

Abstract
Synaptic plasticity in the cerebellum is thought to contribute to motor learning. In particular, long-term depression (LTD) at parallel fiber (PF) to Purkinje neuron (PN) excitatory synapses has attracted much attention of neuroscientists as a primary cellular mechanism for motor learning. In contrast, roles of plasticity at cerebellar inhibitory synapses in vivo remain unknown. Here, we have investigated the roles of long-lasting enhancement of transmission at GABAergic synapses on a PN that is known as rebound potentiation (RP). Previous studies demonstrated that binding of GABAA receptor with GABAA receptor-associated protein (GABARAP) is required for RP, and that a peptide that blocks this binding suppresses RP induction. To address the functional roles of RP, we generated transgenic mice that express this peptide fused to a fluorescent protein selectively in PNs using the PN-specific L7 promoter. These mice failed to show RP, although they showed no changes in the basal amplitude or frequency of miniature IPSCs. The transgenic mice also showed no abnormality in gross cerebellar morphology, LTD, or other excitatory synaptic properties, or intrinsic excitability of PNs. Next, we attempted to evaluate their motor control and learning ability by examining reflex eye movements. The basal dynamic properties of the vestibulo-ocular reflex and optokinetic response, and adaptation of the latter, were normal in the transgenic mice. In contrast, the transgenic mice showed defects in the adaptation of vestibulo-ocular reflex, a model paradigm of cerebellum-dependent motor learning. These results together suggest that RP contributes to a certain type of motor learning.
Introduction
The cerebellum is necessary for fine motor control and synaptic plasticity, which has been considered to contribute to motor learning (Hansel et al., 2001; Boyden et al., 2004; Dean et al., 2010; Gao et al., 2012; Ito, 2012; Hirano, 2013). In particular, long-term depression (LTD) at glutamatergic excitatory synapses between parallel fibers (PFs) and a Purkinje neuron (PN) has been regarded as a critical cellular mechanism of motor learning. However, normal motor learning was observed in some LTD-deficient animals (Welsh et al., 2005; Schonewille et al., 2011), which has made the roles of LTD in motor learning puzzling. Other cerebellar synaptic or intrinsic plasticity mechanisms can also contribute to motor learning (Boyden et al., 2004; Dean et al., 2010; Gao et al., 2012; Hirano, 2013), and one candidate mechanism is rebound potentiation (RP) (Kano et al., 1992; Kawaguchi and Hirano, 2000; Yamamoto et al., 2002).
RP is a long-lasting enhancement of transmission efficiency at GABAergic inhibitory synapses on PNs and is induced by postsynaptic depolarization caused by strong excitatory synaptic inputs, such as those from a climbing fiber (CF). Thus, RP decreases the excitability of a PN depending on the CF activity similarly to LTD, suggesting that RP might contribute to motor learning in concert with LTD. Intensive studies on the molecular mechanisms of RP induction have been performed (Kawaguchi and Hirano, 2000, 2002, 2007; Kitagawa et al., 2009; Kawaguchi et al., 2011), and it was reported that binding of GABAA receptor with GABAA receptor-associated protein (GABARAP) is necessary for the induction and maintenance of RP. GABARAP is a protein that binds to GABAA receptor and tubulin (Wang et al., 1999) and that regulates the number and/or function of cell-surface GABAA receptor in both neurons and heterologous expression systems (Chen et al., 2000; Leil et al., 2004). RP induction is suppressed by a peptide (γ2 peptide) corresponding to an intracellular region of GABAA receptor γ2 subunit that binds to GABARAP (Kawaguchi and Hirano, 2007). Here, to study the roles of RP in motor control and learning, we generated transgenic mice in which γ2 peptide fused to a fluorescent protein is selectively expressed in cerebellar PNs using a PN-specific L7 promoter (Smeyne et al., 1995).
We then examined the ability of the transgenic mice regarding motor control and learning, focusing on two reflex eye movements, vestibulo-ocular reflex (VOR) and optokinetic response (OKR). Both of these reflexes work to stabilize the visual image during head motion (Robinson, 1981). VOR and OKR undergo adaptive modifications in direction so as to reduce the image slip on the retina in various experimental conditions, and these adaptations have been studied as model paradigms of cerebellum-dependent motor learning (Ito, 1982, 2012; du Lac et al., 1995; Boyden et al., 2004). We report here that the transgenic mice showed impaired RP and defects in VOR adaptation.
Materials and Methods
Generation of transgenic mice.
The PN-specific expression construct using the L7 promoter was designed as described previously (Smeyne et al., 1995). The L7 gene was a gift from Dr. A. Kakizuka (Ikeda et al., 1996). The γ2pepV expression vector reported in a previous study (Kawaguchi and Hirano, 2007) was digested with EcoRI and XhoI, and the γ2pepV coding region was blunted with T4 polymerase, followed by insertion into the blunted BamHI site of pL7ΔAUG (Smeyne et al., 1995). Then, the DNA fragment for L7-γ2pepV was isolated by digestion with HindIII and EcoRI. This DNA fragment was purified and used for injection into fertilized eggs of C57BL/6N strain mice to generate γ2pepV transgenic mice (Accession no. CDB0485T: http://www.cdb.riken.jp/arg/TG%20mutant%20mice%20list.html). The transgene was detected by PCR using the following primers: 5′-GGCACTTCTGACTTGCACTTTCCTTGGTCC-3′ and 5′-ATGGCGGACTTGAAGAAGTCGTGCTGCTTC-3′.
Immunohistology.
A male mouse (8–10 weeks old) anesthetized with Somnopentyl (50 mg/kg, Kyoritsu Pharmacy), was perfused transcardially with PBS followed by perfusion of 4% paraformaldehyde in PBS. Its brain was postfixed overnight at 4°C in paraformaldehyde in PBS and then kept in 30% sucrose in PBS overnight. Sagittal sections (50 μm) were cut from the frozen cerebellum with a sliding microtome and stored in PBS. Immunofluorescent staining was performed as described previously (Jiao et al., 1999). The following antibodies were used: rabbit anti-GFP (Invitrogen), rabbit and mouse anti-calbindin (Millipore Bioscience Research Reagents and Swant, respectively), mouse anti-NeuN (Millipore Bioscience Research Reagents), mouse anti-neurogranin (Millipore Bioscience Research Reagents), rabbit anti-parvalbumin (Abcam), and goat anti-rabbit and anti-mouse IgG conjugated with Alexa488 or Alexa568 (Invitrogen). Immunofluorescent images were captured with a confocal microscope (FV1000, Olympus). The width of the molecular or the granule layer was measured using ImageJ (http://rsbweb.nih.gov/ij/) at 10 regions in a transverse slice. Densities of neurons were obtained by counting the number of neurons in each layer and dividing the number by the area of each layer. Ten slices from 3 mice were examined for each case.
Coimmunoprecipitation.
Coimmunoprecipitation experiments were performed as described previously (Mizokami et al., 2007). The cerebellum of control or γ2pepV mouse (8- to 10-week-old, male) was homogenized in buffer A containing 150 mm NaCl, 5 mm EDTA, 0.2% BSA, and 0.5% Triton X-100, 1% protease inhibitor mixture (Nacalai) and 20 mm HEPES-NaOH, pH 7.4. After homogenization, cross-linker solution containing 2.5 mm 3,3′-dithiobis[sulfosuccinimidylpropionate] and 2.5 mm dithiobis[succinimidylpropionate] (Pierce) was added, and the membrane proteins were extracted in 1% Na-deoxycholate for 30 min at 4°C. The cross-linking reaction was performed for 30 min at room temperature and then stopped by adding 50 mm Tris-HCl, pH 7.5.
Then, the protein extract obtained by centrifugation was subjected to immunoprecipitation with ∼10 μg of rabbit anti-γ2 antibody (Millipore), and 20 μl of a 50% slurry of protein A-Sepharose beads (GE Healthcare) was added. The beads were washed twice with PBS, resuspended in 20 μl of glycine buffer containing 50 mm glycine, 150 mm NaCl, 0.1% Triton X-100 titrated to pH 2.5 with HCl. Then, 0.8 μl of 1 m Tris-HCl, pH 9.0, and sample buffer for SDS-PAGE containing 4% SDS, 10% glycerol, 0.001% bromophenol blue, 10 mm dithiothreitol were added, and Western blotting was performed. The antibodies used for immunoblotting were as follows: anti-GABARAP (MBL), anti-β actin (Sigma), and HRP-conjugated secondary antibodies (Millipore Bioscience Research Reagents). Signals were detected using SuperSignal West Femto Maximum Sensitivity Substrate (Pierce) and LAS-3000 plus gel documentation system (Fujifilm).
Electrophysiology.
The methods of whole-cell recording in slice preparations were similar to those described previously (Kashiwabuchi et al., 1995). A mouse of either sex was killed by decapitation, and sagittal slices (250 μm) were prepared from the cerebellum. Most slices were prepared from P14–P18 mice, although P22–P24 or 8- to 10-week-old mice were used in some experiments. Slices from P14–P18 or P22–P24 mice were prepared in a solution containing the following (in mm): 130 NaCl, 4.5 KCl, 2 CaCl2, 5 HEPES, 33 glucose titrated to pH 7.4 with NaOH. Slices from 8- to 10-week-old mice were prepared in a solution containing the following (in mm): 93 N-methyl-d-glucamine, 2.5 KCl, 1.3 NaH2PO4, 30 NaHCO3, 2.0 thiourea, 5.1 Na-ascorbate, 3.1 Na-pyruvate, 0.5 CaCl2, 10 MgCl2, 25 glucose, 20 HEPES, titrated to pH 7.4 with HCl. Then, the slices were transferred to Krebs' solution containing the following (in mm): 124 NaCl, 1.8 KCl, 1.2 KH2PO4, 1.3 MgCl2, 2.5 CaCl2, 26 NaHCO3, and 10 glucose saturated with 95% O2 and 5% CO2, and 37°C heat shock was applied for 45–60 min. After that, slices were maintained at room temperature (22–24°C).
A PN was whole-cell voltage-clamped at −70 or −80 mV with a glass pipette filled with an internal solution containing the following (in mm): 150 CsCl, 0.5 EGTA, 10 HEPES, 2 Mg-ATP (Sigma), and 0.2 Na-GTP (Sigma) titrated to pH 7.3 with CsOH at room temperature, unless otherwise stated. For current-clamp recording, an internal solution containing the following (in mm), 140 d-glucuronic acid, 5 EGTA, 10 HEPES, 155 KOH, 7 KCl, 2 Mg-ATP, and 0.2 Na-GTP, was used. The electrode resistance was 1–4 MΩ, and the pipette was coated with silicon to minimize the stray capacitance. The junction potential between the Krebs' solution and the K glucuronate-based internal solution was 14 mV, which was cancelled in recordings. Input resistance (>150 MΩ) and series resistance (10–25 MΩ) were monitored throughout the experiments by applying an 80 ms −10 mV voltage pulse every 1 min, and the experiment was terminated when either input or series resistance changed by > 20%. Inhibitory postsynaptic currents (IPSCs) were recorded in the presence of 10 μm NBQX (Tocris Bioscience), an AMPA/kainate receptor antagonist. Miniature IPSCs (mIPSCs) were recorded under action potential suppression by 1 μm TTX (Wako). In mIPSC analyses, events >7 pA with appropriate time courses were selected and analyzed with Mini Analysis software (Synaptosoft). The mean mIPSC amplitude was calculated from >200 events in a PN. RP was induced by 5 depolarization pulses (500 ms, 0 mV) at 0.5 Hz. Evoked IPSCs were recorded at a holding potential of −20 mV by applying a 50 μs voltage pulse through a glass electrode, which was filled with Krebs' solution and was placed in the molecular layer in a horizontal cerebellar slice (250 μm).
EPSCs were recorded in the presence of 20 μm bicuculline (Sigma), a GABAAR antagonist. PF-EPSCs were evoked by applying a 50 or 200 μs voltage pulse through a glass electrode placed in the molecular layer. To induce LTD, a 50 ms voltage pulse to 0 mV coupled with a PF stimulation 15 ms after the onset of depolarization was applied 10 times at 1 Hz. PF-EPSCs were monitored at 0.05 Hz. To stimulate CFs, a glass electrode was placed in the granular layer ∼50 μm away from the recorded PN cell body in a slice prepared from a P22–P24 mouse. Most PNs are innervated by a single CF at P22–P24 (Kano and Hashimoto, 2012). CF-EPSCs were evoked by applying a 200 μs voltage pulse and recorded at the holding potential of −20 mV. They were identified by their large amplitude, and all-or-none or stepwise amplitude increase.
The basal firing rate of PN was measured in a current-clamp condition without any current injection for 5 s. A 500 ms constant current injection was applied to obtain the relation between the amplitude of current and the frequency of action potentials. We also performed cell-attached recording of basal firing rate using a glass pipette filled with Krebs' solution.
Eye movement recording.
The methods for reflex eye movement recording were similar to those described previously (Iwashita et al., 2001; Yoshida et al., 2004). A male mouse (8–10 weeks old) was anesthetized with a mixture of 0.9% ketamine and 0.2% xylazine, and a head holder was attached to the skull with small screws using dental cement. The recording was performed 2 d after the surgery. We confirmed that the dynamic properties of eye movements did not change between 2 and 7 d after the surgery. A head-fixed mouse was placed on a turntable surrounded by a rotatable cylindrical screen (60 cm diameter) with vertical black and white stripes (14 degrees interval). The mouse's body was supported with a rubber sheet so that the feet did not reach the turntable. Both the turntable and screen were rotated independently with DC servomotors (RH-14-3002-T, Harmonic Drive) controlled by a personal computer. Sinusoidal oscillations of 10 degrees/s at 0.2–1 Hz were applied to the turntable or screen. To monitor eye movement, the right eye was illuminated by an infrared LED (TLN201, Toshiba), and the frontal image reflected by a hot mirror (DMR, Kenko) was monitored using an infrared-sensitive CCD camera (XC-HR50, Sony). Because the mirror reflects infrared light but transmits visible light, the eye position was monitored without disrupting the mouse's view. The eye image was captured at 200 Hz, and the eye position was analyzed using Geteye software that calculated the pupil centroid (Morita). For VOR recording in the dark, a drop of pilocarpine hydrochloride (Santen) was used to decrease the pupil size. The gain and phase of VOR and OKR were obtained by fitting the eye velocity curve and comparing it with the velocity curve of the turntable or screen. The phase lead of eye velocity was defined as positive, and the phase zero in VOR was defined as the condition in which the head and eye movements were 180 degrees out of phase. The phase zero for OKR occurred when the screen and eye moved synchronously in the same direction. Thus, the phase zero corresponds to the ideal condition for stabilizing the visual image.
In VOR adaptation training, sinusoidal rotation of the turntable at 0.8 Hz, 10 degrees/s for 50 s, 60 times with 10 s intervals, was coupled with that of the screen in-phase (gain-decrease training) at the same amplitude, or out-of-phase (gain-increase training) at half of the amplitude (5 degrees/s). The gain and phase of VOR in the dark were measured 10 min after cessation of the training session and compared with the pretraining values. To induce OKR adaptation, only sinusoidal rotation of the surrounding screen at 0.8 Hz, 10 degrees/s for 50 s, 60 times with 10 s intervals, was applied.
All experimental procedures were performed in accordance with the guidelines regarding the care and use of animals for experimental procedures of the National Institutes of Health and Kyoto University and approved by the local committee for handling experimental animals in the Graduate School of Science, Kyoto University.
Results
Generation of transgenic mice deficient in RP
Our previous study showed that binding of GABARAP to GABAA receptor γ2 subunit is necessary for the induction and maintenance of RP (Kawaguchi and Hirano, 2007). In that study, the expression of a fusion protein (γ2 peptide-Venus; composed of Venus and γ2 peptide) that competitively inhibits the binding of GABAAreceptor γ2 subunit to GABARAP impaired RP without affecting the basal inhibitory synaptic transmission to PNs. We generated transgenic mice (γ2pepV mice) in which the γ2 peptide-Venus transgene was selectively expressed in PNs by using the PN-specific L7 promoter. Three transgenic mice were obtained (Fig. 1A, 2, 8, and 16), although mouse 8 did not generate any offspring. Here, we mainly examined offspring of mouse 2 (line A), but those of mouse 16 (line B) were also studied.
Figure 1.
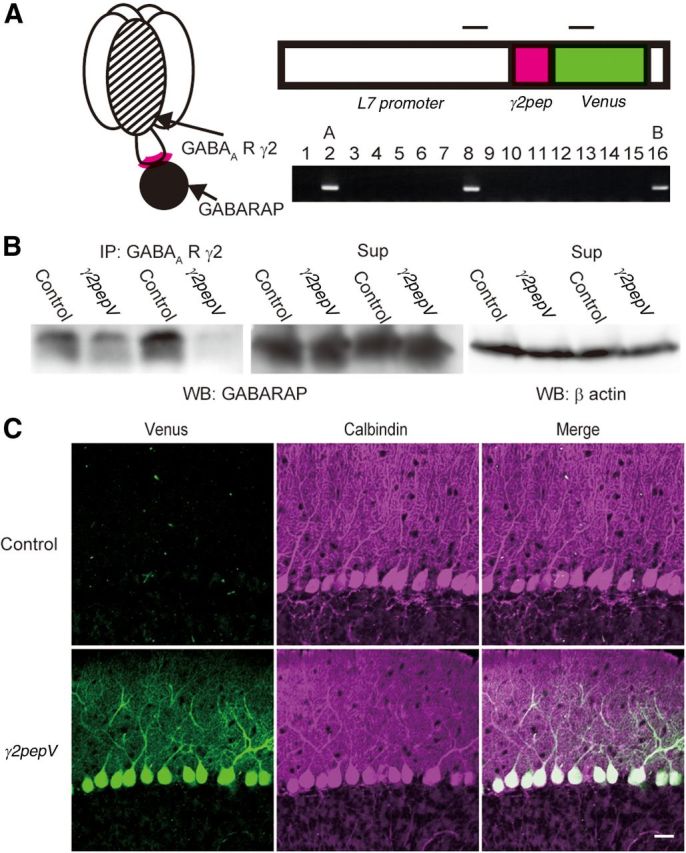
Expression of γ2pepV transgene in heterozygous mice. A, Schematic representation of binding of GABAA receptor γ2 subunit and GABARAP (left). Design of γ2pepV transgene (top) and its detection with PCR (bottom). Insertion of the transgene was examined in 16 mice, and it was detected in mice 2, 8, and 16. Top bars represent PCR primer target regions. The size of the PCR product was 468 base pairs. B, Interaction of GABARAP with GABAA receptor γ2 subunit in cerebellar extracts. The cerebellar extracts were coimmunoprecipitated with an antibody against the GABAA receptor γ2 subunit, followed by SDS-PAGE and Western blotting with an anti-GABARAP antibody. Wild-type (Control) mice showed stronger GABARAP signal than γ2pepV line A mice in the immunoprecipitate. GABARAP signal (middle) and β actin signal (right) in the supernatant are also shown. C, Cerebellar slices of control and line γ2pepV mice. Immunofluorescence for Venus (green) and that for calbindin (PN marker, magenta) are shown. Scale bar, 25 μm.
Figure 2.
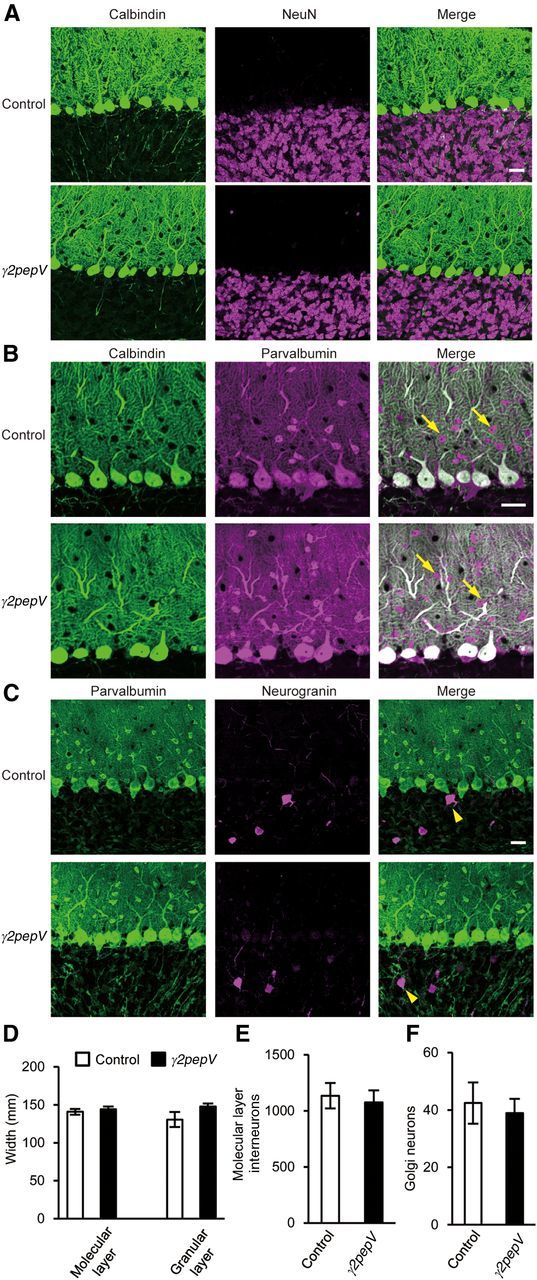
Morphology of the cerebellar cortex. Cerebellar slices stained with antibodies against calbindin (PN marker, green) and NeuN (granule neuron marker, magenta) (A), calbindin (green) and parvalbumin (a marker for molecular layer interneurons and PNs, magenta) (B), or parvalbumin (green) and neurogranin (Golgi neuron marker, magenta) (C). Arrows and arrowheads indicate molecular layer interneurons and Golgi neurons, respectively. Scale bars, 25 μm. D–F, The width of each layer (D) and the cell densities (cell number/mm2) of molecular layer interneurons (E) or Golgi neurons (F) are presented.
Figure 8.
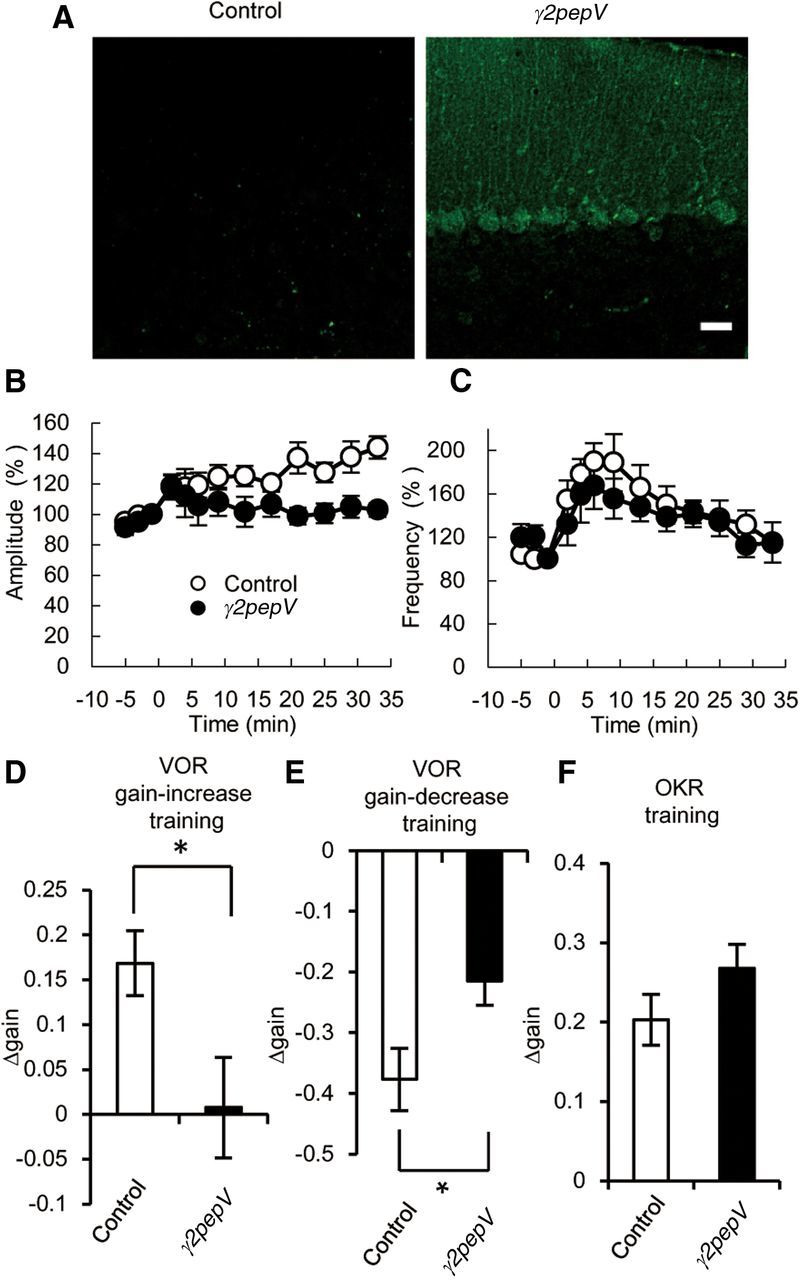
Phenotypes of γ2pepV line B mice. A, Cerebellar slices stained with an antibody against Venus (green). Scale bar, 25 μm. B, C, The time courses of averaged mIPSC amplitudes (B) and frequencies (C) before and after the conditioning depolarization (n = 5 for each). The conditioning depolarization was applied at 0 min. D–F, The differences of gain between before and after the gain-increase VOR training (n = 10 for each) (D), the gain-decrease VOR training (n = 8 for each) (E), or the OKR training (n = 10 for each) (F). Data are mean ± SEM. *p < 0.05.
We examined binding of GABAA receptor and GABARAP in wild-type (control) and γ2pepV line A transgenic mice by coimmunoprecipitation using an antibody against GABAA receptor γ2 subunit. In γ2pepV mice, less GABARAP signal was detected in the immunoprecipitate than in control mice (Fig. 1B; 53 ± 10%, n = 5 for each, p = 0.001, unpaired t test). These results suggest that GABARAP binding to GABAA receptor was suppressed in γ2pepV line A mice.
In line A heterozygous transgenic mice, Venus signal was detected only in PNs (Fig. 1C), and the transgenic mice did not show any gross anatomical abnormality in the cerebellar cortical structure (Fig. 2). The width of granular or molecular layer, densities of neurons, and morphology of PNs and inhibitory interneurons visualized with antibodies against calbindin, NeuN, parvalbumin, and neurogranin were similar in the transgenic and control mice. Calbindin is a marker protein of PNs, and NeuN is a marker of granule neurons. Parvalbumin is expressed in molecular layer interneurons and PNs, whereas neurogranin is expressed in Golgi neurons in the granular layer.
Next, we examined IPSCs and RP in γ2pepV line A mice. First, we recorded mIPSCs and evoked IPSCs from a PN in an acute slice prepared from control or γ2pepV line A mice. The baseline amplitude (Fig. 3A; control, 33 ± 8 pA, n = 5; γ2pepV, 28 ± 3 pA, n = 6, p = 0.54, unpaired t test) and the frequency (Fig. 3B; control, 6.0 ± 2.4 Hz, n = 5; γ2pepV, 5.8 ± 1.5 Hz, n = 6, p = 0.99, unpaired t test) of mIPSCs were not different between the two genotypes. We also found that the amplitude of evoked IPSCs became larger with the increased stimulation intensity similarly in the two genotypes (Fig. 3C; p = 0.36, ANOVA). Thus, the basal inhibitory synaptic transmission to a PN was not significantly altered in line A mice.
Figure 3.

IPSCs and suppression of RP in γ2pepV mice. A, B, Averaged baseline amplitudes (A) and frequencies (B) of mIPSCs recorded from PNs in control (n = 5) and γ2pepV (n = 6) mice. C, Amplitudes of evoked IPSCs versus the stimulus intensity (n = 8 for each). D–F, Representative traces (D), time courses of amplitudes (E), and frequencies (F) of mIPSCs in control (n = 5) and γ2pepV (n = 6) mice before and after the conditioning depolarization (0 min). RP was significantly impaired in γ2pepV line A mice. G, H, Cumulative probabilities of mIPSC amplitudes in control (G, before, 1041 mIPSCs; after, 1051 mIPSCs, 5 cells) and γ2pepV (H, before, 1372 mIPSCs; after, 1401 mIPSCs, 6 cells) mice. In control mice, a significant difference was detected between amplitudes before and 33 min after the RP induction. I, The time courses of amplitudes of mIPSCs in control (n = 4) and γ2pepV (n = 5) mature mice (P8- to P10-week-old) before and after the conditioning depolarization (0 min). Data are mean ± SEM.
Then, we applied conditioning depolarization pulses to induce RP (Fig. 3D–F). The depolarization induced long-lasting increases in mIPSC amplitudes in control mice, but not in γ2pepV mice (Fig. 3D,E,G,H). In control mice, mIPSC amplitudes increased to 175 ± 22% (n = 5, p = 0.042, paired t test) at 33 min after the depolarization. In contrast, mIPSC amplitudes did not significantly increase in γ2pepV mice (90 ± 7%, n = 6, p = 0.11, paired t test). The amplitudes of mIPSCs after the depolarization were significantly different between the genotypes (p < 0.01, unpaired t test). RP was also suppressed in more mature (8- to 10-week-old) line A mice (Fig. 3I; control, 143 ± 9% at 33 min after the depolarization, n = 4, p = 0.026; γ2pepV, 90 ± 9%, n = 5, p = 0.44, paired t test). Thus, RP was suppressed in juvenile and mature γ2pepV line A mice.
A transient increase of mIPSC frequency, corresponding to depolarization-induced potentiation of inhibition (Duguid and Smart, 2004) (DPI), occurred in both genotypes in juvenile mice (Fig. 3F; control, 167 ± 30% at 5 min after the depolarization, n = 5, p = 0.047; γ2pepV, 191 ± 26%, n = 6, p = 0.015, paired t test). The amplitude of DPI was not different between the genotypes (p = 0.55, unpaired t test). Significant DPI was not recorded in 8- to 10-week-old mice of either genotypes (control, 98 ± 8% at 5 min after the depolarization, n = 4, p = 0.25; γ2pepV, 96 ± 4%, n = 5, p = 0.45, paired t test). These data suggest that RP was suppressed without any changes in the basal inhibitory synaptic transmission or DPI in γ2pepV line A mice.
Excitatory synaptic transmission and intrinsic excitability
GABAergic inhibition in PNs was reported to regulate the developmental CF synapse elimination process (Nakayama et al., 2012). Thus, we next examined whether the number of CF innervations to a PN was changed in γ2pepV transgenic mice at P22–P24. A PN in wild-type mouse is innervated by a single CF at this age (Kano and Hashimoto, 2012). Recording of EPSCs induced by CF stimulation showed no significant difference between the two genotypes in the threshold stimulus intensity evoking the EPSCs (control, 15.9 ± 3.6 V, n = 10; γ2pepV, 14.4 ± 3.0 V, n = 10, p = 0.75, unpaired t test), in the number of amplitude steps (Fig. 4A,B; p = 1.0, Fisher's exact probability test), in the maximum EPSC amplitude (Fig. 4C), or in the paired-pulse ratio of EPSC amplitudes (Fig. 4D; p = 0.65, ANOVA). CF-EPSCs showed paired-pulse depression similarly in the two genotypes.
Figure 4.
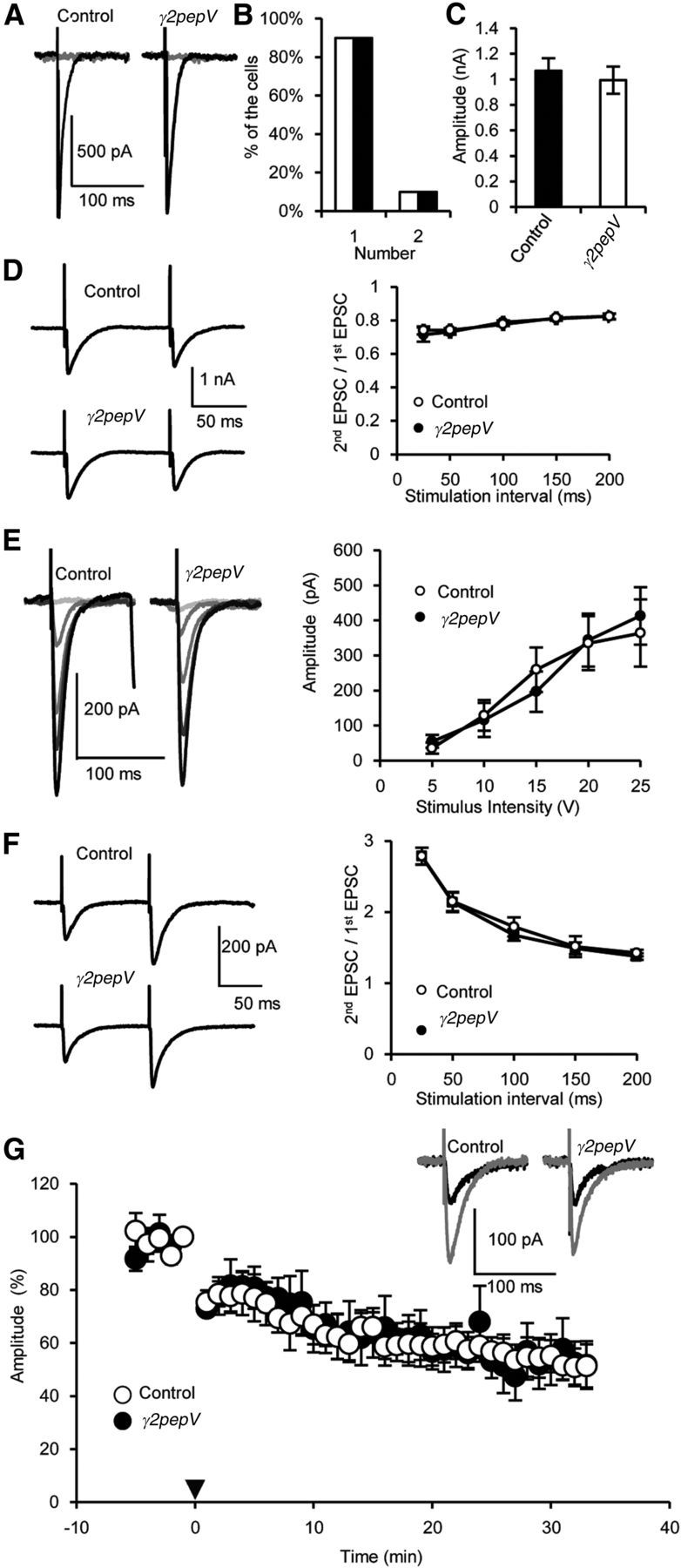
Excitatory synaptic inputs to PNs. A–C, Representative traces (A), the numbers of amplitude steps (B), and the maximum amplitudes of CF-EPSCs (C) (n = 20 for each). D, Representative traces of paired CF-EPSCs and the amplitude of the second EPSC divided by that of the first (n = 10 for each). E, Representative traces and amplitudes of PF-EPSCs versus the stimulus intensity (n = 9 for each). F, Representative traces of paired PF-EPCs and the amplitude of the second EPSC divided by that of the first (n = 10 for each). G, Time courses of averaged PF-EPSC amplitudes before and after the LTD induction (0 min, arrowhead, n = 5 for each). PF-EPSC amplitudes were normalized, taking the mean value between −1 and 0 min as 100%. Representative PF-EPSCs recorded before (gray) and 30 min after (black) the LTD induction were shown. Data are mean ± SEM.
We also observed similar dependence of PF-induced EPSC amplitude on the stimulus intensity (Fig. 4E; p = 0.65, ANOVA). PF-EPSCs showed paired-pulse facilitation similarly in the two genotypes (Fig. 4F; p = 0.45, ANOVA). Furthermore, LTD at PF-PN synapses occurred similarly in the two genotypes (Fig. 4G; control, 52 ± 9% at 33 min, n = 5, p < 0.001; γ2pepV, 52 ± 8%, n = 5; p = 0.022, paired t test). The amplitude of EPSCs 33 min after the LTD induction was not significantly different between the two genotypes (p = 0.98, unpaired t test). Thus, no significant difference was detected in the properties of excitatory synaptic transmissions to a PN in the two genotypes.
We next examined the intrinsic excitability of PNs, and we did not detect a significant difference between the two genotypes (Fig. 5). Firing rate (Fig. 5A,B), firing patterns (Fig. 5C,D), and the relationship between the intensity of injected current and the firing frequency (Fig. 5E; p = 0.27, ANOVA) were similar in the two genotypes. Spontaneous action potential firing was also recorded in a cell-attached condition. The firing rate and pattern were similar in the two recording conditions (data not shown). The height, the 10–90% rise time, and the half-height width of an action potential (Fig. 5F–I), firing threshold (control, 49 ± 2 mV; γ2pepV, 48 ± 2 mV, p = 0.73, unpaired t test), and the amplitude and half-height width of after-hyperpolarization (Fig. 5J,K) were also similar. Further, resting membrane potential and input resistance were not significantly different between the two genotypes (Fig. 5L,M). Thus, no significant difference was detected in the intrinsic excitability of PNs between the two genotypes.
Figure 5.
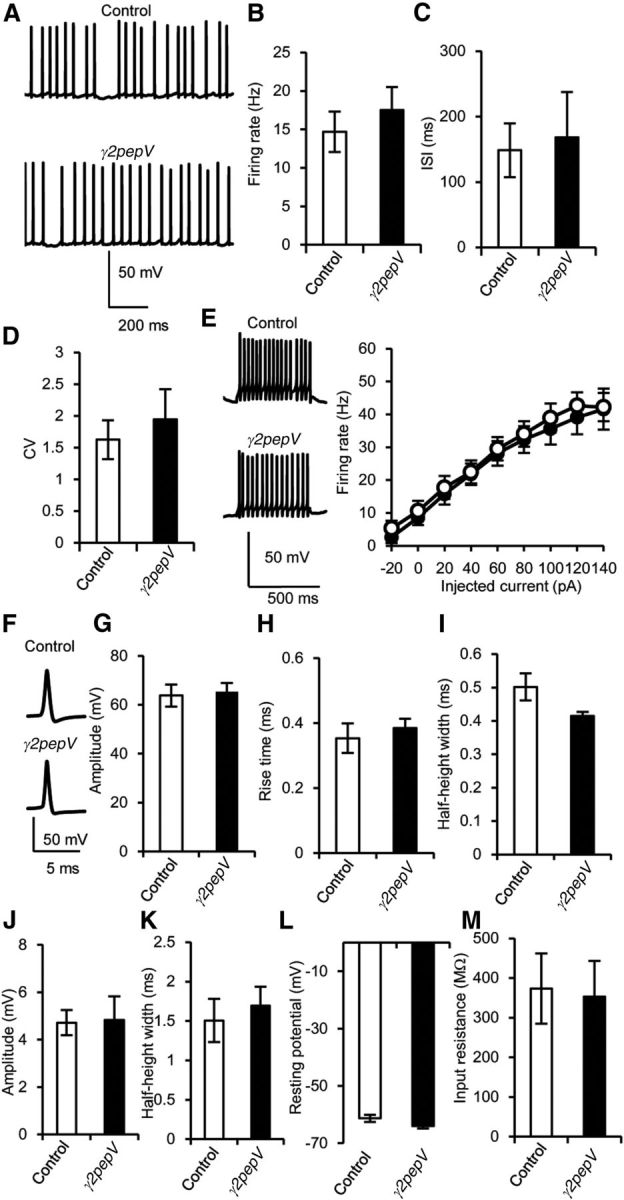
Intrinsic excitability of PNs. A, Representative traces of simple spikes. B–D, Firing rate (B), interspike interval (ISI, C), and coefficient of variation of ISI (CV, D) in current-clamp recordings (n = 20 for each). E, Representative voltage responses to 500 ms constant current injection (140 pA), and the firing rate against the intensity of injected current (n = 10). F–I, Representative traces (F), amplitudes (G), 10–90% rise times (H), and half-height widths (I) of action potential (n = 7 for each). F, Each action potential trace is an average of 10 events. J, K, Amplitudes (J) and half-height widths (K) of after-hyperpolarization (n = 7 for each). L, M, Resting membrane potential (L) and input resistance (M) of PNs. Data are mean ± SEM. There was no significant difference in any of the values between the genotypes (unpaired t test).
Basal dynamics of reflex eye movements
To evaluate how RP deficiency affects the motor control and learning ability, two reflex eye movements, VOR and OKR, were examined. VOR and OKR work to stabilize the visual image on the retina by rotating the eyeballs during head motion. Here, VOR or OKR was induced by sinusoidal horizontal rotation of a head-fixed mouse or by rotating a screen showing vertical black and white stripes surrounding a mouse, respectively. The basal dynamics of VOR and OKR were evaluated using two parameters, gain and phase (Robinson, 1981; Iwashita et al., 2001; Boyden et al., 2004). The gain was calculated by dividing the maximum speed of eyeball rotation by that of the head or screen rotation, and the phase was defined as the timing difference between the eyeball rotation and the head or screen rotation. The gain and phase of VOR in the dark, of VOR in the light, or of OKR were measured at various frequencies of the sinusoidal rotation of the turntable or that of the screen, whereas the maximum velocity of the rotation was kept constant (10 degrees/s).
With an increase of rotation frequency, the gain of VOR in the dark increased similarly in both genotypes (Fig. 6A; p = 0.28, ANOVA), whereas that of VOR in the light showed little change (Fig. 6C; p = 0.32, ANOVA) and that of OKR decreased similarly in the two genotypes (Fig. 6E; p = 0.82, ANOVA). We also found that the dependence of phase on the stimulus frequency in VOR in the dark or in the light and that in OKR were similar between the genotypes (Fig. 6B,D,F; VOR in the dark, p = 0.76; VOR in the light, p = 0.30; OKR, p = 0.28, ANOVA). Thus, the basal dynamics of VOR and OKR were not significantly different between the two genotypes.
Figure 6.
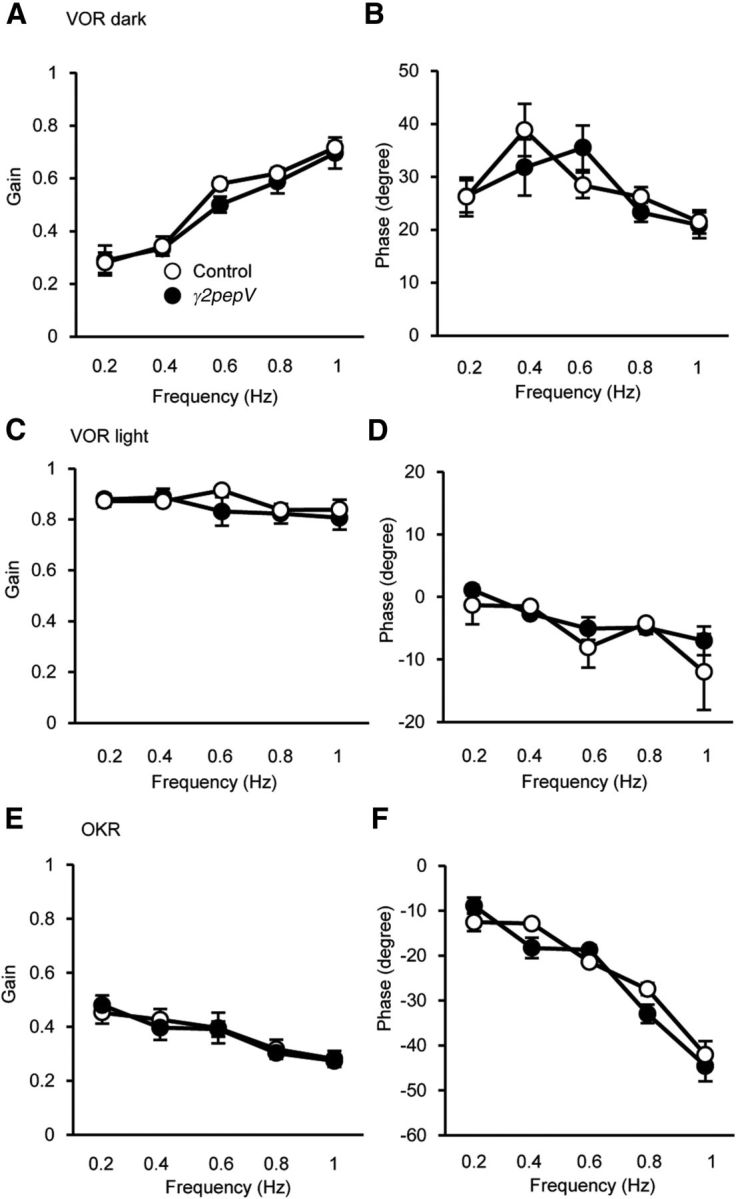
Baseline dynamic properties of VOR and OKR in γ2pepV mice. VOR in the dark (A, B), VOR in the light (C, D), and OKR (E, F) in γ2pepV (n = 5) and control (n = 5) mice. Gain (A, C, E) and phase (B, D, F) values of the eye movement relative to head or screen rotation are plotted versus the stimulus frequency. Data are mean ± SEM.
Defects of VOR adaptation in γ2pepV mice
VOR is known to show adaptive modification that is dependent on the cerebellum (Robinson, 1981; du Lac et al., 1995; Boyden et al., 2004; Ito, 2012). When sinusoidal head rotation of a mouse is coupled to rotation of the external screen in the opposite or in the same direction, the gain of VOR increases or decreases, respectively. These changes are in the direction to reduce the motion of the image on the retina during head motion and thus are adaptive. These VOR adaptations have been studied as paradigms of cerebellum-dependent motor learning. Here, we examined whether these VOR adaptations were affected in γ2pepV line A mice.
After training with the sinusoidal turntable rotation combined with the screen rotation in anti-phase (VOR gain-increase training), the gain of VOR in the dark increased from 0.62 ± 0.03 to 0.80 ± 0.04 (Fig. 7A,B; n = 5, p = 0.023, paired t test), and the phase of VOR in the dark slightly but significantly decreased from 27 ± 2 degrees to 23 ± 2 degrees in control mice (Fig. 7C; n = 5, p < 0.01, paired t test). On the other hand, in γ2pepV mice, the gain of VOR in the dark before and after the training was 0.64 ± 0.05 and 0.63 ± 0.05, respectively (Fig. 7A,B; n = 5, p = 0.90, paired t test), and the phase of VOR in the dark before and after the training was 25 ± 1 degrees and 25 ± 3 degrees, respectively (Fig. 7C; p = 0.78, paired t test). The extent of change of VOR gain by the training was significantly different between the two genotypes (Fig. 7D; control, 0.18 ± 0.05; γ2pepV, −0.01 ± 0.04; p = 0.018, unpaired t test), whereas that of the phase was not significantly different between the two genotypes (Fig. 7E; control, −5 ± 1 degrees; γ2pepV, −1 ± 2 degrees; p = 0.19, unpaired t test). In general, an increase or decrease in the VOR gain tends to be associated with a decrease or increase in the phase lead, respectively, although the gain and the phase can be regulated separately (Katoh et al., 2008).
Figure 7.

Impaired adaptation of VOR in γ2pepV mice. Effects of γ2pepV on the VOR gain-increase (A–E), the VOR gain-decrease (F–J), and the OKR (K–O) trainings. Representative eye-position traces (A, F, K, averages of 10 cycles) before (gray lines) and after (black lines) each training. Changes in the gain (B, D, G, I) and the phase (C, E, H, J) before and after VOR trainings (n = 5 for each), and changes of the gain (L, N) and the phase (M, O) during the OKR training (n = 7 for each). The gain was increased (B) and the phase was decreased (C) significantly by the gain-increase training in control mice but not in γ2pepV mice. The gain-decrease training and the OKR training changed the gain and phase in the two genotypes (G, H). The gain changes after the gain-increase (D) and the gain-decrease (I) training were different between the genotypes. Data are mean ± SEM. B, C, G, H, *p < 0.05 (significant differences between before and after). **p < 0.01 (significant differences between before and after). D, I, *p < 0.05 (significant difference between the two genotypes). **p < 0.01 (significant difference between the two genotypes).
Next, we examined VOR gain-decrease training using a sinusoidal stimulus achieved by combining turntable rotation with screen rotation in the same phase. In control mice, the gain of VOR in the dark decreased from 0.59 ± 0.02 to 0.25 ± 0.03 (Fig. 7F,G; n = 5, p < 0.001, paired t test), and the phase of VOR in the dark increased from 24 ± 2 degrees to 44 ± 2 degrees after the training (Fig. 7H; p < 0.01, paired t test). In γ2pepV mice, the gain of VOR decreased from 0.60 ± 0.03 to 0.38 ± 0.02 (Fig. 7F,G; n = 5, p < 0.001, paired t test), and the phase of VOR in the dark increased from 28 ± 2 degrees to 42 ± 4 degrees (Fig. 7H; p < 0.01, paired t test). Thus, the extent of change of VOR gain by the training in γ2pepV mice was significantly smaller than that in control mice (Fig. 7I; control, −0.34 ± 0.03; γ2pepV, −0.22 ± 0.02, p < 0.01, unpaired t test), whereas that of phase was not significantly different (Fig. 7J; control, 20 ± 3 degrees; γ2pepV, 14 ± 2 degrees, p = 0.17, unpaired t test). Thus, both the gain-increase and the gain-decrease VOR adaptations of gain were suppressed in the γ2pepV mice. In contrast, we did not detect a significant difference in the phase change between the two genotypes.
Normal OKR adaptation in γ2pepV mice
OKR also undergoes adaptive modification that is dependent on the cerebellum. When the sinusoidal rotation of the surrounding screen continues, the gain increases and the phase delay decreases to better follow the image motion (Nagao, 1988; Katoh et al., 2000; Takeuchi et al., 2008). As a result of the 60 min training, the gain of OKR increased from 0.26 ± 0.03 to 0.52 ± 0.06 (Fig. 7K,L; n = 7, p < 0.01, paired t test), and the phase of OKR changed from −27 ± 2 degrees to −18 ± 7 degrees in control mice (Fig. 7M; p < 0.01, paired t test). In γ2pepV mice, the gain of OKR increased from 0.28 ± 0.02 to 0.53 ± 0.02 (Fig. 7K,L; p < 0.001, paired t test), and the phase of OKR changed from −27 ± 2 degrees to −19 ± 1 degrees after the training (Fig. 7M; p < 0.01, paired t test). There was no significant difference between the two genotypes in either the gain increase (Fig. 7N; control, 0.26 ± 0.06; γ2pepV, 0.25 ± 0.03, p = 0.86, unpaired t test) or the decrease in phase delay (Fig. 7O; control, 9 ± 2 degrees; γ2pepV, 8 ± 2 degrees, p = 0.67, unpaired t test) after the training. Thus, the VOR adaptations were significantly affected in the RP-deficient γ2pepV line A mice, but the OKR adaptation was not.
Another transgenic mouse line
Transgenic mouse lines might show different phenotypes depending on the insertion sites of the transgene. Thus, we examined the phenotype of another line (line B) of γ2pepV mice. Line B mice showed much weaker expression of γ2pepV (Fig. 8A). Thus, we used the homozygous transgenic mice of this line for the following analyses.
The amplitude of mIPSCs became 144 ± 7% (Fig. 8B; n = 5, p < 0.01, paired t test) at 33 min after the conditioning depolarization to induce RP in control mice, whereas in γ2pepV line B mice it became 103 ± 5% (Fig. 8B; n = 5, p = 0.14, paired t test). Thus, RP was suppressed in γ2pepV line B mice as in line A. The basal amplitudes and frequencies of mIPSCs were not significantly different between the two genotypes (Table 1), and DPI occurred normally in both control and γ2pepV line B juvenile mice (data not shown).
Table 1.
Statistics of γ2pepV line B mice compared with those of control micea
| Control | γ2pepV | p (unpaired t test) | |
|---|---|---|---|
| mIPSC amplitude | 32 ± 9 pA (n = 5) | 24 ± 4 pA (n = 5) | 0.47 |
| mIPSC frequency | 3.9 ± 0.7 Hz (n = 5) | 3.5 ± 0.9 Hz (n = 5) | 0.76 |
| VOR gain-increase training (gain, before) | 0.67 ± 0.03 (n = 10) | 0.68 ± 0.03 (n = 10) | 0.87 |
| VOR gain-increase training (gain, after) | 0.84 ± 0.06 (n = 10) | 0.69 ± 0.07 (n = 10) | 0.08 |
| VOR gain-increase training (Δgain) | 0.17 ± 0.04 (n = 10) | 0.01 ± 0.06 (n = 10) | 0.03 |
| VOR gain-increase training (phase, before) | 18 ± 1° (n = 10) | 18 ± 1° (n = 10) | 0.85 |
| VOR gain-increase training (phase, after) | 15 ± 1° (n = 10) | 17 ± 2° (n = 10) | 0.44 |
| VOR gain-increase training (Δphase) | −2 ± 1° (n = 10) | −1 ± 1° (n = 10) | 0.45 |
| VOR gain-decrease training (gain, before) | 0.77 ± 0.03 (n = 8) | 0.68 ± 0.03 (n = 8) | 0.07 |
| VOR gain-decrease training (gain, after) | 0.39 ± 0.04 (n = 8) | 0.47 ± 0.04 (n = 8) | 0.20 |
| VOR gain-decrease training (Δgain) | −0.38 ± 0.05 (n = 8) | −0.21 ± 0.04 (n = 8) | 0.03 |
| VOR gain-decrease training (phase, before) | 16 ± 3° (n = 8) | 17 ± 2° (n = 8) | 0.77 |
| VOR gain-decrease training (phase, after) | 24 ± 3° (n = 8) | 27 ± 3° (n = 8) | 0.51 |
| VOR gain-decrease training (Δphase) | 8 ± 2° (n = 8) | 10 ± 1° (n = 8) | 0.54 |
| OKR training (gain, before) | 0.33 ± 0.02 (n = 10) | 0.33 ± 0.02 (n = 10) | 0.96 |
| OKR training (gain, after) | 0.53 ± 0.03 (n = 10) | 0.6 ± 0.03 (n = 10) | 0.15 |
| OKR training (Δgain) | 0.20 ± 0.03 (n = 10) | 0.27 ± 0.03 (n = 10) | 0.16 |
| OKR training (phase, before) | −28 ± 1° (n = 10) | −28 ± 1° (n = 10) | 0.86 |
| OKR training (phase, after) | −19 ± 1° (n = 10) | −18 ± 1° (n = 10) | 0.88 |
| OKR training (Δphase) | 9 ± 1° (n = 10) | 10 ± 1° (n = 10) | 0.83 |
aData are expressed as mean ± SEM.
We also examined reflex eye movements of γ2pepV line B mice. Similarly to the results using line A mice, the baseline dynamics were comparable between the control and line B mice (Table 1). The VOR gain-increase training changed the gain of VOR in the dark from 0.67 ± 0.03 to 0.84 ± 0.06 in control mice (n = 10, p < 0.01, paired t test) and from 0.68 ± 0.03 to 0.69 ± 0.07 in γ2pepV line B mice (n = 10, p = 0.90, paired t test). Thus, γ2pepV line B mice, like line A mice, failed to show the adaptive VOR gain-increase. The extent of the gain change after the VOR gain-increase training was significantly different between the two genotypes (Fig. 8D; Table 1). The VOR gain-decrease training significantly changed the gain of VOR in the dark from 0.77 ± 0.03 to 0.39 ± 0.04 in control mice (n = 8, p < 0.001, paired t test) and from 0.68 ± 0.03 to 0.47 ± 0.04 in γ2pepV line B mice (n = 8, p < 0.001, paired t test). Thus, the VOR gain-decrease adaptation occurred in both genotypes. However, importantly, the extent of gain decrease after the training was significantly different between the two genotypes (Fig. 8E; Table 1). The phase changes in both the gain-increase and gain-decrease trainings were not significantly different between the genotypes. We also examined OKR adaptation and found that it was not significantly impaired in the line B mice (Fig. 8F; Table 1).
Our results indicate that in the γ2pepV transgenic mice RP was specifically suppressed, and the VOR gain adaptations but not the OKR adaptation were impaired. Taking these findings together, we conclude that RP is involved in certain types of motor learning.
Discussion
To study the roles of RP in vivo, we generated transgenic mice in which γ2-peptide fused to Venus was selectively expressed in cerebellar PNs. In these γ2pepV transgenic mice, we first confirmed that RP was suppressed. No abnormality except for that in RP was detected in the electrophysiological properties of PNs or in the cerebellar morphology. The γ2pepV mice showed normal basal dynamics in VOR and OKR and also underwent normal adaptation of OKR. In contrast, both the gain-increase and the gain-decrease adaptations of VOR gain were suppressed in γ2pepV mice. These results indicate that RP, a type of inhibitory synaptic plasticity in the cerebellum, plays essential roles in a certain type of motor learning.
Cerebellar synaptic plasticity and motor learning
Synaptic plasticity in the cerebellar cortex, the cerebellar, and/or vestibular nuclei has been considered to play important roles in motor learning (Hansel et al., 2001; Boyden et al., 2004; Dean et al., 2010; Gao et al., 2012; Ito, 2012; Hirano, 2013). In particular, LTD at PF-PN excitatory glutamatergic synapses has attracted much attention of neuroscientists as a primary candidate mechanism for motor learning. A number of previous studies on mutant mice with defective or facilitated LTD induction showed good correlations between LTD and motor learning ability (Aiba et al., 1994; De Zeeuw et al., 1998; Hansel et al., 2006; Kina et al., 2007; Takeuchi et al., 2008). However, normal motor learning with impaired LTD was also reported (Welsh et al., 2005; Schonewille et al., 2011), which has made the roles of LTD in motor learning puzzling. It has been suggested that some types of synaptic plasticity other than LTD in the cerebellar cortex, such as LTP at PF-PN synapses, LTP and LTD at PF-molecular layer interneuron synapses, and synaptic plasticity at inhibitory synapses on a PN, might also contribute to motor learning (Boyden et al., 2004; Dean et al., 2010; Gao et al., 2012; Hirano, 2013). RP is a candidate synaptic plasticity mechanism that could contribute to motor learning together with other types of synaptic plasticity, in particular with LTD, because the CF activity contributes to the induction of both RP and LTD, and both of them work to suppress the activity of a PN. Synaptic plasticity in the cerebellar or vestibular nuclei, to which a PN sends its inhibitory output, has also been suggested to contribute to motor learning (du Lac et al., 1995; Pugh and Raman, 2006).
Inhibitory synaptic transmission on PNs
A PN receives inhibitory synaptic inputs from molecular layer interneurons (stellate and basket neurons), which are innervated by PFs. Thus, stellate and basket neurons form feedforward inhibitory pathways to PNs. Selective suppression of these inhibitory synaptic transmissions by knock-out of the γ2 subunit of GABAA receptor in PNs impairs certain aspects of VOR adaptation (Wulff et al., 2009). Therefore, these inhibitory synaptic transmissions are necessary for normal motor learning.
At these inhibitory synapses on a PN, three types of plasticity have been reported: depolarization-induced suppression of inhibition, DPI, and RP. Depolarization-induced suppression of inhibition is the short-lasting suppression of presynaptic GABA release mediated by endocannabinoid (Yoshida et al., 2002), whereas DPI is longer-lasting potentiation of GABA release mediated by presynaptic NMDA-type glutamate receptor (Duguid and Smart, 2004). DPI enhances the inhibitory synaptic transmission together with RP, although DPI is shorter-lasting than RP. RP was included in a theoretical model of the cerebellum constructed to analyze the neuronal circuit behavior related to the adaptation of the ocular following response (Yamamoto et al., 2002). However, there have hitherto been no direct experimental data indicating the involvement of any inhibitory synaptic plasticity in motor control or learning.
Here, we showed that the suppression of RP by selective expression of a modified γ2 peptide in PNs of γ2pepV mice affected the gain adaptation of VOR. Thus far, we have detected no abnormality in the morphology of the cerebellum or in electrophysiological properties other than RP in the transgenic mice. We confirmed that DPI and LTD occurred normally in the juvenile transgenic mice. Together, our findings suggest that RP is selectively impaired in the transgenic mice and that it is possible to investigate specific roles of RP in vivo using these transgenic mice.
Differential effects of RP impairment on VOR and OKR adaptations
Adaptive modification of VOR has been extensively studied as a paradigm of motor learning in several animal species (Robinson, 1981; Ito, 1982; du Lac et al., 1995; Boyden et al., 2004; Ito, 2012). The involvement of LTD in VOR adaptation has been suggested, although contributions of other types of synaptic plasticity, such LTP at PF-PN synapses and synaptic plasticity in the vestibular nuclei, have also been suggested (Miles and Lisberger, 1981; Boyden and Raymond, 2003; Schonewille et al., 2010). We report here that RP-impaired γ2pepV mice showed defects in the adaptive gain-changes of VOR after both gain-increase and gain-decrease trainings, suggesting that RP contributes to certain aspects of VOR adaptation. However, it should be noted that VOR gain-decrease adaptation did occur to a certain extent in the γ2pepV mice, indicating other plasticity mechanisms also contribute to this adaptation. How RP changes the activities of PNs during the VOR adaptation trainings is an important problem that should be addressed in the future.
OKR adaptation has been studied mainly in rabbits and rodents (Collewijn and Grootendorst, 1979; Nagao, 1988; Katoh et al., 2000; Shutoh et al., 2002; Hansel et al., 2006; Endo et al., 2009). It has been reported that knock-out mice of proteins involved in LTD induction, such as nitric oxide synthase, metabotropic glutamate receptor mGluR1, Ca2+ and calmodulin-dependent kinase IIα, or G-substrate, show suppressed OKR adaptation. It is also known that delphilin knock-out mice, in which LTD induction is facilitated, show enhanced OKR adaptation (Takeuchi et al., 2008). Thus, OKR adaptation has been correlated with LTD. In this study, we failed to detect defects in OKR adaptation in γ2pepV mice. This result suggests that RP might be specifically involved in VOR adaptation but not in OKR adaptation. These differential effects of RP impairment on VOR adaptation and OKR adaptation suggest that these adaptations might be differently regulated by various forms of synaptic plasticity. Previous studies suggested differential contributions of multiple plasticity mechanisms to adaptations of VOR and OKR (Faulstich et al., 2004). It was also suggested that cerebellar cortical plasticity, such as LTD is more important in short-term adaptation of VOR or OKR than in long-term adaptation (van Alphen and De Zeeuw, 2002; Okamoto et al., 2011). Here, only shot-term adaptations of VOR and OKR have been studied. Thus, it remains possible that RP-deficient mice might not show significant defects in long-term adaptation. A contribution of plasticity in the vestibular nuclei to long-term adaptation of OKR has been suggested (Okamoto et al., 2011).
Roles of inhibitory synaptic plasticity in mammalian CNS
The roles of synaptic plasticity at excitatory synapses in the mammalian CNS have been intensively studied, whereas those at inhibitory synapses have been relatively unexplored. It has been suggested that inhibitory synaptic plasticity contributes to the maintenance of the stability, the wide dynamic range of neuronal circuit activities, and the increases in the computational flexibility of neuronal circuits (Castillo et al., 2011; Maffei, 2011; Kullmann et al., 2012). In the developing visual cortex, enhancement of inhibitory synaptic transmission contributes to the formation of ocular dominance columns (Hensch, 2005). The involvement of inhibitory synaptic plasticity in neuropsychiatric disorders and in the development of addictive behavior has also been suggested (Nugent and Kauer, 2008). Our present study demonstrates that a type of inhibitory plasticity in the cerebellar cortex contributes to a certain form of motor learning.
Footnotes
This work was supported by MEXT, Japan grants to T.H. and S.y.K., the Uehara Memorial Foundation, Global COE program A06 of Kyoto University, and a Grant for Excellent Graduate Schools to Division of Biological Science, Graduate School of Science, Kyoto University from MEXT, Japan. We thank Drs. E. Nakajima and Y. Tagawa for comments on the manuscript.
The authors declare no competing financial interests.
References
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell. 1994;79:377–388. doi: 10.1016/0092-8674(94)90205-4. [DOI] [PubMed] [Google Scholar]
- Boyden ES, Raymond JL. Active reversal of motor memories reveals rules governing memory encoding. Neuron. 2003;39:1031–1042. doi: 10.1016/S0896-6273(03)00562-2. [DOI] [PubMed] [Google Scholar]
- Boyden ES, Katoh A, Raymond JL. Cerebellum-dependent learning: the role of multiple plasticity mechanisms. Annu Rev Neurosci. 2004;27:581–609. doi: 10.1146/annurev.neuro.27.070203.144238. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Chiu CQ, Carroll RC. Long-term plasticity at inhibitory synapses. Curr Opin Neurobiol. 2011;21:328–338. doi: 10.1016/j.conb.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Wang H, Vicini S, Olsen RW. The gamma-aminobutyric acid type A (GABAA) receptor-associated protein (GABARAP) promotes GABAA receptor clustering and modulates the channel kinetics. Proc Natl Acad Sci U S A. 2000;97:11557–11562. doi: 10.1073/pnas.190133497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collewijn H, Grootendorst AF. Adaptation of optokinetic and vestibulo-ocular reflexes to modified visual input in the rabbit. Prog Brain Res. 1979;50:771–781. doi: 10.1016/S0079-6123(08)60874-2. [DOI] [PubMed] [Google Scholar]
- Dean P, Porrill J, Ekerot CF, Jörntell H. The cerebellar microcircuit as an adaptive filter: experimental and computational evidence. Nat Rev Neurosci. 2010;11:30–43. doi: 10.1038/nrn2756. [DOI] [PubMed] [Google Scholar]
- De Zeeuw CI, Hansel C, Bian F, Koekkoek SK, van Alphen AM, Linden DJ, Oberdick J. Expression of a protein kinase C inhibitor in Purkinje cells blocks cerebellar LTD and adaptation of the vestibulo-ocular reflex. Neuron. 1998;20:495–508. doi: 10.1016/S0896-6273(00)80990-3. [DOI] [PubMed] [Google Scholar]
- Duguid IC, Smart TG. Retrograde activation of presynaptic NMDA receptors enhances GABA release at cerebellar interneuron-Purkinje cell synapses. Nat Neurosci. 2004;7:525–533. doi: 10.1038/nn1227. [DOI] [PubMed] [Google Scholar]
- du Lac S, Raymond JL, Sejnowski TJ, Lisberger SG. Learning and memory in the vestibulo-ocular reflex. Annu Rev Neurosci. 1995;18:409–441. doi: 10.1146/annurev.ne.18.030195.002205. [DOI] [PubMed] [Google Scholar]
- Endo S, Shutoh F, Dinh TL, Okamoto T, Ikeda T, Suzuki M, Kawahara S, Yanagihara D, Sato Y, Yamada K, Sakamoto T, Kirino Y, Hartell NA, Yamaguchi K, Itohara S, Nairn AC, Greengard P, Nagao S, Ito M. Dual involvement of G-substrate in motor learning revealed by gene deletion. Proc Natl Acad Sci U S A. 2009;106:3525–3530. doi: 10.1073/pnas.0813341106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulstich BM, Onori KA, du Lac S. Comparison of plasticity and development of mouse optokinetic and vestibulo-ocular reflexes suggests differential gain control mechanisms. Vision Res. 2004;44:3419–3427. doi: 10.1016/j.visres.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Gao Z, van Beugen BJ, De Zeeuw CI. Distributed synergistic plasticity and cerebellar learning. Nat Rev Neurosci. 2012;13:619–635. doi: 10.1038/nrn3312. [DOI] [PubMed] [Google Scholar]
- Hansel C, Linden DJ, D'Angelo E. Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat Neurosci. 2001;4:467–475. doi: 10.1038/87419. [DOI] [PubMed] [Google Scholar]
- Hansel C, de Jeu M, Belmeguenai A, Houtman SH, Buitendijk GH, Andreev D, De Zeeuw CI, Elgersma Y. αCaMKII is essential for cerebellar LTD and motor learning. Neuron. 2006;51:835–843. doi: 10.1016/j.neuron.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–888. doi: 10.1038/nrn1787. [DOI] [PubMed] [Google Scholar]
- Hirano T. Long-term depression and other synaptic plasticity in the cerebellum. Proc Jpn Acad Ser B. 2013;89:183–195. doi: 10.2183/pjab.89.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H, Yamaguchi M, Sugai S, Aze Y, Narumiya S, Kakizuka A. Expanded polyglutamine in the Machado-Joseph disease protein induces cell death in vitro and in vivo. Nat Genet. 1996;13:196–202. doi: 10.1038/ng0696-196. [DOI] [PubMed] [Google Scholar]
- Ito M. Cerebellar control of the vestibulo-ocular reflex: around the flocculus hypothesis. Annu Rev Neurosci. 1982;5:275–296. doi: 10.1146/annurev.ne.05.030182.001423. [DOI] [PubMed] [Google Scholar]
- Ito M. The cerebellum: brain for an implicit self. Upper Saddle River, NJ: FT; 2012. [Google Scholar]
- Iwashita M, Kanai R, Funabiki K, Matsuda K, Hirano T. Dynamic properties, interactions and adaptive modifications of vestibulo-ocular reflex and optokinetic response in mice. Neurosci Res. 2001;39:299–311. doi: 10.1016/S0168-0102(00)00228-5. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Sun Z, Lee T, Fusco FR, Kimble TD, Meade CA, Cuthbertson S, Reiner A. A simple and sensitive antigen retrieval method for free-floating and slide-mounted tissue sections. J Neurosci Methods. 1999;93:149–162. doi: 10.1016/S0165-0270(99)00142-9. [DOI] [PubMed] [Google Scholar]
- Kano M, Hashimoto K. Activity-dependent maturation of climbing fiber to Purkinje cell synapses during postnatal cerebellar development. Cerebellum. 2012;11:449–450. doi: 10.1007/s12311-011-0337-3. [DOI] [PubMed] [Google Scholar]
- Kano M, Rexhausen U, Dreessen J, Konnerth A. Synaptic excitation produces a long-lasting rebound potentiation of inhibitory synaptic signals in cerebellar Purkinje cells. Nature. 1992;356:601–604. doi: 10.1038/356601a0. [DOI] [PubMed] [Google Scholar]
- Kashiwabuchi N, Ikeda K, Araki K, Hirano T, Shibuki K, Takayama C, Inoue Y, Kutsuwada T, Yagi T, Kang Y, Aizawa S, Mishina M. Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long-term depression in GluR δ2 mutant mice. Cell. 1995;81:245–252. doi: 10.1016/0092-8674(95)90334-8. [DOI] [PubMed] [Google Scholar]
- Katoh A, Kitazawa H, Itohara S, Nagao S. Inhibition of nitric oxide synthesis and gene knockout of neuronal nitric oxide synthase impaired adaptation of mouse optokinetic response eye movements. Learn Mem. 2000;7:220–226. doi: 10.1101/lm.7.4.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh A, Chapman PJ, Raymond JL. Disruption of learned timing in P/Q calcium channel mutants. PLoS One. 2008;3:e3635. doi: 10.1371/journal.pone.0003635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi S, Hirano T. Suppression of inhibitory synaptic potentiation by presynaptic activity through postsynaptic GABA(B) receptors in a Purkinje neuron. Neuron. 2000;27:339–347. doi: 10.1016/S0896-6273(00)00041-6. [DOI] [PubMed] [Google Scholar]
- Kawaguchi SY, Hirano T. Signaling cascade regulating long-term potentiation of GABA(A) receptor responsiveness in cerebellar Purkinje neurons. J Neurosci. 2002;22:3969–3976. doi: 10.1523/JNEUROSCI.22-10-03969.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi SY, Hirano T. Sustained structural change of GABA(A) receptor-associated protein underlies long-term potentiation at inhibitory synapses on a cerebellar Purkinje neuron. J Neurosci. 2007;27:6788–6799. doi: 10.1523/JNEUROSCI.1981-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi SY, Nagasaki N, Hirano T. Dynamic impact of temporal context of Ca2+ signals on inhibitory synaptic plasticity. Sci Rep. 2011;1:143. doi: 10.1038/srep00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kina S, Tezuka T, Kusakawa S, Kishimoto Y, Kakizawa S, Hashimoto K, Ohsugi M, Kiyama Y, Horai R, Sudo K, Kakuta S, Iwakura Y, Iino M, Kano M, Manabe T, Yamamoto T. Involvement of protein-tyrosine phosphatase PTPMEG in motor learning and cerebellar long-term depression. Eur J Neurosci. 2007;26:2269–2278. doi: 10.1111/j.1460-9568.2007.05829.x. [DOI] [PubMed] [Google Scholar]
- Kitagawa Y, Hirano T, Kawaguchi SY. Prediction and validation of a mechanism to control the threshold for inhibitory synaptic plasticity. Mol Syst Biol. 2009;5:280. doi: 10.1038/msb.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Moreau AW, Bakiri Y, Nicholson E. Plasticity of inhibition. Neuron. 2012;75:951–962. doi: 10.1016/j.neuron.2012.07.030. [DOI] [PubMed] [Google Scholar]
- Leil TA, Chen ZW, Chang CS, Olsen RW. GABAA receptor-associated protein traffics GABAA receptors to the plasma membrane in neurons. J Neurosci. 2004;24:11429–11438. doi: 10.1523/JNEUROSCI.3355-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A. The many forms and functions of long term plasticity at GABAergic synapses. Neural Plast. 2011;2011:254724. doi: 10.1155/2011/254724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles FA, Lisberger SG. Plasticity in the vestibulo-ocular reflex: a new hypothesis. Annu Rev Neurosci. 1981;4:273–299. doi: 10.1146/annurev.ne.04.030181.001421. [DOI] [PubMed] [Google Scholar]
- Mizokami A, Kanematsu T, Ishibashi H, Yamaguchi T, Tanida I, Takenaka K, Nakayama KI, Fukami K, Takenawa T, Kominami E, Moss SJ, Yamamoto T, Nabekura J, Hirata M. Phospholipase C-related inactive protein is involved in trafficking of γ2 subunit-containing GABA(A) receptors to the cell surface. J Neurosci. 2007;27:1692–1701. doi: 10.1523/JNEUROSCI.3155-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao S. Behavior of floccular Purkinje cells correlated with adaptation of horizontal optokinetic eye movement response in pigmented rabbits. Exp Brain Res. 1988;73:489–497. doi: 10.1007/BF00406606. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Miyazaki T, Kitamura K, Hashimoto K, Yanagawa Y, Obata K, Sakimura K, Watanabe M, Kano M. GABAergic inhibition regulates developmental synapse elimination in the cerebellum. Neuron. 2012;74:384–396. doi: 10.1016/j.neuron.2012.02.032. [DOI] [PubMed] [Google Scholar]
- Nugent FS, Kauer JA. LTP of GABAergic synapses in the ventral tegmental area and beyond. J Physiol. 2008;586:1487–1493. doi: 10.1113/jphysiol.2007.148098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto T, Endo S, Shirao T, Nagao S. Role of cerebellar cortical protein synthesis in transfer of memory trace of cerebellum-dependent motor learning. J Neurosci. 2011;31:8958–8966. doi: 10.1523/JNEUROSCI.1151-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh JR, Raman IM. Potentiation of mossy fiber EPSCs in the cerebellar nuclei by NMDA receptor activation followed by postinhibitory rebound current. Neuron. 2006;51:113–123. doi: 10.1016/j.neuron.2006.05.021. [DOI] [PubMed] [Google Scholar]
- Robinson DA. The use of control systems analysis in the neurophysiology of eye movements. Annu Rev Neurosci. 1981;4:463–503. doi: 10.1146/annurev.ne.04.030181.002335. [DOI] [PubMed] [Google Scholar]
- Schonewille M, Belmeguenai A, Koekkoek SK, Houtman SH, Boele HJ, van Beugen BJ, Gao Z, Badura A, Ohtsuki G, Amerika WE, Hosy E, Hoebeek FE, Elgersma Y, Hansel C, De Zeeuw CI. Purkinje cell-specific knockout of the protein phosphatase PP2B impairs potentiation and cerebellar motor learning. Neuron. 2010;67:618–628. doi: 10.1016/j.neuron.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonewille M, Gao Z, Boele HJ, Veloz MF, Amerika WE, Simek AA, De Jeu MT, Steinberg JP, Takamiya K, Hoebeek FE, Linden DJ, Huganir RL, De Zeeuw CI. Reevaluating the role of LTD in cerebellar motor learning. Neuron. 2011;70:43–50. doi: 10.1016/j.neuron.2011.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutoh F, Katoh A, Kitazawa H, Aiba A, Itohara S, Nagao S. Loss of adaptability of horizontal optokinetic response eye movements in mGluR1 knockout mice. Neurosci Res. 2002;42:141–145. doi: 10.1016/S0168-0102(01)00308-X. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Chu T, Lewin A, Bian F, Sanlioglu S, S-Crisman S, Kunsch C, Lira SA, Oberdick J. Local control of granule cell generation by cerebellar Purkinje cells. Mol Cell Neurosci. 1995;6:230–251. doi: 10.1006/mcne.1995.1019. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Ohtsuki G, Yoshida T, Fukaya M, Wainai T, Yamashita M, Yamazaki Y, Mori H, Sakimura K, Kawamoto S, Watanabe M, Hirano T, Mishina M. Enhancement of both long-term depression induction and optokinetic response adaptation in mice lacking delphilin. PLoS One. 2008;3:e2297. doi: 10.1371/journal.pone.0002297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Alphen AM, De Zeeuw CI. Cerebellar LTD facilitates but is not essential for long-term adaptation of the vestibulo-ocular reflex. Eur J Neurosci. 2002;16:486–490. doi: 10.1046/j.1460-9568.2002.02094.x. [DOI] [PubMed] [Google Scholar]
- Wang H, Bedford FK, Brandon NJ, Moss SJ, Olsen RW. GABA(A)-receptor-associated protein links GABA(A) receptors and the cytoskeleton. Nature. 1999;397:69–72. doi: 10.1038/16264. [DOI] [PubMed] [Google Scholar]
- Welsh JP, Yamaguchi H, Zeng XH, Kojo M, Nakada Y, Takagi A, Sugimori M, Llinás RR. Normal motor learning during pharmacological prevention of Purkinje cell long-term depression. Proc Natl Acad Sci U S A. 2005;102:17166–17171. doi: 10.1073/pnas.0508191102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff P, Schonewille M, Renzi M, Viltono L, Sassoè-Pognetto M, Badura A, Gao Z, Hoebeek FE, van Dorp S, Wisden W, Farrant M, De Zeeuw CI. Synaptic inhibition of Purkinje cells mediates consolidation of vestibulo-cerebellar motor learning. Nat Neurosci. 2009;12:1042–1049. doi: 10.1038/nn.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Kobayashi Y, Takemura A, Kawano K, Kawato M. Computational studies on acquisition and adaptation of ocular following responses based on cerebellar synaptic plasticity. J Neurophysiol. 2002;87:1554–1571. doi: 10.1152/jn.00166.2001. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Hashimoto K, Zimmer A, Maejima T, Araishi K, Kano M. The cannabinoid CB1 receptor mediates retrograde signals for depolarization-induced suppression of inhibition in cerebellar Purkinje cells. J Neurosci. 2002;22:1690–1697. doi: 10.1523/JNEUROSCI.22-05-01690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Katoh A, Ohtsuki G, Mishina M, Hirano T. Oscillating Purkinje neuron activity causing involuntary eye movement in a mutant mouse deficient in the glutamate receptor δ2 subunit. J Neurosci. 2004;24:2440–2448. doi: 10.1523/JNEUROSCI.0783-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]