

Abstract
Niemann-Pick type C1 (NPC1) disease is a rare neurovisceral disorder characterized by intracellular accumulation of unesterified cholesterol, sphingolipids, and other lipids in the lysosomal compartment. A deregulation of lysosomal calcium has been identified as one of the earliest steps of the degenerative process. Since adenosine A2A receptors (A2ARs) control lysosome trafficking and pH, which closely regulates lysosomal calcium, we hypothesized a role for these receptors in NPC1. The aim of this study was to evaluate the effects of the A2AR agonist CGS21680 on human control and NPC1 fibroblasts. We show that CGS21680 raises lysosomal calcium levels and rescues mitochondrial functionality (mitochondrial inner membrane potential and expression of the complex IV of the mitochondrial respiratory chain), which is compromised in NPC1 cells. These effects are prevented by the selective blockade of A2ARs by the antagonist ZM241385. The effects of A2AR activation on lysosomal calcium are not mediated by the cAMP/PKA pathway but they appear to involve the phosphorylation of ERK1/2. Finally, CGS21680 reduces cholesterol accumulation (Filipin III staining), which is the main criterion currently used for identification of a compound or pathway that would be beneficial for NPC disease, and such an effect is prevented by the Ca2+ chelator BAPTA-AM. Our findings strongly support the hypothesis that A2AR agonists may represent a therapeutic option for NPC1 and provide insights on their mechanisms of action.
Introduction
Niemann-Pick type C (NPC) disease is a rare autosomal recessive neurovisceral disorder characterized by hepatosplenomegaly, neurodegeneration, and premature death (Chang et al., 2005). Mutations in the NPC1 gene (which account for 95% of NPC cases; Vanier and Millat, 2003) affect intracellular cholesterol trafficking and homeostasis and lead to intracellular accumulation of unesterified cholesterol, sphingolipids, and other lipids in the endosomal/lysosomal system (Chang et al., 2005). Apart from cases of very early death, all NPC patients develop neurological or psychiatric symptoms such as ataxia, tremor, epilepsy, dystonia, and progressive dementia (Sévin et al., 2007).
Current therapies for NPC are very limited. Despite the many compounds that have been tested, only the drug miglustat (N-butyldeoxynojirimycin, an iminosugar that inhibits glycosphingolipid synthesis) has been approved in the EU for the treatment of progressive neurological manifestations in NPC1 patients. Since the best attainable goals of this treatment are disease stabilization or a reduced rate of disease progression (for review, see Patterson et al., 2012), the identification of new therapeutic targets remains a priority.
Although how lipid storage causes cell death in NPC1 is not yet well understood, a deregulation of lysosomal calcium ions (Ca2+) has been identified as one of the earliest pathogenetic steps (Lloyd-Evans et al., 2008). Indeed, sphingosine storage leads to a decrease in Ca2+ levels in the acidic compartment and, consequently, fusion/trafficking processes are compromised. Furthermore, drugs elevating Ca2+ levels correct the NPC1 phenotype in vitro, reduce neurological disturbances, and prolong survival in a mouse model of NPC1 disease. In addition to the lysosomal compartment, mitochondria have been described as relevant targets of the NPC1 pathogenesis. In particular, a depolarization of inner mitochondrial membrane potential (mMP) has been reported in mice carrying NPC1 genetic mutations (Yu et al., 2005).
Adenosine A2A receptors (A2ARs) are G-protein coupled receptors exerting a variety of physiological effects (Klinger et al., 2002; Fredholm et al., 2011). In the brain, A2ARs are effective modulators of neuronal damage and have attracted much interest as potential targets for neurodegenerative diseases (Popoli et al., 2002; Chen et al., 2007; Popoli, 2008).
Since A2ARs control lysosome trafficking (Carini et al., 2004) and restore altered lysosomal pH (Liu et al., 2008), which closely regulates lysosomal Ca2+ (Christensen et al., 2002), we have hypothesized a role for these receptors in NPC1.
The aim of the present study was to evaluate the effects of A2AR stimulation in NPC1 mutant cells. Primary fibroblasts from subjects carrying disease-associated mutations are considered valuable tools to identify pathological features of neurodegeneration and to investigate new therapeutic strategies (Burbulla and Krüger, 2012). Accordingly, a valuable and widely used model for studying NPC1 is represented by human fibroblasts from NPC1 patients.
We report that a selective adenosine A2AR agonist normalizes intralysosomal calcium levels, rescues mitochondrial functionality, and reduces cholesterol accumulation in human NPC1 fibroblasts.
Materials and Methods
Human fibroblasts.
Human cell lines from individuals of either sex were obtained from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research (Camden, NJ); healthy fibroblasts: GM00043, GM00275, GM00495, GM00321; NPC1 fibroblasts: GM17911, GM17924), GM17926, GM18415. Cells were grown at 37°C in 5% CO2 in Eagle's Minimum Essential Medium containing 2 mm l-glutamine, 15% fetal bovine serum, and 1 U/ml penicillin/streptomycin. Experiments were performed using at least three of the four clones for each group (healthy individuals or NPC1 patients), randomly chosen; actual number of clones used in each experiments is indicated in figure legends.
Fluorescence video-imaging.
An inverted microscope (Axiovert 135; Zeiss), equipped with 40× oil-immersion (1.35 NA; Olympus) and 20× (0.75 NA; Fluar, Zeiss) objectives, was used for fluorescence video-imaging. Excitation wavelengths were applied by means of a monochromator (Polychrome II; Till Photonics), and emission light collected by a cooled digital camera (PCO; Sensicam) and recorded on a PC. The 40× objective allowed the recording of 1280 × 1024 pixels/frame with spatial resolution of 170 nm/pixel.
Mitochondrial inner membrane potential (mMP) was measured by the “redistribution” method (Duchen et al., 2003), using the potentiometric dye tetramethylrhodamine ethyl ester perchlorate (TMRE). Cells were incubated with TMRE [30 nm, 30 min; loading saline solution (in mm): 140 NaCl, 5 KCl, 2.5 CaCl2, 1 MgCl2, 10 glucose, 10 HEPES, pH 7.4] before recording; TMRE was maintained throughout the experiment. Excitation and emission wavelengths were 535 and 560 nm, respectively.
For cytoplasmic Ca2+ analysis, cells were kept for 50 min in loading medium containing the Ca2+-sensitive dye Fura-2-AM (1 μm), washed carefully, and left for further 20–30 min before recording. Excitation and emission wavelengths were 340/380 and 510 nm, respectively. Ca2+ concentrations are represented as the ratio between the emissions at 340 and 380 nm (340/380). The lysosomal Ca2+ content was evaluated by applying the chatepsin C-specific substrate Gly-Phe β-naphthylamide (GPN, 200 μm) to the cells. Since Chatepsin C localizes only in lysosomes, the GPN cleavage causes a lysosomal-specific osmotic permeabilization and Ca2+ efflux from the organelles, confirmed by LisoTracked fluorescence decay due to the efflux of the dye during GPN application. Ca2+ increase from baseline was used as an indication of the lysosomal Ca2+ content (Srinivas et al., 2002).
Data were analyzed by Imaging Workbench 6.0 software package (Indec BioSystems), allowing emission value measurement in regions of interest (Fura-2 experiments) or along line profiles (TMRE experiments), crossing a single mitochondrion. A minimum of two peaks of amplitude were used to calculate the average amplitude of a given mitochondrion. Only clearly distinguishable single mitochondria were considered for analysis. Data were further analyzed by means of Origin Pro 7.5 software (OriginLab).
Immunofluorescence analyses.
After fixation and permeabilization, cells were incubated with 0.1% Triton X-100/3%BSA and then with antibodies against cytochrome-c oxidase (MTCO, 1:100; Abcam) or MnSOD (1:600; Stressgen); or with 10% goat serum/0.25% Triton X-100 and antibodies against Lamp2 (1:100; Abcam). After 2 h at room temperature (RT) and extensive washing, secondary antibodies Cy3R IgG or FITC IgG polyclonal or monoclonal goat antibodies (1:200; Jackson ImmunoResearch) were used. For phospho-ERK (pERK), fibroblasts were incubated with 2.5% horse serum, then with the primary antibody (1:100, 1 h, 37°C; Cell Signaling Technology) and with Alexa Fluor dye 555-conjugated secondary antibody (1:200, 45 min, 37°C; Invitrogen). For Filipin III fluorescence, cells after fixation or after being processed with primary and secondary antibodies and glycine (1.5 mg/ml, PBS) for 10 min (RT) were incubated (30 min in the dark) with 250 μg/ml Filipin III (Sigma). Nuclei were stained with Hoechst-33258 or propidium iodide (Sigma). Coverslips were mounted with Vectashield Mounting Medium (Vector Laboratories) and examined using a DM4000B fluorescence microscope equipped with DFC420C digital camera and Application Suite Software (260RI; all from Leica) for image acquisition. For cell imaging, six different fields in each coverslip were captured, and cells analyzed for their fluorescence content. Immunofluorescence quantification was conducted using NIH ImageJ software (http://rsb.info.nih.gov/ij/), and by means of threshold fluorescence intensity analysis within a region of interest, corresponding to a single cell profile. Colocalization of specific markers was analyzed by Pearson's correlation coefficient (Adler and Parmryd, 2010).
Western blot.
Cells were homogenized according to Manson et al. (2008). Proteins were separated by 8% SDS gels and transferred to PVDF membranes that were incubated (overnight, 4°C) with rabbit anti-MTCO (1:300; Abcam), anti-pERK (1:2000; Cell Signaling Technology), anti-ERK (1:1000; Cell Signaling Technology), or mouse anti-β tubulin (1:20000; Sigma). Bands were revealed by enhanced chemiluminescent substrate onto x-ray films.
Statistical analysis.
Data are expressed as means ± SEM. Statistical significance was evaluated using Student's t test or one-way ANOVA followed by Tukeys post hoc test; p < 0.05 was accepted as statistical significance. Analyses were performed using StataTm 8.1 software (Stata).
Results
Effects of A2AR stimulation on lysosomal Ca2+ content
When fibroblasts were challenged with GPN (200 μm), a slow monophasic and synchronous Ca2+ raise due to ion efflux from the organelles was observed in all cells tested (Fig. 1A). To rule out a possible Ca2+ release from ER, ER depletion was induced by thapsigargin. As Figure 1B shows, ER Ca2+ depletion did not affect the amplitude and kinetic of the GPN-induced Ca2+ signal, confirming the lysosomal source of the Ca2+ raise. The amplitude of GPN-induced Ca2+ signals was evaluated in fibroblasts from three healthy and four NPC1 subjects. In agreement with previous observations (Lloyd-Evans et al., 2008), the mean amplitude of the Ca2+ signal was lower in NPC1 than in healthy fibroblasts (Fig. 1C). To investigate the effect of A2AR stimulation on lysosomal Ca2+, GPN application was preceded by the A2AR agonist CGS21680 (10–100 nm for 3 min). CGS21680 significantly increased the GPN-induced signal in a dose-dependent manner (CGS21680 100 nm +55% vs CGS21680 10 nm, p < 0.001 according to Student's t test) and the effect was prevented by the A2AR antagonist ZM241384 (500 nm; Fig. 1D). During the application of CGS21680 alone, Ca2+ never changed, ruling out a direct effect of A2ARs on Ca2+.
Figure 1.

Effects of A2AR stimulation on lysosomal Ca2+ release in healthy and NPC1 human fibroblasts. A, GPN triggers a slow monophasic and synchronous Ca2+ raise. B, The average amplitude and kinetic of the GPN-induced Ca2+ signal is not affected by thapsigargin. C, Lysosomal Ca2+ contents are reduced in NPC1 versus control fibroblasts (n = 3 healthy and n = 4 NPC1 fibroblast clones; 181–220 fibroblasts for each single clone). D, CGS21680 significantly increased GPN-induced signal in NPC fibroblasts, an effect abolished by ZM241385 (ZM). Data are means ± SEM. *p < 0.05 versus control; **p < 0.05 versus untreated NPC1 fibroblasts.
Effects of A2AR stimulation on mitochondrial parameters (mMP and MTCO)
To test the potential capability of A2AR stimulation to rescue mitochondrial anomalies in NPC1 fibroblasts, we first evaluated mMP by means of the potential-sensitive and lipophilic dye TMRE in video-imaging fluorescence experiments. In fibroblasts from either healthy or NPC1 individuals, mitochondria were uniformly distributed, with an elongated shape oriented along the major axis of the cell. Point-like mitochondria, due to stress-induced mitochondrial fission, were not evidenced (Fig. 2A). However, when comparing the fluorescence intensity of TMRE-loaded mitochondria in healthy and NPC1 fibroblasts, the latter showed a decreased emission intensity, indicating a lower mMP (Fig. 2B). As Figure 2C shows, mMP amplitude was increased by CGS21680 (100 nm for 24 h), and ZM241384 abolished the effect, supporting the specific involvement of A2ARs.
Figure 2.

Effect of A2AR stimulation on mMP and MTCO. A, Photomicrographs showing TMRE-loaded fibroblasts from healthy (CTR, top) or NPC1 (bottom). Scale bar, 15 μm. Right, High-magnificationimages of insets a and b. B, C, Average TMRE-fluorescence intensities as a measure of mMP in healthy and NPC1 fibroblasts; cells were exposed to CGS21680 (CGS) and ZM241385 (ZM) for 24 h (n = 3 clones/group; from n = 57 to n = 362 mitochondria per each single clone). D–G, Healthy and NPC1 fibroblasts were treated for 24 h with CGS21680 with or without cotreatment with ZM241385. Cells were immunolabeled for MTCO (D) or MnSOD (G). Nuclei were labeled with propidium iodide (red) in D and Hoechst 33258 (blue) in G. Scale bar, 20 μm. Mean fluorescence intensities are shown in E and H. Data are mean ± SEM of n = 50–60 cells from each clone, as described in B. F, MTCO Western blot analysis of healthy and NPC1 fibroblasts cultured as described in C. One representative clone for each group is shown. *p < 0.05 versus control fibroblasts; **p < 0.05 versus untreated and CGS+ZM-treated NPC1 fibroblasts; ***p < 0.05 versus CGS; ***a p < 0.05 versus CGS and untreated NPC1.
The expression of the complex IV (MTCO) of the mitochondrial respiratory chain was evaluated by immunofluorescence and Western blotting (Fig. 2D–F). Immunofluorescence of MTCO revealed a similar dotted pattern, with a uniform distribution, in both healthy and NPC1 fibroblasts (Fig. 2D). The A2AR agonist did not affect MTCO expression in healthy fibroblasts (data not shown), but it restored MTCO expression to control levels in NPC1 fibroblasts, as indicated by immunofluorescence intensity analysis (Fig. 2E). Pretreatment with ZM241384 (500 nm) blocked such an effect. These results were confirmed by Western blotting (Fig. 2F). Interestingly, the expression of another important mitochondrial component, MnSOD, did not differ in NPC1 versus control fibroblasts (Fig. 2G,H).
Effects of A2AR stimulation on cholesterol accumulation and lysosomal compartmental storage
Filipin III was used to verify the presence of unesterified cholesterol and its lysosomal localization (Karten et al., 2009). Lysosomes were identified by immunodetection of Lamp2, a marker for late endosomal/lysosomal compartments. In healthy fibroblasts, Filipin III showed an even distribution, whereas in NPC1 fibroblasts the pattern was patchy, with typical vacuole-like formations, suggestive of some association with specific cellular compartments (Fig. 3A). The Lamp2 immunostaining revealed punctuate clusters with a more irregular pattern in NPC1 versus control fibroblasts (Fig. 3A). The analysis of immunofluorescence intensities indicated an increased accumulation of cholesterol in NPC1 fibroblasts (Fig. 3B), which was significantly reduced by CGS21680 (100 nm, 24 h). The A2AR agonist reduced the expression of Lamp2 in both healthy and NPC1 fibroblasts. Interestingly, the highest colocalization was found in NPC1 fibroblasts, where it was reduced to control levels by CGS21680 (Fig. 3A,B).
Figure 3.

Effect of A2AR stimulation on cholesterol accumulation in the lysosomal compartment. A, Fibroblasts from healthy (CTR) or NPC (NCP1) individuals were labeled with Filipin III (blue) and anti-Lamp2 antibody (green). Bottom panels show the colocalization of two markers. Scale bar, 10 μm. B, Mean fluorescence intensity (MFI) values of Filipin III and Lamp2 in fibroblasts treated for 24 h with CGS21680 or vehicle, and Pearson's correlation coefficients (PCC) as a measure of colocalization of the two signals (PCC = 1 maximal colocalization); data are mean ± SEM from 30 to 40 cells from three clones per group. C, Filipin III fluorescence analysis of healthy (CTR) or NPC1 fibroblasts, treated for 24 h with 100 nm CGS21680, 5 μm BAPTA-AM, or their combination. *p < 0.05 versus control fibroblasts; **p < 0.05 versus untreated NPC1 fibroblasts.
To evaluate the involvement of Ca2+ in the effect of A2AR stimulation on cholesterol distribution, control and NPC1 fibroblasts were treated with CGS21680 in the presence of the intracellular Ca2+ chelator BAPTA-AM (5 μm 1 h before and along with CGS21680). As Figure 3C shows, neither CGS21680 nor BAPTA-AM affected cholesterol accumulation (Filipin III fluorescence) in control fibroblasts. On the contrary, BAPTA-AM completely prevented the effect of CGS21680 on cholesterol accumulation in NPC1 fibroblasts.
A2AR-dependent signaling affecting lysosomal Ca2+ content
The main transduction mechanism of A2ARs is the activation of the cAMP/PKA pathway (Fredholm et al., 2011). The effects of CGS21680 on lysosomal Ca2+ content, however, did not depend on the above pathway, since they were not prevented by the specific PKA inhibitor KT5720 (4 μm). KT5720 was ineffective in healthy fibroblasts, while it even potentiated the A2AR-dependent lysosomal Ca2+ increase in NPC1 fibroblasts (Fig. 4A).
Figure 4.
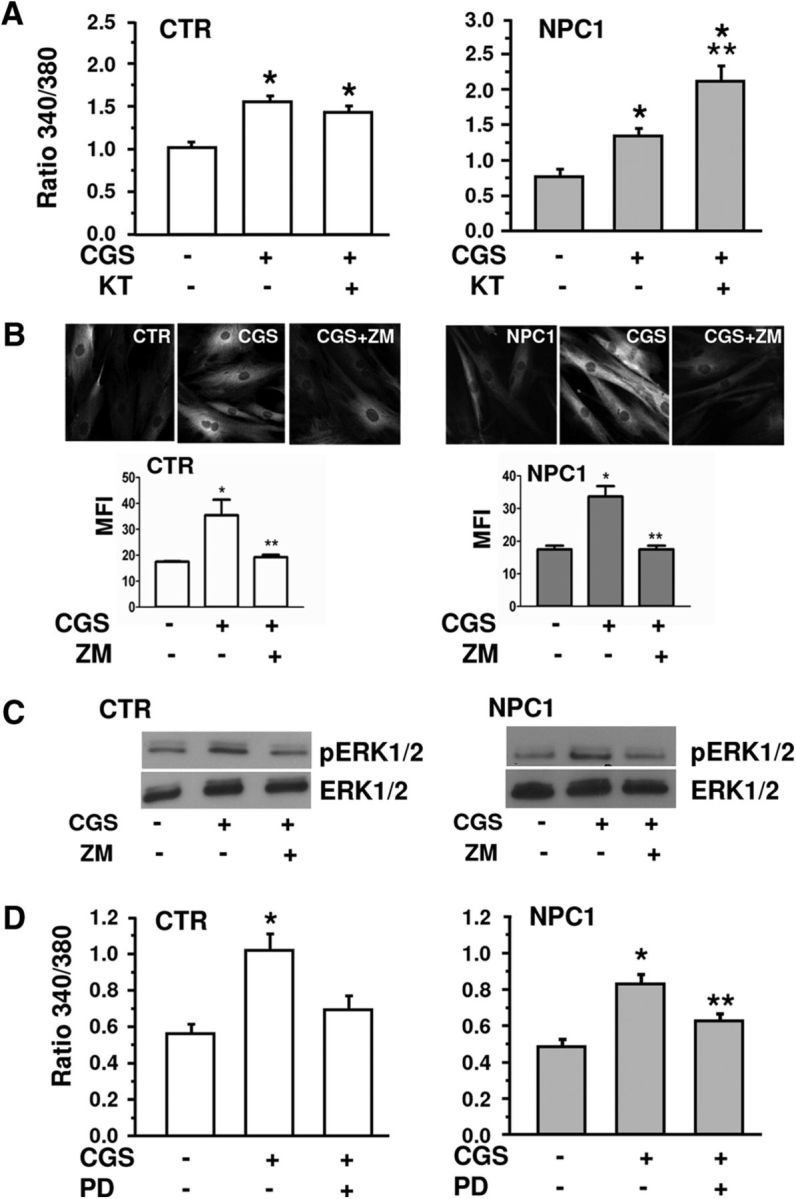
The effects of CGS21680 on lysosomal Ca2+ release are mediated by ERK1/2, but not by cAMP/PKA activation. A, Lysosomal Ca2+ release in healthy (CTR) and NPC1 fibroblasts treated with CGS21680 alone or with the PKA inhibitor KT5720 (KT; 4 μm). Data are means ± SEM from n = 38–100 cells, n = 3 clones/group. B, Representative images of pERK1/2 immunofluorescence in healthy (CTR) and NPC1 fibroblasts, treated with CGS21680 (CGS) as above and with ZM241385 (ZM), and corresponding bar chart of the mean fluorescence intensity (MFI). Means ± SEM from n = 60–105 cells, n = 3 clones/group. C, Western blot analysis for pERK1/2 and total ERK1/2 in healthy (CTR) and NPC1 fibroblasts treated as in B. D, Effect of the ERK1/2 inhibitor PD98059 (PD; 50 μm) applied as for ZM241385 on CGS-mediated Ca2+ increase in healthy and NPC1 cells. Means ± SEM from n = 53–118 cells, n = 3 clones/group. *p < 0.05 versus CTR; **p < 0.05 versus CGS.
As an alternative pathway, the mitogen-activated protein kinases (MAPKs; Klinger et al., 2002) were considered. We first evaluated, by immunofluorescence and Western blot analyses, the ability of CGS21680 to phosphorylate ERK1/2 kinases in healthy and NPC1 fibroblasts. CGS21680 (100 nm for 5 min) increased ERK1/2 phosphorylation to a similar extent in both cell types, and the effect was fully prevented by the A2AR antagonist ZM241385 (Fig. 4B,C). The involvement of ERK1/2 activation in the A2AR-dependent effects on lysosomal Ca2+ content was then assessed by applying the ERK1/2 inhibitor PD98059 (50 μm) to CGS21680-treated fibroblasts. As shown in Fig. 4D, PD98059 significantly reduced CGS21680-mediated Ca2+ increase both in healthy and NPC1 cells.
Finally, we ruled out a possible effect of A2AR activation on NPC1 expression (Gévry et al., 2003) by Western blot experiments showing that NPC1 protein level is not affected by CGS21680 (100 nm, 24 h) in both control and NPC1 fibroblasts (data not shown).
Discussion
Cholesterol has an important role in cellular function (Benarroch, 2008), and in NPC1 disease the altered intracellular cholesterol trafficking and homeostasis severely affect multiple systems, including the CNS.
Intracellular Ca2+ homeostasis deregulation has been observed in a number of primary lysosomal sphingolipid storage disorders. On this basis, Ca2+ homeostasis was recently investigated in NPC1 cell models, where a lower Ca2+ content was observed in the lysosomal compartment.
The known ability of A2AR agonists to restore altered lysosomal pH (Liu et al., 2008), which closely regulates lysosomal calcium (Christensen et al., 2002), led us to hypothesize a role for A2ARs in NPC1. In line with our hypothesis, CGS21680 normalized lysosomal Ca2+ levels in NPC1 cells. The effects were A2AR-depedent, being prevented by ZM241385.
The disruption of lipid trafficking has been associated with alterations of mMP (Yu et al., 2005). In agreement, mitochondria from NPC1 patients showed a lower mMP, although they were morphologically undistinguishable from mitochondria from healthy individuals. In NPC1 fibroblasts, the expression of the complex IV of the mitochondrial respiratory chain was also reduced. Both the above parameters of mitochondrial functionality were rescued by CGS21680 in NPC1 fibroblasts, and the effect of the agonist was prevented by A2AR blockade. Consistent with the apparent normal morphology and density of mitochondria in NPC1 fibroblasts, the expression of the mitochondrial protein MnSOD, crucial for free radical scavenging and for preserving mitochondrial integrity, was comparable in control and NPC1 fibroblasts, thus suggesting a specific deficit in the respiratory chain of NPC1 mitochondria. Importantly, CGS21680 reduced the cholesterol accumulation in late endosomal/lysosomal compartments, a hallmark of the NPC1 pathology. The incomplete colocalization of Filipin III and Lamp2 suggests the probable involvement of other vesicular systems; nonetheless, the high colocalization of the two markers in NPC1 fibroblasts indicates a main accumulation of cholesterol in the lysosomal compartment. Filipin III/Lamp2 colocalization in NPC1 fibroblasts was reduced by CGS21680 treatment, further suggesting that A2AR activation reduces cholesterol accumulation in endosomal/lysosomal compartments. The A2AR agonist also reduced the expression of Lamp2 in healthy fibroblasts, possibly as a result of its ability to reduce lysosomal pH (Liu et al., 2008), thus favoring the lysosomal natural turnover. Consistent with our hypothesis of a Ca2+-dependent endolysosomal rescuing by A2A activation, the CGS21680-induced effect on cholesterol accumulation was prevented by the Ca2+ chelator BAPTA-AM. Altogether, these observations point to Ca2+ homeostasis as central to the mechanisms by which A2A stimulation counteracts cholesterol unbalanced distribution and ameliorates NPC1 pathological phenotype.
Finally, the inability of the PKA inhibitor KT5720 to prevent CGS21680 effects weighs against the involvement of the cAMP/PKA pathway in the A2A-induced Ca2+ raise. A2ARs can select signaling pathways according to the membrane lipid microenvironment and, in particular, changes in cholesterol content blunted A2AR-dependent activation of adenylate cyclase (Charalambous et al., 2008). In our case, however, KT5720 was equally ineffective in preventing CGS21680 effects in NPC1 and control cells, thus ruling out the possibility that alterations in membrane cholesterol content could alter A2AR signaling in NPC1 fibroblasts. Interestingly, while KT5720 was ineffective in control cells, it potentiated the A2A-dependent Ca2+ increase in NPC fibroblasts. This finding suggests that the basal state of activation of the cAMP/PKA pathway is increased in NPC1 versus control cells, and that such an activation contributes to the reduced lysosomal Ca2+ content observed in NPC1 fibroblasts. In agreement with this hypothesis, Manson et al. (2008) reported that in NPC1 fibroblasts, the cAMP pathway is aberrantly increased, and that cholesterol accumulation is reversed by the inhibition of PKA. Our findings indicate that the reduction of intralysosomal Ca2+ may be a crucial step in the induction of cholesterol accumulation, possibly triggered by the aberrant cAMP/PKA pathway (Manson et al., 2008).
In addition to classical second messenger pathways, A2ARs couple to ERKs/MAPKs in a Gs protein-independent manner. In particular, ERK1/2 activation via A2ARs has been demonstrated in different cell types (Seidel et al., 1999; Klinger et al., 2002) and appears to be independent from membrane cholesterol content (Charalambous et al., 2008). Here we report that the A2AR-dependent regulation of lysosomal Ca2+ depends on the activation of ERK1/2 both in healthy and in NPC1 fibroblasts, thus providing a potential mechanism of action for the protective role of A2ARs in NPC1.
In conclusion, our results show that the stimulation of A2ARs normalizes intralysosomal Ca2+ levels through ERK1/2 activation, rescues mitochondrial functionality, and reduces cholesterol accumulation in human NPC1 fibroblasts. Since the main criterion currently used to identify a compound or pathway that would be beneficial for NPC disease is the ability to reduce Filipin III staining in NPC1 fibroblasts (Karten et al., 2009), our findings strongly support the hypothesis that A2AR agonists may represent a therapeutic option for this disease.
Footnotes
This work was supported by Istituto Superiore di Sanità Grant 530F/53.
The authors declare no competing financial interests.
References
- Adler J, Parmryd I. Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander's overlap coefficient. Cytometry A. 2010;77:733–742. doi: 10.1002/cyto.a.20896. [DOI] [PubMed] [Google Scholar]
- Benarroch EE. Brain cholesterol metabolism and neurologic disease. Neurology. 2008;71:1368–1373. doi: 10.1212/01.wnl.0000333215.93440.36. [DOI] [PubMed] [Google Scholar]
- Burbulla LF, Krüger R. The use of primary human fibroblasts for monitoring mitochondrial phenotypes in the field of Parkinson's disease. J Vis Exp. 2012;68:4228. doi: 10.3791/4228. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carini R, Castino R, De Cesaris MG, Splendore R, Démoz M, Albano E, Isidoro C. Preconditioning-induced cytoprotection in hepatocytes requires Ca(2+)-dependent exocytosis of lysosomes. J Cell Sci. 2004;117:1065–1077. doi: 10.1242/jcs.00923. [DOI] [PubMed] [Google Scholar]
- Chang TY, Reid PC, Sugii S, Ohgami N, Cruz JC, Chang CC. Niemann-Pick type C disease and intracellular cholesterol trafficking. J Biol Chem. 2005;280:20917–20920. doi: 10.1074/jbc.R400040200. [DOI] [PubMed] [Google Scholar]
- Charalambous C, Gsandtner I, Keuerleber S, Milan-Lobo L, Kudlacek O, Freissmuth M, Zezula J. Restricted collision coupling of the A2A receptor revisited: evidence for physical separation of two signaling cascades. J Biol Chem. 2008;283:9276–9288. doi: 10.1074/jbc.M706275200. [DOI] [PubMed] [Google Scholar]
- Chen JF, Sonsalla PK, Pedata F, Melani A, Domenici MR, Popoli P, Geiger J, Lopes LV, de Mendonça A. Adenosine A2A receptors and brain injury: broad spectrum of neuroprotection, multifaceted actions and “fine tuning” modulation. Prog Neurobiol. 2007;83:310–331. doi: 10.1016/j.pneurobio.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Christensen KA, Myers JT, Swanson JA. pH-dependent regulation of lysosomal calcium in macrophages. J Cell Sci. 2002;115:599–607. doi: 10.1242/jcs.115.3.599. [DOI] [PubMed] [Google Scholar]
- Duchen MR, Surin A, Jacobson J. Imaging mitochondrial function in intact cells. Methods Enzymol. 2003;361:353–389. doi: 10.1016/S0076-6879(03)61019-0. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE. International union of basic and clinical pharmacology. LXXXI. Nomenclature and classification of adenosine receptors-an update. Pharmacol Rev. 2011;63:1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gévry NY, Lalli E, Sassone-Corsi P, Murphy BD. Regulation of Niemann-Pick C1 gene expression by the 3′5′-cyclic adenosine monophosphate pathway in steroidogenic cells. Mol Endocrinol. 2003;17:704–715. doi: 10.1210/me.2002-0093. [DOI] [PubMed] [Google Scholar]
- Karten B, Peake KB, Vance JE. Mechanisms and consequences of impaired lipid trafficking in Niemann-Pick type C1-deficient mammalian cells. Biochim Biophys Acta. 2009;1791:659–670. doi: 10.1016/j.bbalip.2009.01.025. [DOI] [PubMed] [Google Scholar]
- Klinger M, Freissmuth M, Nanoff C. Adenosine receptors: G protein-mediated signalling and the role of accessory proteins. Cell Signal. 2002;14:99–108. doi: 10.1016/S0898-6568(01)00235-2. [DOI] [PubMed] [Google Scholar]
- Liu J, Lu W, Reigada D, Nguyen J, Laties AM, Mitchell CH. Restoration of lysosomal pH in RPE cells from cultured human and ABCA4(−/−) mice: pharmacologic approaches and functional recovery. Invest Ophthalmol Vis Sci. 2008;49:772–780. doi: 10.1167/iovs.07-0675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14:1247–1255. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- Manson ME, Corey DA, White NM, Kelley TJ. cAMP-mediated regulation of cholesterol accumulation in cystic fibrosis and Niemann-Pick type C cells. Am J Physiol Lung Cell Mol Physiol. 2008;295:L809–L819. doi: 10.1152/ajplung.90402.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT, Wijburg F NP-C Guidelines Working Group. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol Genet Metab. 2012;106:330–344. doi: 10.1016/j.ymgme.2012.03.012. [DOI] [PubMed] [Google Scholar]
- Popoli P. Regulation of brain functions by A2A receptors: implication for therapeutics. Curr Pharm Des. 2008;14:1466–1467. doi: 10.2174/138161208784480135. [DOI] [PubMed] [Google Scholar]
- Popoli P, Pintor A, Domenici MR, Frank C, Tebano MT, Pèzzola A, Scarchilli L, Quarta D, Reggio R, Malchiodi-Albedi F, Falchi M, Massotti M. Blockade of striatal adenosine A2A receptor reduces, through a presynaptic mechanism, quinolinic-acid induced excitotoxicity: a possible relevance to neuroprotective interventions in neurodegenerative diseases of the striatum. J Neurosci. 2002;22:1967–1975. doi: 10.1523/JNEUROSCI.22-05-01967.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel MG, Klinger M, Freissmuth M, Höller C. Activation of mitogen-activated protein kinase by the A(2A)-adenosine receptor via a rap1-dependent and via a p21(ras)-dependent pathway. J Biol Chem. 1999;274:25833–25841. doi: 10.1074/jbc.274.36.25833. [DOI] [PubMed] [Google Scholar]
- Sévin M, Lesca G, Baumann N, Millat G, Lyon-Caen O, Vanier MT, Sedel F. The adult form of Niemann-Pick disease type C. Brain. 2007;130:120–133. doi: 10.1093/brain/awl260. [DOI] [PubMed] [Google Scholar]
- Srinivas SP, Ong A, Goon L, Goon L, Bonanno JA. Lysosomal Ca2+ stores in bovine corneal endothelium. Invest Ophthalmol Vis Sci. 2002;43:2341–2350. [PubMed] [Google Scholar]
- Vanier MT, Millat G. Niemann-Pick disease type C. Clin Genet. 2003;64:269–281. doi: 10.1034/j.1399-0004.2003.00147.x. [DOI] [PubMed] [Google Scholar]
- Yu W, Gong JS, Ko M, Garver WS, Yanagisawa K, Michikawa M. Altered cholesterol metabolism in Niemann-Pick type C1 mouse brains affects mitochondrial function. J Biol Chem. 2005;280:11731–11739. doi: 10.1074/jbc.M412898200. [DOI] [PubMed] [Google Scholar]