

Abstract
The thalamus integrates and transmits sensory information to the neocortex. The activity of thalamocortical relay (TC) cells is modulated by specific inhibitory circuits. Although this inhibition plays a crucial role in regulating thalamic activity, little is known about long-term changes in synaptic strength at these inhibitory synapses. Therefore, we studied long-term plasticity of inhibitory inputs to TC cells in the posterior medial nucleus of the thalamus by combining patch-clamp recordings with two-photon fluorescence microscopy in rat brain slices. We found that specific activity patterns in the postsynaptic TC cell induced inhibitory long-term potentiation (iLTP). This iLTP was non-Hebbian because it did not depend on the timing between presynaptic and postsynaptic activity, but it could be induced by postsynaptic burst activity alone. iLTP required postsynaptic dendritic Ca2+ influx evoked by low-threshold Ca2+ spikes. In contrast, tonic postsynaptic spiking from a depolarized membrane potential (−50 mV), which suppressed these low-threshold Ca2+ spikes, induced no plasticity. The postsynaptic dendritic Ca2+ increase triggered the synthesis of nitric oxide that retrogradely activated presynaptic guanylyl cyclase, resulting in the presynaptic expression of iLTP. The dependence of iLTP on the membrane potential and therefore on the postsynaptic discharge mode suggests that this form of iLTP might occur during sleep, when TC cells discharge in bursts. Therefore, iLTP might be involved in sleep state-dependent modulation of thalamic information processing and thalamic oscillations.
Introduction
The thalamus processes and relays sensory information from the periphery to the cortex. This transfer of information is dynamically modulated by specific thalamic inhibitory circuits. The thalamic nuclei of the rat somatosensory system, the ventral posterior nucleus, and the posterior medial nucleus (PoM) are devoid of local inhibitory interneurons. Instead, GABAergic inhibition is mediated by the thalamic reticular nucleus (nRT) (Crabtree et al., 1998), the zona incerta (Barthó et al., 2002; Trageser and Keller, 2004; Lavallée et al., 2005), and the anterior pretectal nucleus (Bokor et al., 2005; Wanaverbecq et al., 2008). These inhibitory inputs suppress whisker-evoked responses and therefore control the thalamic output to the somatosensory cortex (Bosman et al., 2011). Furthermore, the reciprocal connections between the thalamic relay nuclei and the nRT facilitate the generation of rhythmic activity during sleep states (McCormick and Bal, 1997; Destexhe and Sejnowski, 2003) and can be involved in pathological oscillations during absence epilepsy (McCormick and Contreras, 2001). Although inhibitory synapses are crucial in regulating thalamic activity, little is known about activity-dependent long-term changes of synaptic strength at these synapses.
The cellular learning rules (i.e., the patterns of activity that induce changes in synaptic strength) at inhibitory GABAergic synapses in the brain are quite diverse and differ markedly from glutamatergic synapses (Castillo et al., 2011; Maffei, 2011; Méndez and Bacci, 2011; Woodin and Maffei, 2011; Kullmann et al., 2012). For example, spike-timing-dependent plasticity (STDP), a learning rule that depends on the relative timing of presynaptic and postsynaptic activity (Dan and Poo, 2006; Feldman, 2009), shows a variety of different timing dependencies at GABAergic synapses depending on interneuron cell type (Holmgren and Zilberter, 2001; Lamsa et al., 2010). In contrast to this form of Hebbian plasticity, changes in synaptic strength at GABAergic synapses can also depend solely on postsynaptic activity. Such “non-Hebbian” plasticity, which is induced by postsynaptic action potentials (APs) alone, has been described recently at inhibitory synapses onto cortical pyramidal neurons in visual cortex (Kurotani et al., 2008). The characteristic biophysical membrane properties of TC cells, which result in a bimodal mode of AP discharge depending on the membrane potential and the behavioral state, might therefore be an important determinant for changes in synaptic strength. During slow-wave sleep, TC cells are hyperpolarized and generate bursts of APs that are evoked by low-threshold Ca2+ spikes. In the awake state, TC cells are more depolarized and generate a tonic pattern of single APs with occasional bursts. These distinct patterns of activity might differentially control synaptic plasticity at TC cell synapses.
Here, we studied long-term plasticity of inhibitory inputs onto TC cells in the PoM by patch-clamp recordings and two-photon fluorescence microscopy in acute brain slices from young rats. We investigated the synaptic changes induced by different patterns of activity prevalent in the thalamus and found that the induction of inhibitory long-term potentiation (iLTP) was non-Hebbian. Postsynaptic burst activity evoked by low-threshold Ca2+ spikes was sufficient for the increase in GABAergic synaptic strength. These results suggest that GABAergic inputs can be dynamically modulated, depending on the state of the TC cell. The non-Hebbian iLTP described here might enhance the oscillatory coupling of nRT and thalamic relay nuclei during sleep states, or it might be involved in the regulation of sensory information transfer after episodes of enhanced thalamic activity.
Materials and Methods
Slice preparation.
All experiments were approved by the veterinary office of the canton of Bern, Switzerland. Thalamocortical brain slices were prepared from 17- to 21-d-old Wistar rats of either sex as described previously (Agmon and Connors, 1991). Rats were decapitated, after which the brain was quickly removed and put in ice-cold (0–4°C) artificial CSF containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 1 MgCl2, 2 CaCl2, and 25 glucose; 300-μm-thick slices were cut from the tissue block with a vibratome (Microm) and kept at 35°C for 20 min and then at room temperature until use.
Electrophysiology.
Experiments were performed at 30–34°C. For recording, slices were transferred to a recording chamber perfused with oxygenated artificial CSF. The region of the thalamic nucleus PoM was located on the basis of dark-appearing fibers ascending from nRT into ventral posterior nucleus under bright-field illumination. Whole-cell patch-clamp recordings were made from thalamocortical neurons, which were identified using infrared Dodt gradient contrast using a CCD camera (CoolSnap ES, Roper Scientific) (Nevian and Sakmann, 2004).
Recording electrodes with a resistance of 4–10 MΩ were pulled from borosilicate glass capillaries and filled with intracellular solution containing the following (in mm): 130 K-gluconate, 10 K-HEPES, 10 Na-phosphocreatine, 4 Mg-ATP, 0.3 Na-GTP, 4 NaCl, and 10 Na-gluconate (pH 7.3 with KOH) and biocytin (0.2%, Sigma-Aldrich).
A bipolar glass stimulation electrode (θ borosilicate glass capillaries, Harvard Apparatus) was placed ∼100 μm away from the patched cell soma, on the side toward the nRT. IPSPs were evoked with short (100–200 μs) extracellular stimulation pulses. Recordings were performed using a Dagan BVC-700A amplifier. Data were acquired with an ITC-16 board (Instrutech) using custom-written Igor Pro software (Wavemetrics).
After recordings, most slices were fixed and stained for precise reconstruction of the position of the cell.
iLTP induction protocols.
All experiments were performed in current-clamp mode. During experiments, small fluctuations in membrane potential were manually corrected by current injection. IPSPs with amplitudes between 1 and 4 mV were evoked by extracellular stimulation close to the soma of the patched thalamocortical relay (TC) cell. After obtaining a stable baseline for 10 min at 0.1 Hz stimulation, IPSPs were paired with two postsynaptic suprathreshold current injections at different time intervals: Δt = −25 ms, 25 ms, 500 ms, 5000 ms. For some experiments, IPSP stimulation was halted during iLTP induction. Pairings were repeated 60 times at 0.1 Hz (or at 1 Hz or 5 Hz where indicated). After pairing, IPSPs were recorded for an additional 40 min.
Experiments were discarded if the baseline IPSP peak amplitude was unstable (>20% change between first 15 and last 15 IPSP amplitudes of the baseline period) or if the membrane resistance or membrane potential changed by >30% during the experiment.
Histology.
During experiments, cells were filled with biocytin and preserved in 4% paraformaldehyde for up to 1 week. Then slices were developed with the avidin-biotin-peroxidase method and mounted on cover slides (Feldmeyer et al., 1999; Egger et al., 2008).
Ca2+ imaging.
TC cells were patched with intracellular solution supplemented with the Ca2+-insensitive dye Alexa-594 (50 μm; Invitrogen) and the Ca2+ indicator Fluo-5F (300 μm; Invitrogen). Ca2+ measurements were obtained >15 min after establishment of the whole-cell recording to allow intracellular diffusion and equilibration of the dye into the dendrites.
Two-photon excitation fluorescence was generated with a titanium-sapphire laser (Tsunami, Spectra-Physics) with laser pulses of 100 fs at a wavelength of 800 nm. The laser was coupled to a confocal scanner (TCS SP2, Leica Microsystems) attached to an upright microscope (DMLFS, Leica), equipped with a 40× objective and nondescanned detectors. Red and green fluorescence signals were separated using a dichroic mirror (560DCXR, AHF) and corresponding bandpass filters (HQ520/25 and HQ585/40, AHF) (Nevian and Sakmann, 2006).
Drug application.
During all experiments, d-AP5 (50 μm, Tocris Bioscience and Abcam) and CNQX disodium salt (10 μm, Abcam) were present in the extracellular solution to block excitatory synaptic transmission by NMDA and AMPA/kainate receptors, respectively.
Additionally, where indicated, GABAzine (3 μm, Tocris Bioscience), S-nitroso-N-acetyl-dl-penicillamine (SNAP, 200 μm, Abcam), carboxy-PTIO potassium salt (cPTIO, 50 μm, Sigma-Aldrich), 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, 10 μm, Abcam), TTX (1 μm, Alomone Labs), or nimodipine (10 μm, Alomone Labs) was added to the extracellular solution. BAPTA (10 mm, Sigma-Aldrich) was added to the intracellular solution for some experiments.
Data analysis.
Electrophysiological data were analyzed using custom-written procedures in Igor Pro (Wavemetrics). For iLTP experiments, the change in IPSP peak amplitude was evaluated 20–40 min after the end of the induction period and normalized to the baseline IPSP peak amplitude. For SNAP experiments, the change in IPSP peak amplitude was evaluated 15–25 min after washout.
The coefficient of variation (CV) of IPSP amplitudes was evaluated for the 10 min baseline before and 20–30 min after the induction of LTP. The inverse of the square CV was calculated by 1/CV2 = m2/σ2, with m being the average EPSP amplitude and σ the SD.
For imaging experiments, line scans were made through dendrites at different distances from the soma. Intracellular Ca2+ transients in response to somatically evoked APs were measured. Fluorescence traces over time for the green (Ca2+ sensitive) and red (morphological marker) channel were extracted for each line scan. Relative changes in fluorescence were expressed as ΔG/R = (G(t) − G0)/R, where G(t) is the fluorescence signal from the ROI covering the dendrite in the green channel, G0 is the basal fluorescence averaged for 50 ms before stimulation, and R is the averaged fluorescence of the red channel.
Statistical analysis.
Statistical analysis was performed with paired Student's t test in Excel (Microsoft). Statistical significance was asserted for p < 0.05. Data are presented as mean ± SEM.
Results
LTP of inhibitory synapses onto relay cells is non-Hebbian
To study synaptic plasticity of inhibitory GABAergic synapses onto thalamocortical (TC) relay cells, we first investigated the properties of IPSPs onto these neurons. Whole-cell current-clamp recordings were obtained from TC cells. A depolarizing current step from the resting membrane potential of −60 mV resulted in a characteristic burst discharge of APs on top of a low-threshold Ca2+ spike (Fig. 1B). Inhibitory synapses were isolated in all experiments by bath application of the glutamate receptor antagonists d-AP5 (50 μm) and CNQX (10 μm) to block NMDA and AMPA/kainate receptors, respectively. IPSPs were evoked by extracellular stimulation close to the soma (Fig. 1A). Stimulation strength and position of the stimulation electrode were adjusted so that IPSPs with peak amplitudes between −4 and −1 mV were evoked at membrane potentials of −60 mV. Most inhibitory synaptic inputs showed a biphasic IPSP, suggesting the presence of both GABAA and GABAB receptors at these synapses (Fig. 1C). Indeed, all inhibitory synaptic inputs contained a GABAA component with a fast rise time and a reversal potential near −75 mV, consistent with the predicted chloride reversal potential under our experimental conditions (Fig. 1F,G). Blocking the GABAA component by bath application of the GABAA receptor antagonist GABAzine (3 μm) abolished this fast component, revealing a slow, delayed onset and long-lasting GABAB component (Fig. 1C,D). The GABAB component reached its maximal hyperpolarization ∼230–260 ms after stimulus onset. At this time point, the GABAA component had already decayed to baseline (Fig. 1C). Therefore, we measured the GABAB component of the IPSPs at this time point and normalized it to the peak GABAA component to define the GABAB-to-GABAA ratio (Fig. 1E). Analysis of this ratio revealed that the majority of synapses contained a GABAB component (33 of35 synapses). The GABAB component showed a reversal potential of −80 mV, suggesting the activation of a potassium conductance. Our results are in agreement with previous reports, which showed that the majority of inhibitory inputs onto TC cells in the somatosensory thalamic nuclei contain a slow GABAB component (Landisman and Connors, 2007).
Figure 1.
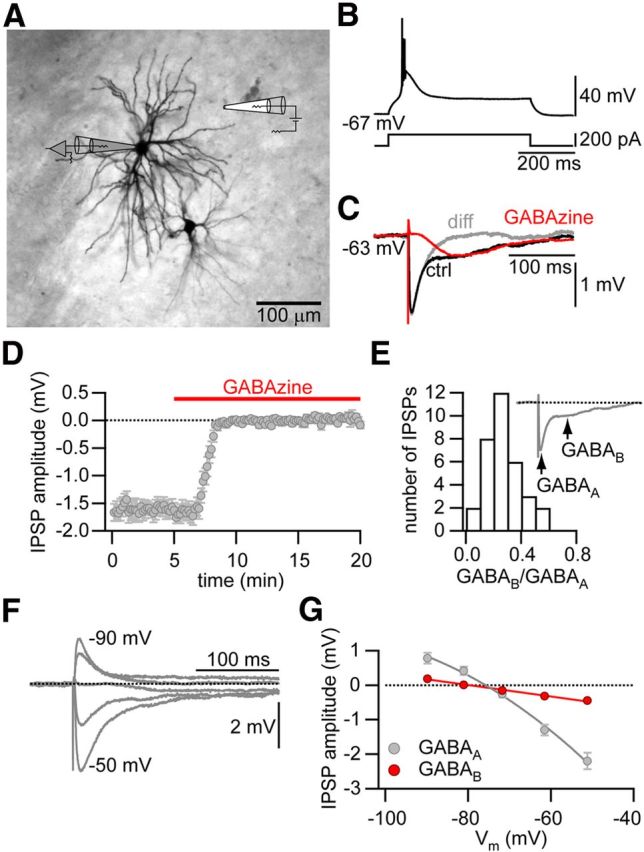
Characterization of GABAergic synapses onto thalamocortical relay cells. A, Schematic representation of the recording configuration. Bright-field image of a biocytin-stained thalomocortical cell in the thalamic nucleus PoM. The recording electrode is indicated in gray and the extracellular stimulation electrode in white. B, Suprathreshold response of the neuron shown in A to a 600-ms-long current injection. C, IPSPs during baseline (black, GABAA and GABAB component) and after bath application of GABAzine (red, isolated GABAB component). Gray trace represents the difference between the IPSP during baseline and in the presence of GABAzine, revealing the isolated GABAA component of the IPSP. D, Averaged IPSP peak amplitude over time. After 5 min baseline recording, GABAzine was applied to the bath. E, Histogram of the distribution of the ratio of the peak amplitude of the GABAB component to the peak amplitude of the GABAA component. Inset, Time points at which the corresponding amplitudes were measured. F, IPSPs recorded at different membrane potentials from −90 mV in steps of 10 mV. G, Plot of the averaged IV curve of the GABAA (black) and GABAB (red) component of the IPSPs.
Next, we investigated whether GABAergic synapses onto TC cells can undergo long-term changes in synaptic efficacy. Synaptic learning rules at GABAergic synapses are quite diverse. The same correlated patterns of presynaptic and postsynaptic activity can lead to different synaptic changes depending on the inhibitory cell types studied. One form of synaptic plasticity is STDP, in which the sign and magnitude of the long-term synaptic change depend on the order and timing of presynaptic and postsynaptic activity (Bi and Poo, 1998; Sjöström et al., 2008). STDP has been well characterized at glutamatergic synapses, but not so much is known about this form of plasticity at inhibitory synapses. To test whether GABAergic synapses onto TC cells undergo STDP, we paired IPSPs with bursts of postsynaptic APs at different time intervals (Nevian and Sakmann, 2006; Frey et al., 2009). After recording IPSPs at 0.1 Hz for a 10 min baseline period, individual IPSPs were paired with two suprathreshold current injections at 100 Hz for 10 min (60 repetitions at 0.1 Hz; Fig. 2A,B). The current injections generated bursts of 2–5 APs on top of a low-threshold Ca2+ spike. After this induction period, IPSPs were recorded for another 40 min to monitor changes in synaptic strength. The membrane potential of the TC cells was held constant at ∼−60 mV for the entire time course of the experiment. A postsynaptic burst following synaptic stimulation by 25 ms (Δt = 25 ms) resulted in LTP of the IPSP amplitude (change in IPSP amplitude, 1.44 ± 0.12, p < 0.05, n = 10; Fig. 2C,D). This potentiation was not the result of the prolonged whole-cell recording configuration because recording IPSPs alone over time did not result in a change in IPSP amplitude (change in IPSP amplitude, 1.04 ± 0.06, p = 0.51, n = 8; Fig. 3A). STDP at glutamatergic synapses is often characterized by the induction of long-term depression when the order of presynaptic and postsynaptic activity is reversed. However, when the postsynaptic burst preceded the IPSP (Δt = −25 ms), IPSP amplitudes were potentiated as well (change in IPSP amplitude, 1.51 ± 0.15, p < 0.002, n = 6; Fig. 2D,E). Substantially increasing the time window between synaptic stimulation and postsynaptic burst firing during the pairing period still resulted in the potentiation of the IPSP amplitude (Δt = 500 ms, 1.47 ± 0.19, p < 0.05, n = 7; and Δt = 5000 ms, 1.45 ± 0.16, p < 0.05, n = 8; Fig. 2D,E). Overall, the induction of iLTP was a robust phenomenon independent of the time interval, as 27 cells from the 31 cells tested in this set of experiments showed a significant increase in inhibitory synaptic strength.
Figure 2.

Non-Hebbian iLTP in thalamic relay neurons. A, Schematic recording configuration. Image represents a bright-field image of a biocytin-stained thalomocortical cell in the thalamic nucleus PoM. Gray represents recording electrode. Recording electrode is indicated in gray, extracellular stimulation electrode in white. Recordings from this cell are shown in B and C. B, Top, Illustration of the pairing protocol. Two postsynaptic current injections at 100 Hz evoking two APs on top of a low-threshold Ca2+ spike were paired with an IPSP (Δt = 25 ms). Bottom, Averaged IPSP during baseline (ctrl, black) and after iLTP induction (after, red). C, Example experiment with IPSP peak amplitude, membrane potential (Vm), and input resistance (Ri) plotted over time. Gray area represents pairing period (Δt = 25 ms). Red bars indicate time period for averaging IPSP amplitude. IPSP amplitudes are displayed as negative values. D, Normalized and averaged IPSP amplitudes over time. iLTP was induced with time delays of Δt = 25 ms, Δt = −25 ms, and Δt = 5 s. Insets, Representative averaged IPSPs during baseline (ctrl, black) and after iLTP induction (after, red). Positive time intervals (Δt) indicate the pairing of IPSPs with a following burst, whereas negative Δt indicate pairing of IPSPs with a preceding burst. E, Normalized change in IPSP amplitude after iLTP induction versus time delay Δt. *Significant LTP (p < 0.05).
Figure 3.
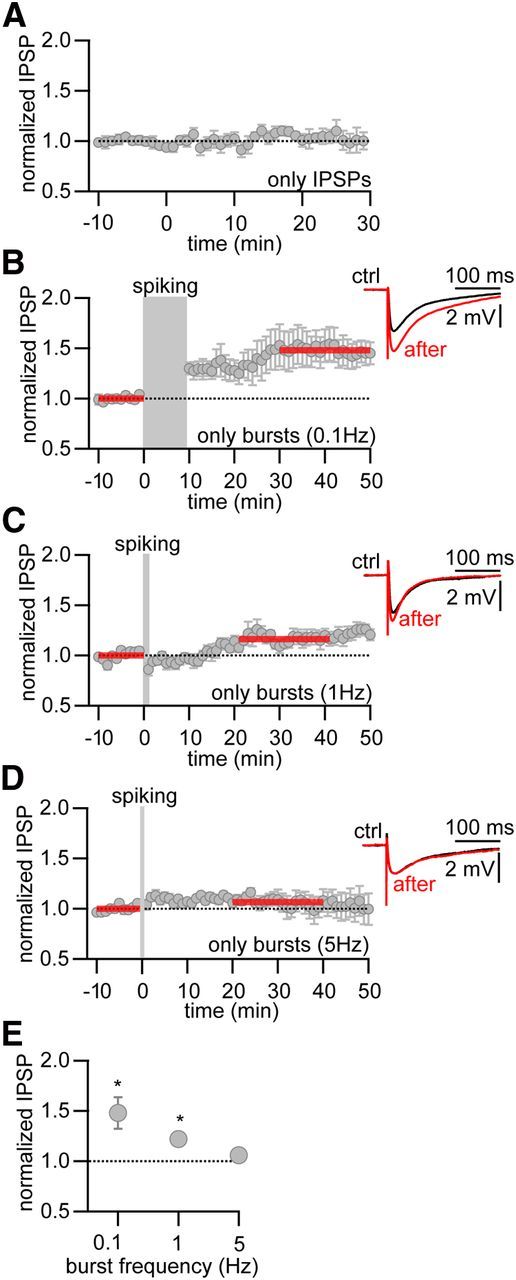
iLTP can be induced by postsynaptic bursting alone. A, Normalized and averaged IPSP amplitudes over time without postsynaptic stimulation. B–D, Normalized and averaged IPSP amplitudes over time. iLTP was induced with postsynaptic stimulation alone (2 current injections at 100 Hz, gray area), in the absence of presynaptic stimulation at different stimulation frequencies. B, 0.1 Hz, 10 min. C, 1 Hz, 1 min. D, 5 Hz, 12 s. Red bars indicate time period used for determining the amount of LTP. Insets, Representative averaged IPSPs during baseline (ctrl, black) and after iLTP induction (after, red). E, Plot of averaged normalized IPSP amplitudes versus burst frequency. *Significant LTP (p < 0.05).
These results suggest that potentiation of GABAergic inputs onto TC cells does not depend on the coincident timing between presynaptic and postsynaptic activity. Therefore, we tested whether presynaptic activity was required at all for the induction of iLTP. For these experiments, presynaptic stimulation was halted during the plasticity induction phase. Postsynaptic bursting alone repeated at 0.1 Hz for 10 min resulted in an increase in IPSP amplitude (change in IPSP amplitude, 1.48 ± 0.16, p < 0.05, n = 9; Fig. 3B,D). Increasing the repetition frequency of postsynaptic bursting to 1 Hz, while keeping the number of bursts unchanged, also resulted in potentiation of the IPSP amplitude (change in amplitude, 1.22 ± 0.06, p < 0.05, n = 11; Fig. 3C). On the other hand, a repetition frequency of 5 Hz resulted in no change in the IPSP amplitudes (1.06 ± 0.06, p = 0.25, n = 7; Fig. 3D,E). In conclusion, our results so far indicate that postsynaptic activity alone is sufficient for the potentiation of inhibitory inputs onto TC cells, thereby suggesting that this form of iLTP is non-Hebbian. Furthermore, iLTP seems to depend on the frequency of postsynaptic stimulation.
iLTP is Ca2+-dependent
LTP is triggered by postsynaptic Ca2+ influx at most synapses in the CNS. To test whether Ca2+ influx is necessary for LTP at inhibitory synapses onto TC cells, we infused the Ca2+ chelator BAPTA (10 mm) into the cells via the patch pipette. For the induction of iLTP, we paired an IPSP with a following postsynaptic burst at Δt = 25 ms. In the presence of intracellular BAPTA, LTP was absent (change in IPSP amplitude, 0.99 ± 0.13, p = 0.89, n = 9; control vs BAPTA; p < 0.05; Fig. 4A,B), thereby showing that Ca2+ plays a crucial role in the induction of iLTP.
Figure 4.
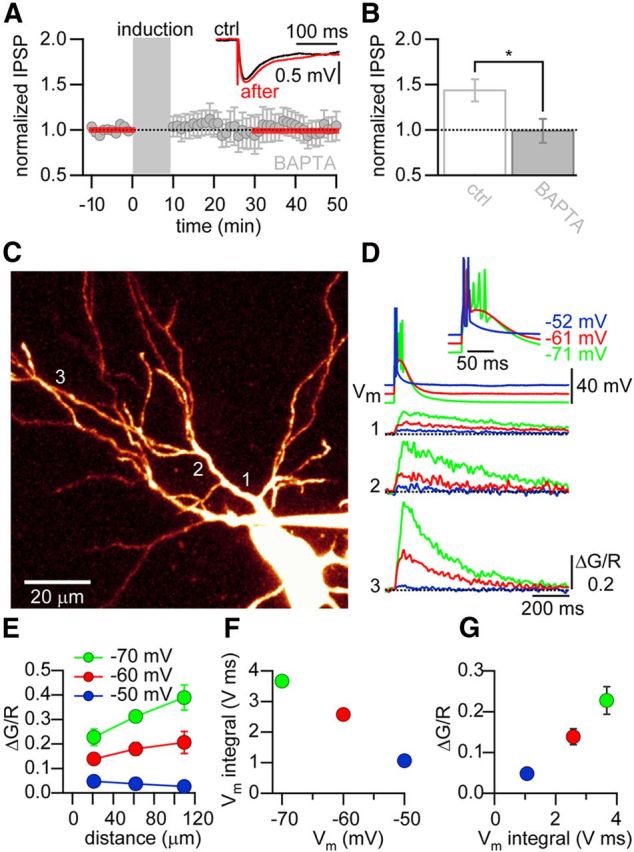
Postsynaptic Ca2+ signals are crucial for iLTP and are membrane potential-dependent. A, Normalized and averaged IPSP amplitudes for cells loaded with the Ca2+ chelator BAPTA (10 mm). Gray area represents pairing period (Δt = 25 ms). Inset, Representative averaged IPSPs during baseline (ctrl, black) and after iLTP induction (after, red). B, Bar graph summary of experiments shown in A. *Significant difference (p < 0.05). C, Two-photon image of a thalamocortical neuron filled with Alexa-594. Imaging locations used in D are indicated with numbers. D, Top, AP firing of the neuron shown in C in response to two current injections at 100 Hz, at different membrane potentials: −52 mV (blue) resulted in tonic firing (single spikes); −61 mV (red) and −71 mV (green) resulted in a burst of APs on top of a low-threshold Ca2+ spike. Inset, Same traces, but magnified. Bottom, Ca2+ transients (ΔG/R) evoked by stimulation at different membrane potentials (same as above). Traces are recorded at different locations: close to the soma (1), at intermediate distance (2), and far from the soma (3). E, Averaged and pooled Ca2+ transients in response to AP firing from the different membrane potentials, plotted against distance from soma. Experiments were pooled in three distance categories (<60, 60–100, and >100 μm). F, Integral of the response to two current injections at 100 Hz plotted against the membrane potential Vm. G, Integral of the response to two current injections at 100 Hz plotted against ΔG/R.
The main source of Ca2+ influx into TC cell dendrites is presumably through T-type voltage-dependent Ca2+ channels (VDCCs) (Errington et al., 2010). These channels are to a large extent inactivated at depolarized membrane potentials and can most efficiently be recruited at hyperpolarized membrane potentials. In the latter case, T-type channels contribute to the generation of a low-threshold Ca2+ spike. The voltage dependence of the inactivation properties of the T-type VDCCs is responsible for the typical tonic AP discharge pattern at depolarized membrane potentials and burst firing at hyperpolarized membrane potentials observed in TC cells. It has been suggested that these discharge modes result in differential dendritic Ca2+ signaling (Crandall et al., 2010; Errington et al., 2010). Because the induction of iLTP requires postsynaptic Ca2+ influx, we measured Ca2+ transients evoked by postsynaptic APs along the dendrites of TC cells with two-photon excitation Ca2+ imaging. Cells were loaded with the green Ca2+ indicator Fluo-5F (300 μm) and the red dye Alexa-594 (50 μm) for visualization of the dendritic arborization (Fig. 4C). Two suprathreshold current injections at 100 Hz were applied at different membrane potentials to evoke the different firing modes of TC cells. From an initial membrane potential of −52 mV, each current injection resulted in the generation of a single AP reminiscent of tonic firing (Fig. 4D). In contrast, current injections from initial membrane potentials of −61 mV or −71 mV led to a low-threshold Ca2+ spike characterized by a slow and long-lasting depolarization that evoked a burst of APs at high frequency (Fig. 4D). Tonic firing of two APs evoked only small Ca2+ transients in the dendrites that decreased with the distance from the soma (Fig. 4D,E). In contrast, burst firing from a more hyperpolarized membrane potential caused a significantly larger Ca2+ influx at all dendritic locations. Under these conditions, the peak amplitude of the Ca2+ transients even increased with the distance from the soma (Fig. 4D,E).
These results suggest that, depending on the membrane potential and the corresponding firing mode of the TC cell, the somatic depolarization differentially propagates into the dendrites resulting in distinct dendritic Ca2+ signals. A similar behavior has been observed in the dendrites of TC cells of the dorsal lateral geniculate nucleus (Errington et al., 2010) and in the dendrites of thalamic reticular neurons (Crandall et al., 2010), suggesting that this might be a general feature of thalamic cells.
The TC cell discharge patterns at the different resting membrane potentials was quantified by calculating the integral of the membrane potential during postsynaptic AP firing (Fig. 4F). The integral increased with decreasing initial membrane potential. This behavior illustrated the growing contribution of the low-threshold Ca2+ spike to the membrane depolarization at hyperpolarized potentials. The integral was linearly correlated with the peak amplitude of the evoked Ca2+ transients at the proximal dendritic location (Fig. 4G).
To separate the contribution of the back-propagating APs to the dendritic Ca2+ transients from the contribution of the low-threshold Ca2+ spike, we measured the Ca2+ transients at different membrane potentials under control conditions and after bath application of TTX (1 μm; Fig. 5). To eliminate differences in the Ca2+ signals evoked by a different number of APs induced at each membrane potential, we matched the number of APs at −50 mV to the number of APs evoked by the low-threshold Ca2+ spike at hyperpolarized potentials in the same cell under control conditions (range, 2–5 APs). After wash-in of TTX, APs were abolished at −50 mV. This was accompanied by a reduction of the peak Ca2+ transient amplitude to 8 ± 4% of control (p < 0.01; n = 9). At more hyperpolarized potentials, the low-threshold Ca2+ spike itself was unaffected by TTX, but the APs riding on the Ca2+ spike were blocked by TTX. This was accompanied by a reduction of the Ca2+ transients to 68 ± 7% (p < 0.05; n = 10) and to 70 ± 4% (p < 0.01; n = 10) for events evoked from membrane potentials of −62 mV and −75 mV, respectively (Fig. 5B,D). The reduction in the Ca2+ transient peak amplitude by TTX was completely accounted for by the missing contribution of the AP-evoked Ca2+ influx, as illustrated by the fact that the linear sum of the Ca2+ transients evoked at −50 mV in control and at hyperpolarized potentials in TTX equaled the Ca2+ transients at hyperpolarized potentials under control conditions (Fig. 5C). This result suggests that the depolarization caused by the low-threshold Ca2+ spike does not activate all dendritic VDCCs.
Figure 5.

iLTP requires Ca2+ influx through L-type VDCCs. A, Two-photon image of a thalamocortical relay cell filled with Alexa-594. Arrow indicates imaging location used in B. B, Membrane potential traces (left) and the corresponding Ca2+ transients (right) for different membrane potentials and after consecutive bath application of TTX (black) and nimodipine (gray). C, Ca2+ transients evoked at −75 mV (green) and −65 mV (red) overlaid with the sum of the Ca2+ transients after bath application of TTX and the control Ca2+ transient evoked by tonic AP firing at −52 mV (black), indicating that the effect of TTX can be completely accounted for by the lack of AP evoked Ca2+ influx. D, Summary of the effect of TTX on the Ca2+ transients evoked from the different membrane potentials. E, Summary of the effect of subsequent bath application of nimodipine. * Significant difference (p < 0.05). F, Normalized and averaged IPSP amplitudes over time in the presence of nimodipine showing that iLTP requires L-type VDCCs. Gray bar indicates pairing period (Δt = 5 s). Inset, Representative averaged IPSPs during baseline (ctrl, black) and after pairing (after, red).
The Ca2+ transient evoked by a low-threshold Ca2+ spike has been suggested to be mediated by T-type VDCCs (Errington et al., 2010). However, it is very likely that other subtypes of VDCCs are also being activated by the underlying membrane depolarization (Suzuki and Rogawski, 1989). We found that bath application of the L-type VDCC blocker nimodipine (10 μm) reduced the low-threshold Ca2+ spike evoked Ca2+ transient in the presence of TTX to 73 ± 5% (p < 0.05; n = 7) at −75 mV and to 49 ± 4% (p < 0.01; n = 7) at −62 mV (Fig. 5B,E). This shows that the relative contribution of L-type VDCCs to the Ca2+ transient was significantly greater at −62 mV compared with −75 mV (p < 0.01; n = 7). Comparing the Ca2+ transients after bath application of both TTX and nimodipine to the control condition without any blockers revealed that at −75 mV only 49 ± 6% of the peak Ca2+ transient amplitude could be accounted for by T-type VDCCs. Ca2+ transients evoked from −62 mV had an even smaller T-type component of 34 ± 6% (p < 0.001, n = 7). Interestingly, the area under the curve of the membrane depolarization caused by the low-threshold Ca2+ spike was significantly reduced in the presence of nimodipine compared with TTX (p < 0.05; n = 6). On the other hand, the initial rising phase of the depolarization was not affected by nimodipine (Fig. 5B). These findings suggest that the low-threshold Ca2+ spike is initiated by the opening of T-type VDCCs and that the resulting depolarization subsequently recruits high-voltage-activated Ca2+ channels (L-type), which in turn contribute to the Ca2+ influx and the duration of the depolarization. A similar sequence of events can be observed during the generation of dendritic Ca2+ spikes in the distal apical dendrite of pyramidal neurons in the somatosensory cortex (Pérez-Garci et al., 2013).
As Ca2+ influx through L-type VDCCs contributed a large fraction to the Ca2+ signal in TC cell dendrites and L-type VDCCs mediate the induction of LTP at many synapses, we tested whether L-type VDCCs were required for iLTP. Bath application of nimodipine had no effect on basal synaptic transmission (IPSPnimodipine/IPSPcontrol = 1.13 ± 0.09, n = 2). In the presence of nimodipine, pairing a burst with an IPSP (Δt = 5 s, −60 mV, 0.1 Hz) blocked the induction of iLTP completely (0.88 ± 0.05, n = 5; Fig. 5F).
The requirement of postsynaptic Ca2+ influx for the induction of iLTP, together with the membrane potential dependence of this Ca2+ influx into the dendrites of TC cells, suggested that iLTP might depend on the membrane potential during the induction period. To test this hypothesis, we varied the membrane potential during the 10 min induction period (Δt = 5 s). In agreement with our hypothesis, induction at membrane potentials of −62 mV (1.45 ± 0.16, p < 0.05, n = 8; Fig. 6B) and of −75 mV (1.21 ± 0.10, p < 0.05, n = 11; Fig. 6C) led to a potentiation of GABAergic synaptic transmission. In contrast, no potentiation was induced at a membrane potential of −50 mV (0.83 ± 0.13, p = 0.23, n = 7; Fig. 6A,D). These results indicate that iLTP depends on the membrane potential and therefore on the firing mode of the TC cell during the induction period.
Figure 6.
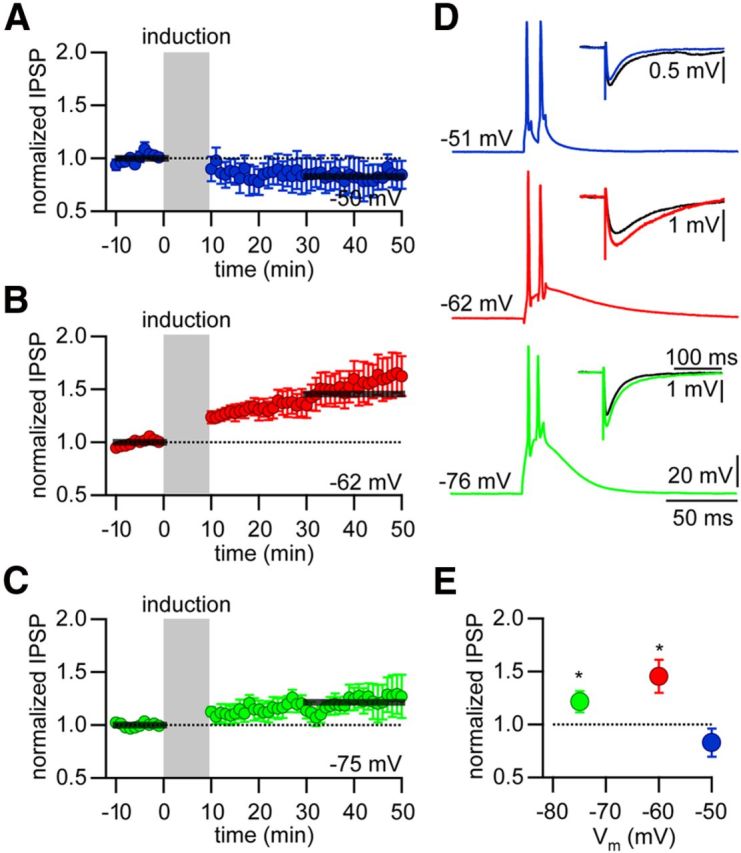
iLTP depends on membrane potential. A–C, Normalized and averaged IPSP peak amplitudes over time. iLTP induction (Δt = 5 s, gray area) was performed at different membrane potentials. A, −50 mV (blue). B, −62 mV (red). C, −75 mV (green). Black bars indicate time period used for determining amount of LTP. D, Representative examples of AP firing during iLTP induction at the different membrane potentials Vm indicated. Insets, Representative averaged IPSPs during baseline (ctrl) and after iLTP induction (after). E, Summary plot of changes in IPSP amplitude versus membrane potential Vm during the induction period. *Significant LTP (p < 0.05).
iLTP requires retrograde nitric oxide (NO) signaling
Finally, we investigated the cellular signaling mechanism that mediated the induction of iLTP downstream from postsynaptic Ca2+ influx. We analyzed the paired pulse ratio (PPR) before and after potentiation induced by pairing an IPSP with a burst at Δt = 25 ms. Under control conditions, two IPSPs evoked at 50 Hz showed short-term depression of their peak amplitudes (PPRbefore = 0.437 ± 0.025). Induction of iLTP significantly increased the degree of short-term depression (PPRafter = 0.381 ± 0.041, normalized change in PPR: 0.860 ± 0.050, p < 0.05, n = 9; Fig. 7A). Accordingly, the ratio of the coefficient of variation (CV−2after/CV−2before) scaled linearly with the change in inhibitory synaptic strength (Fig. 7B). This suggests that iLTP is expressed as an increase in presynaptic release probability. The notion that iLTP is expressed presynaptically was also reflected in an equal potentiation of the GABAA and GABAB component of the IPSPs after pairing. If iLTP was expressed postsynaptically, by a change in the number or properties of postsynaptic GABAA receptors, the GABAB-to-GABAA ratio would be expected to change. However, this ratio remained constant, therefore suggesting that iLTP is expressed as an increase in presynaptic release probability (Fig. 7C).
Figure 7.
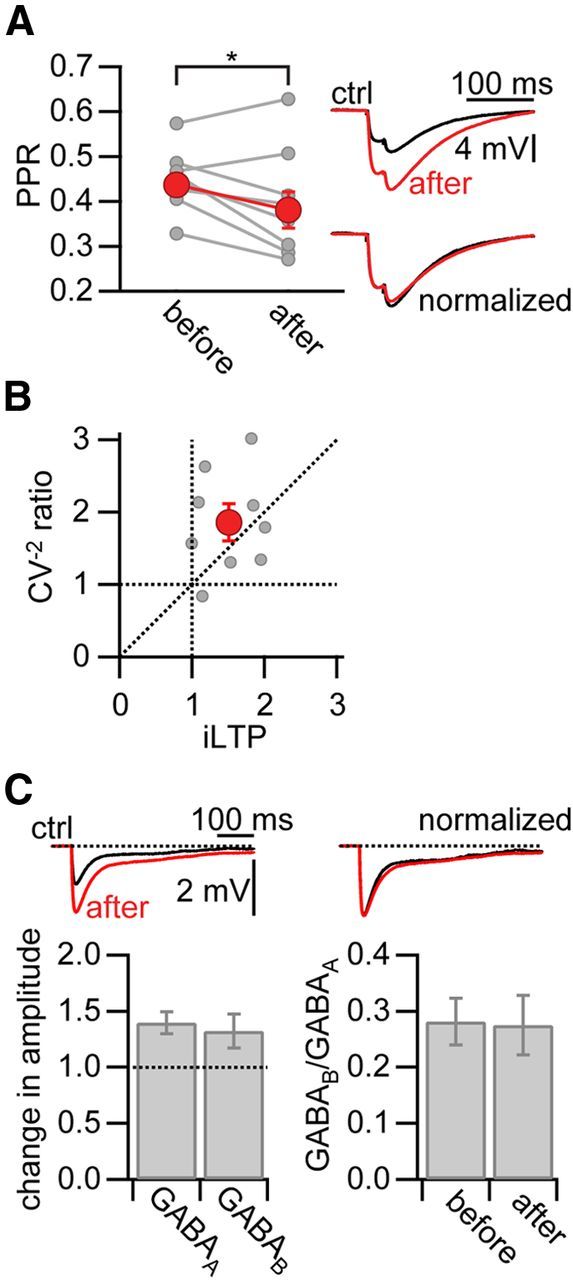
iLTP is expressed presynaptically. A, Left, PPR (amplitude of IPSP1/ IPSP2, 50 Hz) during baseline and after iLTP induction (Δt = 25 ms; data shown in Fig. 2D). Gray circles represent individual experiments. Red circles represent the averaged PPR. Right, Representative averaged IPSPs during baseline (ctrl, black) and after iLTP induction (after, red). *Significant difference (p < 0.05). B, Plot of the ratio of the coefficient of variation (CV−2) after the expression of LTP to the coefficient of variation during baseline versus the change in IPSP amplitude (iLTP). C, Left, Average change in IPSP amplitude for the GABAA and GABAB component (Δt = 25 ms). Right, Average of the GABAB-to-GABAA ratio before and after pairing.
The postsynaptic induction, but presynaptic expression, of iLTP indicates that a retrograde messenger is required to signal from the postsynaptic to the presynaptic site. NO acts as the retrograde messenger in many forms of presynaptically expressed LTP (Castillo et al., 2001; Hardingham and Fox, 2006). Therefore, we tested whether iLTP at inhibitory synapses onto TC cells required NO signaling. Bath application of the NO donor SNAP for 10 min resulted in an immediate continuous increase of the IPSP amplitude. IPSPs remained potentiated after washout of SNAP for the rest of the experiment. The IPSP amplitude was potentiated by a factor of 1.53 ± 0.17 (15–25 min after washout; p < 0.05, n = 8; Fig. 8A). This experiment suggested that NO can indeed induce a long-term increase in IPSP amplitude. To test whether NO was required for iLTP, we performed experiments in the presence of the NO scavenger cPTIO. IPSPs and postsynaptic bursting were evoked with a time delay of Δt = 5 s. In the presence of cPTIO, no potentiation was observed (0.97 ± 0.05, p = 0.57, n = 7; Fig. 8B), suggesting that extracellular NO is necessary for iLTP induction. The presynaptic target of NO might be the NO-dependent guanylyl cyclase. Blocking the activity of guanylyl cyclase by incubating the brain slice with ODQ for the time course of the experiment also blocked the induction of iLTP (1.03 ± 0.06, p = 0.58, n = 7; Fig. 8C,D). These results indicate that iLTP requires retrograde signaling by NO.
Figure 8.
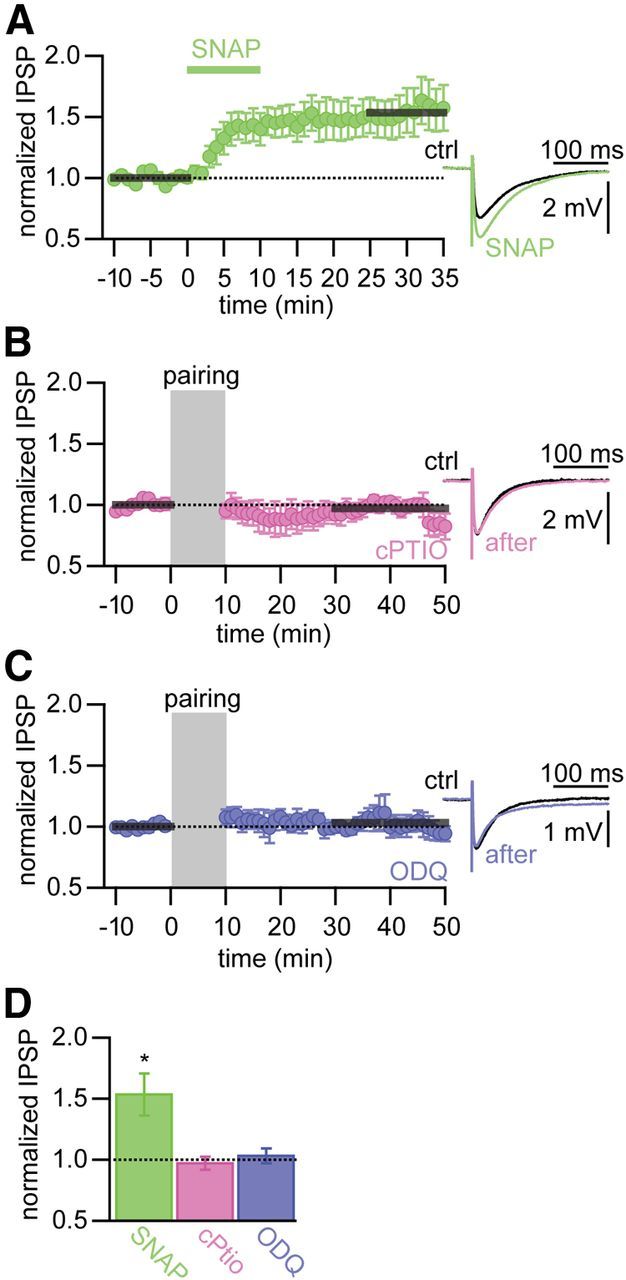
iLTP requires NO signaling. A, Normalized and averaged IPSP amplitudes over time. After a 10 min baseline, the NO donor SNAP was bath applied for 10 min. Right, Representative averaged IPSPs during baseline (ctrl, black) and after SNAP application (green). B, C, Left, Normalized and averaged IPSP amplitudes over time for iLTP induction in the presence of the NO scavenger cPTIO (pink; B) and the NO-dependent guanylyl cyclase inhibitor ODQ (blue; C). Gray area represents iLTP induction period (Δt = 5 s). Gray bars indicate time period used for determining amount of iLTP. Right, Representative averaged IPSPs during baseline (ctrl, black) and iLTP induction (in color). D, Summary of changes in IPSP amplitude for the different pharmacological manipulations in A–C. *Significant LTP (p < 0.05).
Discussion
Our results show that inhibitory inputs onto TC cells can undergo non-Hebbian LTP upon specific postsynaptic activity patterns. iLTP requires low-threshold Ca2+ spike bursting in TC cells. Therefore, iLTP is only induced if the cell is activated from a hyperpolarized membrane potential. The molecular mechanism underlying this form of iLTP requires postsynaptic Ca2+ influx triggering the synthesis of NO. NO acts as a retrograde messenger onto presynaptic inhibitory terminals and stimulates the NO sensitive guanylyl cyclase, leading to the synthesis of cGMP. cGMP is known to activate protein kinases that modify the synaptic release machinery, thereby leading to LTP.
Molecular mechanism of iLTP at thalamic GABAergic synapses
Induction of iLTP requires postsynaptic Ca2+ signaling. Bursts originating from hyperpolarized potentials from ≤−60 mV result in Ca2+ elevations in the dendrites that increase with the distance from the soma. In contrast, APs evoked from depolarized potentials do not propagate well into the dendrites of TC cells, resulting in only a modest increase in dendritic Ca2+. A similar dendritic Ca2+ profile has been described before for TC cells in the lateral geniculate nucleus (Errington et al., 2010) and for reticular neurons (Crandall et al., 2010). The cellular mechanism for this differential Ca2+ signaling depends on the availability of T-type VDCCs. When the membrane potential is depolarized, a large fraction of T-type channels are in an inactivated state and can therefore not be recruited efficiently by depolarization to contribute significantly to dendritic Ca2+ influx. When the membrane potential is more hyperpolarized, more T-type channels can be activated, resulting in the generation of a low-threshold Ca2+ spike that spreads to the most distal portions of the dendritic tree (Williams and Stuart, 2000; Errington et al., 2010). Our experiments suggest that this membrane depolarization subsequently recruits high-voltage-activated Ca2+ channels, like L-type VDCCs. A similar sequence of events can be observed during the generation of a dendritic Ca2+ spike in the distal portion of the apical dendrite of pyramidal neurons in the somatosensory cortex (Pérez-Garci et al., 2013). The activation of L-type VDCCs is required for iLTP as blocking these channels abolished the potentiation of inhibitory synaptic transmission. Furthermore, the differential contribution of L-type VDCCs to the Ca2+ influx at the different membrane potentials might explain the smaller magnitude of iLTP at −75 mV.
Our results indicate that the dendritic Ca2+ elevation activated NO synthase. At glutamatergic synapses, NO synthase is associated with the synaptic protein PSD-95, which ensures close proximity to NMDA receptors to sense Ca2+ influx through this channel. In analogy with this, our findings might suggest a direct functional coupling between L-type VDCCs and NO synthase in TC cell dendrites. NO is a retrograde messenger that is synthesized postsynaptically; and because of its gaseous nature, it can diffuse freely to the presynaptic site to activate presynaptic guanylyl cyclase. The resulting elevated levels of cGMP can activate PKG, which can phosphorylate parts of the presynaptic release machinery or voltage-dependent ion channels (Fourcaudot et al., 2008; Nugent et al., 2009; Yang and Calakos, 2011; Luo et al., 2012), thereby increasing presynaptic release probability of the inhibitory synapses.
Importantly, excitatory synaptic transmission by AMPA/kainate and NMDA receptors was blocked in our experiments. Therefore, the iLTP described here did not depend on the activation of NMDA receptors. Furthermore, presynaptic stimulation was not required for iLTP induction because iLTP can be induced by postsynaptic activity alone. This makes it highly unlikely that other synaptic neurotransmitter receptors (e.g., metabotropic glutamate receptors) play a role in the induction of iLTP as reported for other synapses (Fenselau et al., 2011).
Synaptic plasticity in the thalamus
The thalamus can dynamically adapt to excitatory afferent and feedback input by changes in synaptic efficacy. However, it has been suggested that afferent inputs to the thalamus are less plastic than corticothalamic feedback projections (Krupa et al., 1999; Parker and Dostrovsky, 1999; Fox et al., 2002). This has been confirmed recently by comparing the plasticity rules at corticothalamic and medial lemniscal glutamatergic inputs onto TC cells. Only the former inputs showed NMDA receptor-dependent LTP and LTD, depending on the activation of L-type VDCCs. Interestingly, LTD was induced by postsynaptic tonic AP firing of TC cells from a depolarized membrane potential but was not induced by low-threshold burst firing (Hsu et al., 2010). Therefore, this form of non-Hebbian LTD is exactly opposite to the induction requirements for iLTP. This suggests that TC cells use postsynaptic spiking-induced plasticity mechanisms at different synapses in different transmission states.
Little is known about interthalamic synaptic plasticity so far. Recently, it was shown that the excitatory inputs from TC cells onto nRT cells can undergo LTP during coincident burst firing (Astori and Lüthi, 2013). Furthermore, a postsynaptic activity-dependent increase in the frequency of miniature IPSCs without an increase in amplitude onto TC cells in the lateral geniculate nucleus of mice has been described that depends on postsynaptic NMDA receptors and the synthesis of NO (Bright and Brickley, 2008). However, in contrast to our form of NMDA receptor-independent, long-lasting potentiation of IPSP amplitudes, the reported increase in IPSP frequency lasted only a few minutes.
Non-Hebbian plasticity
We have shown that iLTP is non-Hebbian. In general, non-Hebbian plasticity is induced by either presynaptic (Castillo et al., 1994; Urban and Barrionuevo, 1996; Tsukamoto et al., 2003) or postsynaptic activity alone (Alonso et al., 1990; Kullmann et al., 1992; Aizenman et al., 1998; Berretta et al., 1999; Castro-Alamancos and Calcagnotto, 1999; Kato et al., 2009; Lanté et al., 2011), without the requirement for coincident activity. It is now emerging as a fundamental plasticity mechanism, which might be as important as coincident activity-dependent plasticity (Hebbian plasticity). Non-Hebbian plasticity is implicated in scaling of synaptic inputs onto a cell depending on the total activity of the neuronal network (Ibata et al., 2008; Turrigiano, 2011). Furthermore, non-Hebbian plasticity rules can endow neuronal networks with high-capacity sequence detector capabilities (Granger et al., 1994) or contribute to the resonance properties of neuronal networks (Alonso et al., 1990). In our experiments, iLTP at inhibitory synapses onto TC cells was induced at low repetition rates of TC cell bursting (0.1 Hz as well as 1 Hz), which occur during sleep-related thalamic oscillations (Leresche et al., 1991). This suggests that iLTP might contribute to the oscillatory behavior of the thalamus.
Inhibitory inputs onto layer 5 pyramidal neurons in the visual cortex of rats show non-Hebbian LTD or LTP depending on postsynaptic spiking from a depolarized or hyperpolarized membrane potential, respectively (Kurotani et al., 2008). The differential effect was mediated by activation of L-type VDCCs coupled to GABAA receptor endocytosis or activation of R-type VDCCs and GABAA receptor exocytosis for LTD and LTP, respectively. Our results on iLTP of GABAergic inputs onto TC cells show a similar dependence on the membrane potential and the requirement for the activation of VDCCs.
Physiological implications
During slow-wave sleep, TC cells show bursting activity, whereas in the awake state their membrane potential is more depolarized, resulting mainly in a tonic discharge of APs (Sherman, 2001). Here we have shown that iLTP depended on the membrane potential and the resulting discharge mode of the TC cell. This might suggest that iLTP can be induced during sleep when TC cells are hyperpolarized and fire bursts of APs result from low-threshold Ca2+ spikes. Interestingly, in the visual cortex, it has also been suggested that a non-Hebbian form of LTP at inhibitory synapses onto layer 5 pyramidal neurons is induced during slow-wave sleep (Kurotani et al., 2008).
Importantly, low-threshold Ca2+ spikes are a key component in the induction of slow oscillations in the thalamocortical network (Yang and Cox, 2008; Crunelli et al., 2011). During sleep, oscillations are generated in the thalamocortical and TC cell-nRT feedback loops. Within the thalamus, the excitatory TC cells and the inhibitory nRT cells are reciprocally connected, forming a neuronal oscillator network. One could speculate that synaptic plasticity might play an important role in the amplification of this feedback loop. LTP of excitatory inputs from TC cells onto nRT during coincident burst firing (Astori and Lüthi, 2013) might occur concomitantly with iLTP, which would result in the strengthening of both the excitatory synapses as well as the inhibitory synapses within the feedback loop. Therefore, synaptic plasticity could change the coupling of the thalamic oscillator. Thus, we suggest that non-Hebbian plasticity at inhibitory synapses in the thalamus could contribute to determine the oscillation frequency of the neuronal network during sleep.
Although low-threshold spikes in TC cells have been mainly associated with slow-wave sleep, they can also occur in the awake state under certain conditions (Weyand et al., 2001). Under these conditions, iLTP could influence whisker-related sensory signal processing and transmission. This would allow for a dynamic modulation of the thalamocortical relay. Indeed, iLTP might work as a negative feedback, counteracting increased burst firing in TC cells by increasing the inhibitory input onto these cells.
Footnotes
This work was supported by the Swiss National Science Foundation Grants 3100A0-118395 and PP00P3-128415 to T.N. and an equipment grant of the Bern University Research Foundation to T.N. We thank Dr. Mirko Santello for comments on the manuscript and Natalie Nevian for technical assistance.
The authors declare no competing financial interests.
References
- Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neurosci. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-J. [DOI] [PubMed] [Google Scholar]
- Aizenman CD, Manis PB, Linden DJ. Polarity of long-term synaptic gain change is related to postsynaptic spike firing at a cerebellar inhibitory synapse. Neuron. 1998;21:827–835. doi: 10.1016/S0896-6273(00)80598-X. [DOI] [PubMed] [Google Scholar]
- Alonso A, de Curtis M, Llinás R. Postsynaptic Hebbian and non-Hebbian long-term potentiation of synaptic efficacy in the entorhinal cortex in slices and in the isolated adult guinea pig brain. Proc Natl Acad Sci U S A. 1990;87:9280–9284. doi: 10.1073/pnas.87.23.9280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astori S, Lüthi A. Synaptic plasticity at intrathalamic connections via CaV3.3 T-type Ca2+ channels and GluN2B-containing NMDA receptors. J Neurosci. 2013;33:624–630. doi: 10.1523/JNEUROSCI.3185-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthó P, Freund TF, Acsády L. Selective GABAergic innervation of thalamic nuclei from zona incerta. Eur J Neurosci. 2002;16:999–1014. doi: 10.1046/j.1460-9568.2002.02157.x. [DOI] [PubMed] [Google Scholar]
- Berretta N, Rossokhin AV, Cherubini E, Astrelin AV, Voronin LL. Long-term synaptic changes induced by intracellular tetanization of CA3 pyramidal neurons in hippocampal slices from juvenile rats. Neurosci. 1999;93:469–477. doi: 10.1016/S0306-4522(99)00167-0. [DOI] [PubMed] [Google Scholar]
- Bi GQ, Poo MM. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokor H, Frère SGA, Eyre MD, Slézia A, Ulbert I, Lüthi A, Acsády L. Selective GABAergic control of higher-order thalamic relays. Neuron. 2005;45:929–940. doi: 10.1016/j.neuron.2005.01.048. [DOI] [PubMed] [Google Scholar]
- Bosman LW, Houweling AR, Owens CB, Tanke N, Shevchouk OT, Rahmati N, Teunissen WH, Ju C, Gong W, Koekkoek SK, De Zeeuw CI. Anatomical pathways involved in generating and sensing rhythmic whisker movements. Front Integr Neurosci. 2011;5:53. doi: 10.3389/fnint.2011.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright DP, Brickley SG. Acting locally but sensing globally: impact of GABAergic synaptic plasticity on phasic and tonic inhibition in the thalamus. J Physiol. 2008;586:5091–5099. doi: 10.1113/jphysiol.2008.158576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Weisskopf MG, Nicoll RA. The role of Ca2+ channels in hippocampal mossy fiber synaptic transmission and long-term potentiation. Neuron. 1994;12:261–269. doi: 10.1016/0896-6273(94)90269-0. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Südhof TC, Malenka RC. RIM1α is required for presynaptic long-term potentiation. Nature. 2002;415:327–330. doi: 10.1038/415327a. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Chiu CQ, Carroll RC. Long-term plasticity at inhibitory synapses. Curr Opin Neurobiol. 2011;21:328–338. doi: 10.1016/j.conb.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Calcagnotto ME. Presynaptic long-term potentiation in corticothalamic synapses. J Neurosci. 1999;19:9090–9097. doi: 10.1523/JNEUROSCI.19-20-09090.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree JW, Collingridge GL, Isaac JT. A new intrathalamic pathway linking modality-related nuclei in the dorsal thalamus. Nat Neurosci. 1998;1:389–394. doi: 10.1038/1603. [DOI] [PubMed] [Google Scholar]
- Crandall SR, Govindaiah G, Cox CL. Low-threshold Ca2+ current amplifies distal dendritic signaling in thalamic reticular neurons. J Neurosci. 2010;30:15419–15429. doi: 10.1523/JNEUROSCI.3636-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Errington AC, Hughes SW, Tóth TI. The thalamic low-threshold Ca2+ potential: a key determinant of the local and global dynamics of the slow (<1 Hz) sleep oscillation in thalamocortical networks. Philos Trans A Math Phys Eng Sci. 2011;369:3820–3839. doi: 10.1098/rsta.2011.0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity: from synapse to perception. Physiol Rev. 2006;86:1033–1048. doi: 10.1152/physrev.00030.2005. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Sejnowski TJ. Interactions between membrane conductances underlying thalamocortical slow-wave oscillations. Physiol Rev. 2003;83:1401–1453. doi: 10.1152/physrev.00012.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger V, Nevian T, Bruno RM. Subcolumnar dendritic and axonal organization of spiny stellate and star pyramid neurons within a barrel in rat somatosensory cortex. Cereb Cortex. 2008;18:876–889. doi: 10.1093/cercor/bhm126. [DOI] [PubMed] [Google Scholar]
- Errington AC, Renger JJ, Uebele VN, Crunelli V. State-dependent firing determines intrinsic dendritic Ca2+ signaling in thalamocortical neurons. J Neurosci. 2010;30:14843–14853. doi: 10.1523/JNEUROSCI.2968-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE. Synaptic mechanisms for plasticity in neocortex. Annu Rev Neurosci. 2009;32:33–55. doi: 10.1146/annurev.neuro.051508.135516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmeyer D, Egger V, Lübke J, Sakmann B. Reliable synaptic connections between pairs of excitatory layer 4 neurones within a single barrel of developing rat somatosensory cortex. J Physiol. 1999;521:169–190. doi: 10.1111/j.1469-7793.1999.00169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenselau H, Heinke B, Sandkühler J. Heterosynaptic long-term potentiation at GABAergic synapses of spinal lamina I neurons. J Neurosci. 2011;31:17383–17391. doi: 10.1523/JNEUROSCI.3076-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourcaudot E, Gambino F, Humeau Y, Casassus G, Shaban H, Poulain B, Lüthi A. cAMP/PKA signaling and RIM1α mediate presynaptic LTP in the lateral amygdala. Proc Natl Acad Sci U S A. 2008;105:15130–15135. doi: 10.1073/pnas.0806938105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox K, Wallace H, Glazewski S. Is there a thalamic component to experience-dependent cortical plasticity? Philos Trans R Soc Lond B Biol Sci. 2002;357:1709–1715. doi: 10.1098/rstb.2002.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey MC, Sprengel R, Nevian T. Activity pattern-dependent long-term potentiation in neocortex and hippocampus of GluA1 (GluR-A) subunit-deficient mice. J Neurosci. 2009;29:5587–5596. doi: 10.1523/JNEUROSCI.5314-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger R, Whitson J, Larson J, Lynch G. Non-Hebbian properties of long-term potentiation enable high-capacity encoding of temporal sequences. Proc Natl Acad Sci U S A. 1994;91:10104–10108. doi: 10.1073/pnas.91.21.10104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham N, Fox K. The role of nitric oxide and GluR1 in presynaptic and postsynaptic components of neocortical potentiation. J Neurosci. 2006;26:7395–7404. doi: 10.1523/JNEUROSCI.0652-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren CD, Zilberter Y. Coincident spiking activity induces long-term changes in inhibition of neocortical pyramidal cells. J Neurosci. 2001;21:8270–8277. doi: 10.1523/JNEUROSCI.21-20-08270.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CL, Yang HW, Yen CT, Min MY. Comparison of synaptic transmission and plasticity between sensory and cortical synapses on relay neurons in the ventrobasal nucleus of the rat thalamus. J Physiol. 2010;588:4347–4363. doi: 10.1113/jphysiol.2010.192864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Kato HK, Watabe AM, Manabe T. Non-Hebbian synaptic plasticity induced by repetitive postsynaptic action potentials. J Neurosci. 2009;29:11153–11160. doi: 10.1523/JNEUROSCI.5881-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupa DJ, Ghazanfar AA, Nicolelis MA. Immediate thalamic sensory plasticity depends on corticothalamic feedback. Proc Natl Acad Sci U S A. 1999;96:8200–8205. doi: 10.1073/pnas.96.14.8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Perkel DJ, Manabe T, Nicoll RA. Ca2+ entry via postsynaptic voltage-sensitive Ca2+ channels can transiently potentiate excitatory synaptic transmission in the hippocampus. Neuron. 1992;9:1175–1183. doi: 10.1016/0896-6273(92)90075-O. [DOI] [PubMed] [Google Scholar]
- Kullmann DM, Moreau AW, Bakiri Y, Nicholson E. Plasticity of inhibition. Neuron. 2012;75:951–962. doi: 10.1016/j.neuron.2012.07.030. [DOI] [PubMed] [Google Scholar]
- Kurotani T, Yamada K, Yoshimura Y, Crair MC, Komatsu Y. State-dependent bidirectional modification of somatic inhibition in neocortical pyramidal cells. Neuron. 2008;57:905–916. doi: 10.1016/j.neuron.2008.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamsa KP, Kullmann DM, Woodin MA. Spike-timing dependent plasticity in inhibitory circuits. Front Synaptic Neurosci. 2010;2:8. doi: 10.3389/fnsyn.2010.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landisman CE, Connors BW. VPM and PoM nuclei of the rat somatosensory thalamus: intrinsic neuronal properties and corticothalamic feedback. Cereb Cortex. 2007;17:2853–2865. doi: 10.1093/cercor/bhm025. [DOI] [PubMed] [Google Scholar]
- Lanté F, Toledo-Salas JC, Ondrejcak T, Rowan MJ, Ulrich D. Removal of synaptic Ca2+-permeable AMPA receptors during sleep. J Neurosci. 2011;31:3953–3961. doi: 10.1523/JNEUROSCI.3210-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavallée P, Urbain N, Dufresne C, Bokor H, Acsády L, Deschênes M. Feedforward inhibitory control of sensory information in higher-order thalamic nuclei. J Neurosci. 2005;25:7489–7498. doi: 10.1523/JNEUROSCI.2301-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leresche N, Lightowler S, Soltesz I, Jassik-Gerschenfeld D, Crunelli V. Low-frequency oscillatory activities intrinsic to rat and cat thalamocortical cells. J Physiol. 1991;441:155–174. doi: 10.1113/jphysiol.1991.sp018744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Gangadharan V, Bali KK, Xie RG, Agarwal N, Kurejova M, Tappe-Theodor A, Tegeder I, Feil S, Lewin G, Polgar E, Todd AJ, Schlossmann J, Hofmann F, Liu DL, Hu SJ, Feil R, Kuner T, Kuner R. Presynaptically localized cyclic GMP-dependent protein kinase 1 is a key determinant of spinal synaptic potentiation and pain hypersensitivity. PLoS Biol. 2012;10:e1001283. doi: 10.1371/journal.pbio.1001283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A. The many forms and functions of long term plasticity at GABAergic synapses. Neural Plast. 2011;2011:254724. doi: 10.1155/2011/254724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci. 1997;20:185–215. doi: 10.1146/annurev.neuro.20.1.185. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Contreras D. On the cellular and network bases of epileptic seizures. Annu Rev Physiol. 2001;63:815–846. doi: 10.1146/annurev.physiol.63.1.815. [DOI] [PubMed] [Google Scholar]
- Méndez P, Bacci A. Assortment of GABAergic plasticity in the cortical interneuron melting pot. Neural Plast. 2011;2011:976856. doi: 10.1155/2011/976856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Single spine Ca2+ signals evoked by coincident EPSPs and back-propagating action potentials in spiny stellate cells of layer 4 in the juvenile rat somatosensory barrel cortex. J Neurosci. 2004;24:1689–1699. doi: 10.1523/JNEUROSCI.3332-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J Neurosci. 2006;26:11001–11013. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent FS, Niehaus JL, Kauer JA. PKG and PKA signaling in LTP at GABAergic synapses. Neuropsychopharmacology. 2009;34:1829–1842. doi: 10.1038/npp.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JL, Dostrovsky JO. Cortical involvement in the induction, but not expression, of thalamic plasticity. J Neurosci. 1999;19:8623–8629. doi: 10.1523/JNEUROSCI.19-19-08623.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Garci E, Larkum ME, Nevian T. Inhibition of dendritic Ca2+ spikes by GABAB receptors in cortical pyramidal neurons is mediated by a direct Gi/o-beta-subunit interaction with Cav1 channels. J Physiol. 2013;591:1599–1612. doi: 10.1113/jphysiol.2012.245464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman SM. Tonic and burst firing: dual modes of thalamocortical relay. Trends Neurosci. 2001;24:122–126. doi: 10.1016/S0166-2236(00)01714-8. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Rancz EA, Roth A, Häusser M. Dendritic excitability and synaptic plasticity. Physiol Rev. 2008;88:769–840. doi: 10.1152/physrev.00016.2007. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Rogawski MA. T-type calcium channels mediate the transition between tonic and phasic firing in thalamic neurons. Proc Natl Acad Sci U S A. 1989;86:7228–7232. doi: 10.1073/pnas.86.18.7228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trageser JC, Keller A. Reducing the uncertainty: gating of peripheral inputs by zona incerta. J Neurosci. 2004;24:8911–8915. doi: 10.1523/JNEUROSCI.3218-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto M, Yasui T, Yamada MK, Nishiyama N, Matsuki N, Ikegaya Y. Mossy fibre synaptic NMDA receptors trigger non-Hebbian long-term potentiation at entorhino-CA3 synapses in the rat. J Physiol. 2003;546:665–675. doi: 10.1113/jphysiol.2002.033803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci. 2011;34:89–103. doi: 10.1146/annurev-neuro-060909-153238. [DOI] [PubMed] [Google Scholar]
- Urban NN, Barrionuevo G. Induction of Hebbian and non-Hebbian mossy fiber long-term potentiation by distinct patterns of high-frequency stimulation. J Neurosci. 1996;16:4293–4299. doi: 10.1523/JNEUROSCI.16-13-04293.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanaverbecq N, Bodor AL, Bokor H, Slézia A, Lüthi A, Acsády L. Contrasting the functional properties of GABAergic axon terminals with single and multiple synapses in the thalamus. J Neurosci. 2008;28:11848–11861. doi: 10.1523/JNEUROSCI.3183-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyand TG, Boudreaux M, Guido W. Burst and tonic response modes in thalamic neurons during sleep and wakefulness. J Neurophysiol. 2001;85:1107–1118. doi: 10.1152/jn.2001.85.3.1107. [DOI] [PubMed] [Google Scholar]
- Williams SR, Stuart GJ. Action potential back-propagation and somato-dendritic distribution of ion channels in thalamocortical neurons. J Neurosci. 2000;20:1307–1317. doi: 10.1523/JNEUROSCI.20-04-01307.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodin MA, Maffei A. Inhibitory synaptic plasticity. New York: Springer; 2011. [Google Scholar]
- Yang S, Cox CL. Excitatory and anti-oscillatory actions of nitric oxide in thalamus. J Physiol. 2008;586:3617–3628. doi: 10.1113/jphysiol.2008.153312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Calakos N. Munc13–1 is required for presynaptic long-term potentiation. J Neurosci. 2011;31:12053–12057. doi: 10.1523/JNEUROSCI.2276-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]