cell (see Fig. 2F). These integrated fluorescence intensity values were normalized to the total integrated fluorescence of the cell in each channel and compared in a correlation plot (see Fig. 2H).
Figure 2.
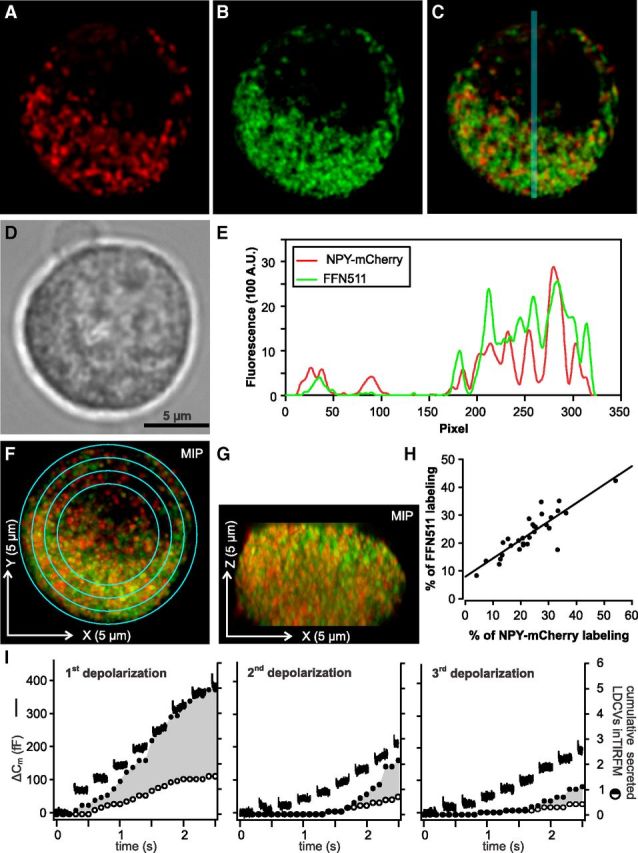
NPY-mCherry loaded LDCVs are not a specialized subset of LDCVs in chromaffin cells. A–C, Midsection of a bovine chromaffin cell transfected with NPY-mCherry (A) and stained with FFN511 (B) visualized with SIM. Both channels are overlayed in C. D, Same cell visualized by transillumination. E, Intensity profile plot along the blue line for NPY-mCherry (red) and FFN511 (green) shown in C. The fluorescence intensities in both channels correlate well. F, G, Maximal intensity projection of the Z-stack acquired at an interval of 200 nm of the same cell as shown in A and B. F and G correspond to the XY and ZX representation of the cell, respectively. H, The integrated fluorescence intensity was measured on single plane images in concentric rings shown in F and it was normalized to the overall integrated fluorescence of the cell. The result for each ring measured in the NPY-mCherry and FFN511 channel was shown in percentage as a dot on the graph. The Pearson's coefficient is 0.87 with p < 0.0001 (ncells = 7, nrings = 4 or 5) indicating a similar spatial cellular distribution of new (red) and old (green) vesicles. I, Secretion of old (marked only with FFN511) and new (containing NPY-mCherry) LDCVs was compared in cells stimulated with trains of depolarizations every 2 min. Secretion was measured via membrane capacitance recording (lines) and visualized with TIRFM (circles). Open circles represent the cumulative secretion of NPY-mCherry containing LDCVs, filled circles show the sum of the cumulative secretion from NPY-mCherry and FFN511 stained LDCVs normalized to the footprint area of the cell (n = 5). Note that the fraction of old versus new secreted vesicles remains constant at 65% for each stimulus.
Immunolabeled cells (see Fig. 7) were visualized at 488 nm (GFP staining), 561 nm (LDCVs), and 647 nm (cis-Golgi). Images were analyzed using software written in house with LabView (National Instruments). An outline of both the PM, following the edge of the GFP signal, and the cis-Golgi was generated manually, and LDCVs were marked manually with a circle. The shortest distance from LDCV center to the PM or the Golgi was calculated automatically.
Figure 7.
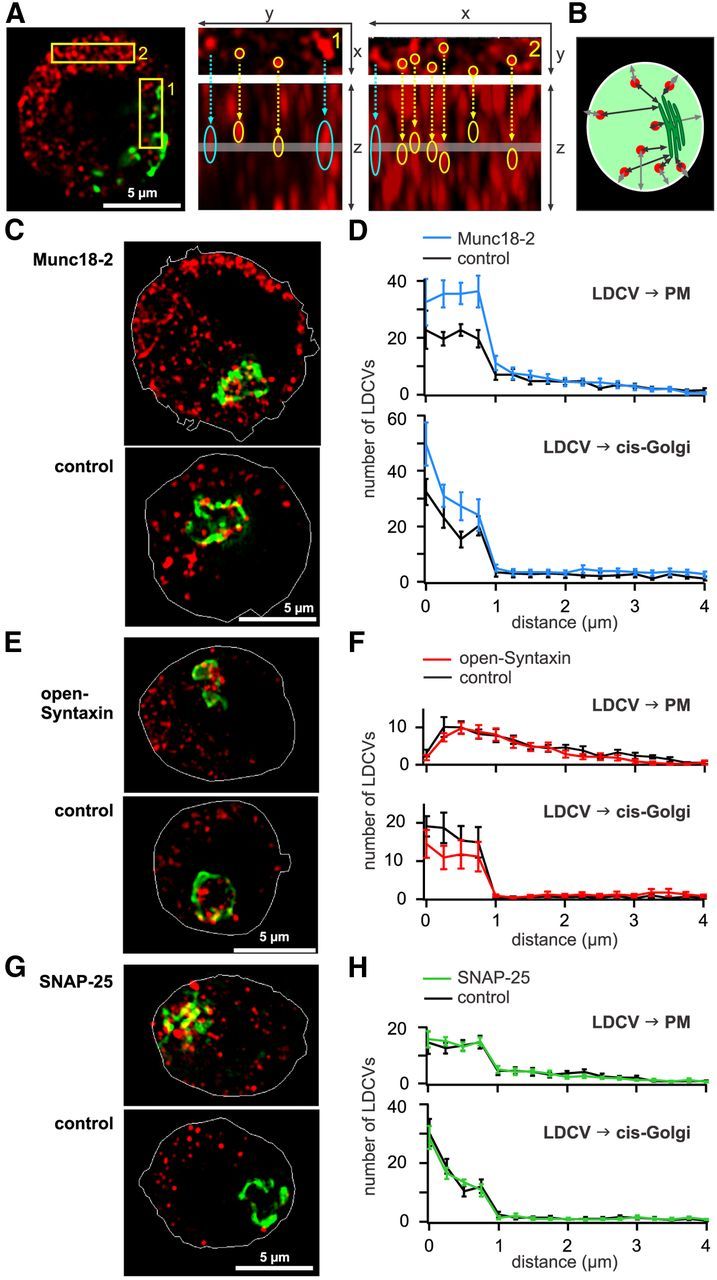
Munc18-2 induces an accumulation of syntaxin in the PM and LDCV close to the PM. A, Study of the influence of overexpression of proteins on the spatial distribution of LDVCs using SIM. Left, Three DIV NPY-mCherry transfected cells fixed and immunolabeled with anti GM130 to stain the Golgi (green). Center, Right, Enlarged view of the outlined areas shown in the picture on the left. XY planes are shown on top and Z planes are shown below. The thickness (200 nm) and position of the XY plane is shown on the Z plane with a gray line. Due to SIM resolution, the maximal LDCVs diameters are 300 and 700 nm in XY and Z direction, respectively. Some LDCVs are identified with a yellow circle, whereas structures that are probably not LDCVs (identified by a blue arrow) have a large round shape on the XY planes and a strongly elongated shape on the Z planes. These structures were not included in the analysis. B, Schematic representation of the analysis method in which the minimal distance between the center of the LDCVs to the PM and the Golgi border is measured. C, SIM images of chromaffin cells transfected for 3 DIV with NPY-mCherry (red) and Munc18-2-mTFP. The outline of the PM (white line) was marked using the picture acquired at 488 nm visualizing the mTFP signal. D, Analysis of the nearest distance between LDCVs and PM (top) or cis-Golgi (bottom), n = 10. E, As in C, SIM images of chromaffin cells transfected for 3 DIV with NPY-mCherry and 6.5 h with open-syntaxin or GFP (control). F, As in D, for cells transfected with open-syntaxin or GFP (control), n = 12 and 10, respectively. G, As in C, SIM images of chromaffin cells transfected for 24 h with NPY-mCherry and 12 h with SNAP-25 or GFP (control). H, As in D, for cells transfected with SNAP-25 or GFP (control), n = 10.
Analysis and statistics.
Secretion visualized in TIRFM was analyzed as described by Becherer et al. (2007) using ImageJ Software (NIH). Shortly, exocytosis was identified as the disappearance of a LDCV from its maximum fluorescence to background fluorescence within 200 ms. All other events, i.e., LDCVs disappearing at slower rate, were classified as undocking. The analyzed number of LDCVs (density, secreted, newcomer or dead-end) was normalized to compensate for variable footprint areas. For this we first normalized the number of LDCVs to the cell's individual footprint area which resulted in a density of LDCVs per μm2. To facilitate comprehension, we then multiplied this density by the average footprint area of all cells measured in each set of experiments independent of their treatment. For Figures 1, 2, and 3 this average footprint area was 44.14 ± 1.81 μm2. In Figures 4 and 5 the overall footprint area of cells measured at 1, 6, and 15 μm Ca2+ was 53.2 ± 3.7 μm2. In Figures 6, 8, and 9 the overall footprint area of control and overexpressing cells was 47.7 ± 3.6 μm2, 78.9 ± 6.4 μm2, 77.9 ± 6.8 μm2, respectively. Membrane capacitance traces were analyzed using IGOR (Wavemetrics).
Figure 1.

Several rounds of depolarizations cannot deplete all LDCVs at the PM. Cells were stimulated with 4 trains of 10 depolarizations to 0 mV each lasting 100 ms at 5 Hz (holding potential: −70 mV) every 2 min. Secretion was measured via membrane capacitance recording and visualized with TIRFM. Shown is the average membrane capacitance increase (ΔCm, lines) and the averaged cumulative number of exocytotic events visualized by TIRFM normalized to the footprint area of the cell in TIRFM (dots). See the Materials and Methods section for more details on the normalization procedure. Pictures show the footprint of one exemplary cell in TIRFM taken immediately before (left row) and after (right row) the stimulus. Arrows mark secreted vesicles upon stimulation (n = 22).
Figure 3.

Flash photolysis of caged Ca2+ does not increase the percentage of released LDCVs. Secretion was induced by four consecutive flashes applied to the cells every 2 min. Exocytosis was measured via membrane capacitance recording and visualized with TIRFM. Shown are the average membrane capacitance increase (line) and the cumulative secreted LDCVs normalized to the footprint area visualized in TIRFM (dots). Pictures show the footprint of one exemplary cell in TIRFM taken immediately before (left row) and after (right row) the stimulus. Arrows mark secreted LDCVs after the stimulus. (n = 21).
Figure 4.

Exocytosis of a majority of LDCVs can be induced by using a long-lasting and strong stimulus. Cells were patched with 1, 6, and 15 μm free [Ca2+] in the pipette solution, shown in A–C, respectively. Displayed are the average membrane capacitance increase (stippled line) and the number of cumulative secreted LDCVs (continuous line) visualized in TIRFM normalized to the footprint area of the cell (n = 16, A, B; n = 17, C). Images show the footprint of exemplary cells in TIRFM taken immediately before reaching whole-cell configuration (left) and after 5 min of perfusion of the cell with high [Ca2+] (right). A, Inset, Membrane capacitance increase induced by 10 consecutive depolarizations applied to the cells after perfusing them for >5 min with 1 μm free Ca2+.
Figure 5.

Approximately 15% of LDCVs are dead-end vesicles. A, B, Kymogram showing the dwell time of all LDCVs for one exemplary cell measured either in presence of 6 (A) or 15 (B) μm free [Ca2+]i. LDCVs are grouped depending whether they were dead-end vesicles, detaching from the PM moving back in the cytoplasm or secreted. C, Survival plot of all LDCVs visualized in the first image of the cells perfused with 6 μm Ca2+ (solid black line, 638 LDCVs in a total of 16 cells). The data were best fitted by double exponential decay (solid gray line) as compared with a mono exponential decay (stippled gray line). The time constant of the fast component was 49 ± 5 s (short stippled black line) and 439 ± 86 s for the slow component (large stippled black line). D, Analysis of TIRFM recording in cells perfused with 6 or 15 μm free [Ca2+]i shown in black and gray, respectively. The average density of LDCVs at the PM was determined at the beginning and at the end of the experiment (i.e., after 5 min). LDCVs visible in TIRFM at the end of the experiment were backtracked to differentiate between newcomers (LDCVs arriving in the evanescent wave during the recording time) and dead-end vesicles (LDCVs already present in the first picture). All values were normalized to the footprint area of the cells (6 μm [Ca2+]i, n = 16; 15 μm [Ca2+]i, n = 17). See Materials and Methods for details on the normalization procedure.
Figure 6.
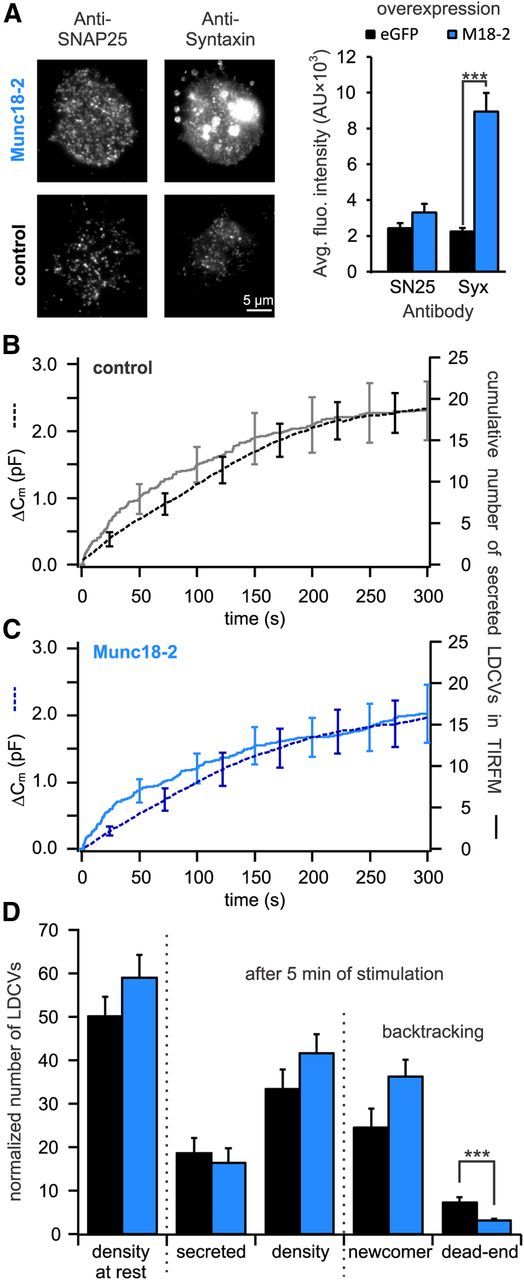
Expression of Munc18-2 decreases the number of dead-end vesicles by >60%. A, Quantitative immunocytochemistry to determine the change in SNAP-25 and syntaxin in the PM upon Munc18-2 overexpression. Left, TIRFM images of anti-syntaxin and anti-SNAP-25 labeled cells either overexpressing Munc18-2 (top) or not (bottom). Right, Quantification of syntaxin and SNAP-25 immunolabeling in the PM. Although Munc18-2 did not affect the [SNAP-25]PM it significantly increased the [syntaxin]PM (***p < 0.001, n = 10 for control and Munc18-2 overexpressing cells, respectively). B, C, Cells transfected with NPY-mCherry alone (B) or together with Munc18-2-TFP (C) were patched with 6 μm free [Ca2+] in the pipette solution. Shown are the average membrane capacitance increase (stippled line) and the cumulative number of secreted LDCVs visualized in TIRFM normalized to the footprint area of the cell (solid line). D, Analysis of TIRFM recording in cells expressing Munc18-2-TFP (blue) or control cells (black). Represented are the average density of LDCVs near the PM at the beginning and the end of the experiment and the average total secretion. LDCVs visible in TIRFM at the end of the experiment were separated between newcomer and dead-end vesicles. All data were normalized to the cell's footprint area (n = 13 for Munc18-2-TFP and control cells; ***p < 0.001). See Materials and Methods for details on the normalization procedure.
Figure 8.

Expression of open-syntaxin increases the number of dead-end vesicles by 70%. A, Quantitative immunocytochemistry to assess the difference in SNAP-25 and syntaxin insertion in the PM upon open-syntaxin overexpression. Left, TIRFM images of anti-syntaxin and anti-SNAP-25 labeled cells either overexpressing open-syntaxin (top) or not (bottom). Right, average fluorescence intensity of syntaxin and SNAP-25 immunolabeling in the PM. Overexpressed open-syntaxin was clearly transported to the PM as the syntaxin immunofluorescence was significantly increased compared with control (***p < 0.001, n = 10 and n = 14 for control and open-syntaxin overexpressing cells, respectively). B, C, Cells transfected with NPY-mCherry and eGFP alone (B) or together with open-syntaxin (C) were patched with 6 μm free [Ca2+] in the pipette solution. Shown are the average membrane capacitance increase (stippled line) and the cumulative number of secreted LDCVs visualized in TIRFM normalized to the footprint area (solid line). D, Analysis of TIRFM recording in cells expressing open-syntaxin (red) or control cells (black). Represented are the average density of LDCVs near the PM at the beginning and the end of the experiment and the average total secretion. LDCVs visible in TIRFM at the end of the experiment were separated between newcomer and dead-end vesicles (n = 16 and n = 15 for open-syntaxin expressing and control cells, respectively; *p < 0.05; **p < 0.01). See Materials and Methods for details on the normalization procedure.
Figure 9.
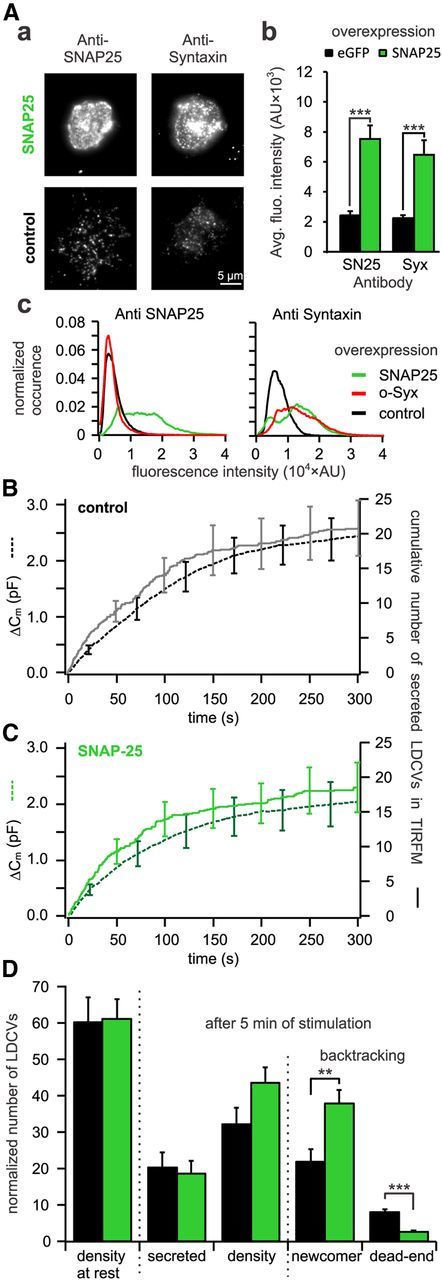
Overexpression of SNAP-25 decreases the number of dead-end vesicles by 67%. A, Quantitative immunocytochemistry to assess the difference in SNAP-25 and syntaxin insertion in the PM upon open-syntaxin overexpression. Aa, TIRFM images of anti-syntaxin and anti-SNAP-25 labeled cells either overexpressing open-syntaxin (top) or not (bottom). Ab, Average fluorescence intensity of syntaxin and SNAP-25 immunolabeling in the PM. For both proteins the fluorescence intensities were significantly increased in SNAP-25 overexpressing cells as compared with control (***p ≤ 0.001, n = 10 for control and SNAP-25 overexpressing cells). Ac, Average fluorescence intensity histograms of the TIRFM picture of the SNAP-25 (left) and syntaxin (right) immunostaining. They were normalized to the surface area of the individual footprints of the cells. Note that upon overexpression of the stained protein the respective histogram is right shifted toward higher values compared with control. Interestingly, upon overexpression of SNAP-25, the histogram of syntaxin immunofluorescence is not only right shifted but changes its shape from a unimodal to a bimodal distribution indicating different protein clustering. B, C, Cells cotransfected with NPY-mCherry and eGFP (B) or together with SNAP-25 (C) were patched with 6 μm free [Ca2+] in the pipette solution. Shown are the average membrane capacitance increase (stippled line) and the cumulative number of secreted LDCVs visualized in TIRFM normalized to the footprint area (solid line). D, Analysis of TIRFM recording in cells overexpressing SNAP-25 (green) or control cells (black). Represented are the average density of LDCVs near the PM at the beginning and the end of the experiment and the average total secretion. LDCVs visible in TIRFM at the end of the experiment were separated between newcomer and dead-end vesicles (n = 14 and n = 13 for SNAP-25 overexpressing and control cells, respectively; **p < 0.01; ***p < 0.001). See Materials and Methods for details on the normalization procedure.
If not stated otherwise, normally distributed data were analyzed using the unpaired Student's t test. Mann–Whitney rank sum test was used when the data were not normally distributed. Statistical analysis was performed in SigmaStat (Systat Software). Results are shown as mean ± SEM.
Results
Multiple rounds of depolarizations deplete the pool of releasable LDCVs but not the number of LDCVs at the PM
Ashery et al. (2000) proposed that repetitive stimulation depletes the pool of docked vesicles. We tested this hypothesis in bovine chromaffin cells by simultaneously measuring the membrane capacitance and visualizing fluorescently labeled LDCVs near the PM with TIRFM (Becherer et al., 2007). This allowed us to record the secretion of the cells and to determine whether this secretion depletes the pool of LDCVs located near the PM at the footprint of the cell. To elicit strong secretion, we used a condition in which priming of LDCVs was favored by high [Ca2+]i (718 ± 39 nm) and stimulated the cells with a train of 10 depolarizations (100 ms at 5 Hz). This stimulus was repeated every 2 min, because previous studies have shown that this delay allows full recovery of the primed pool of LDCVs (von Rüden and Neher, 1993; Smith et al., 1998). This protocol was repeated as long as the cell responded with an increase in membrane capacitance. The first stimulus train elicited a membrane capacitance increase of 307.8 ± 47.7 fF and the release of 0.9 ± 0.3 LDCVs visualized by TIRFM at the footprint of the cell (Fig. 1, n = 21). The response to the following stimuli decreased gradually. By the fourth stimulus, the response measured using membrane capacitance recording was reduced by ∼50% and the number of secreted LDCVs visualized in TIRFM was reduced by ∼80%. A fifth stimulus elicited no more secretion using both measuring methods (data not shown). Two separate phenomena were observed. (1) Comparing the secretory response determined by membrane capacitance and TIRFM revealed a discrepancy between the two measurement methods that develops upon repetitive stimulation. (2) Four trains of depolarizations totally depleted the secretory response of the cells although the number of LDCVs visualized with TIRFM near the PM remained nearly constant throughout the experiment (Fig. 1, see exemplary cells).
In a previous work, we showed that secretion measured using TIRFM is representative of the overall secretion of the cells (Becherer et al., 2007). Among other findings, the authors showed that the time course of secretion was the same using both measurement methods. However, secretion was elicited using a maximum of two rounds of stimulations. The authors also found that secretion detected by TIRFM was ∼10 times lower than secretion measured by membrane capacitance recording. We now investigated the discrepancy between membrane capacitance and TIRFM recording using an array of methods. To rule out that NPY-mCherry labeled LDCVs, i.e., new vesicles generated after the time of transfection, might represent a special subset of LDCVs, we verified these LDCVs displayed the same spatial distribution as old LDCVs. Costaining the cells with the green false fluorescent neurotransmitter FFN511 allow us to visualize all LDCVs because this false fluorescent neurotransmitter is transported via the vesicular mono-amine transporter and accumulates in LDCVs (Gubernator et al., 2009). The cells were visualized by SIM which provides superb spatial resolution and in contrast to TIRFM allows us to reconstruct the cell in 3D (Fig. 2; Gustafsson et al., 2008). NPY-mCherry-positive LDCVs were evenly distributed in the cell like the other LDCVs (Fig. 2H). Thus NPY-mCherry stained LDCVs are not a special subset of vesicles. Another possibility for the relative lack of secretion events in TIRFM may result from a shift in the secretion of new to old LDCVs. In this case the number of NPY-mCherry labeled LDCVs (observable in TIRFM) fusing with the PM would decrease disproportionally compared with the global LDCV release measured by membrane capacitance. To test this hypothesis, we performed the same experiment as shown in Figure 1 using cells in which the LDCVs were marked simultaneously with NPY-mCherry and FFN511. We found that ∼65% of the release was due to old LDCVs but that the fraction of old versus new LDCVs released was similar in each response (Fig. 2I). Thus we can rule out that old LDCVs are preferentially secreted after repetitive stimuli. An alternative explanation, which we cannot test, is that upon stimulation a large amount of vesicular membrane is added to the PM, thereby generating invaginations in the area where the cell is attached to the glass coverslip. LDCVs docked at these invaginations on the PM would be located out of the evanescent wave generated by TIRF and their exocytosis would remain undetected by TIRFM although it occurred at the cell's footprint.
The second phenomenon shown in Figure 1 was the overall gradual decay of secretion although the density of LDCVs was unaltered during the experiment. This is illustrated by the pictures of an exemplary cell acquired just prior and after each train of depolarizations (Fig. 1, left and right). The averaged density of LDCVs at the beginning of the experiments was 0.64 ± 0.03 LDCV · μm−2, compared with 0.62 ± 0.03 LDCV · μm−2 at the end of the experiments (n = 21). Hence, depletion of LDCVs at the PM cannot account for the reduction in the exocytotic response. However, we found that the Ca2+ current measured at steady-state that was elicited by the first depolarization was −29 ± 9 pA and declined to −15 ± 9 pA in the fourth train (p = 0.026, paired t test, n = 19). This Ca2+ current rundown of ∼50% might partially account for the observed decrease in secretion.
To test whether the rundown of Ca2+ currents caused the decrease in secretion, we triggered exocytosis via a stepwise increase of global intracellular [Ca2+]i using UV flash photolysis of NP-EGTA every 2 min (Ellis-Davies and Kaplan, 1994). The [Ca2+]i was 737 ± 50 nm before stimulation and it was consistently elevated to 12.3 ± 1.7 μm upon each flash. The first and second flash raised the membrane capacitance by 309 ± 80 fF and 405 ± 91 fF (n = 22; Fig. 3). The response to subsequent flashes gradually decreased and was almost abolished (76 ± 36 fF) by the fourth stimulus (Fig. 3). Exocytosis visualized by TIRFM was strongly depleted by the third stimulus, verifying our observations on depolarization-induced secretion. Furthermore, as shown in the pictures of an exemplary cell made just prior and after each stimulus, the number of LDCVs at the PM was not depleted (Fig. 3). The density of LDCVs at the PM was 0.72 ± 0.07 LDCV · μm−2 at the beginning and 0.67 ± 0.06 LDCV · μm−2 at the end of the experiments (n = 22). The results show that neither Ca2+ current rundown nor depletion of the number of LDCVs at the PM accounts for the reduced level of secretion triggered by short but strong stimuli. Consequently, it appears that a very high fraction of LDCVs (close to 80%) might be unreleasable under these stimulus conditions.
Perfusing cells with 6 μm [Ca2+]i induces maximal secretion in chromaffin cells
To investigate whether these LDCVs were indeed unreleasable, we examined whether long lasting stimuli could reduce this pool of LDCVs. Chromaffin cells were patched with a pipette solution containing a high concentration of free Ca2+ while exocytosis was measured. We chose three different [Ca2+]i: 1 μm (at which secretion begins), 6 μm (intermediate range), and 15 μm (where the secretion rate is highest; Sørensen et al., 2002; Tian et al., 2005). Immediately after reaching the whole-cell configuration, capacitance measurements and visualization of exocytosis in TIRFM were performed continuously for at least 5 min. Perfusing the cells with 1 μm free Ca2+ for 5 min resulted in a membrane capacitance increase of 1.2 ± 0.2 pF (Fig. 4A). This increase corresponds to the total response after exposure to four rounds of caged Ca2+ photolysis (Fig. 3). The average exocytosis in cells visualized using TIRFM was 6.2 ± 1.6 LDCVs (n = 16). Both membrane capacitance and the number of secreted LDCVs visualized in TIRFM increased linearly with a rate constant of 4.00 ± 0.01 fF · s1 and 0.03 ± 0.01 LDCV · s1, respectively. As shown by an exemplary cell (Fig. 4A, right), the footprint of the cell was not depleted of LDCVs after 5 min of recording. However, the vesicles present at the membrane were still releasable, because a train of depolarizations applied to the cells at the end of the recording period induced a membrane capacitance increase of ∼173.6 ± 39.7 fF and 0.25 ± 0.15 LDCVs in TIRFM (Fig. 4A, inset). Perfusing the cells with 6 μm free Ca2+ for 5 min induced an exocytotic response of the cells that was ∼3.5 times higher than in cells perfused with 1 μm free Ca2+ (Fig. 4B). The maximal secretion rate was 18.9 ± 3 fF · s−1 and 0.19 ± 0.04 LDCV · s−1 (n = 16). This very strong secretion significantly reduced the pool of LDCVs close to the PM (Fig. 4B, right). In contrast to the scenario in which cells were perfused with 1 μm free Ca2+, depolarizing the cells at the end of the recording period did not result in an additional membrane capacitance increase. Thus the remaining LDCVs at the footprint of the cells appeared to be unreleasable.
Next, we perfused the cells with a solution containing 15 μm free Ca2+ to induce maximal secretion. Surprisingly, the resulting membrane capacitance increase was smaller than the response induced by 6 μm free Ca2+ (Fig. 4B,C). This can be attributed to endocytosis which is triggered at very high [Ca2+]i, as revealed by sudden decreases of the membrane capacitance in individual cells (data not shown). These decreases likely correspond to rapid endocytosis that has been shown to occur at an [Ca2+]i >10 μm (Smith and Neher, 1997; Engisch and Nowycky, 1998; Perez Bay et al., 2007). The secretory response measured by TIRFM was slightly but not significantly higher than the response induced by 6 μm free Ca2+ (p = 0.328, n = 17), indicating that secretion in chromaffin cells perfused with 6 μm [Ca2+]i is already maximal.
Fifteen percent of LDCVs are dead-end vesicles
We next investigated the properties of LDCVs that remained at the PM after 5 min perfusion with either 6 or 15 μm [Ca2+]i. Their number after 5 min stimulation was depleted by either 34 or 29% when secretion was induced with 6 or 15 μm Ca2+, respectively (Fig. 5D). Very little residual secretion was measurable (Fig. 4) and the remaining LDCVs at the PM appeared to be unreleasable. We wondered whether these LDCVs were dead-end vesicles. The behavior of LDCVs was very dynamic, many LDCVs docked at and undocked from the PM throughout the experiment (Fig. 5A,B). Some LDCVs present from the beginning of the experiment either undock or are secreted, whereas other LDCVs, which often were secreted at a later time point, arrived and docked at the PM during the time course of the experiment. If those LDCVs, which stay at the PM for very long time, belong to a separate pool of vesicles, then they will appear as a separate component in a survival plot. We analyzed how long the LDCVs, which were visible in the first image of the movie, stayed at the PM before leaving either by exocytosis or through undocking. Then we plotted the number of these LDCV as a function of time (Fig. 5C). We performed this analysis in cells stimulated with 6 μm Ca2+ and we found that the plot was best fit with a double exponential decay. The first component had a time constant of 49 ± 5 s. The second component was ∼10 times slower, with a time constant of 439 ± 86 s (n = 17), indicating the existence of an apparently unreleasable pool of vesicles. We then devised a method to easily identify these vesicles. As can be appreciated after 200 s the pool with the faster time constant is entirely empty (Fig. 5C). Hence all LDCVs remaining at the PM for the entire recording time of 300 s are dead-end vesicles and they can be identified by backtracking all LDCVs visible at the end of the movie. This method is very reliable but underestimates the real pool size of dead-end vesicles. We found that at 6 μm [Ca2+]i ∼75% of the LDCVs which were close to the PM at the end approached the PM during the experiment (labeled as newcomers), whereas 25% of the LDCVs were visible from the beginning and represent dead-end vesicles (Fig. 5D). We then related the number of dead-end vesicles to the number of vesicles at the PM at the start of the experiment and found that the pool of dead-end vesicles corresponded to 14.1 ± 1.5% of all LDCVs. Raising the [Ca2+]i from 6 to 15 μm did not induce their release (16.8 ± 2.1% dead-end vesicles at 15 μm Ca2+). Our results demonstrate for the first time that in wild-type chromaffin cells a number of docked vesicles are reluctant to fuse with the PM independent of the strength of the Ca2+ stimulus.
Expression of Munc18-2 decreased the pool size of dead-end vesicles
We have shown the existence of a new pool of vesicles in chromaffin cells that are located at the PM and are fusion resistant. Yet the molecular identity of this pool of vesicles is unknown. One possibility is that these vesicles correspond to a degradation product of old LDCVs, i.e., lysosomes. A second possibility is that these vesicles are functional but that due to constant stimulation of secretion, the priming factors enabling fusion competence are depleted over time. If this were the case then dwell times of LDCVs before secretion should lengthen with progressively longer stimulation. As shown in Figure 5A,B this dwell time in two exemplary cells did not increase during the experiment. Measuring the average dwell time of vesicles before secretion revealed that at 6 μm [Ca2+]i they resided for 49 ± 7 s and 55 ± 14 s at the PM during the first and the second half of the experiment, respectively (n = 17). Furthermore, no correlation was found between the time of docking and the dwell time of the LDCVs before release (Spearman correlation coefficients were −0.24 or −0.18 and p values were 0.086 or 0.0995 at 6 or 15 μm [Ca2+]i, respectively). Finally, the apparent τundocking was 69.94 ± 0.86 s at 6 μm [Ca2+]i which is much shorter than the acquisition period. It is also unlikely that a shortage of priming factors would lead to a pool of vesicles unable to undock. These results indicate that priming is probably not involved in the generation of dead-end vesicles. A third hypothesis is that some LDCVs are docked to the unproductive 2:1 SNARE acceptor complex. In vitro assays have established that a syntaxin:SNAP-25 ratio of at least 0.1 favors the formation of the 1:1 SNARE acceptor complex (Pobbati et al., 2012). Using quantitative real-time PCR we found that chromaffin cells contain less syntaxin compared with SNAP-25 at mRNA level. In the PM of PC12 cells the syntaxin:SNAP-25 ratio was 0.072 (Knowles et al., 2010). Consequently, few 2:1 SNARE acceptor complexes should be generated in vivo. Furthermore, proteins that are expressed in vivo, such as complexin, Munc13 and Munc18 have been shown to prevent the formation of the 2:1 SNARE acceptor complex (Weninger et al., 2008). Accordingly, the formation of this acceptor complex in vivo has been questioned.
To investigate the role of the 2:1 SNARE acceptor complex in the generation of dead-end vesicles, we aimed to reduce the production of this complex by overexpressing Munc18. Three isoforms of Munc18 have been described in mammals (Tellam et al., 1995). Whereas Munc18-1 is primarily expressed in neurons and neuroendocrine cells, Munc18-2 and -3 can be found in a variety of nonneuronal tissues (Agrawal et al., 2000). Each isoform interacts with a different cognate SNARE but all Munc18 s appear to share most functions except for the priming role of Munc18-1, which cannot be reproduced by Munc18-2 (Gulyás-Kovács et al., 2007). To simplify the interpretation of the phenotype observed by Munc18 overexpression, we chose to overexpress Munc18-2 in chromaffin cells. Cells were electroporated with either NPY-mCherry alone (control) or with the combination of NPY-mCherry and Munc18-2-mTFP, raising the number of Munc18-2 mRNA copies by 6.8 times (Table 1). We stimulated secretion by perfusing the chromaffin cells with the intracellular solution containing 6 μm [Ca2+] because we showed that this solution produced almost maximal secretion. The expression of Munc18-2-mTFP did not lead to changes in secretion as measured by membrane capacitance or TIRFM (Fig. 6B,C). Additionally, the overexpression of Munc18-2-mTFP did not result in a significant increase of the LDCV density at the PM compared with control cells (Fig. 6D). However, Munc18-2 overexpression significantly increased the number of LDCVs at a distance of 0.25–1.0 μm to the PM (p < 0.02, n = 10; Fig. 7C,D). Furthermore, the overexpression of Munc18-2 almost quadrupled the amount of syntaxin in the PM, whereas SNAP-25 level remained unaffected (Fig. 6A; p < 0.001, n = 10 in both conditions), confirming previous observations of Gulyás-Kovács et al. (2007). The number of newcomer LDCVs was increased by ∼50% in Munc18-2-mTFP expressing cells compared with control cells (Fig. 6D). This increase is not significant but does indicate a faster turnover of the LDCVs at the PM. The number of dead-end vesicles was 7.2 ± 1.3 in control cells which corresponds to 14.4 ± 2.7% of all LDCVs visible at the PM before stimulation (n = 13; Fig. 6D). In Munc18-2-mTFP expressing cells the number of dead-end vesicles was reduced to 3.1 ± 0.3 vesicles, accounting for merely 5.4 ± 0.6% of all visible LDCVs at the beginning of the experiment. (p = 0.005, n = 13; Fig. 6D). Consequently, overexpression of Munc18-2-mTFP reduced the proportion of dead-end vesicle by 62.5% compared with control cells (p = 0.001, n = 13) and implies that Munc18-2 functions to prevent their generation.
Expression of open-syntaxin increased the number of dead-end vesicles whereas the overexpression of SNAP-25 induced a decrease
Because Munc18 has a variety of functions throughout exocytosis (de Wit, 2010), we examined whether the reduction of dead-end docking upon Munc18-2 overexpression was indeed due to a destabilization of the 2:1 t-SNARE acceptor complex. We reasoned that by changing the syntaxin:SNAP-25 stoichiometry in the PM, we would affect the 2:1 t-SNARE acceptor complex formation (Pobbati et al., 2012). We first aimed at promoting the formation of the unproductive 2:1 acceptor complex by raising the syntaxin:SNAP-25 ratio through overexpression of syntaxin. However, overexpressed syntaxin1 was predominantly retained in the Golgi because Munc18 becomes a rate limiting factor to process syntaxin to the PM (Rowe et al., 1999). Therefore, we expressed the open form of syntaxin1 (syntaxin1A L165A/E166A), which is transported to the PM independently of Munc18 (Dulubova et al., 1999). This can be appreciated in Figure 8A showing that open-syntaxin expression led to a >4-fold increase in syntaxin in the PM, whereas SNAP-25 levels remained unaltered. Cells expressing open-syntaxin showed a slightly but not significantly reduced density of vesicles at the PM at rest compared with control cells coexpressing NPY-mCherry and eGFP (19%; p = 0.069, n = 16 and 15, control and open-syntaxin expressing cells, respectively; Figs. 8D, 7E,F). This was to be expected because the expression of open-syntaxin lead to a docking defect in wild-type newborn mouse chromaffin cells (Liu et al., 2008). The exocytotic response to the solution containing 6 μm [Ca2+]i was slightly but not significantly reduced in cells expressing open-syntaxin compared with control (Fig. 8B,C). In addition, the number of newcomers was reduced by 32% in open-syntaxin expressing cells compared with controls (p = 0.08, n = 15 and 14, open-syntaxin expressing cells and control cells, respectively; Fig. 8D). This indicates that, in contrast to the phenotype observed in Munc18-2 expressing cells, the expression of open-syntaxin lead to a reduced turnover rate of LDCVs at the PM. Importantly, analyzing the dead-end vesicles revealed that their number was increased by ∼70% from 9.0 ± 0.9 to 15.2 ± 1.7 in control and in open-syntaxin expressing cells, respectively (p < 0.001, n = 15 and n = 16, respectively; Fig. 8D). The simplest explanation for this large increase in the percentage of dead-end vesicles is an increased formation of the unproductive 2:1 acceptor complexes.
To confirm this finding, we increased the concentration of SNAP-25 at the PM in an attempt to decrease the formation of the 2:1 acceptor complex. Cells that expressed NPY-mCherry were cotransfected with either SNAP-25 or eGFP as control. The level of SNAP-25 in the PM was increased by a factor of 3 (Fig. 9Aa,Ab). However, to our surprise the level of syntaxin was elevated as well by a similar factor (Fig. 9Aa,Ab). Only unclustered t-SNAREs participate in exocytosis (Murray and Tamm, 2009; Bar-On et al., 2012). TIRFM with a lateral resolution of 250 nm does not allow the measurement of cluster size of 95–50 nm (Sieber et al., 2007; Bar-On et al., 2012). Hence, we tested whether SNAP-25 and open syntaxin overexpression induced qualitatively the same increase in anti-syntaxin immunofluorescence by generating a fluorescence distribution histogram (Fig. 9Ac). Whereas the overexpression of a syntaxin protein led to a unimodal rightward shift of its own immunofluorescence histogram, the overexpression of SNAP-25 induced a bimodal immunofluorescence distribution. This might indicate a different protein clustering but makes it difficult to predict how the relationship of unclustered syntaxin:SNAP-25 ratio is changed. The overall distribution of LDCVs (Fig. 7G,H) as well as the density of LDCVs at the PM in resting cells and after 5 min of recording (Fig. 9D) were not changed upon SNAP-25 overexpression. Similarly, secretion of LDCVs was nearly identical in cells overexpressing SNAP-25 versus control when visualized in TIRFM or measured with membrane capacitance recordings (Fig. 9B,C). However, the number of newcomer LDCVs was increased by ∼55% in cells overexpressing SNAP-25 compared with control (p < 0.001, n = 14, n = 13 respectively, Fig. 9D), thereby confirming the trend observed in cells expressing Munc18-2-mTFP. Finally, the number of dead-end vesicles was reduced from 8.0 ± 0.7 in control to 2.6 ± 0.4 in SNAP-25 overexpressing cells (p < 0,001, n = 14, n = 13 respectively; Fig. 9D). Thus the overexpression of SNAP-25 leads to a 67% reduction of the number of dead-end vesicles. As expected, these data contrast with the results observed in cells expressing open-syntaxin but reproduce exactly the phenotype of Munc18-2-mTFP expressing cells.
Together these findings support the hypothesis that the unproductive 2:1 acceptor complex is formed in vivo and that LDCVs can dock to this complex thereby preventing their priming.
Discussion
We have characterized dead-end docking and deciphered its molecular mechanism. A strong but short (<2 s) stimulation of exocytosis either through repetitive depolarizations or flash photolysis of caged Ca2+ was not sufficient to deplete the LDCVs that reside near the PM. In contrast, perfusion of the cells with an intracellular solution containing 6 μm free Ca2+ for 5 min induced strong secretion which substantially depleted LDCVs at the PM. Approximately 15% of all LDCVs remained docked throughout the experiment and were defined as dead-end vesicles. These vesicles did not fuse in response to [Ca2+]i as high as 15 μm. In bovine chromaffin cells, ∼10% of all LDCVs remained noticeably immobile at the PM for at least 2 min when [Ca2+]i was maintained between 0.1 and 1 μm (Pasche et al., 2012). The analysis of the dwell-time before secretion did not favor the idea that a priming process is involved and it is unlikely that dead-end vesicles are degradation products, i.e., lysosomes, unable to undergo exocytosis. Dead-end vesicles had a well defined round shape and not the typical elongated shape of lysosomes. Furthermore, overexpression of three different proteins, Munc18-2, open-syntaxin or SNAP-25, affected the proportion of dead-end vesicles while none of these proteins have a known function in the degradation or recycling pathways of LDCVs. Finally, it is possible that dead-end vesicles become trapped at the PM via an unproductive docking reaction. The mechanism of docking involves a variety of molecules such as the t-SNAREs syntaxin and SNAP-25 (de Wit et al., 2006, 2009), Munc18 (Voets et al., 2001), and the vesicular protein synaptotagmin (de Wit et al., 2009). Verhage and Sørensen (2008) postulated that dead-end docking might be mediated by the formation of an unproductive SNARE acceptor complex formed with two syntaxin and one SNAP-25 molecule. This stable complex has been described using biochemical assays (Fasshauer and Margittai, 2004), but has not been observed in living cells. A number of proteins destabilizing the 2:1 complex are expressed in cells, which argues against its existence in vivo (Sørensen et al., 2006). To test whether dead-end vesicles were LDCVs that docked to the unproductive 2:1 complex, we created conditions in which the generation of this complex was either potentially favored or unfavored. Munc18-2 expression and SNAP-25 overexpression possibly promoted the formation of the 1:1 SNARE complex and resulted in a consistent decrease of the dead-end vesicle pool by 66% compared with control. In contrast, when the production of the 2:1 SNARE acceptor complex was likely favored through the expression of open-syntaxin, the fraction of dead-end vesicles was increased by a factor of 2. These results indicate that dead-end vesicles are probably docked to the PM trough the unproductive 2:1 SNARE acceptor complex.
We showed that expression of Munc18-2 strongly reduced the number of dead-end vesicles whereas expression of open-syntaxin increased their number. Both proteins have been shown to be involved in the docking process at various levels independently of the SNARE acceptor complex formation. One of the functions of Munc18 is to dissolve the cortical actin network and thereby to facilitate the movement of LDCVs from the cytoplasm to the PM (Toonen et al., 2006). One could envision that in control cells some LDCVs get trapped in very dense parts of the actin cortex and thus become unreleasable. Although the interaction of Munc18 with actin has been demonstrated for Munc18-1 and not Munc18-2 (Bhaskar et al., 2004), this scenario is compatible with our data. Indeed, expressing Munc18-2 in cells slightly increased the number of newcomer vesicles at the PM which might indicate that the access to the PM is facilitated. However, applying the actin destabilizing agent latrunculin to chromaffin cells or PC12 cells decreased the mobility of LDCVs (Lang et al., 2000; Oheim and Stühmer, 2000), suggesting that the cortical actin does not form a net in which LDCVs become trapped. Hence, the cortical actin network does not seem to be involved in the generation of dead-end vesicles. Additionally, we showed that raising the Munc18-2 concentration in cells increased the amount of syntaxin at the PM which should enhance formation of the unproductive 2:1 SNARE acceptor complex. However, biochemical assays have shown that including Munc18-1 in the reaction of syntaxin with SNAP-25 favors the formation of the 1:1 acceptor complex (Weninger et al., 2008). Thus our data indicate that despite a higher syntaxin concentration, the overexpression of Munc18-2, possibly through binding with syntaxin, shifts the equilibrium of the acceptor complex formation towards the 1:1 t-SNARE complex.
The expression of open-syntaxin in wild-type cells lead to a docking deficiency when compared with control (our own results; Gerber et al., 2008; Liu et al., 2008). This docking deficiency is probably due to the reduced interaction of open-syntaxin with Munc18 (Dulubova et al., 1999; Gerber et al., 2008). However, the effect of open-syntaxin on docking should affect both productive and unproductive docking to the same extent, reducing the pool size of dead-end vesicles, which is opposed to our results. SNAP-25 has also been shown to play a role in docking via its interaction with synaptotagmin, but overexpression of SNAP-25 in wild-type cells did not increase the docking of LDCVs (de Wit et al., 2009). We also showed that SNAP-25 overexpression did not lead to the sole increase in SNAP-25 level in the PM. It was accompanied by a rise in syntaxin level in the PM but not in syntaxin translation (Table 1). If the increase in SNAP-25 and syntaxin at the PM affects the levels of unclustered proteins is unclear and was not addressed. Nevertheless, the simplest explanation for the effects of the overexpression of open-syntaxin or SNAP-25 is a changed syntaxin:SNAP-25 stoichiometry in the PM either favoring or hindering the generation of the unproductive 2:1 SNARE acceptor complex.
The fraction of dead-end vesicles and newcomer vesicles was inversely correlated when either Munc18, open-syntaxin or SNAP-25 were overexpressed. Newcomer vesicles were LDCVs that moved in the evanescent wave during the time course of the experiment and stayed there until the end. We showed that the dead-end undocking time constant, which corresponds to the survival plot's slow time constant, is longer than 7 min (Fig. 7C). Due to this slow undocking of dead-end vesicles it is likely that unproductive dead-end docking sites are saturated throughout the experiment and newcomer vesicles will mainly attach to productive docking sites. It follows that productive and unproductive docking sites are interdependent and likely rely on the same proteins because the size of the depot pool of vesicles is not a limiting factor for docking (Sørensen, 2004). This interdependence of both types of docking sites is consistent with a role for the 2:1 SNARE acceptor complex in dead-end docking.
Several questions beyond the scope of the present study remain to be elucidated. One is to identify the vesicular protein that interacts with the 2:1 SNARE acceptor complex. This acceptor complex forms a tetrameric core complex which precludes the interaction with synaptobrevin (Fasshauer and Margittai, 2004). Possible candidates are synaptotagmin (de Wit et al., 2009) or an unknown vesicular protein which interacts with Munc18 that has been proposed to be involved in docking (Gulyás-Kovács et al., 2007). If this is the case then studying dead-end vesicles may provide a powerful tool for its identification. Another important point is whether dead-end docking is reversible so that dead-end vesicles could constitute a reserve pool for secretion. Our data show that increasing the [Ca2+]i did not lead to their release. Furthermore, we found that the size of dead-end vesicle pool is extremely stable. In four independent sets of experiment in which we stimulated the cells with 6 μm Ca2+ the proportion of dead-end vesicles over all LDCVs visible at the PM before stimulation was invariable (14.1 ± 1.5, 14.4 ± 2.7%, 15.0 ± 1.0% and 15.2 ± 2.0% in Figs. 4, 6, 8, and 9, respectively). Accordingly, it is possible that changes in the cell metabolism might lead to their release. One could imagine that phosphorylation reactions might modulate the size of the dead-end vesicle pool because both SNAP-25 and syntaxin can be phosphorylated by PKA and/or PKC (Risinger and Bennett, 1999). On the other hand, recent work from Murray and Tamm (2009) showed that SNAP-25 and syntaxin interact to form the SNARE acceptor complex only after they diffused out of clusters. Thus, modulating the ratio of free SNAP-25 versus free syntaxin via clustering might be a powerful tool to modulate the formation of the productive or the unproductive SNARE acceptor complexes.
In conclusion, studying dead-end vesicles provides new insights in the docking mechanism. Our study provides the first evidence that the unproductive 2:1 SNARE acceptor complex exists and is functional in vivo. Further examination of dead-end docking will clearly broaden our knowledge of the mechanisms involved in docking modulation.
Footnotes
This work was supported by local funding (HOMFOR, Universität des Saarlandes) and by grants from the Deutsche Forschungsgemeinschaft SFB 530 and 894 (to J.R. and U.B.). We thank David Stevens, Misty Marshall, and Claudia Schirra for helpful comments on the paper, L. Wirtz and U. Kazmaier for synthesizing the FFN511, M. Klose, M. Schneider, K. Sandmeier, C. Bick and A. Ludes for expert technical support.
The authors declare no competing financial interests.
References
- Agrawal A, Adachi R, Tuvim M, Yan XT, Teich AH, Dickey BF. Gene structure and promoter function of murine Munc18-2, a nonneuronal exocytic Sec1 homolog. Biochem Biophys Res Commun. 2000;276:817–822. doi: 10.1006/bbrc.2000.3513. [DOI] [PubMed] [Google Scholar]
- Ashery U, Betz A, Xu T, Brose N, Rettig J. An efficient method for infection of adrenal chromaffin cells using the Semliki Forest virus gene expression system. Eur J Cell Biol. 1999;78:525–532. doi: 10.1016/S0171-9335(99)80017-X. [DOI] [PubMed] [Google Scholar]
- Ashery U, Varoqueaux F, Voets T, Betz A, Thakur P, Koch H, Neher E, Brose N, Rettig J. Munc13-1 acts as a priming factor for large dense-core vesicles in bovine chromaffin cells. EMBO J. 2000;19:3586–3596. doi: 10.1093/emboj/19.14.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-On D, Wolter S, van de Linde S, Heilemann M, Nudelman G, Nachliel E, Gutman M, Sauer M, Ashery U. Super-resolution imaging reveals the internal architecture of nano-sized syntaxin clusters. J Biol Chem. 2012;287:27158–27167. doi: 10.1074/jbc.M112.353250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becherer U, Pasche M, Nofal S, Hof D, Matti U, Rettig J. Quantifying exocytosis by combination of membrane capacitance measurements and total internal reflection fluorescence microscopy in chromaffin cells. PLoS ONE. 2007;2:e505. doi: 10.1371/journal.pone.0000505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar K, Shareef MM, Sharma VM, Shetty AP, Ramamohan Y, Pant HC, Raju TR, Shetty KT. Co-purification and localization of Munc18-1 (p67) and Cdk5 with neuronal cytoskeletal proteins. Neurochem Int. 2004;44:35–44. doi: 10.1016/S0197-0186(03)00099-8. [DOI] [PubMed] [Google Scholar]
- de Wit H. Morphological docking of secretory vesicles. Histochem Cell Biol. 2010;134:103–113. doi: 10.1007/s00418-010-0719-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H, Cornelisse LN, Toonen RF, Verhage M. Docking of secretory vesicles is syntaxin dependent. PLoS One. 2006;1:e126. doi: 10.1371/journal.pone.0000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H, Walter AM, Milosevic I, Gulyás-Kovács A, Riedel D, Sørensen JB, Verhage M. Synaptotagmin-1 docks secretory vesicles to syntaxin-1/SNAP-25 acceptor complexes. Cell. 2009;138:935–946. doi: 10.1016/j.cell.2009.07.027. [DOI] [PubMed] [Google Scholar]
- Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Südhof TC, Rizo J. A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J. 1999;18:4372–4382. doi: 10.1093/emboj/18.16.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis-Davies GC, Kaplan JH. Nitrophenyl-EGTA, a photolabile chelator that selectively binds Ca2+ with high affinity and releases it rapidly upon photolysis. Proc Natl Acad Sci U S A. 1994;91:187–191. doi: 10.1073/pnas.91.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engisch KL, Nowycky MC. Compensatory and excess retrieval: two types of endocytosis following single step depolarizations in bovine adrenal chromaffin cells. J Physiol. 1998;506(Pt 3):591–608. doi: 10.1111/j.1469-7793.1998.591bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasshauer D, Margittai M. A transient N-terminal interaction of SNAP-25 and syntaxin nucleates SNARE assembly. J Biol Chem. 2004;279:7613–7621. doi: 10.1074/jbc.M312064200. [DOI] [PubMed] [Google Scholar]
- Gerber SH, Rah JC, Min SW, Liu X, de Wit H, Dulubova I, Meyer AC, Rizo J, Arancillo M, Hammer RE, Verhage M, Rosenmund C, Südhof TC. Conformational switch of syntaxin-1 controls synaptic vesicle fusion. Science. 2008;321:1507–1510. doi: 10.1126/science.1163174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubernator NG, Zhang H, Staal RG, Mosharov EV, Pereira DB, Yue M, Balsanek V, Vadola PA, Mukherjee B, Edwards RH, Sulzer D, Sames D. Fluorescent false neurotransmitters visualize dopamine release from individual presynaptic terminals. Science. 2009;324:1441–1444. doi: 10.1126/science.1172278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyás-Kovács A, de Wit H, Milosevic I, Kochubey O, Toonen R, Klingauf J, Verhage M, Sørensen JB. Munc18-1: sequential interactions with the fusion machinery stimulate vesicle docking and priming. J Neurosci. 2007;27:8676–8686. doi: 10.1523/JNEUROSCI.0658-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson MG, Shao L, Carlton PM, Wang CJ, Golubovskaya IN, Cande WZ, Agard DA, Sedat JW. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys J. 2008;94:4957–4970. doi: 10.1529/biophysj.107.120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns LM, Levitan ES, Shelden EA, Holz RW, Axelrod D. Restriction of secretory granule motion near the plasma membrane of chromaffin cells. J Cell Biol. 2001;153:177–190. doi: 10.1083/jcb.153.1.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles MK, Barg S, Wan L, Midorikawa M, Chen X, Almers W. Single secretory granules of live cells recruit syntaxin-1 and synaptosomal associated protein 25 (SNAP-25) in large copy numbers. Proc Natl Acad Sci U S A. 2010;107:20810–20815. doi: 10.1073/pnas.1014840107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang T, Wacker I, Wunderlich I, Rohrbach A, Giese G, Soldati T, Almers W. Role of actin cortex in the subplasmalemmal transport of secretory granules in PC-12 cells. Biophys J. 2000;78:2863–2877. doi: 10.1016/S0006-3495(00)76828-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Schirra C, Stevens DR, Matti U, Speidel D, Hof D, Bruns D, Brose N, Rettig J. CAPS facilitates filling of the rapidly releasable pool of large dense-core vesicles. J Neurosci. 2008;28:5594–5601. doi: 10.1523/JNEUROSCI.5672-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Schirra C, Edelmann L, Matti U, Rhee J, Hof D, Bruns D, Brose N, Rieger H, Stevens DR, Rettig J. Two distinct secretory vesicle-priming steps in adrenal chromaffin cells. J Cell Biol. 2010;190:1067–1077. doi: 10.1083/jcb.201001164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray DH, Tamm LK. Clustering of syntaxin-1A in model membranes is modulated by phosphatidylinositol 4,5-bisphosphate and cholesterol. Biochemistry. 2009;48:4617–4625. doi: 10.1021/bi9003217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nofal S, Becherer U, Hof D, Matti U, Rettig J. Primed vesicles can be distinguished from docked vesicles by analyzing their mobility. J Neurosci. 2007;27:1386–1395. doi: 10.1523/JNEUROSCI.4714-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oheim M, Stühmer W. Tracking chromaffin granules on their way through the actin cortex. Eur Biophys J. 2000;29:67–89. doi: 10.1007/s002490050253. [DOI] [PubMed] [Google Scholar]
- Pasche M, Matti U, Hof D, Rettig J, Becherer U. Docking of LDCVs is modulated by lower intracellular [Ca(2+)] than priming. PLoS One. 2012;7:e36416. doi: 10.1371/journal.pone.0036416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattu V, Qu B, Marshall M, Becherer U, Junker C, Matti U, Schwarz EC, Krause E, Hoth M, Rettig J. Syntaxin7 is required for lytic granule release from cytotoxic T lymphocytes. Traffic. 2011;12:890–901. doi: 10.1111/j.1600-0854.2011.01193.x. [DOI] [PubMed] [Google Scholar]
- Perez Bay AE, Ibañez LI, Marengo FD. Rapid recovery of releasable vesicles and formation of nonreleasable endosomes follow intense exocytosis in chromaffin cells. Am J Physiol Cell Physiol. 2007;293:C1509–C1522. doi: 10.1152/ajpcell.00632.2006. [DOI] [PubMed] [Google Scholar]
- Pobbati AV, Chan SW, Lee I, Song H, Hong W. Structural and functional similarity between the Vgll1-TEAD and the YAP-TEAD complexes. Structure. 2012;20:1135–1140. doi: 10.1016/j.str.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Risinger C, Bennett MK. Differential phosphorylation of syntaxin and synaptosome-associated protein of 25 kDa (SNAP-25) isoforms. J Neurochem. 1999;72:614–624. doi: 10.1046/j.1471-4159.1999.0720614.x. [DOI] [PubMed] [Google Scholar]
- Rowe J, Corradi N, Malosio ML, Taverna E, Halban P, Meldolesi J, Rosa P. Blockade of membrane transport and disassembly of the Golgi complex by expression of syntaxin 1A in neurosecretion-incompetent cells: prevention by rbSEC1. J Cell Sci. 1999;112:1865–1877. doi: 10.1242/jcs.112.12.1865. [DOI] [PubMed] [Google Scholar]
- Sanders JD, Yang Y, Liu Y. Differential turnover of syntaxin and SNAP-25 during synaptogenesis in cultured cerebellar granule neurons. J Neurosci Res. 1998;53:670–676. doi: 10.1002/(SICI)1097-4547(19980915)53:6<670::AID-JNR5>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Sieber JJ, Willig KI, Kutzner C, Gerding-Reimers C, Harke B, Donnert G, Rammner B, Eggeling C, Hell SW, Grubmüller H, Lang T. Anatomy and dynamics of a supramolecular membrane protein cluster. Science. 2007;317:1072–1076. doi: 10.1126/science.1141727. [DOI] [PubMed] [Google Scholar]
- Smith C, Neher E. Multiple forms of endocytosis in bovine adrenal chromaffin cells. J Cell Biol. 1997;139:885–894. doi: 10.1083/jcb.139.4.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, Moser T, Xu T, Neher E. Cytosolic Ca2+ acts by two separate pathways to modulate the supply of release-competent vesicles in chromaffin cells. Neuron. 1998;20:1243–1253. doi: 10.1016/S0896-6273(00)80504-8. [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Matti U, Wei SH, Nehring RB, Voets T, Ashery U, Binz T, Neher E, Rettig J. The SNARE protein SNAP-25 is linked to fast calcium triggering of exocytosis. Proc Natl Acad Sci U S A. 2002;99:1627–1632. doi: 10.1073/pnas.251673298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen JB. Formation, stabilisation and fusion of the readily releasable pool of secretory vesicles. Pflugers Arch. 2004;448:347–362. doi: 10.1007/s00424-004-1247-8. [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Wiederhold K, Müller EM, Milosevic I, Nagy G, de Groot BL, Grubmüller H, Fasshauer D. Sequential N- to C-terminal SNARE complex assembly drives priming and fusion of secretory vesicles. EMBO J. 2006;25:955–966. doi: 10.1038/sj.emboj.7601003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speidel D, Bruederle CE, Enk C, Voets T, Varoqueaux F, Reim K, Becherer U, Fornai F, Ruggieri S, Holighaus Y, Weihe E, Bruns D, Brose N, Rettig J. CAPS1 regulates catecholamine loading of large dense-core vesicles. Neuron. 2005;46:75–88. doi: 10.1016/j.neuron.2005.02.019. [DOI] [PubMed] [Google Scholar]
- Steyer JA, Horstmann H, Almers W. Transport, docking and exocytosis of single secretory granules in live chromaffin cells. Nature. 1997;388:474–478. doi: 10.1038/41329. [DOI] [PubMed] [Google Scholar]
- Tellam JT, McIntosh S, James DE. Molecular identification of two novel Munc-18 isoforms expressed in non-neuronal tissues. J Biol Chem. 1995;270:5857–5863. doi: 10.1074/jbc.270.11.5857. [DOI] [PubMed] [Google Scholar]
- Tian JH, Wu ZX, Unzicker M, Lu L, Cai Q, Li C, Schirra C, Matti U, Stevens D, Deng C, Rettig J, Sheng ZH. The role of Snapin in neurosecretion: snapin knock-out mice exhibit impaired calcium-dependent exocytosis of large dense-core vesicles in chromaffin cells. J Neurosci. 2005;25:10546–10555. doi: 10.1523/JNEUROSCI.3275-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen RF, Kochubey O, de Wit H, Gulyas-Kovacs A, Konijnenburg B, Sørensen JB, Klingauf J, Verhage M. Dissecting docking and tethering of secretory vesicles at the target membrane. EMBO J. 2006;25:3725–3737. doi: 10.1038/sj.emboj.7601256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Sørensen JB. Vesicle docking in regulated exocytosis. Traffic. 2008;9:1414–1424. doi: 10.1111/j.1600-0854.2008.00759.x. [DOI] [PubMed] [Google Scholar]
- Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, Südhof TC, Neher E, Verhage M. Munc18-1 promotes large dense-core vesicle docking. Neuron. 2001;31:581–591. doi: 10.1016/S0896-6273(01)00391-9. [DOI] [PubMed] [Google Scholar]
- von Rüden L, Neher E. A Ca-dependent early step in the release of catecholamines from adrenal chromaffin cells. Science. 1993;262:1061–1065. doi: 10.1126/science.8235626. [DOI] [PubMed] [Google Scholar]
- Walter AM, Wiederhold K, Bruns D, Fasshauer D, Sørensen JB. Synaptobrevin N-terminally bound to syntaxin-SNAP-25 defines the primed vesicle state in regulated exocytosis. J Cell Biol. 2010;188:401–413. doi: 10.1083/jcb.200907018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S, Xu T, Ashery U, Kollewe A, Matti U, Antonin W, Rettig J, Neher E. Exocytotic mechanism studied by truncated and zero layer mutants of the C-terminus of SNAP-25. EMBO J. 2000;19:1279–1289. doi: 10.1093/emboj/19.6.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weninger K, Bowen ME, Choi UB, Chu S, Brunger AT. Accessory proteins stabilize the acceptor complex for synaptobrevin, the 1:1 syntaxin/SNAP-25 complex. Structure. 2008;16:308–320. doi: 10.1016/j.str.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Matti U, Melamed R, Hagalili Y, Bruns D, Rettig J, Ashery U. Tomosyn inhibits priming of large dense-core vesicles in a calcium-dependent manner. Proc Natl Acad Sci U S A. 2004;101:2578–2583. doi: 10.1073/pnas.0308700100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilly FE, Sørensen JB, Jahn R, Lang T. Munc18-bound syntaxin readily forms SNARE complexes with synaptobrevin in native plasma membranes. PLoS Biol. 2006;4:e330. doi: 10.1371/journal.pbio.0040330. [DOI] [PMC free article] [PubMed] [Google Scholar]