

Abstract
Glioblastoma multiforme (GBM) is the most aggressive form of brain tumor. In general, tumor growth requires disruption of the tissue microenvironment, yet how this affects glioma progression is unknown. We studied program death-ligand (PD-L)1 in neurons and gliomas in tumors from GBM patients and associated the findings with clinical outcome. Remarkably, we found that upregulation of PD-L1 by neurons in tumor-adjacent brain tissue (TABT) associated positively with GBM patient survival, whereas lack of neuronal PD-L1 expression was associated with high PD-L1 in tumors and unfavorable prognosis. To understand the molecular mechanism of PD-L1 signaling in neurons, we investigated PD-L1 function in cerebellar and cortical neurons and its impact on gliomas. We discovered that neuronal PD-L1-induced caspase-dependent apoptosis of glioma cells. Because interferon (IFN)-β induces PD-L1 expression, we studied the functional consequences of neuronal Ifnb gene deletion on PD-L1 signaling and function. Ifnb−/− neurons lacked PD-L1 and were defective in inducing glioma cell death; this effect was reversed on PD-L1 gene transfection. Ifnb−/− mice with intracerebral isografts survived poorly. Similar to the observations in GBM patients, better survival in wild-type mice was associated with high neuronal PD-L1 in TABT and downregulation of PD-L1 in tumors, which was defective in Ifnb−/− mice. Our data indicated that neuronal PD-L1 signaling in brain cells was important for GBM patient survival. Reciprocal PD-L1 regulation in TABT and tumor tissue could be a prognostic biomarker for GBM. Understanding the complex interactions between tumor and adjacent stromal tissue is important in designing targeted GBM therapies.
Introduction
Glioblastoma multiforme (GBM), a grade IV astrocytoma, is the most common and malignant human primary brain tumor. Even with surgery, radiotherapy, and chemotherapy, most patients die from this rapidly infiltrating tumor within a year of diagnosis (Krex et al., 2007). Developing new therapies requires identifying targets critical for GBM growth control and understanding the interaction of glioblastomas with surrounding stromal cells. In the tissue microenvironment, tumors and surrounding cells communicate by signaling through transmembrane receptors, ligands, and produced factors. This tissue microenvironment can suppress cancer and disruption of the microenvironment is required for tumor progression (Barcellos-Hoff et al., 2009). In glioma tissue, neurons are often closely apposed to tumor areas, suggesting interaction between these two cell types. However, the role of neurons in protecting native tissue from cancer and the mechanisms of that protection are largely unexplored.
Neural progenitor cells can promote tumor regression and in some cases prolong survival in glioma animal models (Benedetti et al., 2000; Herrlinger et al., 2000; Ehtesham et al., 2002; Barresi et al., 2003; Staflin et al., 2004; Walzlein et al., 2008). Endogenous neural precursors exert an antitumor response by specifically targeting stem cell-like tumor cells (Chirasani et al., 2010). However, whether these functions are exclusive to progenitor cells, or whether a mechanism common to other predominant CNS resident cells can limit tumor growth is unknown. Although GBM is among the most common malignant CNS cancers, it is relatively rare (Wrensch et al., 2002). This suggests that the CNS microenvironment is naturally equipped to control proliferative glial and tumor cells and failure of this system can lead to cancer progression.
Program death-ligand (PD-L)1/B7-H1/CD274, a transmembrane receptor ligand and negative regulator of T-cell signaling, is upregulated in a number of tumors, including gliomas (Parsa et al., 2007; Jacobs et al., 2009). PD-L1 might confer immunomodulatory properties that help tumors escape from immune surveillance (Dong et al., 2002). Studies have suggested an association between the malignancy grade of astrocytic tumors and tumor cell PD-L1 expression (Wilmotte et al., 2005; Yao et al., 2009), including upregulation of PD-L1 at the growing edge of tumors (Wilmotte et al., 2005; Yao et al., 2009). Constitutive PD-L1 expression is reported to be important for maintaining tolerance against immune-cell attack in several different tissues (Liang et al., 2003; Keir et al., 2006). Although PD-L1 is not normally highly expressed in CNS residual cells except for blood–brain barrier endothelial cells, it is upregulated in glial cells in CNS inflammation (Liang et al., 2003; Rodig et al., 2003; Salama et al., 2003), and in neurons in viral infection (Lafon et al., 2008). To date, no studies have extensively addressed PD-L1 expression in the brain microenvironment near tumors or investigated whether PD-L1 regulation in brain resident cells is associated with GBM patient outcomes. Given that PD-L1 is crucial for homeostasis in other organs, we asked how PD-L1 expression in brain cells is regulated, and whether regulation has functional consequences for survival in GBM patients.
Materials and Methods
Ethics statement.
All experiments were performed in accordance with and approved by ethical committees in Denmark (Animal Ethics Council, Act on the Use of Animals for Experimental Purposes, No. 2007/561-1364). Experiments using patient tissues were approved by the Local Ethical Board of the University of Lund, Sweden, serial no. LU307-98, and conform to the relevant regulatory standards. Informed consent was obtained. To protect patient anonymity, tumor samples were coded.
Tumor specimens from GBM patients and immunofluorescence histochemistry.
Tumor specimens were surgically removed from 17 patients (6 females and 11 males), which were then confirmed pathologically to be GBM (World Health Organization [WHO] grade IV; WHO grade III) as shown in Table 1. Tissues were stained with antibodies labeled for immunofluorescence (see below) and blindly scored by three independent investigators. At least six cryosections were stained for each staining. Negative controls used were including omitted primary antibody or isotype-matched control antibodies and were included for each patient sample. Some randomly selected patients were investigated at two independent centers included in this study. Tissues were snap-frozen in liquid nitrogen-cooled isopentane (2-metylbutane) and cryosectioned in 7 μm slices and kept at −80°C until staining.
Table 1.
PD-L1 expression in tumor cells and TABT areas in patients with GBM
| Patient no. | Patient ID | Sex | Agea | OSb | Pathology | Treatmentc | Tumor adjacent brain tissue |
Tumor mass |
||
|---|---|---|---|---|---|---|---|---|---|---|
| PD-L1+NF200+ | PD-L1+GFAP+ | PD-L1 | PDL1+GFAP+ | |||||||
| 1 | LU-38 | M | 68 | 534 | GBM | CTMZ + ATMZ | +++ | — | + | + |
| 2 | LU-50 | F | 55 | 390 | GBM | CTMZ + ATMZ | ++ | + | + | — |
| 3 | LU-55 | M | 54 | 692d | GBM | CTMZ + ATMZ + SLCT | +++ | + | + | + |
| 4 | LU-58 | M | 61 | >1898 | GBM | CTMZ + ATMZ + SLCT | +++ | — | — | — |
| 5 | LU-60 | F | 62 | 338 | GBM | CTMZ + ATMZ | ++ | + | + | — |
| 6 | LU-61 | M | 60 | 611 | GBM | CTMZ + ATMZ + SLCT | +++ | — | + | — |
| 7 | LU-63 | F | 59 | 1427 | GBM | CTMZ + ATMZ | ++ | + | + | + |
| 8 | LU-65 | M | 48 | 442 | GBM | CTMZ + ATMZ | + | — | — | — |
| 9 | LU-66 | F | 51 | 640 | GBM | CTMZ + ATMZ + SLCT | ++ | — | + | + |
| 10 | LU-87 | M | 55 | 101 | GBM | RT | — | — | ++ | ++ |
| 11 | LU-91 | M | 49 | 173 | GBM | CTMZ | — | — | ++ | ++ |
| 12 | LU-130 | F | 78 | 299 | GBM | CTMZ + ATMZ | ++ | — | — | — |
| 13 | LU-139 | F | 43 | 452 | GBM | CTMZ + ATMZ + SLCT | + | — | +++ | ++ |
| 14 | LU-142 | M | 65 | 170 | GBM | CTMZ + ATMZ | — | — | +++ | +++ |
| 15 | LU-143 | M | 65 | >790 | GBM | CTMZ + ATMZ | ++ | — | — | — |
| 16 | LU-145 | M | 45 | 392e | GBM | CTMZ + ATMZ + SLCT | + | — | +++ | — |
| 17 | LU-172 | M | 77 | 274 | GBM | RT | — | — | +++ | + |
aAge (years) by the onset of surgery.
bOverall survival (days) by the onset of surgery; >patient alive 20130506.
cAdditional therapy CTMZ, concomitant temozolamide; ATMZ, adjuvant temozolamide, 6 cycles; RT, radiotherapy only; SLCT, second line chemotherapy (e.g., NOC).
dLU-55 tissue is from 3rd operation; OS from 1st operation.
eLU-145 results are from 1st operation. Patient was re-operated at 111229 for new PAD gliosarcoma.
—, Not found; +, <10 positive cells; ++, 10–100 positive cells; +++, many positive cells.
Mice and glioma cell lines.
Ifnb−/− mice were backcrossed to JAX C57BL/6 strains for at least 20 generations. JAX C57BL/6 mice were bred and housed at conventional animal facilities at the University of Copenhagen and the University of Lund. Ten females and five males were included in the survival studies.
GL261 mouse glioma cells of C57BL/6 origin were a kind gift from Dr Geza Safrany (Department of Molecular and Tumor Radiobiology, Frédéric Joliot-Curie National Research Institute for Radiobiology and Radiohygiene, Budapest, Hungary). The GL261 cell line was induced by intracerebral implantation of methylcholanthrene pellets and established as a permanent cell line (Lumniczky et al., 2002). It was selected for this study because of its isogenity to the mouse strain C57BL/6. U87 cells were from a glioblastoma cell line derived from a human glioblastoma (ATCC no. HTB-14). Cells were maintained at 37°C in a humidified and 5% CO2-conditioned atmosphere. Medium was RPMI 1640 supplemented with 1% PS (Invitrogen) and 10% fetal bovine serum (FBS; Biochrome).
Neuronal cultures.
Cerebellar tissue was dissected from 7-d-old mice and cortical tissue was dissected from 1-d-old mice. In vitro culture was as described previously (Liu et al., 2006). Recombinant interferon (IFN)-β (R&D Systems) at 20 U/ml (for 3 d) or 100 U/ml (for 3 or 24 h) was added to neuronal cultures as indicated.
Coculture of neurons with gliomas.
Neurons were seeded at 4 × 105 cells/ml in 96- or 24-well plates with neuronal media for at least 3 d. GL261 or U87 cells were trypsinized and cocultured at 1:5 (GL261 or U87:neuron) for 24 h, unless otherwise stated. For coculturing, neuron and glioma media were used at 1:1. After coculture, glioma cells and neurons were collected by scraping with a pipette tip. When indicated, a pan-caspase inhibitor (Z-VAD-FMK; Sigma-Aldrich), or different blocking antibodies and fusion proteins; fab antibodies for B7.1 or B7.2 or CTLA-4Ig/blocks both B7.1 and B7.2, anti-PD-1, anti-PD-L1, or their fusion proteins were used.
Fluorescence-activated cell sorting.
Fluorescence-activated cell sorting (FACS) was performed as described previously (Liu et al., 2006) using the following reagents: PE-anti-PD-L1 (MIH5), Bio-rat IgG2a (R35–95), IgG1 (MPC-11), FITC-anti-active caspase-3 (C92–605), PE-anti-Ki-67 (B56), and FITC-anti-PD-1 (J43) all from eBioscience; anti-cleaved caspase 3 and nuclear dye 7AAD (both from Cell Signaling Technology). Cells were analyzed using a four-color Becton Dickinson FACSCalibur. Positive staining was evaluated by comparison to samples processed with isotype-matched control antibodies. FlowJo 8.8.6 was used for data analysis (Tree Star). Experiments were repeated three times with similar results.
Propidium iodide cell cycle staining.
Single-cell suspensions were prepared in buffer (PBS + 2% FBS; PBS + 0.1% bovine serum albumin), washed twice, and resuspended at 1 × 106 cells/ml. One milliliter aliquots were distributed in 15 ml polypropylene, V-bottomed tubes and 3 ml cold absolute ethanol was added. Cells were fixed for at least 1 h on ice and 1 ml of propidium iodide (PI)/RNase Staining Buffer (BD Biosciences) was added to the cell pellet and thoroughly mixed before FACS analysis.
Carboxyfluorescein diacetate succinimidyl ester labeling.
GL261 cells were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen) as previously described (Liu et al., 2006).
For GL261 Western blots, CFSE+ GL261 cells were purified with FACSaria before and after 24 h of coculture with 3 d in vitro (DIV) with cerebellar granular neurons (CGNs).
Western blot.
Proteins were extracted in 40 μl SDS loading buffer (Invitrogen) from GL261 cells and 15 μl of protein lysate was separated by 4–12% SDS-PAGE and blotted onto Hybond-C extra nitrocellulose membranes (GE Healthcare). Membranes were blocked in 5% milk in PBS-Tween-20 (0.05%) for 1 h at room temperature and incubated with primary antibodies: rabbit-anti-HSP90 (BioVision), goat-anti-PD-L1 (B7-H1, AF1019; R&D Systems), mouse anti-caspase 8 1:1000 (Cell Signaling Technology; CST 1C-12), rabbit anti-BAX (Cell Signaling Technology; CST 2774), or rabbit anti-GAPDH 1:20,000 (Abcam; EPR1977Y) overnight at 4°C with rocking. Membranes were washed three times in PBS-Tween-20, and incubated with anti-rabbit, anti-mouse, or anti-goat antibodies labeled with horseradish peroxidase for 1 h at room temperature and developed using an ECL technique (Millipore).
Thymidine incorporation.
Neurons were cultured as described above. After 72 h, neuron media was replaced with 100 μl neurobasal media and 100 μl R10 containing 2 × 105 GL261 cells/ml. Coculturing was for 18–20 h before a 4 h, 1 μCi [3H]thymidine pulse. Pulsed cells were measured on a β scintillation counter to determine incorporated radioactive thymidine. A proliferation index was calculated for each experiment by dividing incorporated thymidine by average GL261 thymidine incorporation. Results were multiplied by 100 for percentage thymidine incorporation compared with GL261.
BrdU cell proliferation assay.
BrdU incorporation and europium signal were measured with a DELFIA Cell Proliferation Kit (PerkinElmer), according to the manufacturer's instructions. After coculture with neurons, GL261 cells were pulsed with BrdU for 1 h with 100 nm BrdU and incorporation was measured on a FLUOstar OPTIMA plate reader with europium filters (BMG Labtech).
PD-L1 plasmid construction.
The PD-L1 open reading frame was amplified by PCR from a mouse PD-L1 cDNA clone (Clontech) using a forward primer flanked by an NheI site (5′-atatatgctagctcgccaccatgccaggctgcacttgcac-3′) and reverse primer flanked by a Cfr9 I site (5′-atatatcccgggttacgtctcctcgaattgtg-3′). PCR was 95°C 2 min and 30 cycles of 95°C 30 s, 58°C 30 s, and 70°C 1 min. The PCR product was subcloned in-frame into pIRES2-EGFP (Clontech), and PD-L1 was verified by sequencing (Eurofins MWG).
Transfection of cerebellar granular neurons.
Ifnb−/− mouse neurons (5 × 106) were transfected with 10 μg pIRES2-EGFP-PD-L1 or pIRES2-EGFP control using the Amaxa nucleofector technique (program G13) according to the manufacturer's instructions (Clontech). Western blot and FACS were used to evaluate transfection efficiency.
Transduction of GL261 cells with short hairpin RNA.
GL261 cells were transduced with lentiviral short hairpin RNAs to knock down PD-L1 expression (shPD-L1). Nonsilencing shpLKO (shCtrl) was used as the negative control. At 72 h post-transduction, transduced cells were selected with 1 μg/ml puromycin, cryopreserved at passages 5–6, and used for experiments at passages 8–15. Transduction efficiency was confirmed by Western blot.
CGN pdl1 silencing with siRNA.
Accell SMART pool small-interfering RNA (siRNA), which combines four different siRNAs, was from Dharmacon (Thermo Scientific). Predesigned pdl1 SMART pool siRNA was introduced to CGNs in Accell delivery medium according to the manufacturer's protocol and incubated for 72 h before coculturing. Delivery efficiency and siRNA specificity were examined by FACS for PD-L1. The control was Accell nontargeting control siRNA.
Experimental murine glioma model.
Ifnb−/− and Ifnb+/+ mouse glioma models were induced by anesthetizing mice with isoflurane (Forene; 3.0%, airflow 250) before inoculation into the right frontal lobe with 1000 GL261 cells in 5 μl culture medium on day 0. For intracerebral injections, the head of the mouse was fixed in a stereotactic frame, a small hole was drilled into the skull, and a Hamilton syringe needle was inserted. Coordinates for intracerebral injections were 1.5 mm to the right and 1.0 mm in front of the bregma, 2.75 mm deep. The needle was left in the brain for 5 min and slowly withdrawn to diminish backflow through the insertion canal. The hole was sealed with bone wax.
For intracerebellar injections, 3000 GL261 cells were injected into the cerebellum of mice anesthetized as above. The reason for the increased number of GL261 cells in this model was that the death rate for wild-type mice was much lower for injection in the cerebellum than for intracerebral injection if 1000 GL261 was injected.
Tumor-bearing intracerebrally injected mice were killed when neurological symptoms were detected and brains were examined for remnant tumors with immunofluorescence staining on coronal sections as indicated.
Immunofluorescence staining.
GL261 cells were cultured alone or in coculture with 3 d in vitro (DIV) neurons from Ifnb+/+ or Ifnb−/− mice at 1:5 (tumor:neuron) for 24 h on 8-well LabTek chamber slides (Nunc). Cells were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 and stained with Ki-67-Alexa 555 antibody (BD Biosciences). Hoechst 33342 (Invitrogen) was used to stain nuclei. To quantify Ki-67+ cells, 24 fields (63× objective) per well were counted systematically for a total of ∼500 Ki-67+ cells. For each experimental condition, triplicates were counted.
Immunofluorescence histochemistry on glioma mouse model brains.
Brains were dissected, immediately embedded in OTC Compound (Sakura Finetek), and snap-frozen in isopentane on dry ice. Tissues were cryosectioned in 10 μm slices and kept at −80°C until hematoxylin staining or immunostaining. For immunostaining, sections were incubated with rabbit anti-NF200 (Sigma-Aldrich), anti-GFAP (Millipore), and biotinylated anti-PD-L1 (MIH5, eBioscience), followed by fluorescent-conjugated secondary antibodies. Hoechst 33342 was used for nuclear staining. Negative controls were omission of primary antibody or isotype-matched control antibodies. Slides were mounted in DAPI-Pro-Long Gold anti-fade mounting medium (Invitrogen) and visualized with a Zeiss fluorescent microscope.
Immunofluorescence histochemistry on GBM patient tumor specimens.
Antibodies used were as follows: rabbit anti-NF200 (Sigma-Aldrich), mouse anti-human PD-L1 (AbD; Serotec), purified mouse anti-human PD-L1 (Biolegend), FITC mouse anti-human PD-L1 (558065; BD PharMingen), mouse anti-human GFAP (Cy3, catalog #ab49874; Abcam). Isotype controls were mouse lgG1 (catalog #557351; BD PharMingen), mouse lgG2b (catalog #13-4714-85, eBioscience), and rabbit lgG (eBioscience). Secondary antibodies were Alexa Fluor 488 donkey anti-rabbit, Alexa Fluor 568 goat anti-mouse (Invitrogen), goat-anti rabbit Alexa Fluor 594, or goat-anti-mouse Alexa Fluor 488 (Invitrogen).
Affymetrix data Analysis.
RNA was prepared in biological triplicates from 3-d-old cultures of Ifnb+/+, Ifnb−/−, IFN-β-treated Ifnb−/−, or IFN-β-treated Ifnb+/+ postnatal day 6–7 CGNs using TRIzol (Sigma-Aldrich), followed by DNAseI (Invitrogen) digestion, and TRIzol RNA purification. Spectrophotometry was used to determine RNA purity. Gene expression analysis used Affymetrix 430 2.0 microarray chips (SCIBLU; Affymetrix Core Laboratory, Lund University) with data analyzed with Arraystar 3 software (DNASTAR). Data were quantile normalized and processed by the RMA (Affymetrix) algorithm. Intensity values were log2-transformed and normally distributed data were tested in unpaired, two-tailed Student's t tests assuming equal variance, set to filter for differential regulation confidence 95% (p < 0.05).
Statistical analysis.
Statistical evaluation was performed using GraphPad Prism software. Comparison of PD-L1 expression with survival in GBM patients was performed using a log-rank correlation test. Student's unpaired t test (with Welch's correction) or ANOVA with post hoc Bonferroni's multiple-comparison test (when more than two groups were compared) was used for in vitro studies. Survival curves for experimental glioma models were compared using a log-rank test. p < 0.05 was considered significant.
Results
Neuronal PD-L1 expression in tumor-adjacent brain tissue is associated with GBM prognosis
PD-L1 expression by gliomas limits T-cell activation, helps cells escape immune surveillance, and correlates with high-grade tumors (Wilmotte et al., 2005; Parsa et al., 2007; Yao et al., 2009). However, whether PD-L1 expression by stromal cells in the tumor microenvironment affects GBM outcome is unknown. We investigated PD-L1 regulation in CNS resident cells using surgically removed tumor tissue samples encompassing tumor-adjacent brain tissue (TABT). TABT is a relative term because gliomas from tumor masses often diffusely infiltrate the brain with individual glioma cells intermingling with surrounding brain tissue. As a result, the infiltrating regions of gliomas are not characterized by an abrupt border but by a gradual decrease in the relative abundance of infiltrating tumor cells in the immediately adjacent area.
This study included 17 GBM patients whose characteristics are summarized in Table 1 with clinical survival in Figure 1. We microscopically examined TABT in all samples from patients. GFAP is expressed by reactive glial cells and a subpopulation of high-grade gliomas. Therefore, to identify tumor cells and reactive glial cells, antibodies against GFAP were used and to identify neuronal cells, NF-200 was used. We found GFAP+ tumor cells with high PD-L1 expression in the tumor mass (TM) in some of the investigated GBM patients (Table 1; Fig. 1A–F, two GBM patients), consistent with earlier reports (Wilmotte et al., 2005; Yao et al., 2009). A generally high degree of PD-L1 expression (>10 PD-L1+ cells; Table 1) in the TM significantly correlated with poor prognosis compared with GBM patients with no or lower PD-L1 expression (1–9 PD-L1+ cells) in the TM, shown in Kaplan–Meier survival curves (Fig. 1O). In a group of GBM patients with high numbers of PD-L1+ tumor cells (Fig. 1G,H), PD-L1-expressing brain cells, such as neurons (defined as positive for the neuronal marker NF200), were rare in TABT. PD-L1+ astrocytes were also rare in the TABT even if signs of astrogliosis were detected in some cases (Fig. 1I–J; Table 1). In contrast in another group of GBM patients, we found NF200+ neurons that broadly expressed PD-L1 (>10 NF200+PD-L1+ cells) in vicinity of the TM (Table 1; Fig. 1K–N). We also detected that highly upregulated PD-L1 in neurons and their processes in TABTs to be associated with longer survival times after operation in this group. As shown in Kaplan–Meier survival curves, the number of PD-L1+NF200+ neurons (PD-L1highNF200+, N = 10) in TABTs was significantly associated with prolonged postoperative survival days. Conversely, a low number of PD-L1+ neurons (<10 PD-L1+ neurons/PD-L1lowNF200+, N = 7) negatively correlated with patient survival (Fig. 1P). No significant association was found between survival and degree of PD-L1 positivity in resident glial cells (data not shown).
Figure 1.
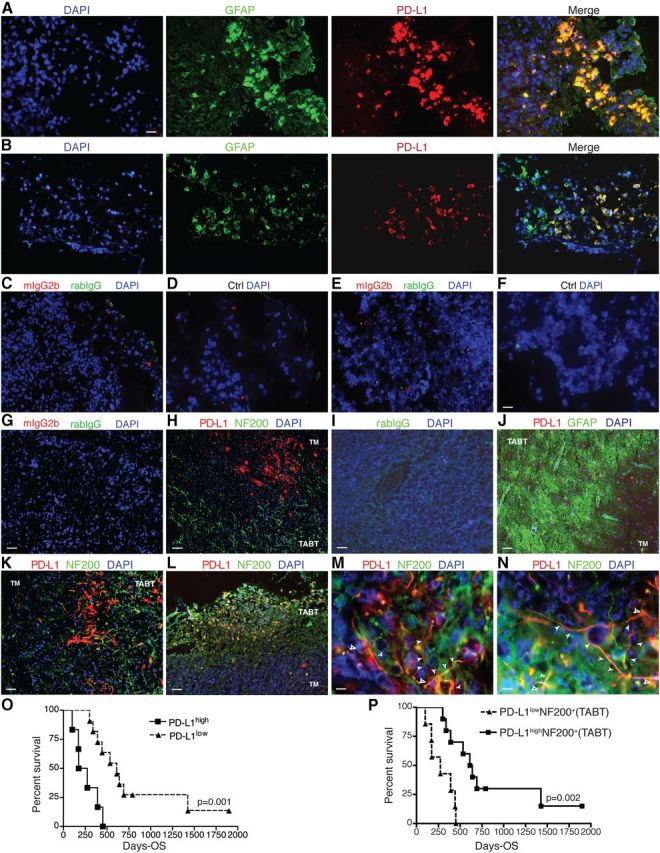
Neuronal expression of PD-L1 in tumor adjacent brain tissue confers better prognosis; PD-L1 expression in tumors negatively associates with GBM patient survival. Immunofluorescence staining of GBM patient brain tissue. TABT areas are designated based on findings of relative intact NF200+ neuronal staining with normal or high degree of GFAP+/astrogliosis. A, B, GFAP (Alexa Fluor 488/green), PD-L1 (Alex Fluor 568/red), and DAPI (blue) staining of PD-L1 expression in gliomas on the edge of tumors. C–F, Isotype-matched controls or omitted primary antibodies (Ctrl) combined with DAPI (blue) on the adjacent tissues of patients shown in A, B, respectively. G–N, Isotype-matched controls, NF200 (Alexa Fluor 488/green), or GFAP (Alexa Fluor 488/green), PD-L1 (Alex Fluor 568/red), and DAPI (blue). Scale bars: A–F, I, 31 μm; G, H, J–L, 62 μm; M, N, 10 μm. Open white arrows, soma of neurons; filled white arrows, neuronal processes. O, Kaplan–Meier survival curve showing significant negative association between high PD-L1 expression by gliomas and GMB patient survival. P, Kaplan–Meier survival curve showing significant positive association between high PD-L1 expression by NF200+ neurons in TABT and survival of GMB patients. N = 17. Log-rank test was used for statistic analysis. p < 0.05 was considered significant.
These data indicated that resident CNS cells, in particular TABT neurons close to the TM, had upregulated PD-L1 expression. TABT neuronal PD-L1 expression was inversely related to PD-L1 expression in tumor cells, and strongly associated with favorable prognosis of GBM patients compared to GBM patients with high PD-L1 expression in tumor cells. This suggested that upregulation of PD-L1 in native brain neurons was a negative feedback signal for downregulation of PD-L1 expression by tumor cells. A defect in such a regulatory pathway might underlie the sustained or upregulated PD-L1 expression in tumor cells that allows tumor cells to evade immune surveillance and correlates with GBM aggressiveness (Wintterle et al., 2003; Wilmotte et al., 2005; Yao et al., 2009).
Neurons induce cell death of murine and human gliomas
We investigated the function of neurons in limiting glioma progression compared with CNS glial cells. We used in vitro cultures of CGNs, cortical neurons, and mixed primary glial cells, evaluating their regulation of GL261, a murine glioma cell line capable of inducing experimental glioma in mice after isograft. Gliomas were labeled and gated with CFSE and also detected by their large size in FACS analysis (Fig. 2A). NF200 was used as a marker of neurons. The culture media was controlled during cocultures using the same for only GL261 and coculture with neurons.
Figure 2.

Neurons induce proliferative arrest and cell death of GL261 and U87 glioma cells. A, FACS dot plot gating on CFSE-labeled GL261 cells after coculture with cerebellar granule neurons. B, DNA synthesis assessed by BrdU incorporation in GL261 cells in coculture with neurons from 3 DIV culture of CGNs, or cortex, cultured primary glial cells compared with GL261 cells alone. Neurons and glial cells were cocultured with GL261 cells at 5 to 1. Data are mean ± SD from three separate experiments; ***p < 0.001. C, Representative FACS histogram with PI staining (gated on sub-G1 phase) of GL261 cells alone and after coculturing with neurons. D, DNA synthesis by BrdU incorporation of U87 cells in coculture with neurons compared with U87 cells alone. Three DIV CGNs cocultured with U87 cells at 10 to 1. Data are mean ± SD; **p < 0.01. E, Representative FACS histogram of PI staining (gated on sub-G1 phase) of U87 cells alone and after coculturing with 3 DIV CGNs at 10:1 (CGN to U87).
As shown in Figure 2B, both CGNs and cortical neurons significantly inhibited glioma cell proliferation measured by BrdU incorporation, but glial cells did not limit GL261 cell proliferation. Of note, both types of neurons also induced cell death in GL261 cells, detected by PI FACS analysis of sub-G1 phase (Fig. 2C). However, cortical neurons had significantly lower effects on GL261 cell proliferation and induction of cell death than CGNs. Therefore, we focused on CGNs to determine the mechanism of GL261 inhibition.
To study the killing function of CGNs on human glioblastoma cells, we used the U87 human glioblastoma cell line, which is tumorigenic in vivo and highly expresses PD-L1 because of a phosphatase and tensin homolog (PTEN) mutation (Parsa et al., 2007). U87 cells were cocultured with CGNs and U87 proliferation and cell death were measured. Neurons significantly reduced U87 cell proliferation and increased cell death (Fig. 2D,E), indicating that adult postmitotic neurons induced glioma cell death.
Ifnb−/− cerebellar granule neurons lack PD-L1 expression and the capacity to kill gliomas
Neurons that have reduced IFN-β production also have lower PD-L1 expression upon viral infection, and recombinant IFN-β induces PD-L1 expression (Lafon et al., 2008). We studied whether Ifnb gene defects in neurons affected PD-L1 regulation and the capacity of neurons to regulate gliomas. We obtained CGNs from IFN-β knock-out (Ifnb−/−) mice and wild-type littermates (Ifnb+/+) and analyzed their PD-L1 expression using immunocytochemistry and FACS. Figure 3A,B shows that Ifnb−/− CGNs lacked PD-L1 expression in contrast to Ifnb+/+ CGNs. However, stimulation of Ifnb−/− CGNs with recombinant IFN-β restored PD-L1 expression. Microarray analysis of Ifnb+/+ CGNs and Ifnb−/− CGNs revealed no significant differences in mRNA levels for PD-L1 (Table 2) indicating that PD-L1 production was post-translationally regulated and required intact endogenous IFN-β signaling. Although stimulation of Ifnb−/− CGNs with recombinant IFN-β significantly induced PD-L1 mRNA, similar treatment of Ifnb+/+ CGNs resulted in significantly higher induction of PD-L1, demonstrating that a functional Ifnb gene was required for proper regulation of PD-L1 in neurons.
Figure 3.
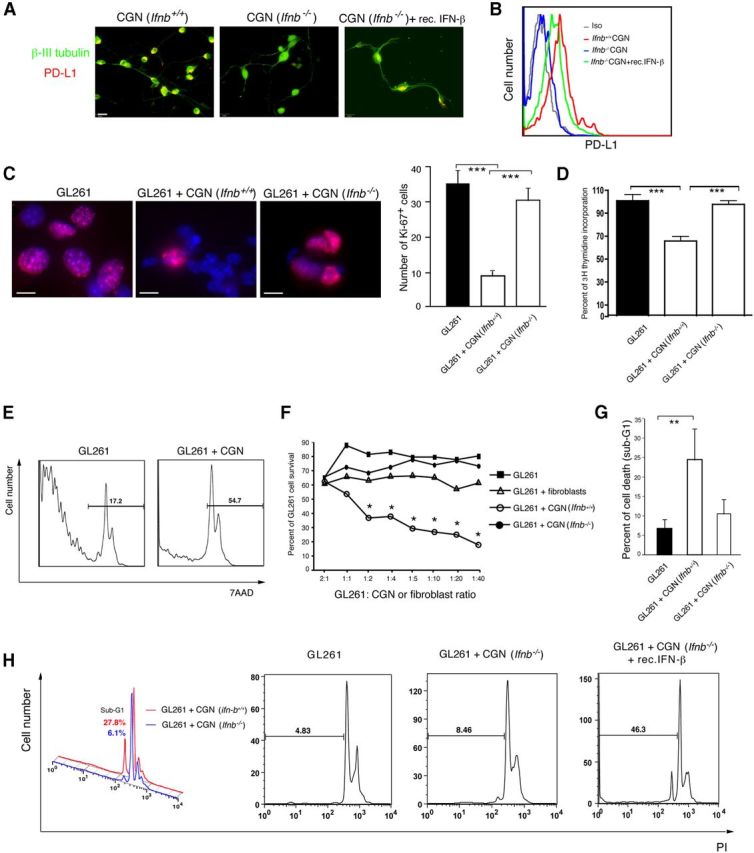
Ifnb−/− CGNs lack PD-L1 expression and capacity to kill gliomas. A, Micrographs of PD-L1 (red) and β III tubulin (green) immunofluorescent cytochemistry staining of CGNs. Scale bar, 31 μm. B, FACS histograms of PD-L1 staining versus isotype-matched controls for Ifnb+/+ CGNs, Ifnb−/− CGNs, and Ifnb−/− CGNs treated with recombinant IFN-β (100 U/ml) for 24 h. C, Immunostaining of Ki-67 (Alexa Fluor 555/red) in GL261 cells alone and in coculture with Ifnb+/+ or Ifnb−/− 3 DIV CGNs. Blue, Hoechst nuclei. Scale bars, 25 μm. Left, Cell counting of Ki-67 in GL261 after coculture with Ifnb+/+ CGNs or Ifnb−/− CGNs compared with GL261 cells alone (right). One representative experiment is shown from two to three individual experiments. Data are mean ± SD from triplicates; ***p < 0.001. D, Thymidine incorporation assay on GL261 cells with cocultured with 3 DIV Ifnb+/+ CGNs or Ifnb−/− CGNs compared with GL261 cells alone. Data are mean ± SEM from five replicates from one representative of three experiments; ***p < 0.001. E, Representative FACS histograms of 7AAD-stained gated CFSE-labeled GL261 cells, before and after coculture with Ifnb+/+ CGNs. F, 7AAD in GL261 cells at different ratios in coculture with 3 DIV Ifnb+/+ CGNs or Ifnb−/− CGNs compared with GL261 cells alone; control, GL261 cultured with fibroblasts. Data are mean ± SD from three individual experiments. p < 0.05 for GL261 cells after coculture with Ifnb+/+ CGNs compared with GL261 cells only. G, PI staining (gated on sub-G1) of GL261 cells alone or after coculturing with 3 DIV Ifnb+/+ CGNs or Ifnb−/− CGNs. Data are mean ± SD from two to three individual experiments. **p < 0.01. H, Representative histograms of PI staining (gated on sub-G1) of cultured GL261 cells alone and after coculturing with Ifnb+/+ CGNs or Ifnb−/− CGNs (with or without treatment with recombinant IFN-β 100 U/ml for 3 h).
Table 2.
PD-L1 mRNA in CGNs after treatment with recombinant (rec) IFN-β
| Groups | Fold expression of PD-L1 | p value |
|---|---|---|
| Ifnb−/− + rec IFN-β/Ifnb−/− | 5.773 | 0.001 |
| Ifnb+/+/Ifnb−/− | 1.193 | 0.162 |
| Ifnb+/+ + rec IFN-β/Ifnb−/− | 15.45 | 0.001 |
| Ifnb+/+/Ifnb−/−>+ rec IFN-β | 0.2067 | 0.002 |
| Ifnb+/+ + rec IFN-β/Ifnb−/− + rec IFN-β | 2.677 | 0.072 |
| Ifnb+/+ + rec IFN-β/Ifnb+/+ | 12.95 | 0.001 |
Affymetrix gene expression analysis of PD-L1 on 3 DIV CGNs from Ifnb+/+ or Ifnb−/− mice, nontreated or treated with IFN-β (100 U/ml) for 3 h. RNA from triplicate CGN cultures analyzed on affymetrix mouse 430 2.0 arrays. Student's t test was used to calculate p values.
We then studied the functionality of CGNs in regard to limiting gliomas. We cultured Ifnb+/+ CGNs and Ifnb−/− CGNs with GL261 murine glioma cells. We observed that wild-type CGNs not only have capacity to inhibit glioma proliferation, they induce glioma death. In contrast to Ifnb+/+ CGNs, Ifnb−/−, CGNs did not inhibit glioma cell proliferation as assessed by Ki-67 staining and verified with a thymidine incorporation assay (Fig. 3C,D). Unlike wild-type CGNs, Ifnb−/− CGNs were defective in triggering cell death of GL261 cells at all investigated ratios, with dead cells measured by 7AAD (Fig. 3E,F) and PI staining gated on sub-G1 population of GL261 (Fig. 3G,H). Of note, treating Ifnb−/− CGNs with recombinant IFN-β rescued their ability to kill GL261 cells (Fig. 3H). Ifnb−/− CGNs were pretreated with recombinant IFN-β and washed before coculturing with GL261. Figure 3H shows percentage of PI staining in sub-G1 population; GL261 alone 7.6 ± 2.5%; Group 2, CGN Ifnb−/− + GL261 12% ± 3.3%; Group 3, recombinant IFN-β-treated CGNs Ifnb−/− + GL261 36.5 ± 6.9%; p = 0.223 Group 1 versus 2; 0.0053 Group 1 versus 3; 0.0107 Group 2 versus 3. These data indicated that neuronal IFN-β had a central role in regulating cell proliferation and cell death of gliomas.
Pdl1 gene silencing in CGNs leads to defective glioma-killing whereas ectopic expression of pdl1 in Ifnb−/− CGNs conveys glioma-killing capacity
We investigated whether PD-L1 expression by neurons is responsible for the observed glioma-killing capacity. We blocked neuronal PD-L1 with antibody before coculturing with glioma cells. As shown in Figure 4A, blocking neuronal PD-L1 abrogated the capacity of neurons to kill gliomas. To verify the neuronal PD-L1 requirement, we silenced pdl1 in Ifnb+/+ CGNs using siRNA (Fig. 4B). Loss of neuronal pdl1 resulted in loss of capacity to kill gliomas (Fig. 4C).
Figure 4.

Cerebellar granule neuronal PD-L1 is required for glioma-killing capacity. A, Analysis of FACS staining with 7AAD-stained (dead) GL261 cells after coculture with 3 DIV CGNs (untreated or pretreated with anti-PD-L1 antibody) compared with GL261 cells alone. Ratios are GL261:CGNs. Data are mean ± SD from three individual experiments; *p < 0.05, **p < 0.01, ***p < 0.001. B, Transfection efficiency of siRNA into CGNs by FACS staining for PD-L1. C, PI staining of GL261 cells after coculture with pdl1 siRNA-transfected CGNs and control siRNA-transfected CGNs. Representative data are from two independent experiments. D, Western blot and FACS (E) of 3 DIV Ifnb−/− CGNs either mock transfected (no plasmid) or transfected with pIRES2-EGFP or pIRES2-EGFP-PD-L1. F, DNA synthesis by BrdU incorporation of GL261 cells alone or in coculture with 3 DIV Ifnb−/− CGNs transfected with pIRES2-EGFP or pIRES2-EGFP-PD-L1. Data are mean ± SD from three individual experiments; ***p < 0.001. G, PI staining (gated on sub-G1, dead cells) of GL261 cells alone or after coculture with 3 DIV mock-transfected Ifnb−/− CGNs, or Ifnb−/− CGN transfected with pIRES2-EGFP or pIRES2-EGFP-PD-L1. Representative data are from two independent experiments.
These data indicated that neuronal induction of GL261 cell death required IFN-β-mediated upregulation of cell-surface PD-L1. To directly investigate whether the lack of PD-L1 was solely responsible for the inability of Ifnb−/− CGNs to regulate GL261 cells, Ifnb−/− CGNs lacking PD-L1 expression were transfected with a PD-L1 expression plasmid with expression confirmed by Western blot (Fig. 4D). FACS analysis of CGNs (Fig. 4E) gated for living cells showed a similar trend for PD-L1 expression as detected by WB. However, FACS sensitivity was different because only a small population of CGNs was included (gated on live cells and NF200-positive cells). For FACS analysis, CGNs were trypsin treated before staining. As CGNs are highly sensitive to an intact neuronal network, trypsin treatment often leads to death for a large proportion of neurons. Therefore, gating only for live cells showed successful transfection of the CGNs. However, the sensitivity of FACS detection was not directly comparable to PD-L1 expression by Western blot, which detected total protein production by untrypsinized CGNs. Microscopic examination of CGNs for coexpression of a fluorescent protein from the PD-L1 expression plasmid showed a 35% transfection rate.
Ifnb−/− CGNs ectopically expressing PD-L1 had glioma-killing function (Fig. 4F,G). Coculture of Ifnb−/− CGNs ectopically expressing PD-L1 with GL261 cells inhibited glioma cell proliferation as assessed by BrdU incorporation (Fig. 4F). As shown in Figure 4G, ectopic expression of PD-L1 was sufficient for Ifnb−/− CGNs to induce GL261 cell death as measured by percentage of cells in sub-G1 phase. These results indicated that ectopic expression of PD-L1 in Ifnb−/− CGNs conveyed the capacity to inhibit glioma proliferation and that neuronal PD-L1 was essential for CGN killing of glioma cells.
Neuronal PD-L1 induces caspase-dependent apoptosis of glioma cells
We next investigated the molecular signals by which neuronal PD-L1 induced glioma cell death. Wild-type CGNs were cocultured with GL261 cells, and CFSE-labeled GL261 cells before and after coculturing were purified using FACSaria and analyzed by Western blot and FACS for Bax and active caspase 8 and 3. Bax is a proapoptotic Bcl-2-family protein that activates caspase 8 and downstream caspases including caspase 3 in response to apoptotic signals. CGNs activated Bax and the downstream caspases in GL261 gliomas, as determined by increases in Bax p25 and p18 fragments, cleaved caspase 8 fragments; p41/43 and p18, and cleaved caspase 3 (Fig. 5A,B). Next, we studied whether neuronal PD-L1 was responsible for induction of caspase-dependent glioma apoptosis. Preventing direct CGN-GL261 cell interaction prevented neuron induction of activated caspase 3 in GL261 cells. In addition, blocking PD-L1 on neurons with antibody before coculturing with GL261 cells prevented activation of caspase 3 (Fig. 5C,D). To verify these results, pdl1 was silenced in CGNs using siRNA before coculturing with GL261 cells. In agreement with the previous results, lack of neuronal PD-L1 abrogated induction of cleaved caspase 3 (Fig. 5E). We also investigated whether induction of active cleaved caspase 3 via PD-L1 led to GL261 cell death. The results confirmed that neuronal PD-L1 signaling was essential for caspase-dependent killing of gliomas (Fig. 5F,G). Using a pan-caspase inhibitor (Z-VAD-FMK), to block caspase activity of GL261 cells abrogated activation of caspases (measured by cleaved caspase 3) and blocked the CGN capacity to kill gliomas. These data collectively indicated that neuronal PD-L1 was required to trigger a caspase-dependent apoptotic signal and cell death of gliomas.
Figure 5.
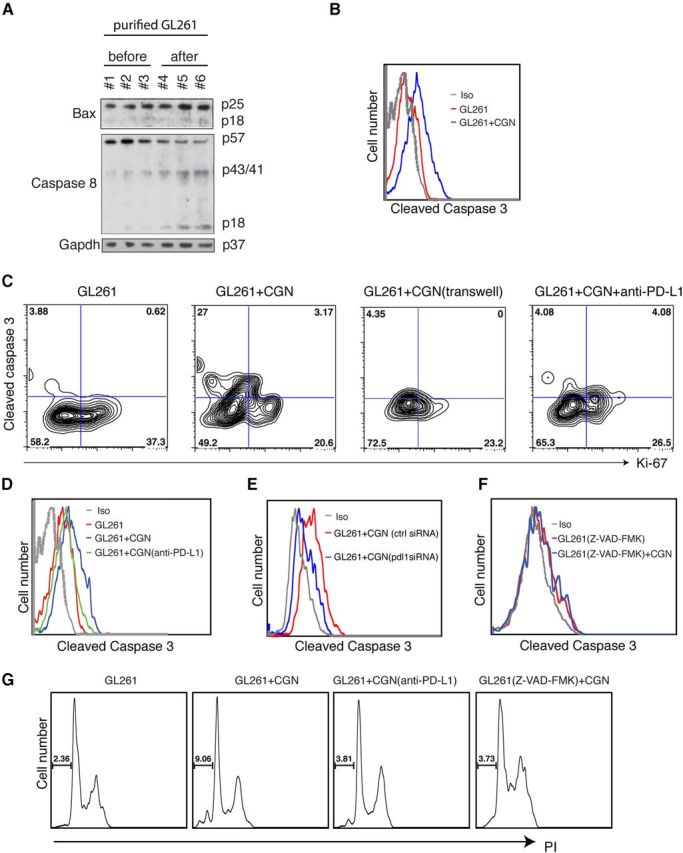
Neuronal PD-L1 kills glioma cells via caspase-dependent apoptosis. A, GL261 cells (CFSE labeled) purified with FACS after coculture with CGNs. Western blot of BAX, caspase 8, and GAPDH in GL261 cells before and after coculture with CGNs. GAPDH was the loading control. B, FACS histogram of cleaved caspase 3 in GL261 cells before and after coculture with CGNs (gated on CFSE). C, FACS staining for Ki-67 and caspase-3 in GL261 cells without and after coculture with 3 DIV CGNs (or blocking with anti-PD-L1 antibody). GL261 cells and CGNs were separated with a transwell membrane. One representative of three experiments is shown. D, Cleaved caspase 3 in gated GL261 cells before and after coculture with CGNs; anti-PD-L1 (20 μg/ml) was applied to CGNs for 30 min before coculturing. E, Cleaved caspase 3 in gated GL261 cells before and after coculture with control-transfected CGNs (ctrl siRNA) or pdl1-silenced CGNs (pdl1 siRNA). F, Caspase inhibitor (Z-VAD-FMK, 20 μm) was added to GL261 cells 2 h before coculturing. G, PI staining of GL261 cells before and after coculture with CGNs, with anti-PD-L1 added to CGNS or Z-VAD-FMK to GL261 cells.
Of note, glioma cell proliferation measured by Ki-67 positivity was stable when CGNs and glioma cells were separated by insertion of a membrane (Fig. 5C), indicating that PD-L1 signaling mainly induced caspase-mediated cell death in gliomas and lack of glioma proliferation was at least in part the consequence of cell-death induction by PD-L1. The results also suggested that soluble factors, most likely IFN-β, triggered some degree of proliferation arrest in gliomas but not cell death at physiological levels.
Neuronal PD-L1 binds an unknown glioma receptor to exert its killing function
PD-L1 negatively regulates proliferation and induces apoptosis of T-lymphocytes via the B7.1 and PD-1 receptors (Dong et al., 1999; Butte et al., 2007). We investigated whether GL261 cells expressed any known lymphocyte receptors and whether engagement of these receptors was involved in GL261 cell death. Additionally, we investigated effects of PD-L1 and B7.2 on GL261 cells, because the receptors on GL261 cells responsible for mediating cell-death effects of neuronal PD-L1 might be different from other cells. We detected no expression of PD-1 when PD-L1 and B7.1 were expressed in GL261 cells (Fig. 6A–C). To determine whether any receptors on GL261 cells bound neuronal PD-L1 and led to neuronal-mediated killing, we used antibodies and fusion proteins to block B7.1, B7.2, PD-L1, and PD-1 on GL261 cells before or during coculturing with neurons. Cell death percentages were unchanged by receptor blocking, except if neuronal PD-L1 was blocked (Fig. 6D). These results suggested that B7.1, B7.2, PD-L1, and PD-1 were not the GL261 receptors for cell death mediated by neuronal PD-L1. Our results therefore point to existence of an unknown GL261 receptor that bound neuronal PD-L1 to trigger caspase-dependent apoptosis.
Figure 6.
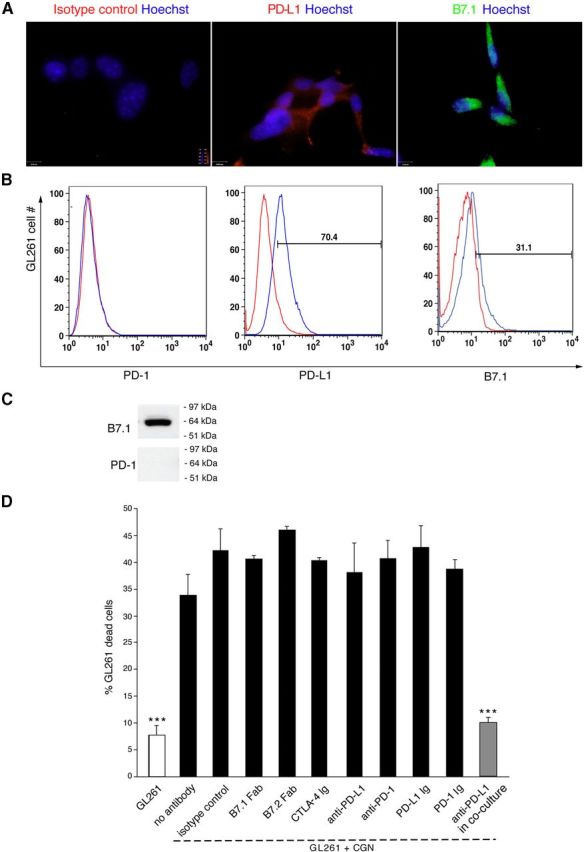
Neuronal PD-L1 binds an unknown receptor on gliomas for apoptotic function. A, Immunostaining of in vitro cultured GL261 cells with isotype control, PD-L1 (Cy3-red), or B7.1 (FITC) and Hoechst. Scale bar, 25 μm. B, FACS for PD-1, PD-L1, and B7.1 in cultured GL261 cells. Red line, isotype controls. Data are representative of three independent experiments. C, Western blot of PD-1 and B7.1 proteins in GL261 cells. D, Analysis of percentage of dead GL261 cells using FACS for 7AAD of GL261 cells alone or after coculture with 3 DIV CGNs. Blocking antibodies or fusion protein (20 μg/ml) were used before coculturing unless indicated. Data are mean ± SD from three individual experiments; ***p < 0.001.
Silencing glioma-PD-L1 does not affect neuronal capacity to limit gliomas
GL261 cells, similar to many other aggressive and tumorigenic gliomas, expressed PD-L1. We investigated the function of PD-L1 in GL261 cells by silencing PD-L1 using lentiviral shRNAs, rather than using antibodies that might induce a signal. Inhibition of PD-L1 synthesis was confirmed by Western blot (Fig. 7A). Even when pdl1 was silenced, CGNs prevented GL261 proliferation. Hence, lack of PD-L1 in GL261 cells did not influence the capacity of neurons to prevent GL261 growth (Fig. 7B) indicating that glioma PD-L1 was not required for neuron-mediated inhibition of glioma cells.
Figure 7.
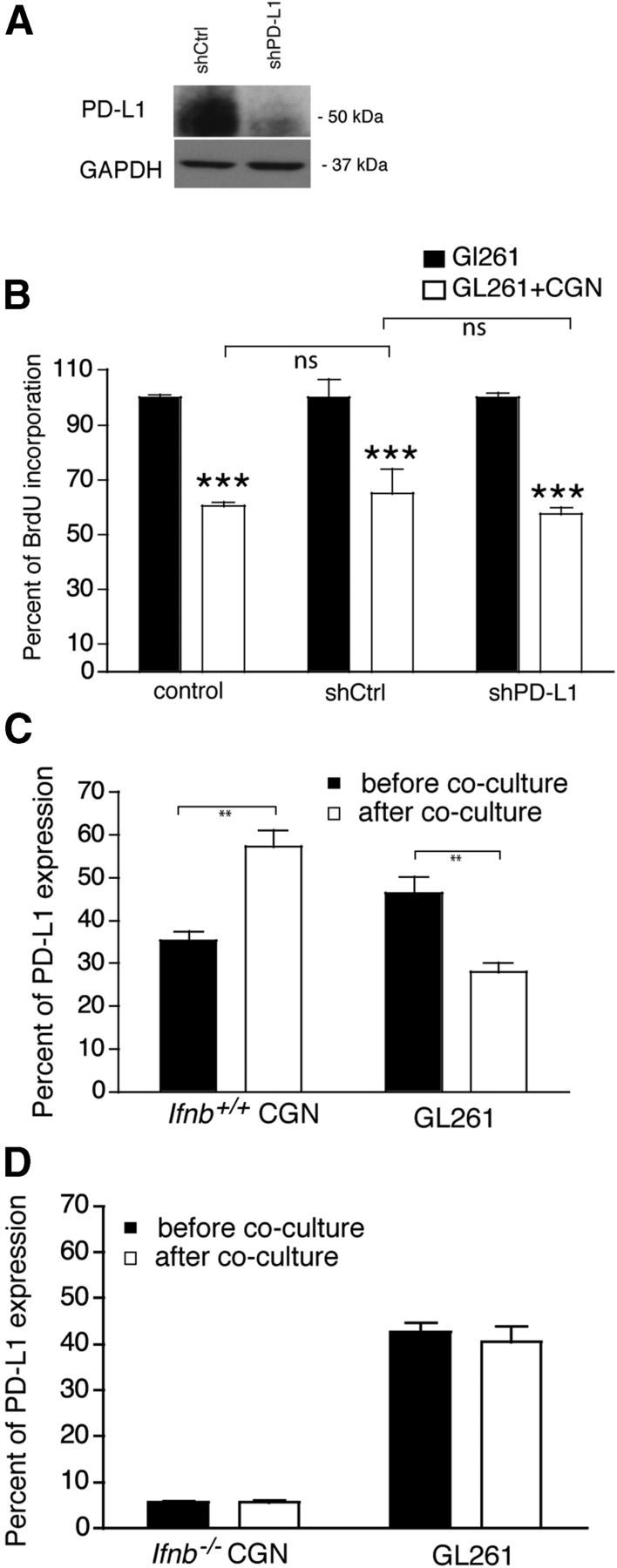
GL261 PD-L1 is not involved in neuron-mediated glioma killing. A, Western blot of PD-L1. PD-L1 was knocked down in GL261 cells using lentiviral shRNA to PD-L1 (shPD-L1) or nonsilencing shRNA control (shCtrl). B, DNA synthesis by BrdU incorporation of shPD-L1 GL261 cells or control cells (shCtrl), and native GL261 cells after coculture with 3 DIV Ifnb+/+ CGNs at 1:2 (GL261:CGN). Shown is a representative of two independent experiments. Data are mean ± SEM of five replicates, ***p < 0.001. C, FACS of percentage of PD-L1+ cells in 3 DIV Ifnb+/+ CGNs before and after coculture with GL261 cells, and percentage of PD-L1+ in GL261 cells before and after coculture with 3 DIV Ifnb+/+ CGNs. Data are mean ± SD from three individual experiments; **p < 0.01. D, FACS of percentage of PD-L1+ cells in 3 DIV Ifnb−/− CGNs before and after coculture with GL261 cells, and percentage of PD-L1+ in GL261 cells before and after coculture with 3 DIV Ifnb−/− CGNs.
Unexpectedly, although GL261 cells expressed PD-L1 constitutively in vitro (Fig. 6A,B, 7A), when cocultured with Ifnb+/+ CGNs, GL261 cells significantly downregulated PD-L1 expression (Fig. 7C). In contrast, PD-L1 expression was significantly upregulated in Ifnb+/+ CGNs in vitro (Fig. 7C). PD-L1 expression was substantially reduced in Ifnb−/− CGNs (Fig. 3A,B, 7D) with no increase in expression upon coculturing with glioma cells. Glioma PD-L1 expression also did not change after coculturing with Ifnb−/− CGNs (Fig. 7D). These findings were consistent with the findings on prognosis in GBM patients reported above.
Survival is poor for Ifnb−/− mice isografted with GL261 cells and associated with lack of PD-L1 downregulation in tumor cells
The findings that IFN-β-mediated neuronal PD-L1 was essential for regulation of glioma progression prompted us to examine the glioma-limiting capacity of the tumor microenvironment in mouse brains. Mice were injected intracranially with GL261 cells, inducing a murine glioma model highly similar to human GBM, including infiltrative and angiogenic capacity (Szatmári et al., 2006). Understanding the molecular basis for stromal cell interaction with tumors is possible only with syngenic tumors, so we injected GL261 cells into the cerebellum or cerebrum of Ifnb+/+ and Ifnb−/− C57BL/6 mice to investigate the effect of different microenvironments in different areas of the brain on outcomes. Due to different take frequency in preliminary experiments 3000 GL261 cells were injected in the cerebellum (Fig. 8A) and 1000 GL261 cells in the cerebrum (Fig. 8B).
Figure 8.

Ifnb+/+ Mice injected with GL261 cells have significantly better survival than Ifnb−/− mice with associated neuronal PD-L1 expression. After inoculation, mice were checked daily for survival and induction of neurological deficits. Ifnb−/− and Ifnb+/+ mice were inoculated (A) in the cerebellum with 3000 GL261 cells, N = 4–5 mice per group; or (B) intracerebrally with 1000 GL261 cells, N = 10 mice per group. **p < 0.01. C, Hematoxylin staining of a mouse brain section injected intracerebrally with GL261 glioma cells. Black square indicates the area included in further immunostaining analysis. Immunostaining of brain tissue sections of a glioma-bearing Ifnb+/+ mouse (E–J) and Ifnb−/− mouse (K–N). D, Isotype control and Hoechst. Scale bar, 62 μm. E, NF200 (Alexa Fluor 488/green), GFAP (Cy3/red), and Hoechst, showing tumor edge and brain. Scale bar, 62 μm. F, PD-L1 (Cy3/red) and Hoechst. Scale bar, 62 μm. G, PD-L1 (Cy3/red) of GL261 cells after establishment of solid tumor in Ifnb+/+ mouse brain. Scale bar, 10 μm. H, Isotype controls and Hoechst. Scale bar, 62 μm. I, PD-L1 (Cy3), GFAP (Alexa Fluor 488/green), and Hoechst. Scale bar, 31 μm. J, PD-L1 (Cy3/red), NF200 (Alexa Fluor 488/green), and Hoechst. Scale bar, 31 μm. K–M, Ifnb−/− tumor-bearing brain sections stained for PD-L1 (Cy3/red), NF200 (Alexa Fluor 488/green), and Hoechst shows PD-L1 staining in glioma but no NF200+ neurons by the tumor edge, and infiltrating gliomas. Scale bars: K, 31 μm; L, 62 μm; M, 31 μm; N, 10 μm.
Survival was reduced for Ifnb−/− mice compared with age- and gender-matched Ifnb+/+ mice. The lack of the Ifnb gene in the tumor microenvironment led to similar results regardless of the GL261 injection location (Fig. 8A,B), indicating the essential regulatory function of IFN-β in limiting cancer progression in the brain environment.
In vivo engrafted GL261 tumors form a solid mass from which neoplastic cells disseminate into adjacent brain tissue. Coronal sections of tumor-bearing brains from intracerebrally injected mice depicted the histologically investigated area/black frame (Fig. 8C). The edges of the tumor areas were often surrounded by NF200+ neurons (Fig. 8D,E). Although a majority of GL261 cells expressed PD-L1 in culture (Fig. 6A,B), when injected in the brains of Ifnb+/+ mice, GL261 cells downregulated PD-L1 expression. Intracranially injected GL261 cells showed particularly downregulated PD-L1 expression on the TM edge in the vicinity of TABT (Fig. 8F–J). PD-L1 expression in naive mouse brains with no challenge was very low (data not shown); however, when GL261 cells were injected into Ifnb+/+ mice to establish experimental glioma, PD-L1 expression was strongly induced in the brain in neurons (Fig. 8I), and to a lesser degree in morphologically detected GFAP+ reactive astrocytes in TABT areas (Fig. 8J). Whereas PD-L1 expression was highly induced in brain cells adjacent to tumors in Ifnb+/+ mice (Fig. 8F,I), PD-L1 expression in the TM was markedly reduced (Fig. 8F–J). Moreover, although some GFAP+ astrocytes expressed PD-L1 (Fig. 8J), NF200+ neurons in healthy tissue on the TM edge were the predominant PD-L1-expressing cells in Ifnb+/+ mouse brains (Fig. 8E,F, sequential sections, I). PD-L1 was highly expressed in the TM in Ifnb−/− mice, with almost no PD-L1 positivity in less-affected brain tissue (Fig. 8K–N). These findings were similar to our findings on GBM patients. Collectively, our data showed that endogenous IFN-β-mediated PD-L1 could be important for the capacity of brain stromal cells, in particular neurons, to inhibit tumor growth in their microenvironment. This activity could play a key role in prolonged survival with GBM.
Discussion
Treatment of CNS tumors is exceptionally complex. This situation is underscored by GBM, a highly invasive tumor characterized by rapid growth, dismal prognoses, and resistance to standard treatments. New targets for successful GBM treatment are urgently needed. PD-L1 is expressed by many cancer cell types (Dong et al., 2002) including glioblastomas (Wilmotte et al., 2005), and could be involved in the induction of apoptosis, resulting in immunosuppression of cancer-antigen specific T cells. Therefore, targeting PD-1 on T cells is a potential therapy for many cancers. The discovery of altered regulation of PD-L1 in GBM patients with a PTEN mutation (Parsa et al., 2007) identified this patient group as suboptimal candidates for PD-L1 directed immunotherapy (Waldron et al., 2010).
Few reports address the role of tissue-specific PD-L1 expression in the tumor microenvironment and its plausible role in cancer suppression. The possibility that CNS resident cells might upregulate PD-L1 expression (Liang et al., 2003; Lafon et al., 2008) prompted us to investigate PD-L1 expression in CNS stromal cells adjacent to tumors in GBM patients. PD-L1 expression might affect the tumor microenvironment and thereby tumor growth. We found that PD-L1 expression in surgically resected tumors from GBM patients showed a remarkable pattern. In a subgroup of GBM patients, neurons and some activated astrocytes highly expressed PD-L1 cells in TABT. Another group of GBM patients highly expressed PD-L1 in tumor cells. Notably, we found that neuronal PD-L1 was significantly associated with a favorable prognosis in that GBM patient subgroup, whereas PD-L1 expression in tumor cells was associated with poor prognosis. These findings have strong implications for future immunotherapies and might be developed into biomarkers for therapeutic selection.
Because neuronal PD-L1 was associated with better outcomes in GBM patients and the tissue microenvironment can suppress tumor growth (Barcellos-Hoff et al., 2009), we investigated the ability of neurons to limit glioma progression and the related functionality of neuronal PD-L1. For the first time, we report here that adult postmitotic neurons are capable of killing murine and human gliomas. Understanding this phenomenon could help explain why GBMs, which are among the most lethal primary cancers, are relatively rare among newly diagnosed adult cancers (Wrensch et al., 2002). In addition, cerebellar GBM is rare in humans (Weber et al., 2006). We found that CGNs had more potent killing function than cortical neurons and induction of experimental gliomas in the cerebellum required higher numbers of cancer cells. The finding that neurons in general and cerebellar granular neurons in particular exert strict protective effects that prevent the growth of gliomas could explain the relative rarity of GBM primary cancers in humans.
Understanding and identifying targets for effective therapeutics requires dissecting the basic molecular mechanisms central to the intrinsic regulatory functions of neurons. Our discovery that neurons are capable of killing gliomas led us to identify the factors important for this regulation. Little information is available about genomic polymorphisms related to susceptibility to gliomas. Nonetheless, defects in the Ifnb gene and interferon regulatory genes in gliomas have received attention. Early reports identified absence of the Ifnb gene in a large proportion of human malignant glioma cell lines (Miyakoshi et al., 1990). A pharmacological role for IFN-β in the suppression of human gliomas is reported (James et al., 1991). Previous studies on different cancers support Ifnb as a cancer suppressor (Diaz et al., 1988; Mizuno et al., 1990; Sugawa et al., 1993). We reported that neurons produce IFN-β (Teige et al., 2003). Defects in the Ifnb gene might underlie tissue-specific failure to regulate cancer progression. Recombinant IFN-β induces PD-L1 expression in neurons (Lafon et al., 2008). Hence, we examined whether neuronal capacity to inhibit glioma progression was mediated by endogenous IFN-β-mediated PD-L1 regulation. We found that endogenous production of IFN-β was critical for post-transcriptional regulation of PD-L1 production and expression by neurons. In addition, Ifnb deletion in CGNs abrogated their capacity to induce cell death in glioma cells and addition of IFN-β to Ifnb−/− CGNs rescued this defect. In agreement, the general antitumor effects of pharmacological levels of IFN-β include a direct tumor-suppressor function via inhibition of cell proliferation (Kaynor et al., 2002; Chawla-Sarkar et al., 2003). However, in our study, blocking cell–cell interaction while allowing transport of soluble factors, such as IFN-β, produced by neurons resulted in inhibition of neuronal caspase-dependent killing of gliomas. This finding indicated that physiological levels of IFN-β produced by neurons are not sufficient for glioma-killing function. We instead found that direct neuronal-killing function is mediated by neuronal expression of PD-L1. Treatment of IFN-β-deficient cells with IFN-β or pdl1 gene transfection of Ifnb−/− CGNs rescued neuronal capacity to kill gliomas, whereas pdl1 knockdown of wild-type neurons led to loss of killing function. Addressing direct in vivo effects of neuronal PD-L1 on the regulation of glioma by using neuron-specific deletion of PD-L1, to exclude impact of massive immunological defects, would prove valuable.
Given the importance of the Ifnb gene to glioma etiology, and in light of our findings, we propose that IFN-β functions in the native tissue environment of the CNS to protect against tumor development by regulating neuronal PD-L1. We investigated whether a lack of IFN-β in the CNS microenvironment influenced the outcome of glioma progression and PD-L1 expression. We found that mice lacking the Ifnb gene had poor survival after glioma induction. Similarly, a recent study showed that the type I IFN signaling pathway is important in the pathogenesis and clinical course of experimental gliomas, as Ifnar1-deficient mice experienced accelerated gliomagenesis (Fujita et al., 2010). This suggests that the environment of IFN-deficient hosts allows the growth of gliomas that would not survive in an immunologically competent CNS microenvironment. We found upregulation of PD-L1 in neurons in the vicinity of established tumors. In agreement, we found pathological findings in Ifnb+/+ and Ifnb−/− mice to resemble the subgroups of GBM patients that we investigated. Ifnb+/+ mice particularly upregulated PD-L1 in healthy neurons adjacent to tumor cells, whereas Ifnb−/− neurons showed a lack of neuronal PD-L1 expression. PD-L1 expression by IFN-β-competent hosts was associated with reciprocal downregulation of PD-L1 by tumor cells, which was also defective in the Ifnb−/− brain.
The poor efficacy and many side effects of IFN-β treatment could be due to the wide expression of its receptor, and systemic versus site-specific effects. Our data support the importance of site- and cell-specific effects of IFN-β. Additionally, past problems in using IFN-β for cancer treatment could result from the inclusion of wide and heterogeneous patient groups. A vast fraction of patients with GBM or other types of cancer might have defects in type I interferon signaling, making treatment with type I interferons nonproductive. In addition, certain therapies, if given to GBM patients with PTEN mutations, might induce unwanted PD-L1 upregulation in tumor cells. Thus, understanding the mode of action of endogenous IFN-β in the tissue microenvironment and the cell-specific post-transcriptional regulation of PD-L1 is important. We found that neuronal PD-L1 is required for tumor killing but PD-L1 expression by tumors was not. Also relevant is that not all tissue-specific PD-L1 is involved in limiting glioma growth, as coculture of glial cells capable of expressing PD-L1 with GL261 cells did not result in tumor killing. This indicated additional glial factors that prevent tumor growth inhibition, or complementary factors of neurons that contribute to tumor prevention.
Our findings shed light on plausible drawbacks in past trials with type I interferons, and highlight the potential side effects of anti-PD-L1 therapy that could result from disrupting the host environment or factors normally important for limiting tumor progression. Optimism over therapeutics based on PD-L1 blockade should be tempered by the caveat that upsetting the delicate balance of immune stimulation and inhibition could result in adverse outcomes. Our current findings also indicate that biological markers that detect alterations in the IFNB and PDL1 genes might be useful for characterizing patients as suitable candidates for specific therapies.
Footnotes
The project has received support from The Danish Cancer Society, The Novo Nordisk Foundation, The Danish Research Council-Medicine, and The Lundbeck Foundation to the principal investigator (S. I.-N). We thank Dr Reidar Albrechtsen for his valuable technical support in cell imaging.
The authors declare no competing financial interests.
References
- Barcellos-Hoff MH, Newcomb EW, Zagzag D, Narayana A. Therapeutic targets in malignant glioblastoma microenvironment. Semin Radiat Oncol. 2009;19:163–170. doi: 10.1016/j.semradonc.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barresi V, Belluardo N, Sipione S, Mudò G, Cattaneo E, Condorelli DF. Transplantation of prodrug-converting neural progenitor cells for brain tumor therapy. Cancer Gene Ther. 2003;10:396–402. doi: 10.1038/sj.cgt.7700580. [DOI] [PubMed] [Google Scholar]
- Benedetti S, Pirola B, Pollo B, Magrassi L, Bruzzone MG, Rigamonti D, Galli R, Selleri S, Di Meco F, De Fraja C, Vescovi A, Cattaneo E, Finocchiaro G. Gene therapy of experimental brain tumors using neural progenitor cells. Nat Med. 2000;6:447–450. doi: 10.1038/74710. [DOI] [PubMed] [Google Scholar]
- Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, Borden EC. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. 2003;8:237–249. doi: 10.1023/A:1023668705040. [DOI] [PubMed] [Google Scholar]
- Chirasani SR, Sternjak A, Wend P, Momma S, Campos B, Herrmann IM, Graf D, Mitsiadis T, Herold-Mende C, Besser D, Synowitz M, Kettenmann H, Glass R. Bone morphogenetic protein-7 release from endogenous neural precursor cells suppresses the tumourigenicity of stem-like glioblastoma cells. Brain. 2010;133:1961–1972. doi: 10.1093/brain/awq128. [DOI] [PubMed] [Google Scholar]
- Diaz MO, Ziemin S, Le Beau MM, Pitha P, Smith SD, Chilcote RR, Rowley JD. Homozygous deletion of the alpha- and beta 1-interferon genes in human leukemia and derived cell lines. Proc Natl Acad Sci U S A. 1988;85:5259–5263. doi: 10.1073/pnas.85.14.5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, costimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- Ehtesham M, Kabos P, Gutierrez MA, Chung NH, Griffith TS, Black KL, Yu JS. Induction of glioblastoma apoptosis using neural stem cell-mediated delivery of tumor necrosis factor-related apoptosis-inducing ligand. Cancer Res. 2002;62:7170–7174. [PubMed] [Google Scholar]
- Fujita M, Scheurer ME, Decker SA, McDonald HA, Kohanbash G, Kastenhuber ER, Kato H, Bondy ML, Ohlfest JR, Okada H. Role of type 1 IFNs in antiglioma immunosurveillance: using mouse studies to guide examination of novel prognostic markers in humans. Clin Cancer Res. 2010;16:3409–3419. doi: 10.1158/1078-0432.CCR-10-0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrlinger U, Woiciechowski C, Sena-Esteves M, Aboody KS, Jacobs AH, Rainov NG, Snyder EY, Breakefield XO. Neural precursor cells for delivery of replication-conditional HSV-1 vectors to intracerebral gliomas. Mol Ther. 2000;1:347–357. doi: 10.1006/mthe.2000.0046. [DOI] [PubMed] [Google Scholar]
- Jacobs JF, Idema AJ, Bol KF, Nierkens S, Grauer OM, Wesseling P, Grotenhuis JA, Hoogerbrugge PM, de Vries IJ, Adema GJ. Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro Oncol. 2009;11:394–402. doi: 10.1215/15228517-2008-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James CD, He J, Carlbom E, Nordenskjold M, Cavenee WK, Collins VP. Chromosome 9 deletion mapping reveals interferon alpha and interferon beta-1 gene deletions in human glial tumors. Cancer Res. 1991;51:1684–1688. [PubMed] [Google Scholar]
- Kaynor C, Xin M, Wakefield J, Barsoum J, Qin XQ. Direct evidence that IFN-beta functions as a tumor-suppressor protein. J Interferon Cytokine Res. 2002;22:1089–1098. doi: 10.1089/10799900260442511. [DOI] [PubMed] [Google Scholar]
- Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, Koulmanda M, Freeman GJ, Sayegh MH, Sharpe AH. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203:883–895. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krex D, Klink B, Hartmann C, von Deimling A, Pietsch T, Simon M, Sabel M, Steinbach JP, Heese O, Reifenberger G, Weller M, Schackert G. Long-term survival with glioblastoma multiforme. Brain. 2007;130:2596–2606. doi: 10.1093/brain/awm204. [DOI] [PubMed] [Google Scholar]
- Lafon M, Mégret F, Meuth SG, Simon O, Velandia Romero ML, Lafage M, Chen L, Alexopoulou L, Flavell RA, Prehaud C, Wiendl H. Detrimental contribution of the immuno-inhibitor B7-H1 to rabies virus encephalitis. J Immunol. 2008;180:7506–7515. doi: 10.4049/jimmunol.180.11.7506. [DOI] [PubMed] [Google Scholar]
- Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, Freeman GJ, Sharpe AH. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol. 2003;33:2706–2716. doi: 10.1002/eji.200324228. [DOI] [PubMed] [Google Scholar]
- Liu Y, Teige I, Birnir B, Issazadeh-Navikas S. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med. 2006;12:518–525. doi: 10.1038/nm1402. [DOI] [PubMed] [Google Scholar]
- Lumniczky K, Desaknai S, Mangel L, Szende B, Hamada H, Hidvegi EJ, Safrany G. Local tumor irradiation augments the antitumor effect of cytokine-producing autologous cancer cell vaccines in a murine glioma model. Cancer Gene Ther. 2002;9:44–52. doi: 10.1038/sj.cgt.7700398. [DOI] [PubMed] [Google Scholar]
- Miyakoshi J, Dobler KD, Allalunis-Turner J, McKean JD, Petruk K, Allen PB, Aronyk KN, Weir B, Huyser-Wierenga D, Fulton D. Absence of IFNA and IFNB genes from human malignant glioma cell lines and lack of correlation with cellular sensitivity to interferons. Cancer Res. 1990;50:278–283. [PubMed] [Google Scholar]
- Mizuno M, Yoshida J, Sugita K, Inoue I, Seo H, Hayashi Y, Koshizaka T, Yagi K. Growth inhibition of glioma cells transfected with the human beta-interferon gene by liposomes coupled with a monoclonal antibody. Cancer Res. 1990;50:7826–7829. [PubMed] [Google Scholar]
- Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, Mischel PS, Stokoe D, Pieper RO. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, Greenfield EA, Liang SC, Sharpe AH, Lichtman AH, Freeman GJ. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol. 2003;33:3117–3126. doi: 10.1002/eji.200324270. [DOI] [PubMed] [Google Scholar]
- Salama AD, Chitnis T, Imitola J, Ansari MJ, Akiba H, Tushima F, Azuma M, Yagita H, Sayegh MH, Khoury SJ. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J Exp Med. 2003;198:71–78. doi: 10.1084/jem.20022119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staflin K, Honeth G, Kalliomäki S, Kjellman C, Edvardsen K, Lindvall M. Neural progenitor cell lines inhibit rat tumor growth in vivo. Cancer Res. 2004;64:5347–5354. doi: 10.1158/0008-5472.CAN-03-1246. [DOI] [PubMed] [Google Scholar]
- Sugawa N, Ekstrand AJ, Ueda S, Collins VP. Frequency of IFN beta 1 gene loss in 47 primary human gliomas. Noshuyo Byori. 1993;10:161–163. [PubMed] [Google Scholar]
- Szatmári T, Lumniczky K, Désaknai S, Trajcevski S, Hídvégi EJ, Hamada H, Sáfrány G. Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci. 2006;97:546–553. doi: 10.1111/j.1349-7006.2006.00208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teige I, Treschow A, Teige A, Mattsson R, Navikas V, Leanderson T, Holmdahl R, Issazadeh-Navikas S. IFN-beta gene deletion leads to augmented and chronic demyelinating experimental autoimmune encephalomyelitis. J Immunol. 2003;170:4776–4784. doi: 10.4049/jimmunol.170.9.4776. [DOI] [PubMed] [Google Scholar]
- Waldron JS, Yang I, Han S, Tihan T, Sughrue ME, Mills SA, Pieper RO, Parsa AT. Implications for immunotherapy of tumor-mediated T-cell apoptosis associated with loss of the tumor suppressor PTEN in glioblastoma. J Clin Neurosci. 2010;17:1543–1547. doi: 10.1016/j.jocn.2010.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walzlein JH, Synowitz M, Engels B, Markovic DS, Gabrusiewicz K, Nikolaev E, Yoshikawa K, Kaminska B, Kempermann G, Uckert W, Kaczmarek L, Kettenmann H, Glass R. The antitumorigenic response of neural precursors depends on subventricular proliferation and age. Stem Cells. 2008;26:2945–2954. doi: 10.1634/stemcells.2008-0307. [DOI] [PubMed] [Google Scholar]
- Weber DC, Miller RC, Villà S, Hanssens P, Baumert BG, Castadot P, Varlet P, Abacioglu U, Igdem S, Szutowicz E, Nishioka H, Hofer S, Rutz HP, Ozsahin M, Taghian A, Mirimanoff RO. Outcome and prognostic factors in cerebellar glioblastoma multiforme in adults: a retrospective study from the rare cancer network. Int J Radiat Oncol Biol Phys. 2006;66:179–186. doi: 10.1016/j.ijrobp.2006.04.035. [DOI] [PubMed] [Google Scholar]
- Wilmotte R, Burkhardt K, Kindler V, Belkouch MC, Dussex G, Tribolet Nd, Walker PR, Dietrich PY. B7-homolog 1 expression by human glioma: a new mechanism of immune evasion. Neuroreport. 2005;16:1081–1085. doi: 10.1097/00001756-200507130-00010. [DOI] [PubMed] [Google Scholar]
- Wintterle S, Schreiner B, Mitsdoerffer M, Schneider D, Chen L, Meyermann R, Weller M, Wiendl H. Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res. 2003;63:7462–7467. [PubMed] [Google Scholar]
- Wrensch M, Minn Y, Chew T, Bondy M, Berger MS. Epidemiology of primary brain tumors: current concepts and review of the literature. Neuro Oncol. 2002;4:278–299. doi: 10.1093/neuonc/4.4.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Tao R, Wang X, Wang Y, Mao Y, Zhou LF. B7-H1 is correlated with malignancy-grade gliomas but is not expressed exclusively on tumor stem-like cells. Neuro Oncol. 2009;11:757–766. doi: 10.1215/15228517-2009-014. [DOI] [PMC free article] [PubMed] [Google Scholar]