

Abstract
Inflammation has been implicated in the progression of neurological disease, yet precisely how inflammation affects neuronal function remains unclear. Tumor necrosis factor-α (TNFα) is a proinflammatory cytokine that regulates synapse function by controlling neurotransmitter receptor trafficking and homeostatic synaptic plasticity. Here we characterize the mechanisms through which TNFα regulates inhibitory synapse function in mature rat and mouse hippocampal neurons. Acute application of TNFα induces a rapid and persistent decrease of inhibitory synaptic strength and downregulation of cell-surface levels of GABAARs containing α1, α2, β2/3, and γ2 subunits. We show that trafficking of GABAARs in response to TNFα is mediated by neuronally expressed TNF receptor 1 and requires activation of p38 MAPK, phosphatidylinositol 3-kinase, protein phosphatase 1 (PP1), and dynamin GTPase. Furthermore, TNFα enhances the association of PP1 with GABAAR β3 subunits and dephosphorylates a site on β3 known to regulate phospho-dependent interactions with the endocytic machinery. Conversely, we find that calcineurin and PP2A are not essential components of the signaling pathway and that clustering of the scaffolding protein gephyrin is only reduced after the initial receptor endocytosis. Together, these findings demonstrate a distinct mechanism of regulated GABAAR endocytosis that may contribute to the disruption of circuit homeostasis under neuroinflammatory conditions.
Introduction
Immune molecules expressed in the CNS, such as cytokines, class I major histocompatibility complex, and complement cascade components, are active in a variety of contexts ranging from synapse development to cell survival and synaptic plasticity (Yirmiya and Goshen, 2011). In particular, the proinflammatory cytokine tumor necrosis factor-α (TNFα) is an important regulator of synapse function implicated in neurotransmitter receptor trafficking (Beattie et al., 2002; Ogoshi et al., 2005; Stellwagen et al., 2005), homeostatic synaptic plasticity (Stellwagen and Malenka, 2006; Kaneko et al., 2008; Steinmetz and Turrigiano, 2010), and regulation of gliotransmission (Santello et al., 2011). TNFα is a proteolytically cleaved transmembrane protein that signals through TNF receptor 1 (TNFR1) and TNFR2 (MacEwan, 2002). Expression of TNFα in the CNS is upregulated by glia in response to inflammatory neurological insults, such as stroke, infection, and neurodegenerative diseases (Tonelli and Postolache, 2005; Frankola et al., 2011), and in response to prolonged neuronal activity blockade (Stellwagen and Malenka, 2006). Neurons respond to elevated levels of TNFα by rapidly increasing excitatory synaptic strength (Tancredi et al., 1992; Beattie et al., 2002; Stellwagen et al., 2005) and weakening inhibitory synaptic strength, resulting in a higher excitatory/inhibitory ratio (Stellwagen et al., 2005). Although the increase in excitatory synaptic strength is known to occur through TNFR1, phosphatidylinositol 3-kinase (PI3K) activity and cell-surface delivery of Ca2+-permeable AMPA receptors (AMPARs; Ogoshi et al., 2005; Stellwagen et al., 2005; Yin et al., 2012), the detailed molecular mechanisms underlying the weakening of inhibitory synaptic strength induced by TNFα remain essentially unknown.
GABAA receptors (GABAARs) are the primary mediators of fast inhibitory neurotransmission in the brain, exerting tight control over the excitatory/inhibitory balance (Mann and Paulsen, 2007; Jacob et al., 2008). Accordingly, selective disruption of GABAAR function leads to homeostatic modifications in circuit development (Duveau et al., 2011; Pallotto et al., 2012), epilepsy (DeLorey et al., 1998), and anxiety-like behavior (Crestani et al., 1999). A pivotal point in the regulation of GABAergic synaptic strength is the dynamic trafficking of GABAARs (Luscher et al., 2011). In particular, endocytosis of GABAARs is regulated by phosphorylation-dependent binding of the clathrin adaptor protein 2 (AP2) adaptor complex to motifs in the intracellular domains of GABAAR β and γ subunits (Arancibia-Cárcamo and Kittler, 2009). Phosphoresidues within these motifs are subject to regulation by kinases (e.g., PKA and PKC) and phosphatases (e.g., protein phosphatase 1 [PP1]/PP2A and calcineurin; Luscher et al., 2011). The effect of TNFα on phospho-dependent regulation of GABAAR trafficking remains unexplored.
Here, using multiple experimental approaches, we characterize the mechanisms through which TNFα downregulates GABAergic neurotransmission in hippocampal neurons. TNFα signals through neuronal TNFR1, p38 MAPK, PI3K, and PP1 to rapidly reduce inhibitory synaptic strength and surface levels of synaptic GABAARs. These effects persist over time and are followed by a reduction in clustering of the postsynaptic scaffolding protein gephyrin. Moreover, we show that TNFα increases the association of PP1 with GABAAR β3, dephosphorylates β3 S408/409, and reduces inhibitory synaptic strength via dynamin GTPase activity. Together, these results describe a mechanism of regulated GABAAR endocytosis via which GABAergic inhibition is downregulated under conditions associated with elevated TNFα levels.
Materials and Methods
Hippocampal neuron culture.
Animal procedures were performed in accordance with the guidelines of the Canadian Council for Animal Care and the Montreal General Hospital Facility Animal Care Committee. All cell culture experiments (except those in Fig. 4C,D) made use of mixed neuron–glia cultures prepared based on established protocols (Kaech and Banker, 2006) from E17–E18 Sprague Dawley rat hippocampus (of either sex). Briefly, dissociated cells were plated at a density of 2.7 × 104/cm2 on either 12 mm poly-d-lysine-coated coverslips (Thermo Fisher Scientific) for immunostaining and electrophysiology experiments or on 10 cm poly-d-lysine-coated tissue culture plates for immunoprecipitation and Western blotting experiments. Culture media consisted of Neurobasal medium supplemented with B-27 and Glutamax (Life Technologies). Glial proliferation was inhibited at 12 d in vitro with 8 μm AraC. Experiments were performed at 14–22 d in vitro. For experiments in Figure 4, C and D, glia derived from P1–P2 mouse forebrain (of either sex) were grown to confluence on poly-d-lysine-coated 24-well plates. Neurons were dissociated from P0 mouse hippocampus [of either sex, C57BL/6J wild type (WT; The Jackson Laboratory) or TNFR1−/− (kindly provided by Dr. P. Brodt, McGill University, Montréal, Québec, Canada)], seeded at a density of 3.7 × 104/cm2 on 12 mm poly-d-lysine-coated coverslips for 2 h, transferred to the glial culture plate wells, and grown in the same media used for rat cultures; additional glial proliferation was inhibited with AraC.
Figure 4.

TNFα signals through neuronal TNFR1 to downregulate GABAergic neurotransmission. A, Representative dendritic regions of surface immunostaining for GABAAR γ2 subunit, from untreated (Ctrl) or TNFα-treated cultured neurons, preincubated with either TNFR1 neutralizing antibody (R1nAb) or TNFR2 neutralizing antibody (R2nAb). Scale bar, 3 μm. B, Quantification data of γ2 cluster area corresponding to conditions in A, showing that blocking TNFR1 function blocks the effects of TNFα (gray bars), whereas blocking TNFR2 does not (n = 135–191 images in each condition; two-way ANOVA, Bonferroni's post hoc test, *p < 0.05, ***p < 0.001; n.s., not significant). C, Representative traces of mIPSC recordings from untreated (Ctrl) or TNFα-treated WT or TNFR1−/− neurons cocultured with WT glia or TNFR1−/− glia. D, Group data of average mIPSC amplitude indicating that TNFα-induced downregulation is absent from TNFR1−/− neurons cocultured with WT glia but present in WT neurons cocultured with WT or TNFR1−/− glia (n = 12–22 cells in each condition; two-way ANOVA, Bonferroni's post hoc test, *p < 0.05, **p < 0.01; n.s., not significant).
Culture electrophysiology and treatments.
Miniature IPSCs (mIPSCs) were recorded at room temperature using the whole-cell patch-clamp method from neurons superfused with ACSF containing the following (in mm): 115 NaCl, 5 KCl, 23 glucose, 26 sucrose, 4.2 HEPES, 2.5 CaCl2, and 1.3 MgCl2, pH 7.2 (osmolality, 295–305 mOsm). Recording pipettes were pulled from borosilicate glass (King Precision Glass) to an open-tip resistance of 3–6 MΩ and filled with an internal solution of the following (in mm): 127 CsCl, 8 NaCl, 1 CaCl2, 10 HEPES, 10 EGTA, 0.3 Na3-GTP, and 2 Mg-ATP, pH 7.2 (osmolality, 280–290 mOsm). To isolate mIPSCs, 200 nm TTX, 10 μm NBQX, and 25 μm d-APV (Tocris Bioscience) were added to the external solution, and recordings were obtained at Vh = −70 mV. Signals were amplified and filtered (1 kHz) using an Axopatch 200B amplifier, sampled at 5 kHz using a Digidata 1440A, and recorded with Clampex 10.3 (Molecular Devices). Analyses of mIPSC amplitude, frequency, 10–90% rise time, and decay tau were performed using Clampfit 10.3 (Molecular Devices). Only cells with a stable series resistance of <25 MΩ throughout the recording period were included in the analysis. Treatments were performed by adding recombinant mouse TNFα [at the indicated concentrations in Fig. 1 and at 100 ng/ml (6 nm) for all other figures; Kamiya Biomedical] to a defined volume of conditioned growth media originating from the wells being treated and returning the plate to the incubator (37°C, 5% CO2) for 45 min. Coverslips were then transferred to the recording chamber, and recordings were obtained within 75 min. For treatments with pharmacological inhibitors (Tocris Bioscience) of dynamin GTPase (dynasore, 80 μm), PP1/PP2A (okadaic acid, 0.5 μm), PP1 (tautomycetin, 10 nm), and PP2A (fostriecin, 10 nm), the inhibitor or its corresponding solvent was applied as above but for 55 min (i.e., 10 min before TNFα). For experiments examining multiple treatment conditions, control (Ctrl) and treated cells were recorded on the same day. Data from multiple recording days were then pooled to obtain group data for each experiment.
Figure 1.
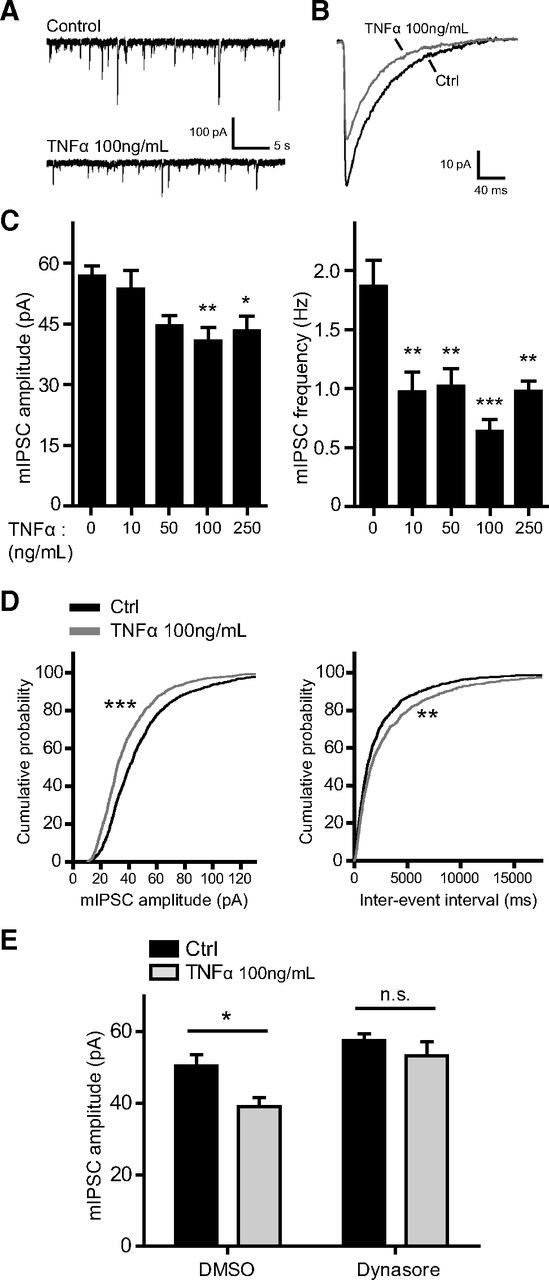
TNFα downregulates inhibitory synaptic strength via dynamin GTPase activity. A, Representative traces of mIPSC recordings from untreated (Control) and TNFα-treated cultured neurons (100 ng/ml, 45 min). B, Superimposed average mIPSC traces from an untreated neuron (Ctrl, black trace) and a TNFα-treated neuron (gray trace), showing a reduction in mIPSC amplitude in response to TNFα (100 ng/ml, 45 min). C, Group data showing a reduction of average mIPSC amplitude (left) and mIPSC frequency (right) in response to various concentrations of TNFα applied for 45 min (n = 14–23 cells in each condition; one-way ANOVA, Tukey's post hoc test, *p < 0.05, **p < 0.01, ***p < 0.001). D, Cumulative distribution plots of mIPSC amplitudes (left) and interevent intervals (right) from untreated (black traces) and TNFα-treated (100 ng/ml, 45 min; gray traces) neurons (∼1200 events from each condition; **p < 0.005, ***p < 0.0001, Kolmogorov–Smirnov test). E, Pretreatment with the dynamin GTPase inhibitor dynasore (80 μm) blocks the TNFα-induced (100 ng/ml, 45 min) reduction of mIPSC amplitude (n = 16–18 cells in each condition; two-way ANOVA, Bonferroni's post hoc test, *p < 0.05; n.s., not significant).
Immunocytochemistry and treatments.
For surface GABAAR immunostaining, cultured cells were live labeled by washing with ice-cold ACSF and incubating with primary antibody in ACSF on ice. The following primary antibodies were used to target the extracellular (N-terminal) domains of GABAAR subunits: anti-GABAAR α1 (1:400; catalog #AGA-001; Alomone Labs), anti-GABAAR α2 (1:500; catalog #224103; Synaptic Systems), anti-GABAAR α5 (1:1000; kind gift from Dr. J.-M. Fritschy, University of Zürich, Zürich, Switzerland), anti-GABAAR β2/3 (1:100; catalog #MAB341; Millipore), and anti-GABAAR γ2 (1:100; catalog #AGA-005; Alomone Labs). Cells were then washed three times with ice-cold ACSF and fixed in 2% paraformaldehyde solution for 15 min. Appropriate fluorescent secondary antibodies (1:1000; Alexa Fluor; Life Technologies) were then applied for 1 h at room temperature in a PBS solution containing 3% bovine serum albumin (BSA) and 2% normal goat serum (NGS). For double immunostaining of gephyrin and microtubule-associated protein 2 (MAP2), or glutamic acid decarboxylase 65 (GAD65) and MAP2, cells were washed with ice-cold ACSF, fixed in 2% paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100 for 3 min, blocked with PBS/3% BSA/2% NGS solution for 10 min, and incubated with primary antibodies in PBS/3% BSA/2% NGS for 2 h at room temperature. The following primary antibodies were used: anti-gephyrin (1:400; catalog #147011; Synaptic Systems), anti-GAD65/67 (1:500; GAD-6; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), and anti-MAP2 (1:1000; catalog #188004; Synaptic Systems). For determining the percentage of cells expressing high levels of GABAAR α1 that are GABAergic interneurons, surface α1 subunit was immunolabeled as above; cells were then fixed, permeabilized, and immunostained with anti GAD65/67 (GAD-6) as above. Treatments with TNFα (100 ng/ml) and pharmacological inhibitors were performed as for electrophysiology experiments. Inhibitors (Tocris Bioscience) were as follows: PKA (KT 5720 [(9S,10R,12R)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylicacid hexyl ester]; 1 μm), PKC [Gö 6983 (2-[1-(3-dimethylaminopropyl)-5-methoxyindol-3-yl]-3-(1H-indol-3-yl)maleimide); 1 μm], PI3K (wortmannin, 100 nm), p38 MAPK (SB 202190 [4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1H-imidazole]; 10 μm), calcineurin (cyclosporin A, 20 μm), and PP1/PP2A (okadaic acid, 0.05 or 0.5 μm). TNFα receptor function was blocked by starting with a 10 min pretreatment with either anti-TNFR1 neutralizing antibody (5 μg/ml; catalog #MAB430; R&D Systems) or anti-TNFR2 neutralizing antibody (2 μg/ml; catalog #AF726; R&D Systems), before adding TNFα (100 ng/ml) for an additional 45 min.
Image acquisition and analysis.
All images were acquired blind to the experimental conditions, using identical acquisition parameters across all coverslips in each experiment. Images of surface GABAAR immunostaining were captured using a CCD camera (Hamamatsu Orca-R2) mounted on an epifluorescence microscope [63×, 1.42 numerical aperture (NA); Olympus BX61]. All other images were acquired as single optical sections using a confocal microscope (63×, NA 1.42; Olympus FV1000). Each experimental condition was represented by two to three coverslips per experiment, and ∼10–20 images were acquired per coverslip. Image analysis was performed essentially as described previously (Stellwagen et al., 2005). Briefly, unadjusted whole images were analyzed using NIH ImageJ software, applying identical analysis parameters across all conditions within each experiment. For surface GABAAR immunostaining, the area selected by a low threshold was used to approximate somatodendritic area, and a high threshold was used to select receptor puncta. High-threshold selections were analyzed using the “Analyze Particles” function, with particle size restricted to 1 μm2 to exclude selections of large bright areas that do not represent receptor puncta. The total particle area of each image was divided by its corresponding low-threshold area to obtain values normalized to somatodendritic area. The resulting values were then expressed as percentage control to obtain “cluster area” as reported in Results. The same analysis was used for experiments in which cells were permeabilized and double immunostained for gephyrin plus MAP2 or GAD65 plus MAP2, except that (1) particle size was restricted to 2 μm2 for gephyrin images and 4 μm2 for GAD65 images and (2) somatodendritic area was determined from corresponding MAP2 images. Cluster density was obtained by normalizing particle count to somatodendritic area for each image and expressing as percentage control. “Average cluster size” is average particle size expressed as percentage control. Because cluster area is a measure of the summated puncta area per image, it represents a combined measure of cluster density and average cluster size.
Culture cell-surface biotinylation, immunoprecipitation, and Western blotting.
For surface biotinylation assays, cells were treated with TNFα (100 ng/ml) for 45 min in conditioned growth media, washed three times with ice-cold ACSF, and incubated for 30 min with 0.2 mg/ml Sulfo-NHS-SS-Biotin (Thermo Fisher Scientific) diluted in ice-cold ACSF. Cells were then washed three times with ice-cold ACSF containing 100 mm glycine and lysed in 20 mm HEPES, pH 7.4, 150 mm NaCl, 1% Triton X-100, 0.1% SDS, 1 mm EDTA, 1 mm PMSF, 50 mm NaF, 1 mm Na3VO4, and protease inhibitor mixture (BioShop Canada). Biotinylated proteins were isolated by incubating lysates with NeutrAvidin agarose resin (Thermo Fisher Scientific) for 1 h at 4°C. Biotinylated proteins were eluted with 2× SDS sample buffer and analyzed by SDS-PAGE along with corresponding total lysates.
For immunoprecipitation experiments cells were treated with TNFα (100 ng/ml) for 30 min, washed two times with ice-cold ACSF, and lysed in 20 mm HEPES, pH 7.4, 150 mm NaCl, 1% NP-40, 1 mm EDTA, 1 mm PMSF, 50 mm NaF, 1 mm Na3VO4, and protease inhibitor mixture (BioShop Canada). Lysates were precleared with protein A/G agarose beads (Thermo Fisher Scientific) and incubated with 2 μg of anti-GABAAR β3 (catalog #75-149; Neuromab) per milligram of lysate protein. Precipitates were eluted with 2× SDS sample buffer and analyzed by SDS-PAGE together with corresponding unprecipitated lysates.
Gels were transferred onto PVDF membrane and immunoblotted with anti-GABAAR β3 (1:500; catalog #75-149; Neuromab), anti-GABAAR β3 (1:400; catalog #NB100-66169; Novus Biologicals), anti-GluA1 (1:1000; catalog #AB1504; Millipore), anti-phospho-p38 MAPK (1:1000; catalog #9215; Cell Signaling Technology), anti-total p38 MAPK (1:1000; catalog #9212; Cell Signaling Technology), anti-phospho Akt (1:2000; catalog #4060; Cell Signaling Technology), anti-total Akt (1:1000; catalog #4685; Cell Signaling Technology), anti-α-tubulin (1:5000; catalog #05-289; Millipore), anti-GABAAR β3 phospho-S408/409 (1:250; catalog #612-401-D51; Rockland), anti-PP1α (1:1000; catalog #1950-1; Epitomics), and anti-PP2A (1:1000; catalog #1512-1; Epitomics). Quantitative analyses of Western blots were performed by determining band intensities on autoradiography film (Denville Scientific), using NIH ImageJ software. For surface biotinylation assays, “surface” quantifications depict surface/total ratios, and “total” quantifications depict total/α-tubulin ratios. For immunoprecipitation experiments quantifications depict coimmunoprecipitation PP1/β3 or PP2A/β3 ratios.
Acute hippocampal slice cell-surface biotinylation and Western blotting.
Coronal slices (300 μm thick) of the dorsal hippocampus were prepared using a vibratome (Leica VT1200S), from 6- to 9-week-old C57BL/6J WT mice (of either sex) and allowed to recover at ∼32°C for 30 min in ACSF containing 119 mm NaCl, 26.2 mm NaHCO3, 11 mm glucose, 2.5 mm KCl, 1 mm NaH2PO4, 2.5 mm CaCl2, and 1.3 mm MgCl2, saturated with 95% O2/5% CO2. Slices were then incubated in the same solution, at room temperature, with TNFα (100 ng/ml, 1 h). Next, slices were incubated for 45 min in ice-cold ACSF solution containing 1 mg/ml Sulfo-NHS-SS-Biotin (Thermo Fisher Scientific). Slices were then washed three times with ice-cold ACSF containing 100 mm glycine and lysed in 20 mm HEPES, pH 7.4, 150 mm NaCl, 1% Triton X-100, 0.1% SDS, 1 mm EDTA, 1 mm PMSF, 50 mm NaF, 1 mm Na3VO4, and protease inhibitor mixture (BioShop Canada). Biotinylated proteins were isolated, SDS-PAGE was performed, PVDF membranes were immunoblotted, and quantifications were performed as for culture experiments.
Acute slice electrophysiology and treatments.
Coronal slices of the dorsal hippocampus were prepared and treated with TNFα as for cell-surface biotinylation experiments, from 5- to 8-week-old C57BL/6J WT mice (of either sex). DMSO (0.1%) or tautomycetin (10 nm, in DMSO) were added to the treatment chamber 20 min before adding TNFα. Slices were placed in a submersion-type recording chamber and perfused (∼1.5 ml/min) at 30°C with ACSF containing 119 mm NaCl, 26.2 mm NaHCO3, 11 mm glucose, 2.5 mm KCl, 1 mm NaH2PO4, 2.5 mm CaCl2, and 1.3 mm MgCl2, saturated with 95% O2/5% CO2. Whole-cell patch-clamp recordings were obtained from hippocampal CA1 pyramidal neurons under visual guidance by infrared differential interference microscopy (Nikon Eclipse FN1). mIPSCs were pharmacologically isolated, recorded, and analyzed as for culture experiments.
Statistical analyses.
Statistical analysis and plotting were performed with Prism 5 (GraphPad Software). All data were obtained from a minimum of three independent experiments and presented as mean ± SEM.
Results
TNFα rapidly downregulates inhibitory synaptic strength via dynamin GTPase activity
To study the acute effects of TNFα on inhibitory neurotransmission, we recorded mIPSCs from hippocampal neuron cultures treated with various concentrations of TNFα for 45 min (Fig. 1A–D). Consistent with previous findings (Stellwagen et al., 2005), TNFα treatment significantly reduced average mIPSC amplitude, exerting a gradual effect that reached a plateau by 100 ng/ml (Ctrl, 56.8 ± 2.6 pA; TNFα at 10 ng/ml, 53.7 ± 4.6 pA; TNFα at 50 ng/ml, 44.52 ± 2.5 pA; TNFα at 100 ng/ml, 40.8 ± 3.4 pA; TNFα at 250 ng/ml, 43.3 ± 3.7 pA; Fig. 1C, left). In contrast, mIPSC frequency was significantly reduced starting at 10 ng/ml (Ctrl, 1.86 ± 0.22 Hz; TNFα at 10 ng/ml, 0.97 ± 0.17 Hz; TNFα at 50 ng/ml, 1.02 ± 0.15 Hz; TNFα at 100 ng/ml, 0.64 ± 0.10 Hz; TNFα at 250 ng/ml, 0.98 ± 0.09 Hz; Fig. 1C, right), suggesting that, at least in a mixed population of cultured hippocampal neurons, TNFα may act presynaptically to regulate neurotransmitter release probability at inhibitory synapses. For subsequent experiments, we focused on the 100 ng/ml dose because it exerts the strongest effect on quantal amplitude and it is the most commonly used concentration when studying TNFα-dependent receptor trafficking at hippocampal synapses (Beattie et al., 2002; Ogoshi et al., 2005; Stellwagen et al., 2005; Stellwagen and Malenka, 2006; Leonoudakis et al., 2008). We further determined that mIPSC 10–90% rise time (Ctrl, 2.06 ± 0.08 ms; TNFα, 2.26 ± 0.18 ms; Student's t test, p = 0.30) and decay time (Ctrl, 41.6 ± 0.95 ms; TNFα, 40.3 ± 1.10 ms; Student's t test, p = 0.40) were unaffected by TNFα treatment (100 ng/ml, 45 min), implying no substantial change in the subunit composition of synaptic GABAARs. As an additional control, we recorded cells treated with denatured (boiled) TNFα and observed no significant differences compared with untreated control cells (Ctrl, 55.4 ± 3.7 pA, 1.56 ± 0.28 Hz, n = 11 cells; boiled TNFα at 250 ng/ml, 53.8 ± 5.7 pA, 1.48 ± 0.27 Hz, n = 8 cells; Student's t test, p = 0.81 for amplitude and p = 0.86 for frequency).
The rapid reduction of mIPSC amplitude in response to TNFα could be attributed to reduced GABAAR delivery to the synapse, reduced receptor stability at the synapse, or increased receptor endocytosis (Luscher et al., 2011). Because treatment with TNFα is known to cause a nearly twofold increase in the internalization of antibody-labeled GABAARs (Stellwagen et al., 2005), we suspected that the majority of the reduction in inhibitory synaptic strength would be endocytosis dependent. Endocytosis of GABAARs occurs primarily via an AP2/clathrin/dynamin-dependent mechanism (Kittler et al., 2000, 2005, 2008; Kanematsu et al., 2007); therefore, in a separate set of experiments, we pretreated cultures with the dynamin GTPase inhibitor dynasore (Macia et al., 2006) and measured mIPSC amplitudes in response to TNFα (Fig. 1E). The TNFα-induced reduction of mIPSC amplitude was blocked by dynasore (DMSO pretreatment: Ctrl, 50.4 ± 3.2 pA, 0.84 ± 0.15 Hz; TNFα, 39.1 ± 2.5 pA, 0.75 ± 0.10 Hz; dynasore pretreatment: Ctrl, 57.5 ± 1.9 pA, 3.51 ± 0.68 Hz; TNFα, 53.3 ± 3.9 pA, 4.05 ± 0.73 Hz), suggesting that TNFα reduces the strength of hippocampal inhibitory synapses primarily through GABAAR endocytosis.
TNFα rapidly downregulates cell-surface levels of GABAARs
GABAARs consist of heteropentameric assemblies of diverse subunit composition, resulting in receptor subtypes that differ in expression, cell-surface localization, pharmacology, and function (Pirker et al., 2000; Möhler et al., 2004; Farrant and Nusser, 2005; Fritschy et al., 2012). At hippocampal synapses, GABAARs are composed of combinations of two α1–α3 subunits, two β1–β3 subunits, and one γ2 subunit (Brünig et al., 2002; Michels and Moss, 2007; Olsen and Sieghart, 2009; Kasugai et al., 2010). We used cell-surface immunostaining to determine whether TNFα preferentially endocytoses certain receptor subtypes (Fig. 2A). TNFα treatment (100 ng/ml, 45 min) reduced the surface cluster area of α1, α2, β2/3, and γ2 subunits by 25–30% compared with untreated Ctrl cells (α1, 75.9 ± 4.7% of Ctrl; α2, 71.4 ± 1.5% of Ctrl; β2/3, 71.7 ± 2.3% of Ctrl; γ2, 69.7 ± 2.1% of Ctrl; Fig. 2B). There was no obvious difference in the specificity of the effect among these subunits, likely because they are trafficked in tandem as common components of synaptically localized receptors. Consistent with previous reports (Fritschy et al., 1994; Brünig et al., 2002), we observed a differential basal expression of the α1 subunit, with ∼20% of cells expressing markedly higher levels of surface α1 subunit in our cultures. Consequently, we independently assessed the effects of TNFα on cells expressing high levels of the α1 subunit. We found that levels of surface α1 on high expressors were unaltered after TNFα treatment (102.1 ± 3.5% of Ctrl), suggesting that α1 high expressors define a subset of neurons unresponsive to TNFα. Previous reports have suggested that, in the hippocampus, higher expression of the α1 subunit is observed in interneurons (Fritschy et al., 1994; Brünig et al., 2002; Yu et al., 2006); however, based on colabeling of α1 and GAD, we observed that only ∼13% of high α1-expressing cells also expressed GAD, indicating that high α1-expressing cells in our cultures consisted of a mixed population of GABAergic interneurons and other cell types.
Figure 2.

TNFα downregulates cell-surface levels of GABAARs. A, Representative images of surface immunostaining for α1, α2, α5, β2/3, and γ2 GABAAR subunits in untreated (Control) and TNFα-treated (100 ng/ml, 45 min) cultures. Scale bars: low magnification, 20 μm; high magnification, 3 μm. B, Quantification data of subunit cluster area corresponding to subunits represented in A, showing a significant reduction in surface GABAARs in response to TNFα treatment (gray bars), for all subunits examined (α1, n = 119–120, **p < 0.01; α2, n = 189–261, ***p < 0.001; α5, n = 110–116, *p < 0.05; β2/3, n = 210–258, ***p < 0.001; γ2, n = 316–420, ***p < 0.001; all comparisons by two-tailed Student's t test). C, Representative Western blots of surface biotinylation assays comparing control and TNFα-treated (100 ng/ml, 45 min) cultures. D, Quantification of surface biotinylation assays showing a decrease in surface GABAAR β3 after TNFα treatment but no change in total β3 levels (n = 11 independent experiments; Mann–Whitney U test, *p < 0.05).
We also assessed the effects of TNFα treatment on surface levels of α5 subunit-containing receptor clusters, which are mainly located at extrasynaptic sites and primarily mediate tonic inhibition (Brünig et al., 2002; Caraiscos et al., 2004; Prenosil et al., 2006). We observed a much smaller but significant decrease in surface α5 cluster area (92.4 ± 1.98% of Ctrl; Fig. 2B), suggesting that tonic inhibition mediated by α5-containing receptors may not be reduced as robustly as phasic inhibition.
To further determine whether TNFα exerts its effects by increasing receptor endocytosis rather than by solely reducing receptor clustering, we used cell-surface biotinylation assays to measure cell-surface levels of GABAAR β3 (Fig. 2C,D). TNFα treatment (100 ng/ml, 45 min) decreased surface GABAAR β3 without affecting total levels (surface, 70.8 ± 12.9% of Ctrl; total, 99.3 ± 15.5% of Ctrl), providing additional evidence for increased receptor endocytosis.
A reduction of gephyrin clustering follows the reduction of surface GABAARs in response to TNFα
Next, we determined whether the effects of TNFα on GABAAR trafficking persisted over a short time course and whether other functionally important components of the GABAergic synapse were also altered over this time course. Using cell-surface immunostaining, we observed that surface GABAAR γ2 cluster area gradually decreased in the presence of TNFα, reaching a minimum at 3 h (62.0 ± 3.3% of Ctrl), followed by a partial recovery at 6 h (79.5 ± 4.4% of Ctrl; Fig. 3A,B). This indicated that the effects of TNFα were rapid and persistent but that a recovery may eventually occur, possibly as a result of ligand-induced TNFR internalization (Mosselmans et al., 1988; Higuchi and Aggarwal, 1994) or proteolytic degradation of the exogenously supplied TNFα (Vey et al., 1996). To determine whether over this time course TNFα reduces surface levels of GABAARs by mechanisms other than receptor trafficking (e.g., via reduced receptor protein synthesis or increased receptor degradation), we measured total levels of GABAAR β3 after 3 and 6 h of TNFα treatment. We observed no significant differences compared with untreated control cells (TNFα at 100 ng/ml for 3 h, 97.0 ± 0.8% of Ctrl; TNFα at 100 ng/ml for 6 h, 96.8 ± 3.5% of Ctrl; n = 3 independent experiments; Kruskal–Wallis one-way ANOVA, p = 0.24).
Figure 3.

TNFα-induced downregulation of surface GABAARs precedes a downregulation of gephyrin clustering, with no change in GAD65 puncta. A, Representative images from time course experiments with cultured neurons undergoing TNFα treatment ranging from 10 min to 6 h in duration. Top row shows a rapid downregulation in surface levels of the γ2 subunit in response to TNFα. Middle row represents a matching set of experiments in which cells were permeabilized and immunostained for gephyrin (white) and MAP2 (blue), showing a slower downregulation of cluster area than for γ2. Bottom row represents another matching set of experiments in which cells were permeabilized and immunostained for GAD65 (white) and MAP2 (blue), showing no change in puncta area. Scale bars: low magnification, 20 μm; high magnification, 1.5 μm. B, Quantification data of surface γ2 cluster area, over the 6 h time course of TNFα treatment (n = 120–158 images per time point; one-way ANOVA, Tukey's post hoc test, **p < 0.01, ***p < 0.001). C, Quantification data of gephyrin cluster area normalized to MAP2 area (white boxes). MAP2 area (dark circles) was unaltered (n = 65–82 images per time point; one-way ANOVA, Tukey's post hoc test, *p < 0.05, ***p < 0.001). D, Quantification data of GAD65 puncta area normalized to MAP2 area (white boxes). MAP2 area (dark circles) was unaltered (n = 80–112 images per time point; one-way ANOVA, Tukey's post hoc test, no significant differences).
We hypothesized that exposure to TNFα may also affect clustering of gephyrin—a postsynaptic scaffolding protein present at most inhibitory synapses (Tretter et al., 2012). Multiple lines of evidence indicate that gephyrin contributes significantly to the stabilization of GABAARs at postsynaptic sites (Kneussel et al., 1999; Lévi et al., 2004; Yu et al., 2007; Tretter et al., 2008; Mukherjee et al., 2011). We observed a significant downregulation of gephyrin cluster area at 3 h (84.5 ± 4.1% of Ctrl) and 6 h (66.6 ± 3.2% of Ctrl) of TNFα treatment, whereas shorter treatments of 10 and 45 min elicited only small, nonsignificant decreases (96.4 ± 3.6% of Ctrl at 10 min; 93.0 ± 4.2% of Ctrl at 45 min; Fig. 3A,C). This effect was primarily attributable to reduced cluster density (100.3 ± 3.5% of Ctrl at 10 min; 95.7 ± 4.2% of Ctrl at 45 min; 85.2 ± 3.9% of Ctrl at 3 h; 70.1 ± 1.2% of Ctrl at 6 h) rather than reduced average cluster size (96.4 ± 1.2% of Ctrl at 10 min; 97.5 ± 1.5% of Ctrl at 45 min; 99.5 ± 3.3% of Ctrl at 3 h; 95.7 ± 2.4% of Ctrl at 6 h). Together, these findings suggest that GABAAR clusters are initially lost mainly as a result of endocytosis but that, after longer exposures to TNFα (>3 h), a reduction in gephyrin clustering may also destabilize synaptic GABAARs.
Because we observed a decrease in mIPSC frequency (Fig. 1C), we asked whether correlates of GABA synthesis or the number of GABA release sites could be altered by TNFα. To address this possibility, we immunostained for GAD65—the rate-limiting enzyme in the production of GABA from glutamate at nerve terminals (Buddhala et al., 2009). GAD65 is aggregated in puncta at inhibitory presynaptic terminals and its expression is subject to activity-dependent homeostatic regulation (Rannals and Kapur, 2011; Lau and Murthy, 2012). Over the same 6 h time course of TNFα treatment as for surface GABAAR γ2 and gephyrin, we observed no significant changes in GAD65 puncta area (95.1 ± 3.8% of Ctrl at 10 min; 99.4 ± 3.3% of Ctrl at 45 min; 105.3 ± 4.4% of Ctrl at 3 h; 94.6 ± 4.1% of Ctrl at 6 h; Fig. 3A,D), nor any significant changes in puncta density (95.9 ± 3.6% of Ctrl at 10 min; 106.2 ± 3.7% of Ctrl at 45 min; 114.1 ± 4.6% of Ctrl at 3 h; 105.7 ± 4.7% of Ctrl at 6 h) or average puncta size (102.0 ± 2.5% of Ctrl at 10 min; 97.9 ± 2.3% of Ctrl at 45 min; 95.0 ± 1.9% of Ctrl at 3 h; 94.1 ± 2.4% of Ctrl at 6 h). Therefore, the mIPSC frequency decrease caused by TNFα is unlikely to be due to a decrease in the number of GABA release sites. As well, because average puncta size was not affected, TNFα does not appear to affect the production of GABA at synaptic sites.
We costained for MAP2 to normalize gephyrin and GAD65 measurements to somatodendritic area, as well as to assess somatodendritic integrity over a prolonged exposure to TNFα. We observed no significant differences in MAP2 area in either set of experiments (Fig. 3C,D, plotted as “MAP2 area only”), indicating that TNFα does not exert any overt deleterious effects on neuronal structure and integrity under our conditions.
TNFα signals through neuronal TNFR1, p38 MAPK, PI3K, and PP1 to downregulate surface GABAAR levels and GABAergic neurotransmission
To identify signal transduction mechanisms, we first examined the relative contribution of TNFR1 versus TNFR2 signaling to TNFα-dependent GABAAR trafficking. Both TNFRs are constitutively expressed throughout the CNS by neurons and glia (Dopp et al., 1997; Zhang et al., 2010; Brambilla et al., 2011). Knowing that AMPAR trafficking in response to TNFα occurs via neuronal TNFR1 (Stellwagen et al., 2005; He et al., 2012) and that soluble TNFα preferentially signals through TNFR1 (McCoy and Tansey, 2008), we predicted that TNFR1 would be required in this case. Indeed, when selectively blocking either TNFR1 or TNFR2 using neutralizing antibodies, we observed that blockade of TNFR1 signaling prevented the TNFα-induced downregulation of surface GABAAR γ2 cluster area (109.2 ± 4.7% of TNFR1 control), whereas blockade of TNFR2 did not (81.4 ± 4.0% of TNFR2 control; Fig. 4A,B). Because TNFα can also control the release of glutamate from astrocytes (Domercq et al., 2006; Santello et al., 2011), we assessed whether the effect occurred via direct activation of neuronally expressed TNFR1 or via glial TNFR1 activation followed by release of a glial factor that would control GABAAR trafficking. To this end, we prepared Banker cocultures (Kaech and Banker, 2006) using WT or TNFR1−/− glial feeder layers in combination with either WT or TNFR1−/− neurons. When using a WT glial feeder layer, mIPSC amplitude was reduced in WT neurons after TNFα treatment (Ctrl, 59.5 ± 5.2 pA; TNFα, 31.4.0 ± 3.6 pA) but was unaffected in TNFR1−/− neurons (Ctrl, 59.3 ± 7.1 pA; TNFα, 58.2 ± 4.0 pA; Fig. 4C,D). When using a TNFR1−/− glial feeder layer, we still observed a reduction in mIPSCs recorded from WT neurons (Ctrl, 56.7 ± 3.6 pA; TNFα, 36.8 ± 3.2 pA; Fig. 4C,D). Therefore, we conclude that neuronal TNFR1 controls GABAAR trafficking.
Signal transduction via TNFR1 involves assembly of adaptor proteins in a signaling complex around the intracellular “death domain” of the receptor, resulting in activation of various downstream targets and various cellular responses, depending on cell type and cell state (Sriram and O'Callaghan, 2007; McCoy and Tansey, 2008; Zhang et al., 2009). However, little is known about the intracellular substrates of TNFR1 that control receptor trafficking in neurons; only PI3K is known to be required for increasing surface levels of AMPARs (Stellwagen et al., 2005). Therefore, using pharmacological inhibitors, we sought to identify such signaling components, aiming to find a link between TNFα signaling and mechanisms mediating GABAAR endocytosis. Given the rapid effects of TNFα on surface levels of GABAARs, we focused on inhibitors of kinases and phosphatases known to regulate GABAAR trafficking (Luscher et al., 2011) or known to be downstream of TNFR1 (Grivennikov et al., 2006; Zhang et al., 2010). We found that blocking PKA (with KT 5720) or PKC (with Gö 6983)—kinases that phosphorylate GABAARs (McDonald et al., 1998; Brandon et al., 2000, 2003)—did not alter the effects of TNFα (KT 5720 alone, 100.0 ± 3.9% of DMSO alone; KT 5720 + TNFα, 59.1 ± 2.8% of DMSO alone; Gö 6983 alone, 100.0 ± 6.6% of DMSO alone; Gö 6983 + TNFα, 73.7 ± 3.3% of DMSO alone; Fig. 5A,B). However, preincubation with inhibitors of PI3K (wortmannin) or p38 MAPK (SB 202190) blocked the TNFα-induced reduction in surface γ2 subunit cluster area (wortmannin alone, 73.5 ± 5.4% of DMSO alone; wortmannin + TNFα, 83.1 ± 3.9% of DMSO alone; SB 202190 alone, 68.3 ± 6.6% of DMSO alone; SB 202190 + TNFα, 71.7 ± 4.1% of DMSO alone; Fig. 5A,B). Because wortmannin alone and SB 202190 alone reduced γ2 cluster area, it is not clear whether an additional decrease in response to TNFα was occluded in these cases. We targeted PP1 and PP2A using two different concentrations of okadaic acid (0.05 vs 0.5 μm), because lower concentrations are known to preferentially block PP2A, whereas higher concentrations also block PP1 (Cohen et al., 1989; Surmeier et al., 1995; Suh et al., 2013). Interestingly, we found that we could block the effect of TNFα with 0.5 μm okadaic acid but not with 0.05 μm, suggesting that PP1 but not PP2A is required (0.05 μm okadaic acid alone, 103.2 ± 5.9% of DMSO alone; 0.05 μm okadaic acid + TNFα, 77.5 ± 5.5% of DMSO alone; 0.5 μm okadaic acid alone, 96 ± 5.8% of DMSO alone; 0.5 μm okadaic acid + TNFα, 98.3 ± 4.7% of DMSO alone; Fig. 5A,B). Lastly, we found that calcineurin activity (blocked with cyclosporin A) was not required in this signaling pathway (cyclosporin A alone, 90.2 ± 9.1% of DMSO alone; cyclosporin A + TNFα, 60.2 ± 3.3% of DMSO alone). Because PP1/PP2A activity has been implicated previously in GABAAR endocytosis (Jovanovic et al., 2004; Kanematsu et al., 2006, 2007; Lin et al., 2011) whereas calcineurin activity has been implicated in GABAAR cluster dispersal (Bannai et al., 2009; Muir et al., 2010), our findings are again consistent with an endocytosis-dependent effect.
Figure 5.

Downregulation of surface GABAAR γ2 levels in response to TNFα is blocked by inhibitors of PI3K, p38 MAPK, and PP1/PP2A. A, Representative dendritic regions of surface immunostaining for GABAAR γ2 subunit, from control (Ctrl) versus TNFα-treated (100 ng/ml, 45 min) cultured neurons, preincubated with DMSO (solvent, 0.1%) or inhibitors of PKA [KT 5720 (KT), 1 μm], PKC [Gö 6983 (Gö), 1 μm], PI3K [wortmannin (Wortm), 100 nm], p38 MAPK [SB 202190 (SB), 10 μm], calcineurin [cyclosporin A (CsA), 20 μm], and PP1/PP2A (okadaic acid (OKA), 0.05 or 0.5 μm). Scale bar, 3 μm. B, Quantification data corresponding to conditions in A, indicating that TNFα-induced downregulation of surface γ2 cluster area is blocked by preincubation with wortmannin, SB 2022190, and 0.5 μm okadaic acid but not KT 5720, Gö 6983, cyclosporin A, or 0.05 μm okadaic acid (n = 60–228 images in each condition; two-way ANOVA, Bonferroni's post hoc test, *p < 0.05, **p < 0.01, ***p < 0.001; n.s., not significant).
To define which phosphatase is required for TNFα-dependent trafficking of synaptic GABAARs, we assessed the effects of TNFα on mIPSC amplitude after simultaneous blockade of PP1 and PP2A (with 0.5 μm okadaic acid) versus selective blockade of PP1 (with tautomycetin; Mitsuhashi et al., 2001) or PP2A (with fostriecin; Walsh et al., 1997). Consistent with immunostaining experiments, inhibition of PP1/PP2A blocked the TNFα-induced decrease in mIPSC amplitude (DMSO alone, 56.8 ± 3.9 pA; DMSO + TNFα, 43.3 ± 3.8 pA; okadaic acid alone, 62.3 ± 4.7 pA; okadaic acid + TNFα, 58.1 ± 4.4 pA; Fig. 6A,B). Selective inhibition of PP1 also blocked the effects of TNFα (tautomycetin alone, 44.4 ± 4.8 pA; tautomycetin + TNFα, 55.4 ± 4.0 pA), whereas selective inhibition of PP2A did not [dimethyl formamide (DMF) alone, 50.5 ± 3.2 pA; DMF + TNFα, 35.2 ± 2.2 pA; fostriecin alone, 51.3 ± 5.1 pA; fostriecin + TNFα, 32.5 ± 2.8 pA]. Interestingly, the effects of TNFα on mIPSC frequency were reversed by okadaic acid and tautomycetin but unaffected by fostriecin (DMSO alone, 0.80 ± 0.10 Hz; DMSO + TNFα, 0.59 ± 0.07 Hz; okadaic acid alone, 1.45 ± 0.18 Hz; okadaic acid + TNFα, 1.79 ± 0.23 Hz; tautomycetin alone, 0.84 ± 0.14 Hz; tautomycetin + TNFα, 1.42 ± 0.22 Hz; DMF alone, 1.35 ± 0.24 Hz; DMF + TNFα, 0.84 ± 0.19 Hz; fostriecin alone, 1.39 ± 0.21 Hz; fostriecin + TNFα, 0.78 ± 0.13 Hz). We conclude that TNFα downregulates GABAergic neurotransmission through PP1 but not PP2A activity.
Figure 6.

TNFα downregulates GABAergic neurotransmission through PP1 but not PP2A. A, mIPSCs were recorded from control or TNFα-treated (100 ng/ml, 45 min) cultured neurons (preincubated with solvent alone or inhibitor). DMSO (0.1%) was used as a solvent for okadaic acid (OKA) and tautomycetin (TMT); DMF (0.1%) was used as a solvent for fostriecin (FOST). Shown are representative traces for control with DMSO preincubation, TNFα-treated with DMSO or DMF preincubation, TNFα-treated with okadaic acid preincubation (PP1/PP2A inhibitor, 0.5 μm), TNFα-treated with tautomycetin preincubation (PP1 inhibitor, 10 nm), and TNFα-treated with fostriecin preincubation (PP2A inhibitor, 10 nm). B, Group data of average mIPSC amplitudes for all conditions, showing that preincubation with okadaic acid or tautomycetin blocks the TNFα-induced downregulation of mIPSC amplitude, whereas preincubation with fostriecin does not (n = 16–25 cells in each condition; two-way ANOVA, Bonferroni's post hoc test, *p < 0.05, **p < 0.01, ***p < 0.001; n.s., not significant).
TNFα dephosphorylates GABAAR β3 S408/409 by sequentially activating p38 MAPK and PI3K while enhancing the association of PP1α with β3
To further characterize the signaling pathway linking neuronal TNFR1 to GABAAR trafficking, we focused on the three molecular components we had identified using pharmacological inhibition: p38 MAPK, PI3K, and PP1. We used Western blots to assess activation of p38 and PI3K using antibodies specific for phospho-p38 MAPK and phospho-Akt—a downstream target of PI3K activation. Phosphorylation of both p38 MAPK and Akt was significantly upregulated in response to TNFα (100 ng/ml, 45 min; phospho-p38: DMSO + TNFα, 150.0 ± 9.8% of DMSO alone; phospho-Akt: DMSO + TNFα, 182.5 ± 34.6% of DMSO alone; Fig. 7A,B), indicating that both signaling components are activated. In addition, using pharmacological blockade of p38 MAPK (using SB 202190) or PI3K (using wortmannin), we determined that TNFα-induced phosphorylation of p38 MAPK is unaffected by wortmannin (wortmannin alone, 108.0 ± 2.6% of DMSO alone; wortmannin + TNFα, 160.5 ± 8.5% of DMSO alone; Fig. 7A,B), whereas TNFα-induced phosphorylation of Akt is blocked by SB 202190 (SB alone, 83.5 ± 12.5% of DMSO alone; SB + TNFα, 59.6 ± 21.9% of DMSO alone; Fig. 7A,B), indicating that p38 MAPK activation occurs upstream of PI3K activation in this signaling pathway.
Figure 7.

TNFα dephosphorylates GABAAR β3 S408/409 by sequentially activating p38 MAPK and PI3K and enhancing the association of PP1α with β3 subunits. A, Representative Western blots of phospho- p38 MAPK, total p38 MAPK, phospho-Akt, and total Akt in lysates from control or TNFα-treated (100 ng/ml, 45 min) cultures, preincubated with DMSO (solvent, 0.1%) or inhibitors of PI3K [wortmannin (Wm), 100 nm] and p38 MAPK [SB 202190 (SB), 10 μm]. B, Quantification data showing an increase in phospho-p38 MAPK with TNFα treatment, unaffected by PI3K inhibition (left), and an increase in phosphorylation of the PI3K downstream effector Akt, blocked by p38 MAPK inhibition (right) (n = 4–5 independent experiments; Mann–Whitney U test, *p < 0.05, **p < 0.01; n.s., not significant). C, Representative Western blots of phospho-S408/409 GABAAR β3, total GABAAR β3, and α-tubulin in lysates from control or TNFα-treated (100 ng/ml, 45 min) cultures, preincubated with DMSO (solvent, 0.1%) or inhibitors of PI3K [wortmannin (Wm), 100 nm], p38 MAPK [SB 202190 (SB), 10 μm], PP1/PP2A [okadaic acid (OKA), 0.5 μm], PP1 [tautomycetin (TMT), 10 nm], and PP2A [fostriecin (FOST), 10 nm]. D, Quantification data showing a decrease in GABAAR β3 phospho-S408/409, blocked by inhibition of PI3K, p38 MAPK, and PP1, but not by inhibition of PP2A (n = 5 independent experiments; Mann–Whitney U test, **p < 0.01, ***p < 0.001; n.s., not significant). E, Representative Western blots of coimmunoprecipitation experiments in which the GABAAR β3 subunit was immunoprecipitated (IP) from untreated (Ctrl) or TNFα-treated (100 ng/ml, 30 min) cultures; blots were then probed for GABAAR β3 and for the catalytic subunits of PP1 (PP1α) and PP2A (PP2Ac). F, Quantification showing that TNFα treatment enhances the association of PP1α but not PP2Ac with β3 subunits (n = 5–6 independent experiments; Mann–Whitney U test, **p < 0.01; n.s., not significant).
Knowing that PP1 can localize to inhibitory synapses (Bausen et al., 2010) and that dephosphorylation of GABAAR β3 should favor receptor endocytosis (Kittler et al., 2005; Smith et al., 2012), we hypothesized that PP1 acts downstream of p38 MAPK and PI3K to directly dephosphorylate GABAAR β3. We assessed the phosphorylation state of β3 subunits using a phospho-specific antibody detecting phosphorylated β3 S408/409 (Fig. 7C). We focused on this site for two reasons. First, whereas other serine sites on GABAARs (e.g., γ2 S327) are known to be regulated by calcineurin (Bannai et al., 2009; Muir et al., 2010), β3 S408/409 is the only site known to be regulated by PP1 (for review, see Luscher et al., 2011). Second, this site is located in the β1–β3 intracellular domains, in a basic patch sorting motif that directly binds the μ2 subunit of the clathrin adaptor AP2 to mediate dynamin-dependent endocytosis (Kittler et al., 2005; Smith et al., 2012). Phosphorylation of this site disrupts the interaction with μ2–AP2 and increases synaptic GABAAR content (Kittler et al., 2005). We observed a significant reduction in phospho-S408/409 in response to TNFα treatment (100 ng/ml, 45 min; DMSO + TNFα, 62.3 ± 3.7% of DMSO alone; Fig. 7C,D), consistent with an increased rate of GABAAR endocytosis and a loss of surface GABAAR β3. However, pretreatment with either 0.5 μm okadaic acid or tautomycetin, but not with fostriecin, blocked TNFα-induced β3 S408/409 dephosphorylation (Fig. 7C,D), indicating that this is a PP1-mediated dephosphorylation (okadaic acid + TNFα, 91.0 ± 6.7% of okadaic acid alone; tautomycetin + TNFα, 113.6 ± 14.5% of tautomycetin alone; fostriecin + TNFα, 55.4 ± 16.1% of fostriecin alone; Fig. 7D). Furthermore, inhibition of p38 MAPK or PI3K also blocked TNFα-induced β3 S408/409 dephosphorylation (Fig. 7C,D), indicating that activation of p38 MAPK and PI3K is required for receptor dephosphorylation (wortmannin + TNFα, 112.5 ± 7.2% of wortmannin alone; SB 202190 + TNFα, 101.6 ± 5.8% of SB 202190 alone; Fig. 7D).
Previous studies have suggested that an increased abundance of protein phosphatase associated with the GABAAR protein complex correlates with increased dephosphorylation of β3 and subsequent endocytosis of GABAARs (Jovanovic et al., 2004; Kanematsu et al., 2006). The association of PP1 and PP2A with the β3 subunit is facilitated by the adaptor/regulatory proteins PRIP-1 and PRIP-2, which selectively bind to the intracellular domains of β1–β3 subunits but not α1–α3 or γ2 subunits (Terunuma et al., 2004; Kanematsu et al., 2006, 2007; Yanagihori et al., 2006). We used coimmunoprecipitation to determine whether the association of β3 with the catalytic subunits of PP1 (PP1α) or PP2A (PP2Ac) was regulated by TNFα (Fig. 7E,F). Consistent with mIPSC recordings showing a requirement of PP1 but not PP2A activity, we observed an increase in the association of PP1α with β3 but no change in the association of PP2Ac with β3 (PP1α, 180.7 ± 28.3% of Ctrl; PP2Ac, 94.6 ± 13.1% of Ctrl; Fig. 7F). This suggests that, in response to TNFα, PP1 is targeted to the intracellular domain of the β3 subunit, which may enable it to dephosphorylate key residues that regulate interactions with the endocytic machinery.
A similar TNFα signaling pathway regulates GABAergic synapses in acute hippocampal slices
To extend our findings from cultured hippocampal neurons to a more intact hippocampal preparation, we treated acute hippocampal slices from young adult mice with TNFα (100 ng/ml, 1 h) and assessed activation of the p38 MAPK–PI3K–β3 S408/409 signaling pathway. In agreement with our results from culture experiments, we observed a significant increase in phospho-p38 MAPK (179.8 ± 20.2% of Ctrl), a trend for increased phospho-Akt (144.3 ± 20.7% of Ctrl) and a significant decrease in β3 S408/409 phosphorylation (68.6 ± 8.9% of Ctrl; Fig. 8A,B). Similarly, surface GABAAR β3 was decreased (67.5 ± 12.9% of Ctrl), whereas, in accordance with previous findings (Stellwagen et al., 2005), surface GluA1 was increased (150.1 ± 20.53% of Ctrl; Fig. 8A,B). We observed no changes in total p38 MAPK, total Akt, total GABAAR β3, or total GluA1 (p38 MAPK, 102.7 ± 6.1% of Ctrl; Akt, 87.5 ± 9.7% of Ctrl; β3, 103.7 ± 6.2% of Ctrl; GluA1, 108.9 ± 9.4% of Ctrl; n = 8–9 animals in each condition; Mann–Whitney U test, p > 0.05 for each). To determine whether in acute hippocampal slices TNFα (100 ng/ml, >1 h) decreases synaptic GABAAR content in a PP1-dependent manner, we recorded mIPSCs from CA1 pyramidal neurons, in slices pretreated with either DMSO or the PP1-specific inhibitor tautomycetin (Fig. 8C). Consistent with results from culture recordings (Fig. 6), TNFα treatment caused a significant decrease in mIPSC amplitude that was blocked by pretreatment with tautomycetin (DMSO alone, 51.8 ± 3.2 pA; DMSO + TNFα, 39.8 ± 2.7 pA; tautomycetin alone, 48.4 ± 2.0 pA; tautomycetin + TNFα, 47.1 ± 2.5 pA; Fig. 8C,D). However, we did not observe any differences in mIPSC frequency (DMSO alone, 10.6 ± 0.9 Hz; DMSO + TNFα, 10.8 ± 1.4 Hz; tautomycetin alone, 8.1 ± 0.7 Hz; tautomycetin + TNFα, 8.5 ± 0.7 Hz; n = 10–13 cells; two-way ANOVA, Bonferroni's post hoc test, p > 0.05), suggesting that the effect observed in culture (Fig. 1) is either culture specific or does not occur at GABAergic synapses onto CA1 pyramidal neurons.
Figure 8.
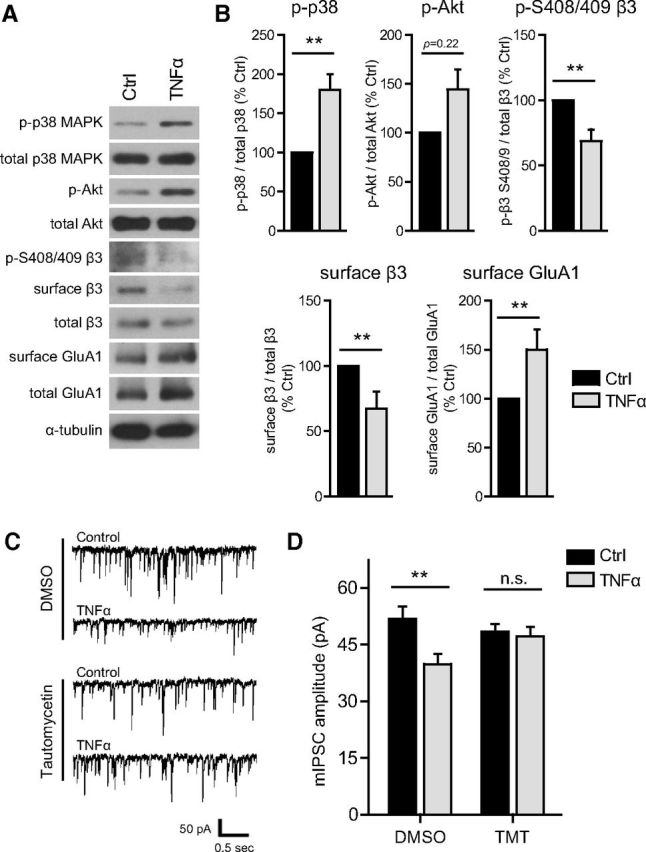
A similar TNFα signaling pathway regulates inhibitory synapses in acute hippocampal slices. A, Representative Western blots of phospho-p38 MAPK, total p38 MAPK, phospho-Akt, total Akt, phospho-S408/409 GABAAR β3, surface (biotinylated) GABAAR β3, total lysate GABAAR β3, surface (biotinylated) GluA1, total lysate GluA1, and α-tubulin, from acute slices of mouse dorsal hippocampus, either untreated (Ctrl) or treated with TNFα (100 ng/ml, 1 h). B, Quantification data corresponding to experiments represented in A, showing a TNFα-induced increase in phospho-p38 MAPK, an increase in phospho-Akt, a decrease in phospho-S408/409 β3, a decrease in surface levels of GABAAR β3, and an increase in surface levels of GluA1 (n = 8–9 animals in each condition; Mann–Whitney U test, **p < 0.01). C, Sample traces of mIPSCs recorded from acute hippocampal slice CA1 pyramidal neurons, either control or TNFα-treated (100 ng/ml, >1 h), preincubated with either DMSO (0.1%) or tautomycetin (TMT, 10 nm in DMSO). D, Group data of average mIPSC amplitudes, showing that preincubation with tautomycetin blocks the TNFα-induced mIPSC amplitude reduction (n = 10–13 cells in each condition; two-way ANOVA, Bonferroni's post hoc test, **p < 0.01; n.s., not significant).
Discussion
In this study, we characterized the effects of TNFα at GABAergic hippocampal synapses, defining the signaling pathway through which it controls GABAAR endocytosis. We demonstrated that TNFα rapidly downregulates GABAergic synaptic strength and surface GABAARs via neuronal TNFR1 activation, p38 MAPK, PI3K, PP1, and dynamin GTPase activity. This is accompanied by an enhanced association of PP1α with GABAAR β3 and dephosphorylation of β3 S408/409. Furthermore, on a longer timescale, gephyrin cluster density is also reduced. Together, these findings support a model whereby TNFα rapidly and persistently tunes down inhibition by enhancing GABAAR endocytosis through PP1-dependent dephosphorylation of β3 subunits, which is then compounded by a loss of synaptic scaffolding support for GABAARs (Fig. 9).
Figure 9.

Proposed model for TNFα-induced downregulation of GABAergic neurotransmission at hippocampal synapses. Compared with normal conditions (left), conditions associated with elevated levels of TNFα (right) lead to diminished postsynaptic responses to GABA as a result of lower levels of synaptic GABAARs. Activation of TNFR1 signals through p38 MAPK, PI3K, and PP1, which in turn dephosphorylates β3 S408/409 to enhance GABAAR endocytosis. A reduction in synaptic GABAARs is followed by a reduction in gephyrin clustering. TNFα may also affect presynaptic function by reducing synaptic vesicle release probability at some synapses, without affecting GABA synthesis.
A large number of functionally distinct GABAAR subtypes are assembled from various subunit combinations (Olsen and Sieghart, 2009). Our results indicate that receptors containing subunits α1, α2, β2/3, and γ2—which are preferentially localized to synapses (Mangan et al., 2005; Michels and Moss, 2007)—are all reduced by 25–30% on the cell surface. Therefore, despite the differential subcellular distribution of α1- and α2-containing receptors in vivo (Prenosil et al., 2006; Thomson and Jovanovic, 2010; Panzanelli et al., 2011), TNFα has similar effects on both receptor subtypes. In agreement with this, we observed no significant difference in mIPSC decay kinetics, which would be expected to change with an altered ratio of α1- to α2-containing receptors remaining at the synapse (Okada et al., 2000; Bosman et al., 2005; Schneider Gasser et al., 2007). Furthermore, we observed a full shift in the cumulative distribution of mIPSC amplitudes, suggesting that all synapses were affected to a similar extent. Such a broad effect on synaptic receptor subtypes is probably achieved by controlling endocytosis through common components, such as the β subunits. However, because we observed that surface levels of clustered α5-containing receptors—which also coassemble with β subunits (Glykys and Mody, 2007)—are only modestly decreased, it is possible that some level of specificity is achieved at the subcellular level by differential activation of signaling molecules present at synaptic versus extrasynaptic compartments. Furthermore, some cell-type specificity of the effect is also likely because TNFα did not reduce surface levels of α1 subunits in a subpopulation of cells expressing high levels of α1. Although we are not certain what neuron subtype(s) this population represents, it probably consists of a mixture of hippocampal interneurons and a small fraction of subicular neurons introduced during the dissection procedure; both of these neuron types are known to express higher levels of the α1 subunit (Brünig et al., 2002; Yu et al., 2006; Schneider Gasser et al., 2007). It is possible that such neuron subtypes do not express some necessary component of the signaling pathway linking TNFα to GABAAR trafficking.
In addition to GABAAR content at the synapse, TNFα also regulated clustering of gephyrin, a scaffolding protein that helps maintain a high density of GABAARs at the inhibitory synapse by directly interacting with GABAAR subunits (Tretter et al., 2012; Kowalczyk et al., 2013). We determined that, whereas surface γ2 clusters were rapidly reduced within 45 min, gephyrin clustering was reduced more slowly, reaching a minimum after 3 h. Because loss of gephyrin is known to disrupt GABAAR clustering by increasing receptor lateral mobility (Jacob et al., 2005; Yu et al., 2007), the TNFα-induced slow reduction in gephyrin clustering is likely to destabilize synaptic GABAAR clusters, thus serving as a second mechanism via which TNFα weakens inhibitory synapses. We suspect that the decrease in gephyrin clustering occurs in response to the initial reduction in surface levels of GABAARs. This would be consistent with reports documenting a loss of gephyrin clusters in α1–α3 and γ2 knock-out mice (Essrich et al., 1998; Schweizer et al., 2003; Studer et al., 2006; Patrizi et al., 2008; Panzanelli et al., 2011). However, we cannot exclude the possibility that a separate TNFα signaling pathway directly regulates gephyrin cluster density, perhaps by regulating phosphorylation of gephyrin (Tyagarajan et al., 2011).
TNFα is known to traffic AMPARs to the surface via neuronal TNFR1 (Stellwagen et al., 2005), whereas astrocytic TNFR1 can control the release of glutamate from astrocytes (Domercq et al., 2006). We now show that activation of neuronally expressed TNFR1 is necessary for the effects of TNFα on GABAAR trafficking, whereas astrocytic TNFR1 does not play a role in this pathway. Therefore, it is apparent that TNFα controls neurotransmitter receptor trafficking by acting directly on neurons rather than by acting on glial TNFR1 to induce the release of a glial factor that would subsequently signal to neurons. Furthermore, the divergence in signaling pathways controlling AMPARs exocytosis versus GABAAR endocytosis must occur downstream of neuronal TNFR1. We provide evidence that p38 MAPK and PI3K are sequentially activated by TNFα and are required for TNFα-dependent GABAAR endocytosis. Through activation of Akt, PI3K is known to regulate insulin-dependent translocation of GABAARs to the neuronal cell surface (Wang et al., 2003). However, PI3K can also control PP1 activity (Ragolia and Begum, 1998; De Luca et al., 1999), suggesting that it can indirectly lead to receptor dephosphorylation.
Using several experimental approaches, we identified PP1 as a mediator of TNFα-dependent GABAAR trafficking in cultured hippocampal neurons and acute hippocampal slice CA1 pyramidal neurons. In addition, we have shown that, in response to TNFα treatment, GABAAR β3 is dephosphorylated at S408/409 via a signaling pathway that requires activation of p38 MAPK, PI3K, and PP1 but not PP2A. These data support a model of regulated endocytosis occurring via PP1-dependent dephosphorylation of β3 subunits, which enhances the proportion of β3 subunits bound to AP2 and consequently enhances dynamin-dependent endocytosis of GABAARs. Therefore, TNFα appears to engage a mechanism shown previously to mediate constitutive endocytosis of GABAARs through phospho-dependent regulation of the β3–AP2 interaction (Kittler et al., 2005; Smith et al., 2012). A similar mechanism of constitutive endocytosis has been described based on phosphorylation of the γ2 subunit at Y365/367 (Kittler et al., 2008). Although we do not rule out TNFα-dependent regulation of γ2 phosphorylation, we suspect that this does not play a major role because we can completely block the effects of TNFα via serine/threonine phosphatase inhibition with okadaic acid, whereas phosphorylation of Y365/367 is regulated by tyrosine phosphatases (Brandon et al., 2001). Similarly, although phosphorylation of GABAARs can also affect channel gating (Brandon et al., 2002) and therefore could account for the decrease in mIPSC amplitude, we suspect this does not play a major role because the majority of the effect requires dynamin GTPase activity.
In culture experiments, we observed a decrease in mIPSC frequency in response to TNFα treatment, yet immunostaining for GAD65 revealed no effect on puncta density or size, suggesting no change in the number of GABA release sites or in GABA synthesis. We conclude that the decrease in event frequency is likely to be regulated by a decrease in release probability and perhaps to some extent by low-amplitude events falling below the detection threshold at higher concentrations of TNFα (>50 ng/ml). Although we did not observe an effect of TNFα treatment on the frequency of mIPSCs recorded from CA1 pyramidal neurons in acute slices, it is possible that the effect is nonetheless present at inhibitory synapses onto other hippocampal neuron subtypes. For example, TNFα controls astrocyte-mediated presynaptic modulation at excitatory synapses onto dentate gyrus neurons (Santello et al., 2011).
TNFα is an important mediator of homeostatic synaptic plasticity and is upregulated during prolonged periods of neuronal inactivity, when it is required for the inverse regulation of AMPAR and GABAAR trafficking (Stellwagen and Malenka, 2006). Our findings imply that such homeostatic downregulation of synaptic GABAAR content is mechanistically distinct from the heterosynaptic depression of inhibitory synapses (inhibitory long-term depression; Castillo et al., 2011), which requires either calcineurin (Lu et al., 2000) or PP2A (Lu et al., 2010) rather than PP1 to dephosphorylate the receptor.
TNFα is strongly upregulated in a number of neurological disorders, including epilepsy, autism spectrum disorder (ASD), and neurodegenerative diseases (Vezzani and Granata, 2005; Collins et al., 2012; Montgomery and Bowers, 2012; Onore et al., 2012). Mice lacking TNFR1 display reduced seizure susceptibility in the hippocampus, suggesting that TNFR1-dependent signaling enhances excitability in pathological states (Balosso et al., 2005). Similarly, impairment in GABAAR function achieved by knocking out the β3 subunit produces electroencephalographic abnormalities and seizures (DeLorey et al., 1998; Liljelund et al., 2005), and epilepsy is associated with a downregulation of gephyrin and GABAARs (González et al., 2013). Deficits in GABAergic neurotransmission are also implicated in ASD (Chao et al., 2010; Han et al., 2012), which is often accompanied by immune dysregulation and elevated levels of TNFα (Chez et al., 2007; Malik et al., 2011). It has been proposed that an elevation of the excitatory/inhibitory ratio, which is regulated by TNFα (Stellwagen et al., 2005), is a major cause of ASD (Rubenstein and Merzenich, 2003; Yizhar et al., 2011). In summary, we propose that our findings provide a mechanistic link between the inflammatory component of certain neurological diseases and phenotypes associated with deficits in GABAergic neurotransmission.
Footnotes
This work was supported by the Canadian Institutes of Health Research, the Natural Sciences and Engineering Research Council of Canada, the National Alliance for Research on Schizophrenia and Depression Young Investigator Award (D.S.), an Alexander Graham Bell Canada Graduate Doctoral Scholarship (H.P.), and a Doctoral Training Award from the Fonds de la recherche en santé du Québec (H.P.). We thank members of the Stellwagen laboratory and Drs. Keith Murai and Edward Ruthazer for helpful discussions and Meggie Stainforth-Dubois and Erin Downing for technical assistance.
The authors declare no competing financial interests.
References
- Arancibia-Cárcamo IL, Kittler JT. Regulation of GABA(A) receptor membrane trafficking and synaptic localization. Pharmacol Ther. 2009;123:17–31. doi: 10.1016/j.pharmthera.2009.03.012. [DOI] [PubMed] [Google Scholar]
- Balosso S, Ravizza T, Perego C, Peschon J, Campbell IL, De Simoni MG, Vezzani A. Tumor necrosis factor-alpha inhibits seizures in mice via p75 receptors. Ann Neurol. 2005;57:804–812. doi: 10.1002/ana.20480. [DOI] [PubMed] [Google Scholar]
- Bannai H, Lévi S, Schweizer C, Inoue T, Launey T, Racine V, Sibarita JB, Mikoshiba K, Triller A. Activity-dependent tuning of inhibitory neurotransmission based on GABAAR diffusion dynamics. Neuron. 2009;62:670–682. doi: 10.1016/j.neuron.2009.04.023. [DOI] [PubMed] [Google Scholar]
- Bausen M, Weltzien F, Betz H, O'Sullivan GA. Regulation of postsynaptic gephyrin cluster size by protein phosphatase 1. Mol Cell Neurosci. 2010;44:201–209. doi: 10.1016/j.mcn.2010.02.007. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Bosman LW, Heinen K, Spijker S, Brussaard AB. Mice lacking the major adult GABAA receptor subtype have normal number of synapses, but retain juvenile IPSC kinetics until adulthood. J Neurophysiol. 2005;94:338–346. doi: 10.1152/jn.00084.2005. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Ashbaugh JJ, Magliozzi R, Dellarole A, Karmally S, Szymkowski DE, Bethea JR. Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain. 2011;134:2736–2754. doi: 10.1093/brain/awr199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon NJ, Delmas P, Kittler JT, McDonald BJ, Sieghart W, Brown DA, Smart TG, Moss SJ. GABAA receptor phosphorylation and functional modulation in cortical neurons by a protein kinase C-dependent pathway. J Biol Chem. 2000;275:38856–38862. doi: 10.1074/jbc.M004910200. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Delmas P, Hill J, Smart TG, Moss SJ. Constitutive tyrosine phosphorylation of the GABA(A) receptor gamma 2 subunit in rat brain. Neuropharmacology. 2001;41:745–752. doi: 10.1016/S0028-3908(01)00121-6. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Jovanovic JN, Moss SJ. Multiple roles of protein kinases in the modulation of gamma-aminobutyric acid(A) receptor function and cell surface expression. Pharmacol Ther. 2002;94:113–122. doi: 10.1016/S0163-7258(02)00175-4. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Jovanovic JN, Colledge M, Kittler JT, Brandon JM, Scott JD, Moss SJ. A-kinase anchoring protein 79/150 facilitates the phosphorylation of GABA(A) receptors by cAMP-dependent protein kinase via selective interaction with receptor beta subunits. Mol Cell Neurosci. 2003;22:87–97. doi: 10.1016/S1044-7431(02)00017-9. [DOI] [PubMed] [Google Scholar]
- Brünig I, Scotti E, Sidler C, Fritschy JM. Intact sorting, targeting, and clustering of gamma-aminobutyric acid A receptor subtypes in hippocampal neurons in vitro. J Comp Neurol. 2002;443:43–55. doi: 10.1002/cne.10102. [DOI] [PubMed] [Google Scholar]
- Buddhala C, Hsu CC, Wu JY. A novel mechanism for GABA synthesis and packaging into synaptic vesicles. Neurochem Int. 2009;55:9–12. doi: 10.1016/j.neuint.2009.01.020. [DOI] [PubMed] [Google Scholar]
- Caraiscos VB, Elliott EM, You-Ten KE, Cheng VY, Belelli D, Newell JG, Jackson MF, Lambert JJ, Rosahl TW, Wafford KA, MacDonald JF, Orser BA. Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by alpha5 subunit-containing gamma-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A. 2004;101:3662–3667. doi: 10.1073/pnas.0307231101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Chiu CQ, Carroll RC. Long-term plasticity at inhibitory synapses. Curr Opin Neurobiol. 2011;21:328–338. doi: 10.1016/j.conb.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JL, Noebels JL, Rosenmund C, Zoghbi HY. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chez MG, Dowling T, Patel PB, Khanna P, Kominsky M. Elevation of tumor necrosis factor-alpha in cerebrospinal fluid of autistic children. Pediatr Neurol. 2007;36:361–365. doi: 10.1016/j.pediatrneurol.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Cohen P, Klumpp S, Schelling DL. An improved procedure for identifying and quantitating protein phosphatases in mammalian tissues. FEBS Lett. 1989;250:596–600. doi: 10.1016/0014-5793(89)80803-8. [DOI] [PubMed] [Google Scholar]
- Collins LM, Toulouse A, Connor TJ, Nolan YM. Contributions of central and systemic inflammation to the pathophysiology of Parkinson's disease. Neuropharmacology. 2012;62:2154–2168. doi: 10.1016/j.neuropharm.2012.01.028. [DOI] [PubMed] [Google Scholar]
- Crestani F, Lorez M, Baer K, Essrich C, Benke D, Laurent JP, Belzung C, Fritschy JM, Lüscher B, Mohler H. Decreased GABAA-receptor clustering results in enhanced anxiety and a bias for threat cues. Nat Neurosci. 1999;2:833–839. doi: 10.1038/12207. [DOI] [PubMed] [Google Scholar]
- DeLorey TM, Handforth A, Anagnostaras SG, Homanics GE, Minassian BA, Asatourian A, Fanselow MS, Delgado-Escueta A, Ellison GD, Olsen RW. Mice lacking the beta3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J Neurosci. 1998;18:8505–8514. doi: 10.1523/JNEUROSCI.18-20-08505.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca JP, Garnache AK, Rulfs J, Miller TB., Jr Wortmannin inhibits insulin-stimulated activation of protein phosphatase 1 in rat cardiomyocytes. Am J Physiol. 1999;276:H1520–H1526. doi: 10.1152/ajpheart.1999.276.5.H1520. [DOI] [PubMed] [Google Scholar]
- Domercq M, Brambilla L, Pilati E, Marchaland J, Volterra A, Bezzi P. P2Y1 receptor-evoked glutamate exocytosis from astrocytes: control by tumor necrosis factor-alpha and prostaglandins. J Biol Chem. 2006;281:30684–30696. doi: 10.1074/jbc.M606429200. [DOI] [PubMed] [Google Scholar]
- Dopp JM, Mackenzie-Graham A, Otero GC, Merrill JE. Differential expression, cytokine modulation, and specific functions of type-1 and type-2 tumor necrosis factor receptors in rat glia. J Neuroimmunol. 1997;75:104–112. doi: 10.1016/S0165-5728(97)00009-X. [DOI] [PubMed] [Google Scholar]
- Duveau V, Laustela S, Barth L, Gianolini F, Vogt KE, Keist R, Chandra D, Homanics GE, Rudolph U, Fritschy JM. Spatiotemporal specificity of GABAA receptor-mediated regulation of adult hippocampal neurogenesis. Eur J Neurosci. 2011;34:362–373. doi: 10.1111/j.1460-9568.2011.07782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essrich C, Lorez M, Benson JA, Fritschy JM, Lüscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat Neurosci. 1998;1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Frankola KA, Greig NH, Luo W, Tweedie D. Targeting TNF-alpha to elucidate and ameliorate neuroinflammation in neurodegenerative diseases. CNS Neurol Disord Drug Targets. 2011;10:391–403. doi: 10.2174/187152711794653751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschy JM, Paysan J, Enna A, Mohler H. Switch in the expression of rat GABAA-receptor subtypes during postnatal development: an immunohistochemical study. J Neurosci. 1994;14:5302–5324. doi: 10.1523/JNEUROSCI.14-09-05302.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschy JM, Panzanelli P, Tyagarajan SK. Molecular and functional heterogeneity of GABAergic synapses. Cell Mol Life Sci. 2012;69:2485–2499. doi: 10.1007/s00018-012-0926-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Mody I. Activation of GABAA receptors: views from outside the synaptic cleft. Neuron. 2007;56:763–770. doi: 10.1016/j.neuron.2007.11.002. [DOI] [PubMed] [Google Scholar]
- González MI, Cruz Del Angel Y, Brooks-Kayal A. Down-regulation of gephyrin and GABA(A) receptor subunits during epileptogenesis in the CA1 region of hippocampus. Epilepsia. 2013;54:616–624. doi: 10.1111/epi.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Kuprash DV, Liu ZG, Nedospasov SA. Intracellular signals and events activated by cytokines of the tumor necrosis factor superfamily: From simple paradigms to complex mechanisms. Int Rev Cytol. 2006;252:129–161. doi: 10.1016/S0074-7696(06)52002-9. [DOI] [PubMed] [Google Scholar]
- Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, Rubenstein JL, Scheuer T, de la Iglesia HO, Catterall WA. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature. 2012;489:385–390. doi: 10.1038/nature11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He P, Liu Q, Wu J, Shen Y. Genetic deletion of TNF receptor suppresses excitatory synaptic transmission via reducing AMPA receptor synaptic localization in cortical neurons. FASEB J. 2012;26:334–345. doi: 10.1096/fj.11-192716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Aggarwal BB. TNF induces internalization of the p60 receptor and shedding of the p80 receptor. J Immunol. 1994;152:3550–3558. [PubMed] [Google Scholar]
- Jacob TC, Bogdanov YD, Magnus C, Saliba RS, Kittler JT, Haydon PG, Moss SJ. Gephyrin regulates the cell surface dynamics of synaptic GABAA receptors. J Neurosci. 2005;25:10469–10478. doi: 10.1523/JNEUROSCI.2267-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob TC, Moss SJ, Jurd R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat Rev Neurosci. 2008;9:331–343. doi: 10.1038/nrn2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Thomas P, Kittler JT, Smart TG, Moss SJ. Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABA(A) receptor phosphorylation, activity, and cell-surface stability. J Neurosci. 2004;24:522–530. doi: 10.1523/JNEUROSCI.3606-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kaneko M, Stellwagen D, Malenka RC, Stryker MP. Tumor necrosis factor-alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron. 2008;58:673–680. doi: 10.1016/j.neuron.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanematsu T, Yasunaga A, Mizoguchi Y, Kuratani A, Kittler JT, Jovanovic JN, Takenaka K, Nakayama KI, Fukami K, Takenawa T, Moss SJ, Nabekura J, Hirata M. Modulation of GABA(A) receptor phosphorylation and membrane trafficking by phospholipase C-related inactive protein/protein phosphatase 1 and 2A signaling complex underlying brain-derived neurotrophic factor-dependent regulation of GABAergic inhibition. J Biol Chem. 2006;281:22180–22189. doi: 10.1074/jbc.M603118200. [DOI] [PubMed] [Google Scholar]
- Kanematsu T, Fujii M, Mizokami A, Kittler JT, Nabekura J, Moss SJ, Hirata M. Phospholipase C-related inactive protein is implicated in the constitutive internalization of GABAA receptors mediated by clathrin and AP2 adaptor complex. J Neurochem. 2007;101:898–905. doi: 10.1111/j.1471-4159.2006.04399.x. [DOI] [PubMed] [Google Scholar]
- Kasugai Y, Swinny JD, Roberts JD, Dalezios Y, Fukazawa Y, Sieghart W, Shigemoto R, Somogyi P. Quantitative localisation of synaptic and extrasynaptic GABAA receptor subunits on hippocampal pyramidal cells by freeze-fracture replica immunolabelling. Eur J Neurosci. 2010;32:1868–1888. doi: 10.1111/j.1460-9568.2010.07473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Delmas P, Jovanovic JN, Brown DA, Smart TG, Moss SJ. Constitutive endocytosis of GABAA receptors by an association with the adaptin AP2 complex modulates inhibitory synaptic currents in hippocampal neurons. J Neurosci. 2000;20:7972–7977. doi: 10.1523/JNEUROSCI.20-21-07972.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Chen G, Honing S, Bogdanov Y, McAinsh K, Arancibia-Carcamo IL, Jovanovic JN, Pangalos MN, Haucke V, Yan Z, Moss SJ. Phospho-dependent binding of the clathrin AP2 adaptor complex to GABAA receptors regulates the efficacy of inhibitory synaptic transmission. Proc Natl Acad Sci U S A. 2005;102:14871–14876. doi: 10.1073/pnas.0506653102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Chen G, Kukhtina V, Vahedi-Faridi A, Gu Z, Tretter V, Smith KR, McAinsh K, Arancibia-Carcamo IL, Saenger W, Haucke V, Yan Z, Moss SJ. Regulation of synaptic inhibition by phospho-dependent binding of the AP2 complex to a YECL motif in the GABAA receptor gamma2 subunit. Proc Natl Acad Sci U S A. 2008;105:3616–3621. doi: 10.1073/pnas.0707920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneussel M, Brandstätter JH, Laube B, Stahl S, Müller U, Betz H. Loss of postsynaptic GABAA receptor clustering in gephyrin-deficient mice. J Neurosci. 1999;19:9289–9297. doi: 10.1523/JNEUROSCI.19-21-09289.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk S, Winkelmann A, Smolinsky B, Förstera B, Neundorf I, Schwarz G, Meier JC. Direct binding of GABA(A) receptor beta2 and beta3 subunits to gephyrin. Eur J Neurosci. 2013;37:544–554. doi: 10.1111/ejn.12078. [DOI] [PubMed] [Google Scholar]
- Lau CG, Murthy VN. Activity-dependent regulation of inhibition via GAD67. J Neurosci. 2012;32:8521–8531. doi: 10.1523/JNEUROSCI.1245-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonoudakis D, Zhao P, Beattie EC. Rapid tumor necrosis factor alpha-induced exocytosis of glutamate receptor 2-lacking AMPA receptors to extrasynaptic plasma membrane potentiates excitotoxicity. J Neurosci. 2008;28:2119–2130. doi: 10.1523/JNEUROSCI.5159-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévi S, Logan SM, Tovar KR, Craig AM. Gephyrin is critical for glycine receptor clustering but not for the formation of functional GABAergic synapses in hippocampal neurons. J Neurosci. 2004;24:207–217. doi: 10.1523/JNEUROSCI.1661-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljelund P, Handforth A, Homanics GE, Olsen RW. GABAA receptor beta3 subunit gene-deficient heterozygous mice show parent-of-origin and gender-related differences in beta3 subunit levels, EEG, and behavior. Brain Res Dev Brain Res. 2005;157:150–161. doi: 10.1016/j.devbrainres.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Lin HC, Tseng YC, Mao SC, Chen PS, Gean PW. GABAA receptor endocytosis in the basolateral amygdala is critical to the reinstatement of fear memory measured by fear-potentiated startle. J Neurosci. 2011;31:8851–8861. doi: 10.1523/JNEUROSCI.0979-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Cheng PL, Lim BK, Khoshnevisrad N, Poo MM. Elevated BDNF after cocaine withdrawal facilitates LTP in medial prefrontal cortex by suppressing GABA inhibition. Neuron. 2010;67:821–833. doi: 10.1016/j.neuron.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YM, Mansuy IM, Kandel ER, Roder J. Calcineurin-mediated LTD of GABAergic inhibition underlies the increased excitability of CA1 neurons associated with LTP. Neuron. 2000;26:197–205. doi: 10.1016/S0896-6273(00)81150-2. [DOI] [PubMed] [Google Scholar]
- Luscher B, Fuchs T, Kilpatrick CL. GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron. 2011;70:385–409. doi: 10.1016/j.neuron.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacEwan DJ. TNF receptor subtype signalling: differences and cellular consequences. Cell Signal. 2002;14:477–492. doi: 10.1016/S0898-6568(01)00262-5. [DOI] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Malik M, Sheikh AM, Wen G, Spivack W, Brown WT, Li X. Expression of inflammatory cytokines, Bcl2 and cathepsin D are altered in lymphoblasts of autistic subjects. Immunobiology. 2011;216:80–85. doi: 10.1016/j.imbio.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Mangan PS, Sun C, Carpenter M, Goodkin HP, Sieghart W, Kapur J. Cultured hippocampal pyramidal neurons express two kinds of GABAA receptors. Mol Pharmacol. 2005;67:775–788. doi: 10.1124/mol.104.007385. [DOI] [PubMed] [Google Scholar]
- Mann EO, Paulsen O. Role of GABAergic inhibition in hippocampal network oscillations. Trends Neurosci. 2007;30:343–349. doi: 10.1016/j.tins.2007.05.003. [DOI] [PubMed] [Google Scholar]
- McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation. 2008;5:45. doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald BJ, Amato A, Connolly CN, Benke D, Moss SJ, Smart TG. Adjacent phosphorylation sites on GABAA receptor beta subunits determine regulation by cAMP-dependent protein kinase. Nat Neurosci. 1998;1:23–28. doi: 10.1038/223. [DOI] [PubMed] [Google Scholar]
- Michels G, Moss SJ. GABAA receptors: properties and trafficking. Crit Rev Biochem Mol Biol. 2007;42:3–14. doi: 10.1080/10409230601146219. [DOI] [PubMed] [Google Scholar]
- Mitsuhashi S, Matsuura N, Ubukata M, Oikawa H, Shima H, Kikuchi K. Tautomycetin is a novel and specific inhibitor of serine/threonine protein phosphatase type 1, PP1. Biochem Biophys Res Commun. 2001;287:328–331. doi: 10.1006/bbrc.2001.5596. [DOI] [PubMed] [Google Scholar]
- Möhler H, Fritschy JM, Crestani F, Hensch T, Rudolph U. Specific GABA(A) circuits in brain development and therapy. Biochem Pharmacol. 2004;68:1685–1690. doi: 10.1016/j.bcp.2004.07.025. [DOI] [PubMed] [Google Scholar]
- Montgomery SL, Bowers WJ. Tumor necrosis factor-alpha and the roles it plays in homeostatic and degenerative processes within the central nervous system. J Neuroimmune Pharmacol. 2012;7:42–59. doi: 10.1007/s11481-011-9287-2. [DOI] [PubMed] [Google Scholar]
- Mosselmans R, Hepburn A, Dumont JE, Fiers W, Galand P. Endocytic pathway of recombinant murine tumor necrosis factor in L-929 cells. J Immunol. 1988;141:3096–3100. [PubMed] [Google Scholar]
- Muir J, Arancibia-Carcamo IL, MacAskill AF, Smith KR, Griffin LD, Kittler JT. NMDA receptors regulate GABAA receptor lateral mobility and clustering at inhibitory synapses through serine 327 on the gamma2 subunit. Proc Natl Acad Sci U S A. 2010;107:16679–16684. doi: 10.1073/pnas.1000589107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee J, Kretschmannova K, Gouzer G, Maric HM, Ramsden S, Tretter V, Harvey K, Davies PA, Triller A, Schindelin H, Moss SJ. The residence time of GABA(A)Rs at inhibitory synapses is determined by direct binding of the receptor alpha1 subunit to gephyrin. J Neurosci. 2011;31:14677–14687. doi: 10.1523/JNEUROSCI.2001-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogoshi F, Yin HZ, Kuppumbatti Y, Song B, Amindari S, Weiss JH. Tumor necrosis-factor-alpha (TNF-alpha) induces rapid insertion of Ca2+-permeable alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)/kainate (Ca-A/K) channels in a subset of hippocampal pyramidal neurons. Exp Neurol. 2005;193:384–393. doi: 10.1016/j.expneurol.2004.12.026. [DOI] [PubMed] [Google Scholar]
- Okada M, Onodera K, Van Renterghem C, Sieghart W, Takahashi T. Functional correlation of GABA(A) receptor alpha subunits expression with the properties of IPSCs in the developing thalamus. J Neurosci. 2000;20:2202–2208. doi: 10.1523/JNEUROSCI.20-06-02202.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RW, Sieghart W. GABA A receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology. 2009;56:141–148. doi: 10.1016/j.neuropharm.2008.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onore C, Careaga M, Ashwood P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav Immun. 2012;26:383–392. doi: 10.1016/j.bbi.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallotto M, Nissant A, Fritschy JM, Rudolph U, Sassoè-Pognetto M, Panzanelli P, Lledo PM. Early formation of GABAergic synapses governs the development of adult-born neurons in the olfactory bulb. J Neurosci. 2012;32:9103–9115. doi: 10.1523/JNEUROSCI.0214-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzanelli P, Gunn BG, Schlatter MC, Benke D, Tyagarajan SK, Scheiffele P, Belelli D, Lambert JJ, Rudolph U, Fritschy JM. Distinct mechanisms regulate GABAA receptor and gephyrin clustering at perisomatic and axo-axonic synapses on CA1 pyramidal cells. J Physiol. 2011;589:4959–4980. doi: 10.1113/jphysiol.2011.216028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrizi A, Scelfo B, Viltono L, Briatore F, Fukaya M, Watanabe M, Strata P, Varoqueaux F, Brose N, Fritschy JM, Sassoè-Pognetto M. Synapse formation and clustering of neuroligin-2 in the absence of GABAA receptors. Proc Natl Acad Sci U S A. 2008;105:13151–13156. doi: 10.1073/pnas.0802390105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G. GABA(A) receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience. 2000;101:815–850. doi: 10.1016/S0306-4522(00)00442-5. [DOI] [PubMed] [Google Scholar]
- Prenosil GA, Schneider Gasser EM, Rudolph U, Keist R, Fritschy JM, Vogt KE. Specific subtypes of GABAA receptors mediate phasic and tonic forms of inhibition in hippocampal pyramidal neurons. J Neurophysiol. 2006;96:846–857. doi: 10.1152/jn.01199.2005. [DOI] [PubMed] [Google Scholar]
- Ragolia L, Begum N. Protein phosphatase-1 and insulin action. Mol Cell Biochem. 1998;182:49–58. doi: 10.1023/A:1006827227162. [DOI] [PubMed] [Google Scholar]
- Rannals MD, Kapur J. Homeostatic strengthening of inhibitory synapses is mediated by the accumulation of GABAA receptors. J Neurosci. 2011;31:17701–17712. doi: 10.1523/JNEUROSCI.4476-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183X.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santello M, Bezzi P, Volterra A. TNFalpha controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron. 2011;69:988–1001. doi: 10.1016/j.neuron.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Schneider Gasser EM, Duveau V, Prenosil GA, Fritschy JM. Reorganization of GABAergic circuits maintains GABAA receptor-mediated transmission onto CA1 interneurons in alpha1-subunit-null mice. Eur J Neurosci. 2007;25:3287–3304. doi: 10.1111/j.1460-9568.2007.05558.x. [DOI] [PubMed] [Google Scholar]
- Schweizer C, Balsiger S, Bluethmann H, Mansuy IM, Fritschy JM, Mohler H, Lüscher B. The gamma 2 subunit of GABA(A) receptors is required for maintenance of receptors at mature synapses. Mol Cell Neurosci. 2003;24:442–450. doi: 10.1016/S1044-7431(03)00202-1. [DOI] [PubMed] [Google Scholar]
- Smith KR, Muir J, Rao Y, Browarski M, Gruenig MC, Sheehan DF, Haucke V, Kittler JT. Stabilization of GABAA receptors at endocytic zones is mediated by an AP2 binding motif within the GABAA receptor β3 subunit. J Neurosci. 2012;32:2485–2498. doi: 10.1523/JNEUROSCI.1622-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram K, O'Callaghan JP. Divergent roles for tumor necrosis factor-alpha in the brain. J Neuroimmune Pharmacol. 2007;2:140–153. doi: 10.1007/s11481-007-9070-6. [DOI] [PubMed] [Google Scholar]
- Steinmetz CC, Turrigiano GG. Tumor necrosis factor-alpha signaling maintains the ability of cortical synapses to express synaptic scaling. J Neurosci. 2010;30:14685–14690. doi: 10.1523/JNEUROSCI.2210-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer R, von Boehmer L, Haenggi T, Schweizer C, Benke D, Rudolph U, Fritschy JM. Alteration of GABAergic synapses and gephyrin clusters in the thalamic reticular nucleus of GABAA receptor α3 subunit-null mice. Eur J Neurosci. 2006;24:1307–1315. doi: 10.1111/j.1460-9568.2006.05006.x. [DOI] [PubMed] [Google Scholar]
- Suh YH, Park JY, Park S, Jou I, Roche PA, Roche KW. Regulation of metabotropic glutamate receptor 7 (mGluR7) internalization and surface expression by Ser/Thr protein phosphatase 1. J Biol Chem. 2013;288:17544–17551. doi: 10.1074/jbc.M112.439513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Bargas J, Hemmings HC, Jr, Nairn AC, Greengard P. Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron. 1995;14:385–397. doi: 10.1016/0896-6273(95)90294-5. [DOI] [PubMed] [Google Scholar]
- Tancredi V, D'Arcangelo G, Grassi F, Tarroni P, Palmieri G, Santoni A, Eusebi F. Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci Lett. 1992;146:176–178. doi: 10.1016/0304-3940(92)90071-E. [DOI] [PubMed] [Google Scholar]
- Terunuma M, Jang IS, Ha SH, Kittler JT, Kanematsu T, Jovanovic JN, Nakayama KI, Akaike N, Ryu SH, Moss SJ, Hirata M. GABAA receptor phospho-dependent modulation is regulated by phospholipase C-related inactive protein type 1, a novel protein phosphatase 1 anchoring protein. J Neurosci. 2004;24:7074–7084. doi: 10.1523/JNEUROSCI.1323-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson AM, Jovanovic JN. Mechanisms underlying synapse-specific clustering of GABA(A) receptors. Eur J Neurosci. 2010;31:2193–2203. doi: 10.1111/j.1460-9568.2010.07252.x. [DOI] [PubMed] [Google Scholar]
- Tonelli LH, Postolache TT. Tumor necrosis factor alpha, interleukin-1 beta, interleukin-6 and major histocompatibility complex molecules in the normal brain and after peripheral immune challenge. Neurol Res. 2005;27:679–684. doi: 10.1179/016164105X49463. [DOI] [PubMed] [Google Scholar]
- Tretter V, Jacob TC, Mukherjee J, Fritschy JM, Pangalos MN, Moss SJ. The clustering of GABAA receptor subtypes at inhibitory synapses is facilitated via the direct binding of receptor alpha 2 subunits to gephyrin. J Neurosci. 2008;28:1356–1365. doi: 10.1523/JNEUROSCI.5050-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter V, Mukherjee J, Maric HM, Schindelin H, Sieghart W, Moss SJ. Gephyrin, the enigmatic organizer at GABAergic synapses. Front Cell Neurosci. 2012;6:23. doi: 10.3389/fncel.2012.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagarajan SK, Ghosh H, Yévenes GE, Nikonenko I, Ebeling C, Schwerdel C, Sidler C, Zeilhofer HU, Gerrits B, Muller D, Fritschy JM. Regulation of GABAergic synapse formation and plasticity by GSK3beta-dependent phosphorylation of gephyrin. Proc Natl Acad Sci U S A. 2011;108:379–384. doi: 10.1073/pnas.1011824108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vey E, Burger D, Dayer JM. Expression and cleavage of tumor necrosis factor-alpha and tumor necrosis factor receptors by human monocytic cell lines upon direct contact with stimulated T cells. Eur J Immunol. 1996;26:2404–2409. doi: 10.1002/eji.1830261021. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46:1724–1743. doi: 10.1111/j.1528-1167.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- Walsh AH, Cheng A, Honkanen RE. Fostriecin, an antitumor antibiotic with inhibitory activity against serine/threonine protein phosphatases types 1 (PP1) and 2A (PP2A), is highly selective for PP2A. FEBS Lett. 1997;416:230–234. doi: 10.1016/S0014-5793(97)01210-6. [DOI] [PubMed] [Google Scholar]
- Wang Q, Liu L, Pei L, Ju W, Ahmadian G, Lu J, Wang Y, Liu F, Wang YT. Control of synaptic strength, a novel function of Akt. Neuron. 2003;38:915–928. doi: 10.1016/S0896-6273(03)00356-8. [DOI] [PubMed] [Google Scholar]
- Yanagihori S, Terunuma M, Koyano K, Kanematsu T, Ho Ryu S, Hirata M. Protein phosphatase regulation by PRIP, a PLC-related catalytically inactive protein–implications in the phospho-modulation of the GABAA receptor. Adv Enzyme Regul. 2006;46:203–222. doi: 10.1016/j.advenzreg.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Yin HZ, Hsu CI, Yu S, Rao SD, Sorkin LS, Weiss JH. TNF-alpha triggers rapid membrane insertion of Ca(2+) permeable AMPA receptors into adult motor neurons and enhances their susceptibility to slow excitotoxic injury. Exp Neurol. 2012;238:93–102. doi: 10.1016/j.expneurol.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P, Deisseroth K. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Jiang M, Miralles CP, Li RW, Chen G, de Blas AL. Gephyrin clustering is required for the stability of GABAergic synapses. Mol Cell Neurosci. 2007;36:484–500. doi: 10.1016/j.mcn.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ZY, Wang W, Fritschy JM, Witte OW, Redecker C. Changes in neocortical and hippocampal GABAA receptor subunit distribution during brain maturation and aging. Brain Res. 2006;1099:73–81. doi: 10.1016/j.brainres.2006.04.118. [DOI] [PubMed] [Google Scholar]
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- Zhang H, Nei H, Dougherty PM. A p38 mitogen-activated protein kinase-dependent mechanism of disinhibition in spinal synaptic transmission induced by tumor necrosis factor-α. J Neurosci. 2010;30:12844–12855. doi: 10.1523/JNEUROSCI.2437-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]