

Abstract
In Alzheimer's disease (AD), soluble amyloid-β oligomers (AβOs) trigger neurotoxic signaling, at least partially, via the cellular prion protein (PrPC). However, it is unknown whether other ligands of PrPC can regulate this potentially toxic interaction. Stress-inducible phosphoprotein 1 (STI1), an Hsp90 cochaperone secreted by astrocytes, binds to PrPC in the vicinity of the AβO binding site to protect neurons against toxic stimuli. Here, we investigated a potential role of STI1 in AβO toxicity. We confirmed the specific binding of AβOs and STI1 to the PrP and showed that STI1 efficiently inhibited AβO binding to PrP in vitro (IC50 of ∼70 nm) and also decreased AβO binding to cultured mouse primary hippocampal neurons. Treatment with STI1 prevented AβO-induced synaptic loss and neuronal death in mouse cultured neurons and long-term potentiation inhibition in mouse hippocampal slices. Interestingly, STI1-haploinsufficient neurons were more sensitive to AβO-induced cell death and could be rescued by treatment with recombinant STI1. Noteworthy, both AβO binding to PrPC and PrPC-dependent AβO toxicity were inhibited by TPR2A, the PrPC-interacting domain of STI1. Additionally, PrPC–STI1 engagement activated α7 nicotinic acetylcholine receptors, which participated in neuroprotection against AβO-induced toxicity. We found an age-dependent upregulation of cortical STI1 in the APPswe/PS1dE9 mouse model of AD and in the brains of AD-affected individuals, suggesting a compensatory response. Our findings reveal a previously unrecognized role of the PrPC ligand STI1 in protecting neurons in AD and suggest a novel pathway that may help to offset AβO-induced toxicity.
Introduction
Neuronal dysfunction in Alzheimer's disease (AD) is related to accumulation of soluble oligomers of the amyloid-β peptide (AβOs; Lambert et al., 1998; Walsh et al., 2002; Ferreira and Klein, 2011; Mucke and Selkoe, 2012). Interaction of these toxic particles with several distinct types of receptors in neurons (Wang et al., 2000; Xie et al., 2002; Laurén et al., 2009; Decker et al., 2010) triggers glutamate excitotoxicity, synaptic dysfunction, inhibition of long-term potentiation (LTP), and neuronal death (Querfurth and LaFerla, 2010; Paula-Lima et al., 2013). The exact mechanisms underlying each of these effects are not fully understood, but toxic actions of AβOs seem to depend, at least in part, on the cellular prion protein (PrPC; Laurén et al., 2009; Gimbel et al., 2010; Bate and Williams, 2011; Kudo et al., 2012).
PrPC is a master regulator of cellular signaling (Martins et al., 2010), likely by scaffolding distinct ligands and neuronal transmembrane receptors (Linden et al., 2008; Beraldo et al., 2010, 2011; Santos et al., 2013). Interaction of AβO with the PrPC region comprising amino acid residues 95–105 appears critical for neuronal toxicity (Laurén et al., 2009; Chung et al., 2010; Barry et al., 2011; Freir et al., 2011). Accordingly, disrupting AβO binding to PrPC seems to alleviate PrPC-dependent AβO toxicity. For example, antibodies targeting PrPC prevent synaptic plasticity deficits induced by AβOs (Chung et al., 2010; Barry et al., 2011; Freir et al., 2011). However, PrPC antibodies can lead to toxicity by triggering neuronal signaling (Solforosi et al., 2004). Therefore, endogenous physiological ligands of PrPC may provide an alternative means of modulating AβO-induced toxicity.
Stress-inducible phosphoprotein 1 (STI1) is a cochaperone secreted by astrocytes that can interact with and signal via PrPC (Zanata et al., 2002; Lopes et al., 2005; Lima et al., 2007; Caetano et al., 2008; Roffé et al., 2010; Hajj et al., 2013). STI1 binds to PrPC at residues 113–128 (Chiarini et al., 2002; Zanata et al., 2002), adjacent to the AβO binding site, leading to reciprocal conformational changes in both proteins (Romano et al., 2009). Extracellular STI1 forms a signaling complex with PrPC in hippocampal neurons that promotes calcium influx through α7 nicotinic acetylcholine receptors (α7nAChR; Beraldo et al., 2010). This in turn triggers several signaling pathways that protect neurons from apoptosis (Lopes et al., 2005; Caetano et al., 2008; Beraldo et al., 2010; Roffé et al., 2010). Importantly, both PrPC and α7nAChR are recognized targets of Aβ peptides (Wang et al., 2000, 2009; Magdesian et al., 2005; Laurén et al., 2009; Um et al., 2012). Interestingly, recent system biology approaches have implicated differential expression of the STI1 gene (STIP1) in AD (Zhang et al., 2013). Moreover, a loss-of-function STI1 mutation increases Tau toxicity in a fly model of tauopathy (Ambegaokar and Jackson, 2011).
Here, we provide evidence supporting a role for STI1-regulated pathways in AD. We find that STI1 inhibited AβO binding to PrP and to cells expressing PrPC. In addition, toxic effects mediated by AβO could be prevented by STI1 in a PrPC and α7nAChR-dependent way. Our results suggest that altered levels of STI1 in individuals with AD may influence AβO-induced neuronal toxicity.
Materials and Methods
Mouse lines.
Genetically modified STI1−/+ mice were generated by standard homologous recombination techniques (Prado et al., 2006), using C57BL/6j ES cells, as described previously (Beraldo et al., 2013). In mammals, elimination of STI1 causes early embryonic lethality; hence, STI1−/+ neurons were used here (Beraldo et al., 2013). Prnp−/− mice in a C57BL/6j background were kindly donated by Dr. Frank Jirik (University of Calgary, Calgary, Alberta, Canada) (Tsutsui et al., 2008). APPswe/PS1dE9 (Jankowsky et al., 2001) and α7nAChR−/− (Orr-Urtreger et al., 1997) mice in a C57BL/6j background were obtained from The Jackson Laboratory. Procedures were conducted in accordance with approved animal use protocols at the University of Western Ontario (2008/127) and the A. C. Camargo Hospital (037/09) following Canadian Council of Animal Care and National Institutes of Health guidelines.
Preparation of proteins and peptides.
Recombinant mouse PrP with an N-terminal His tag was produced in Escherichia coli strain BL21(DE3). For this, bacteria were transformed with pRSET/PrP plasmid DNA kindly provided by Prof. Kurt Wüthrich (ETH Zürich, Zürich, Switzerland). PrP was expressed in inclusion bodies that were solubilized in 8 m urea and 20 mm Tris-HCl, pH 8.0, and purified using Ni2+-affinity chromatography. After that, PrP was refolded by dialysis against 10 mm NaOAc, 10 mm 2-mercaptoethanol, and 1 mm EDTA, pH 5.0, and stored for <10 d at 4°C. Recombinant mouse PrP(112–231) peptide was provided by the PrP5 PrioNet facility (University of Alberta, Edmonton, Canada). Recombinant STI1 was produced as a (His)6–SUMO-tag-fused protein by cloning STI1 cDNA into pE–SUMO vector (Lifesensors) and transforming E. coli strain BL21(DE3) with the obtained pE–SUMO–STI1 plasmid DNA. After initial purification using Ni2+-affinity chromatography, the (His)6–SUMO tag was cleaved off using SUMO Pro enzyme (Lifesensors), and untagged STI1 was obtained by a second Ni2+-affinity purification step. Pure STI1 was stored for less than a week at 4°C or fast-frozen in liquid nitrogen and stored at −80°C for 1–2 months. TPR2A and its 230–245 amino acid deletion variant (TPR2AΔ230–245) were produced as N-terminal His-tag constructs in E. coli strain BL21(DE3) transformed with pDEST17 vector (Invitrogen) containing the correspondent gene. After cleavage with His–TEV protease, (His)6 tag and the protease were removed by Ni2+-affinity purification, and the untagged peptides were stored for <10 d at 4°C. Quality of protein preparations, including Hsp90 (Cayman Chemical) and lysozyme (Sigma-Aldrich), was routinely checked using 4–20% SDS-PAGE gels (Lonza) stained for protein bands with RapidStain reagent (EMD Millipore), which allows 100 ng resolution. Circular dichroism (CD) spectra measured as described previously (Ostapchenko et al., 2008) were used to assess the quality of recombinant proteins. The presence of lipopolysaccharides in protein preparations was tested using ToxinSensor Endotoxin Assay Kit (GenScript); no more than 0.2 endotoxin units (EU) of E. coli endotoxin equivalent was present in our preparations. AβOs were prepared from Aβ1–42 peptide (rPeptide) similarly to a previously described procedure (Caetano et al., 2011). Briefly, the peptide was monomerized in hexafluoroisopropanol, dried in a SpeedVac centrifuge, restored in DMSO to 1 mm solution, and diluted in PBS (CD and LTP experiments) or F-12 medium (all other experiments) to the final concentration of 100 μm (hereafter monomer concentration used as AβO concentration). After incubation for 24 h at 4°C, AβOs were cleared by centrifugation when needed and either used immediately or stored at −80°C for no more than a few weeks. Peptide preparation quality was checked by several methods. Western blot with 6E10 (1:2000; Covance) antibody was done by a standard technique after peptide separation on 13.5% Tris-tricine SDS-PAGE and electrotransfer to polyvinyl difluoride membrane. CD spectra were obtained from 25 μm AβO using a J810 spectropolarimeter (Jasco) equipped with a 1 mm cuvette, with five scans averaged for each resulting spectra. Size-exchange chromatography was done using ÄKTA-FPLC (GE Healthcare) equipped with a Superdex 75 column (GE Healthcare) following the procedure described previously (Larson et al., 2012). For atomic force microscopy (AFM), AβO preparations were diluted to 0.1 μm, deposited on a freshly cleaved piece of mica for 10 min, and dried under a nitrogen stream. Images were acquired in tapping mode using a Cypher AFM (Asylum Research) mounted with silicon tips (AC160TS; from Olympus; nominal spring constant of 40 N/m). Section analyses were performed using the AFM software to determine the height of the species imaged. Their corresponding molecular weight was determined via a calibration curve describing the AFM heights of proteins of known molecular weight. Scrambled Aβ1–42 peptide (rPeptide) was prepared following the same procedure as for the AβO preparation.
Surface plasmon resonance.
Surface plasmon resonance (SPR) experiments were performed using the Biacore X system (GE Healthcare) equipped with either a nitrilotriacetic acid (NTA) or CM5 sensor chip. The NTA chip was first charged with nickel ions and then uniformly covered with either PrP or STI1 bearing (His)6 tags with SPR signal of ∼10,000 resonance units (RU). The CM5 chip was prepared by a standard amine-coupling procedure (Fischer, 2010). All ligands were injected in 25 mm HEPES, 150 mm NaCl, and 10 mm imidazole, pH 7.0, at 5 μl/min, and on-kinetics were registered for 6 min. After each injection, off-kinetics were followed for 2 min. The chip surface was regenerated between injections by a short injection of 10 mm HCl. SPR curves for STI1 and AβO binding to PrP were analyzed using a simple bimolecular binding model with GraphPad Software Prism linear (for initial binding rates), exponential decay (for off-kinetics), and “one-site binding” (to determine RUmax and KD from the Langmuir equation for the STI1–PrP complex) regressions (Balducci et al., 2010).
AβO binding to cells.
HEK293T cells were transfected with pH-sensitive GFP–PrPC vector (pHFP–PrPC) using a modified calcium phosphate method as described previously (Caetano et al., 2011). pHFP–PrPC was generated on the basis of pEGFP–PrPC vector (Lee et al., 2001) with GFP nucleotide sequence exchanged for that of pHFP (pHluorin; Miesenbock et al., 1998). Fluorescent AβOs were prepared from HiLyte Fluor 555-tagged Aβ1–42 (Anaspec) following the procedure described above. Three hundred nanomolar HiLyte Fluor 555–AβOs alone or mixed with 500 nm STI1 or 1000 nm TPR2A were added to cultures for 15 min, after which cells were washed with Krebs–Ringer–HEPES (KRH) buffer (in mm: 50 HEPES, 115 NaCl, 5.9 KCl, 1 MgCl2, 2 CaCl2, and 10 glucose, pH 7.4) and immediately imaged on a LSM510 confocal microscope (Carl Zeiss) equipped with a 63×/1.4 numerical aperture (NA) oil-immersion objective. Data were collected from at least eight images taken for each treatment in three independent experiments. Bound HiLyte Fluor 555–AβO was quantified for at least 20 cells for each experimental condition as mean fluorescence per cell area and normalized to nontransfected cells using NIH ImageJ software.
Primary cultures of hippocampal neurons from E17 mouse embryos were obtained as described previously (Beraldo et al., 2013). Neuronal cultures hereafter were derived from embryos of either sex. Cultures were maintained on poly-lysine-coated coverslips in Neurobasal medium with 2% B-27 supplement (Invitrogen). On day 4, cytosine arabinoside (2 μm; Sigma) was added to prevent astrocyte growth. Half of the culture medium was changed every 2–3 days. On day 15, neurons were treated for 15 min with 200 nm AβO alone or mixed with 500 nm STI1, washed with KRH buffer (in mm: 125 NaCl, 5 KCl, 5 HEPES, 2.6 MgSO4, and 10 glucose, pH 7.2). For γ-tubulin and AβO immunostaining, cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.5% Triton X-100 in PBS for 5 min, and blocked with 5% BSA (Sigma) in PBS for 1 h. After that, coverslips were incubated with anti-γ-tubulin (1:500; Abcam) and 6E10 (against amyloid-β 1–16 epitope; 1:350; Covance) antibodies overnight at 4°C, followed by secondary Alexa Fluor-488 (for γ-tubulin) and Alexa Fluor-633 (for AβO) antibodies (Invitrogen) for 1 h at 4°C. For colocalization analysis, the PrP antibody 8H4 (epitope 145–180; Abcam) and 6E10 antibodies were labeled with Alexa Fluor-488 and Alexa Fluor-633, respectively, using Zenon Mouse Labeling Kit (Invitrogen). Briefly, 1 μg of each primary antibody was incubated at room temperature for 5 min with 5 μl of the corresponding Zenon coupling reagent, after which the reaction was stopped by 5 min incubation with the blocking reagent. Labeled antibodies were diluted immediately in KRH buffer (1:350 for both antibodies), added to neurons treated with AβO, AβO/STI1, or vehicle, as described above, and incubated for 30 min at 37°C. Subsequently, cultures were washed with KRH buffer and imaged on an LSM510 confocal microscope equipped with a 63×/1.4 NA oil-immersion objective or a SP5 II confocal microscope (Leica) equipped with a 63×/1.47 NA oil-immersion objective. AβOs, γ-tubulin, and PrPC were quantified in at least three independent experiments. At least five Z-stack images were taken randomly from each coverslip representing a single treatment of neurons derived from a single embryo, and the corresponding fluorescence was integrated using NIH ImageJ software. Neurites from at least 20 cells were analyzed with cell bodies excluded from the quantification. AβO–PrPC colocalization was determined as percentage of AβO fluorescence volume colocalized with PrPC fluorescence using the NIH ImageJ colocalization plug-in.
Expression of synaptophysin.
For these experiments, primary cultured hippocampal neurons were obtained as indicated previously (Roffé et al., 2013). Cytosine β-d-arabinofuranoside at 1 μm was added on day 2, and cultures were maintained with no media replacement. On day 20, cells that were preincubated with or without 100 nm STI1 for 30 min and were treated with 500 nm AβO for 1 or 4 h unless otherwise indicated. For Western blots, cells were lysed in RIPA buffer (50 mm Tris-Cl, pH 7.4, 150 mm NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS) and analyzed by SDS-PAGE, followed by transfer to PVDF membrane and blotting with anti-synaptophysin (1:10,000; Santa Cruz Biotechnologies) and anti-GAPDH (1:1000; Sigma) antibodies. For immunofluorescence, cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.2% Triton X-100 in PBS for 5 min, and blocked with 5% BSA (Sigma) in PBS for 1 h. Anti-synaptophysin (1:100; Santa Cruz Biotechnologies) diluted in 1% BSA in PBS was added for 1 h, followed by anti-mouse Alexa Fluor-488 (1:1000; Invitrogen) for 1 h. Twenty images were analyzed per experiment with the NIH ImageJ histogram tool in at least three independent experiments for each experimental treatment, using a Nikon TE2000 microscope in epifluorescence mode. Images taken from cells labeled with secondary antibody only were used to set the threshold for the experiment. Cell bodies were excluded from the analysis.
Electrophysiology in hippocampal tissue slices.
Field EPSPs (fEPSPs) were recorded from hippocampal slices derived from wild-type mice that were between 21- and 30 d-old as described previously (Martyn et al., 2012). Slices were pretreated for 15- 30 min with or without 0.5 or 1 μm STI1, followed by 30–60 min treatment with vehicle or 1 μm AβO. No difference was observed between these two time points and protein concentrations used, and, therefore, the results of these experiments were pooled together.
Cell death and viability assays for AβO.
Neuronal cultures (1 × 105 cells per 16 mm dish) were prepared as described previously (Beraldo et al., 2013). On day 11, neurons were treated with different proteins or peptides for 48 h. Cell death was evaluated using the LIVE/DEAD Viability/Cytotoxicity Kit for mammalian cells (Invitrogen) as per the instructions of the manufacturer. Eight images from random fields containing at least 300 cells were taken for each experimental treatment of neurons prepared from at least five embryos on an LSM-510 confocal microscope equipped with 10×/0.45 NA objective and appropriate filters. Live (calcein-stained, green channel) and dead (ethidium-stained, red channel) cell counting was done using NIH ImageJ Cell Counter plug-in and calculated as percentage of dead cells [number dead cells/(number of dead cells + viable cells) × 100]. For the lactate dehydrogenase (LDH) release assay, neuronal cultures were prepared in the same way but using phenol red-free medium. LDH release in cultured media was analyzed with LDH Activity Assay kit (Sigma) following the instructions of the manufacturer. For this, cultured media (400 μl in a 16 mm dish) were concentrated to 100 μl using Nanosep 10K centrifugal devices (Pall Life Sciences) and mixed with 200 μl of LDH substrate mix. After 30 min incubation, LDH activity was measured by OD450 on an iMark Microplate Absorbance Reader (Bio-Rad) and normalized to total protein concentration in the samples.
Cell death and viability assay for staurosporine.
Neuronal cultures (1 × 105 cells per 16 mm dish) from wild-type or α7nAChR−/− mice were prepared as described above. Primary hippocampal neurons were treated with staurosporine (50 nm) in the presence or absence of 1 μm STI1 for 16 h as described previously (Beraldo et al., 2010). The cell death assay was performed using LIVE/DEAD Viability/Cytotoxicity Kit as described above. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay (Sigma) was conducted according to the protocol of the manufacturer. MTT stock solution (5 mg/ml) was added to hippocampal neurons as of the culture medium volume and incubated for 4 h. After that, the medium was removed, and cells were solubilized with isopropanol/0.1 N HCl, after which absorbance of reduced dye was measured at 570 nm with background subtraction at 650 nm.
Calcium signaling.
Primary hippocampal neurons were obtained as described for experiments with AβO binding, and calcium imaging was performed as described previously (Beraldo et al., 2010), by loading neurons with either 10 μm fura-2 AM for 40 min or 5 μm Fluo-4 AM (Invitrogen) for 30 min at 37°C in Neurobasal medium supplemented with 1 mm CaCl2. For fura-2 AM experiments, data acquisition was performed using a DMI6000 B microscope (Leica) equipped with a 40×/0.75 NA dry objective and 340 nm/380 nm (excitation) and 510 nm (emission) filters. Fluorescence ratio (340/380) was normalized using Leica AF6000 software. For Fluo-4 AM, data acquisition was performed on an LSM-510 confocal microscope with excitation at 488 nm and emission at 505–530 nm. Fluorescence was normalized as F1/F0 (in which F1 is maximal fluorescence and F0 is basal fluorescence). For each experimental condition, at least three different neuronal cultures from independent pups were used, and 30–40 cells were analyzed.
Human postmortem brain tissue.
Parietal cortical tissues from age- and sex-matched controls (n = 6, 3 females and 3 males) and AD-affected individuals (n = 6, 3 females and 3 males) were provided by the Institute for Brain Aging and Dementia Tissue Repository/University of California, Irvine. AD diagnosis was confirmed by pathological and clinical criteria (McKhann et al., 1984; Khachaturian, 1985; Michalski and Fahnestock, 2003). Cortical samples were homogenized in RIPA buffer supplemented with protease inhibitor cocktail III (Calbiochem). STI1 levels were analyzed by SDS-PAGE, followed by Western blot analysis with anti-recombinant mouse STI1 antibody raised in rabbits (Zanata et al., 2002; Beraldo et al., 2013; purified IgG, 0.2 μg/ml, generated by Bethyl Laboratories) using β-actin levels as a control.
Mouse brain tissue.
Cortical tissues from APPswe/PS1dE9 or wild-type control male mice were collected and homogenized in RIPA buffer as described above. STI1 levels were analyzed by SDS-PAGE, followed by Western blot analysis with rabbit anti-STI1 antibody (Zanata et al., 2002; Beraldo et al., 2013) using β-actin levels as a control.
Results
STI1 prevents AβO binding to PrPC
AβOs and STI1 bind to adjacent regions of PrPC, to residues 95–105 (Laurén et al., 2009) and 113–128, respectively (Zanata et al., 2002). To determine whether binding of these two PrPC ligands can occur simultaneously or whether they are mutually exclusive, we used SPR. We optimized standard procedures to obtain highly pure recombinant proteins (>95% according to SDS-PAGE analysis; Fig. 1A) and to produce well defined AβOs with substantial presence of low-order oligomers (Townsend et al., 2006; Hung et al., 2008; Larson et al., 2012; Figueiredo et al., 2013). Western blot analysis of AβO preparations showed 5–10% low-molecular-weight oligomers (2-, 3-, 4-mers) along with small amounts of higher-molecular-weight (HMW) components but no fibrils (Fig. 1B). Importantly, the size-exclusion chromatography profile of these oligomers was similar to that of AD brain-derived amyloid-β species (Larson et al., 2012) and contained peaks corresponding to monomers, dimers, and trimers, with small amounts of HMW AβO (Fig. 1C). AFM analysis confirmed the abundance of low-order oligomers in our AβO preparation, represented as 0.3–1 nm high round dots and a small amount of larger dots with their height (>1 nm) corresponding to HMW AβO (Fig. 1D). In addition, CD measurements demonstrated β-sheet structure in oligomer preparations and showed characteristic spectra for recombinant PrP (Ostapchenko et al., 2008) and STI1 (Romano et al., 2009; Fig. 1E).
Figure 1.

SPR studies of AβO binding to PrP. A–E, Characterization of protein and peptide preparations performed as described in Materials and Methods. A, SDS-PAGE analysis of recombinant proteins. B, Western blot of AβO preparation with 6E10 antibody. Lane 1, Freshly prepared AβOs; lane 2, 1 μm AβOs after 48 h incubation in Neurobasal medium/2% B-27 at 37°C. C, Size-exchange chromatogram of AβOs; peaks for Aβ1–42 monomers, dimers, and trimers and HMW aggregates are shown by arrows. D, A representative AFM image of AβO preparations showing monomers (∼0.3 nm high), dimers/trimers/tetramers (0.6–1.0 nm high), and a few HMW aggregates (>1 nm high). Scale bar, 100 nm. E, CD spectra of recombinant proteins and AβOs. F–M, SPR kinetics. F, Binding of AβOs (nanomolar) to full-length PrP and to its N-terminal mutant PrP(112–231) on an NTA chip. G, Binding of scrambled Aβ and AβO (both 2.5 μm) to PrP. H, Binding of AβOs (2.5 μm) to PrP in the presence of increasing concentrations of STI1 (nanomolar). Inset shows an inhibition curve for AβO binding to PrP obtained in multiple experiments (errors are smaller than symbols). I, Similar to H, but experiments were done in the presence of 0.2 EU E. coli endotoxin (amount detected in recombinant STI1). J, Similar to H, but experiments were done in the presence of 500 nm lysozyme. K, Binding of STI1 (nanomolar) to PrP immobilized on a CM5 chip. L, Binding of AβOs and Hsp90 to STI1 immobilized on a CM5 chip. M, Effects of TPR2A (1 μm) and TPR2AΔ230–245 (1 μm) on AβO (2.5 μm) binding to PrP. SPR data are representative of at least three independent experiments.
Initial experiments demonstrated that AβOs bind specifically to PrP in a dose-dependent manner (Fig. 1F,G). We used a simple bimolecular binding model to analyze SPR data and estimate kinetic constants of AβO–PrP binding. Considering that dimers and trimers, the main PrP-binding species in this preparation, represent ∼9% of the AβOs in our preparation, we estimated KD = 15 nm, with kon = 3500 M−1 s−1 and koff = 5.4 × 10−5 s−1, which is consistent with previous studies (Balducci et al., 2010). Of note, approximating off-kinetics with exponential decay gave a high error estimate in the koff measurement (∼50%), probably attributable to the fact that SPR signal noise and thermal drift magnitude were of the same order as the total SPR signal change during off-kinetics. Consequently, the calculated KD, as koff/kon, fell in the range of 7–30 nm. PrP lacking the N-terminal region [PrP(112–231)] was unable to interact with AβOs (Fig. 1F). As a control, scrambled Aβ did not bind to full-length PrP (Fig. 1G).
Recombinant STI1 impaired the binding of AβOs to immobilized PrP with an IC50 of ∼70 nm (Fig. 1H). To ensure absence of nonspecific effects, we determined the binding of AβOs premixed with either lipopolysaccharide (amount equivalent to that present in 500 nm recombinant STI1; Fig. 1I) or an irrelevant protein (500 nm lysozyme; Fig. 1J) to PrP. Neither of them altered AβO binding to PrP. STI1 showed dose-dependent binding to PrP (Fig. 1K) with KD = 550 ± 150 nm, kon = 2.0 ± 0.6 × 105 M−1 s−1, and koff = 11.0 ± 0.6 × 10−2 s−1. The measured KD value is in the same order of magnitude of values determined using different methodologies (Zanata et al., 2002; Romano et al., 2009). AβOs did not interact directly with STI1, although STI1 was able to interact with Hsp90 as a positive control under the same conditions (Fig. 1L). The TPR2A domain of STI1 (containing PrPC binding motif amino acids 230–245) decreased binding of AβO to PrP (IC50 of ∼300 nm), whereas TPR2AΔ230–245, which lacks the PrP binding site, had no effect (Fig. 1M). Together, these results suggest that STI1 interferes with AβO–PrP binding by impairing AβO binding to PrP and not because of a direct interaction between STI1 and AβO.
STI1 prevents AβO binding to cells expressing PrPC
To investigate whether STI1 affects AβO binding to PrPC on membranes of living cells, we initially used HEK293T cells. AβOs bound only marginally to nontransfected cells, whereas HEK293T cells expressing pHFP–PrPC displayed abundant coating with AβOs (Fig. 2A,B). In the presence of 500 nm STI1, AβO binding to pHFP–PrPC-transfected HEK293T cells was significantly decreased (Fig. 2A,B). TPR2A (1 μm) also decreased AβO binding to cells (Fig. 2A,B).
Figure 2.

STI1 and TPR2A inhibit AβO binding to HEK293T cells expressing pHFP–PrPC. A, Representative images of HEK293T cells in differential interference contrast (DIC) channel (column 1), green channel (pHFP fluorescence, column 2), red channel (HiLyte Fluor 555–AβO fluorescence, column 3), and merged (column 4). Row 1 shows nontransfected cells, and rows 2–5 show cells transfected with pHFP–PrPC in the absence (row 2) or presence (row 3) of AβO, AβO premixed with 500 nm STI1 (row 4) or 1 μm TPR2A (row 5). B, Quantification of data from A. Total AβO bound to pHFP–PrPC-transfected cells was normalized by cell size and by the amount of AβO bound to nontransfected cells. Data collected from at least 20 cells in three independent experiments were analyzed with one-way ANOVA with Tukey's post hoc test. ***p < 0.001.
In cultured hippocampal neurons, AβO binding showed a punctate pattern mainly localized to neurites (Fig. 3A,C) as described previously (De Felice et al., 2007, 2009). As observed in HEK293T cells, AβO binding to hippocampal neurons in culture was significantly decreased by STI1 when compared with cells treated with vehicle (Fig. 3A–D). Additionally, colocalization analysis indicated that ∼50% of AβO puncta were colocalized with PrPC (Fig. 3C,D). In the presence of STI1, colocalization between AβO puncta and PrPC was significantly decreased in hippocampal neurons (Fig. 3C,D).
Figure 3.

STI1 decreases AβO binding to PrPC in hippocampal neuronal cultures. A, Representative images showing γ-tubulin (left column) and AβO (right column) staining of 15 d in vitro neurons treated with AβO (top row) or AβO/STI1 mix (bottom row) as described in Materials and Methods. B, Quantification of A. The amount of bound AβOs was normalized by γ-tubulin levels and presented relative to the treatment with AβO alone. C, Representative images showing PrPC (left column), AβO (middle column), and merged (right column) staining of 15 d in vitro neurons treated with AβO (top row) or AβO/STI1 mix (bottom row) as described in Materials and Methods. White arrows indicate colocalized staining. D, Quantification of C. Bound AβO was quantified as percentage of image area. Colocalization with PrPC was quantified as described in Materials and Methods. Scale bars, 10 μm. Data were collected from at least three independent experiments from neurites of at least 25 cells for each condition and analyzed with Student's t test. **p < 0.01, ***p < 0.001.
STI1 prevents AβO-induced synaptic loss
AβO treatment of human brain tissue downregulates several genes involved in synaptic transmission, including synaptophysin (Sebollela et al., 2012). Moreover, AβOs elicit PrPC-dependent synaptic loss (Um et al., 2012). Treatment of hippocampal neurons in culture with AβOs for 60 min led to a decrease in synaptophysin levels (Fig. 4A–C). In contrast, exposure of hippocampal neurons to STI1 (100 nm) increased the levels of synaptophysin (Fig. 4B,C). In the presence of STI1, the toxic effect of AβOs on synaptophysin levels was prevented (Fig. 4B,C). Importantly, neither AβO or STI1 altered the levels of synaptophysin in hippocampal neurons cultured from Prnp−/− embryos (Fig. 4D), indicating that these effects of AβOs and STI1 depend on the presence of PrPC.
Figure 4.

Synaptophysin levels in neurons treated with AβOs and STI1. A, Synaptophysin level in cultured hippocampal wild-type neurons before and after 5, 10, 15, and 30 min treatment with 500 nm AβOs. Cells were lysed, and Western blots against synaptophysin (Syp) and GAPDH were performed. B, Representative images for wild-type (Prnp+/+) neuronal cultures treated with AβOs (500 nm, 1 h), STI1 (100 nm, 30 min), or both (100 nm STI1 for 30 min, followed by 500 nm AβOs for 1 h) and immunolabeled against synaptophysin. Scale bars, 10 μm. C, Quantification of B. D, The same as C but for Prnp−/− neuronal cultures. At least three independent experiments were done for each condition. Data were collected from 20 images containing neurites from at least 60 cells for each experiment and analyzed with one-way ANOVA with Tukey's post hoc test. *p < 0.05.
STI1 rescues AβO-induced inhibition of LTP in hippocampal slices
It has been shown that impairment of LTP in hippocampal slices by AβOs is mediated by PrPC (Laurén et al., 2009; Barry et al., 2011; Freir et al., 2011). Our results indicate that AβOs, but not a preparation of scrambled Aβ, decreased LTP at Schaffer collateral–CA1 synapses (Fig. 5A,B). When slices were previously treated with STI1 (0.5–1 μm), before being exposed to AβOs (1 μm), no decrease in LTP was observed, suggesting that treatment with STI1 prevents AβO-induced LTP inhibition (Fig. 5C,D). Interleaved recordings from control slices treated with AβOs alone confirmed the neurotoxic potency of AβO in these experiments, whereas STI1 alone did not modify LTP (data not shown).
Figure 5.

LTP measurements in hippocampal slices. A, LTP in mouse hippocampal slices, treated with AβO or scrambled Aβ as described in Materials and Methods. fEPSPs were recorded for 60 min after LTP induction. Inset shows typical pre-high-frequency stimulation (pre-HFS, dotted line) and post-HFS (solid lines) fEPSP traces. B, Bar graph summarizing averaged fEPSP slope values recorded at the endpoint (i.e., 80 min) of A. C, The same as A but treated with AβOs alone or with STI1. D, Bar graph summarizing averaged fEPSP slope values recorded at the endpoint (i.e., 80 min) of C. fEPSP slopes are presented as mean ± SEM of at least five slices, relative to preinduction values, and analyzed by one-way ANOVA with Tukey's post hoc test. *p < 0.05, **p < 0.01.
STI1 protects neurons against cellular injury induced by AβO
It has been shown that AβOs cause neuronal cell death in a PrPC-dependent manner (Resenberger et al., 2011; Kudo et al., 2012). Corroborating this observation, we showed that wild-type but not Prnp−/− cultured hippocampal neurons displayed decreased viability, measured using the LIVE/DEAD Viability/Cytotoxicity assay, when exposed to 1 μm AβO for 48 h (Fig. 6A). Of note, incubation of AβOs in culture medium did not produce measurable amounts of fibrils or prefibrillar aggregates (Fig. 1B), arguing that the observed toxicity is caused by low-molecular-weight Aβ species. Next, we checked whether levels of endogenous STI1 could influence the neurotoxic effect of AβOs. STI1−/+ neurons, shown previously to have 50% of wild-type STI1 protein levels (Beraldo et al., 2013), presented increased sensitivity to AβO exposure (Fig. 6B–D). Treatment of cultured neurons with STI1 prevented the toxic effects of AβOs in both STI1 mutant and wild-type neurons (Fig. 6B,D). Neither STI1 by itself nor scrambled Aβ had any effect on neuronal viability (Fig. 6B,D). Moreover, treatment of cultured neurons with TPR2A, the PrPC binding domain of STI1, also decreased the toxicity of AβOs (Fig. 6E). We also used LDH release as an indicator of cell death. Treatment with recombinant STI1 rescued neuronal death induced by AβOs in both genotypes, confirming the results obtained with the LIVE/DEAD Viability/Cytotoxicity assay (Fig. 6F). In these experiments AβO-induced LDH release appeared higher in STI1−/+ neurons, but this difference failed to reach statistical significance (Fig. 6F). Together, our results indicate that STI1 decreases the binding of AβOs to PrPC and prevents several toxic activities of AβOs in hippocampal neurons.
Figure 6.
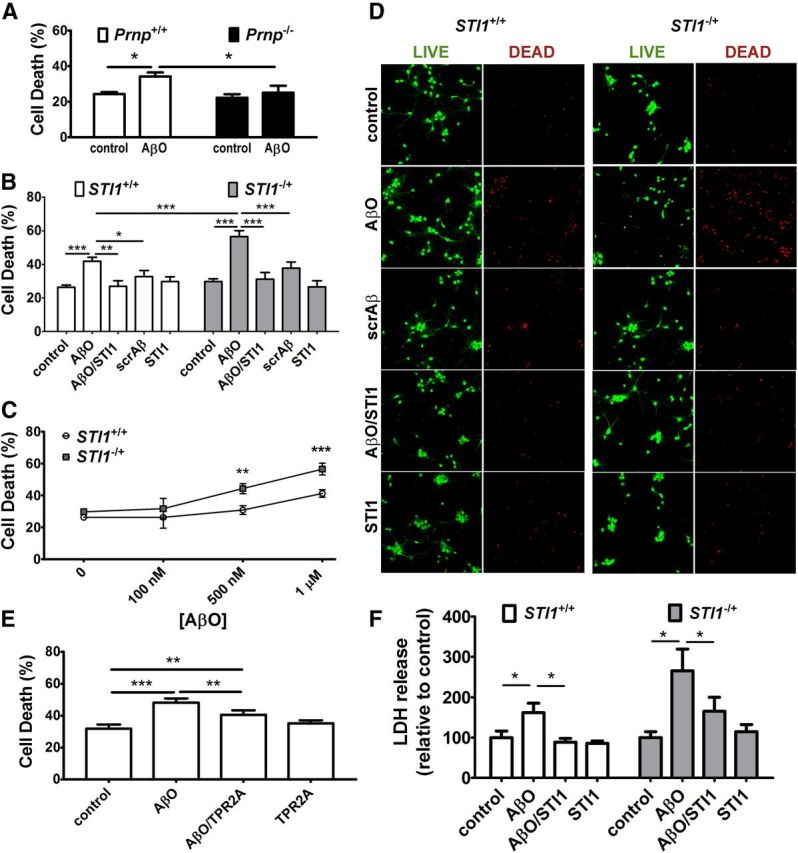
STI1 and TPR2A effect on AβO-induced cell death in hippocampal neurons. A–E, LIVE/DEAD assay. A, Comparison of cell death in Prnp+/+ and Prnp−/− neuronal cultures after 48 h treatment with 1 μm AβO. B, Comparison of cell death in STI1+/+ and STI1−/+ neurons after 48 h treatment with 1 μm scrambled Aβ, 1 μm AβOs, 1 μm STI1, or 1 μm AβOs/1 μm STI1 mix. C, The same as in B but only for different concentrations of AβOs. D, Representative images for B. Left two columns, Live (green) and dead (red) STI1+/+ neurons, nontreated (top row) or treated with 1 μm AβOs, 1 μm AβOs/1 μm STI1, 1 μm scrambled Aβ, or 1 μm STI1 (rows 2–5, respectively); right two columns, the same for STI1−/+ neurons. E, Comparison of cell death in wild-type neuronal cultures after 48 h treatment with 1 μm AβOs, 1 μm TPR2A, or their mix. F, LDH release in STI1+/+ and STI1−/+ neuronal cultures after 48 h treatment with 1 μm AβOs, 1 μm STI1, or their mix. At least five independent experiments were done for each genotype and condition. Experiments with different genotypes were analyzed by two-way ANOVA, followed by Bonferroni's post hoc test, and within the same genotype by one-way ANOVA, followed by Tukey's post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001.
STI1 induces intracellular Ca2+ increase and neuronal protection via α7nAChRs
We demonstrated previously that PrPC forms a biochemical and functional complex with α7nAChRs and that signaling and neuronal protection by STI1 was blocked by α-bungarotoxin, a selective α7nAChR-specific antagonist (Beraldo et al., 2010). Aβ1–42 has been shown to interact with α7nAChRs (Wang et al., 2000; Magdesian et al., 2005; Snyder et al., 2005), which is thought to play an important role in AD (Hernandez and Dineley, 2012). To test whether neuroprotection by STI1 might involve α7nAChRs, we cultured neurons from α7nAChR−/− mice and investigated the effect of STI1. We used neurons labeled with either fura-2 or Fluo-4 in independent experiments and found that Ca2+ increase induced by STI1 was abolished in α7nAChR−/− neurons (Fig. 7A–E). Moreover, the TPR2A peptide also increased intracellular Ca2+ in an α7nAChR-dependent way.
Figure 7.

STI1 neuroprotection and effect on intracellular calcium in wild-type (WT) and α7nAChR−/− (α7KO) neurons. A, Representative kinetics of intracellular calcium levels in wild-type and α7nAChR−/− hippocampal neurons treated with STI1. Intracellular Ca2+ was measured by fura-2 AM fluorescence as described in Materials and Methods. B, Calcium levels from A averaged from at least 30 cells. C, The same as A but measured with Fluo-4 AM fluorescence as described in Materials and Methods. D, The same as C but neurons were treated with TPR2A. E, Calcium levels from C and D averaged from at least 30 cells. F, MTT assay of cell viability in wild-type and α7nAChR−/− neuronal cultures, treated for 16 h with 50 nm staurosporine, 1 μm STI1, or their mix, as described in Materials and Methods. G, The same as in F but measured by LIVE/DEAD Viability/Cytotoxicity assay, as described in Materials and Methods. Data represent four independent experiments analyzed by two-way ANOVA with Bonferroni's post hoc test. *p < 0.01, **p < 0.001, ***p < 0.0001. H, Comparison of cell death in α7nAChR−/− neuronal cultures after 48 h treatment with 1 μm AβO, 1 μm STI1, or their mix, measured by LIVE/DEAD Viability/Cytotoxicity assay. Neuronal cultures were obtained from 13 independent embryos, and the data were analyzed by one-way ANOVA with Tukey's post hoc test. ***p < 0.0001.
Neuroprotection by STI1 against apoptosis induced by staurosporine (100 nm) was observed in wild-type neurons but not in α7nAChR−/− neurons, as determined by either the MTT reduction assay or the LIVE/DEAD Viability/Cytotoxicity assay (Fig. 7F,G). Similarly to wild-type neurons, AβOs induced ∼15–20% increase in cell death in α7nAChR−/− neurons; surprisingly, however, addition of STI1 did not rescue those neurons from AβO-induced cell death (Fig. 7H). Of note, α7nAChR−/− neuronal cultures showed an increased background level of cell death [∼40 vs ∼20% for wild-type neurons (Fig. 6B)], suggesting that expression of α7nAChR is important for cell viability in neuronal cultures.
STI1 levels are increased in AD
STI1 is part of the cellular stress response, and we showed recently that STI1 knock-out cells are less resilient to stress (Beraldo et al., 2013). Moreover, network analysis suggests that STIP1, the STI1 gene, may be a critical biological node for regulation of the unfolded protein response in AD cerebral cortex (Zhang et al., 2013). To determine whether STI1 levels change in AD, we fist performed analysis of the APPswe/PS1dE9 transgenic mouse model. These experiments revealed a 50% increase in cortical STI1 levels in 12-month-old APPswe/PS1dE9 but not in 9-month-old mice compared with wild-type controls (Fig. 8A,B). Importantly, STI1 levels were also increased in AD brains when compared with age-matched controls (cohorts described in Table 1; Michalski and Fahnestock, 2003; Fig. 8C).
Figure 8.

STI1 levels in APPswe/PS1dE9 mice and AD brains. A, Comparison of STI1 levels in 9-month-old APPswe/PS1dE9 and wild-type (WT) mice by Western blot. B, Similar analysis for 12-month-old mice. Data collected from at least six animals were normalized by actin levels and analyzed by Student's t test. *p < 0.05. C, Comparison of STI1 levels in AD (Alz) and age-matched control brains. Data were collected from three male and three female AD brains and age-matched control brains, normalized by β-actin levels and analyzed by Student's t test. *p < 0.05.
Table 1.
Human parietal cortex samples
| Diagnosis | Age | PMI | Sex |
|---|---|---|---|
| Pair 1 | F | ||
| Control | 82 | 7.25 | |
| AD | 82 | 3.00 | |
| Pair 2 | F | ||
| Control | 87 | 6.50 | |
| AD | 88 | 4.25 | |
| Pair 3 | M | ||
| Control | 64 | 4.50 | |
| AD | 66 | 3.00 | |
| Pair 4 | M | ||
| Control | 78 | 6.00 | |
| AD | 79 | 2.75 | |
| Pair 5 | M | ||
| Control | 80 | 7.00 | |
| AD | 80 | 2.75 | |
| Pair 6 | F | ||
| Control | 93 | 8.00 | |
| AD | 79 | 3.25 |
Samples were taken from control and AD postmortem brains with indicated postmortem interval (PMI, hours) and age and sex matched in pairs (except for pair 6) for Western blot analysis of STI1 levels.
Discussion
Here we show that the PrPC ligand STI1 prevents deficits of synaptic plasticity and increased neuronal death induced by toxic AβO species. Mechanistically, both interference with AβO binding to neurons and α7nAChR activation play a role in neuroprotection induced by STI1. Our data also demonstrate that STI1 levels are increased in AD. Although the biological significance of this change in STI1 levels is not understood, it is possible that, without this compensatory response, toxic effects of AβOs could be more prominent. These results open a novel avenue in AD research indicating that endogenous PrPC ligands can regulate toxicity by AβOs.
STI1 interferes with the AβO–PrPC interaction
AβOs have been shown to interact with several synaptic molecules (Ferreira and Klein, 2011), but its interaction with PrPC is one of the best characterized. Despite initial controversy (Balducci et al., 2010; Benilova and De Strooper, 2010; Kessels et al., 2010), a number of observations supported the notion that interaction of AβOs with PrPC activates toxic signaling in neurons (Laurén et al., 2009; Gimbel et al., 2010; Bate and Williams, 2011; Resenberger et al., 2011; Kudo et al., 2012; Um et al., 2012). We used well characterized synthetic AβOs (Fig. 1) to mimic the effects of toxic AD-related Aβ species. In our conditions, AβO preparations typically contained ∼7% trimers and ∼2% dimers, in addition to tetramers and small levels of higher-order oligomers. Dimers and trimers are thought to be among the most toxic assemblies of Aβ (Townsend et al., 2006; Hung et al., 2008; Figueiredo et al., 2013) and have been shown to bind PrPC and to induce PrPC-dependent toxic effects (Larson et al., 2012).
We confirmed the specificity of AβO binding to purified PrP using SPR and also demonstrated that expression of PrPC in cells increased AβO binding substantially. These results are consistent with several other publications showing interaction between AβOs from different sources and PrPC both in vitro and in vivo (Laurén et al., 2009; Balducci et al., 2010; Chen et al., 2010; Kessels et al., 2010; Larson et al., 2012). The AβO–PrPC complex is poorly understood at the molecular and structural levels, and its formation, based on SPR kinetic curves, is not likely to be a one-step process. It appears to start with a relatively slow binding phase with kon of just 3500 M−1 s−1, but then the two proteins associate tightly (koff of ∼5 × 10−5 s−1), resulting in a high-affinity complex with KD of ∼15 nm. Therefore, it is reasonable to hypothesize that binding occurs in more than one step, and the initial lower-affinity interaction is followed by a rearrangement step leading to formation of a strong complex. Of note, the KD for on-chip STI1 binding to PrP (550 nm) is higher than that observed for AβOs, but its initial binding rate (kon = 2 × 105 M−1 s−1) is much faster, suggesting the possibility that STI1 could prevent the formation of this hypothetical initial low-affinity AβO–PrPC complex. The molecular mechanism of this competition is probably related to the adjacent binding sites for STI1 and AβO on the PrPC N-terminal domain. Thus, binding of STI1 (or the TPR2A domain of STI1, which contains the motif responsible for STI1 binding to PrP) to amino acid residues 113–128 on PrP possibly makes adjacent regions sterically unavailable to other ligands. This could explain why TPR2A alone, a less bulky molecule compared with STI1, shows weaker inhibition of AβO binding to PrP. Alternatively, conformational changes on PrP induced by STI1 (Romano et al., 2009) could also affect the interactions between PrP and AβO.
STI1 prevents toxic effects of AβOs
Interaction of AβOs with neurons leads to multiple neurotoxic effects, and although the underlying mechanisms have not been completely delineated, NMDA receptor-mediated excitotoxicity and abnormal activation of Fyn kinase have been implicated (Larson et al., 2012; Um et al., 2012). Abnormal activation of NMDAR and Fyn kinase seem to connect toxic actions of AβOs to altered Tau function (Ittner et al., 2010). PrPC seems to interact directly with NMDA receptors and to regulate their desensitization by providing a source of copper, which can be disrupted by increased Aβ1–42 (You et al., 2012). In agreement with these toxic effects, AβOs disrupt synaptic plasticity, including LTP, which has been shown to be an effect dependent on PrPC (Laurén et al., 2009; Barry et al., 2011; Freir et al., 2011). We found that, in neuronal cultures, AβOs decreased the levels of the presynaptic marker synaptophysin, similar to findings in AβO-treated human cortical slices (Sebollela et al., 2012). Conversely, STI1 increased immunoreactivity for synaptophysin, consistent with its known effect of increasing neuronal protein synthesis (Roffé et al., 2010). Additionally, STI1 was able to protect hippocampal neurons from the toxic effect of AβOs on synaptophysin levels. Importantly, the effects of both STI1 and AβOs on synaptophysin levels were lost in cultures from PrPC-null mice, indicating that PrPC is involved in these signaling pathways. Moreover, STI1 prevented inhibition of LTP induced by AβOs. These results suggest that increased extracellular levels of STI1, a PrPC ligand that is secreted by astrocytes (Beraldo et al., 2013; Hajj et al., 2013), can mitigate AβO-mediated synaptic toxicity.
We also showed that neurons haploinsufficient for STI1 are more sensitive to AβO-induced cell death, a result consistent with our recent findings that cells are less resilient in the absence of STI1 (Beraldo et al., 2013). Hence, the differential expression of STI1 in AD brains may have physiological significance. Interestingly, flies harboring an STI1 mutation showed increased toxicity in a model of tauopathy (Ambegaokar and Jackson, 2011), suggesting that STI1 may be a critical regulator of distinct pathological signatures in AD. The increased neuronal death induced by AβOs in STI1-mutant hippocampal neurons could be prevented by extracellular recombinant STI1. We showed previously that extracellular recombinant STI1 reproduces the effect of secreted STI1 (Caetano et al., 2008). Moreover, the TPR2A STI1 domain, which lacks cochaperone activity because it is unable to bind both Hsp90 and Hsp70 (Brinker et al., 2002), could also prevent AβO-mediated neuronal death, suggesting that the neuroprotective effects of STI1 may not be related to a cochaperone mechanism.
Mechanism for prevention of AβO-mediated toxicity
Although STI1 decreases the binding of AβOs to PrPC in vitro, it is also possible that the protein could regulate AβO-mediated toxicity by activating neuroprotective signaling pathways (Lopes et al., 2005; Caetano et al., 2008; Beraldo et al., 2010; Roffé et al., 2010). We found that STI1-mediated Ca2+ influx was abolished in neurons from α7nAChR-null mice and that STI1-mediated neuroprotection is impaired in these mutants. Importantly, prevention of AβO-induced neuronal death by STI1 was not observed in α7nAChR-null neurons. This result suggests that, in the presence of STI1, residual AβO complexes with PrPC or other targets may still initiate toxic responses. Nonetheless, STI1 activation of the PrPC/α7nAChR pathway seems to prevent these effects. Together, these results argue that decrease of AβO interaction with PrPC in addition to activation of α7nAChR-mediated neuroprotection pathways may participate in the effects of STI1.
Our work is consistent with previous studies showing neuroprotective roles of α7nAChR in AD (Dineley et al., 2001; Hernandez et al., 2010; Shen et al., 2010). Indeed, Aβ actions via α7 nAChRs may also affect hippocampal LTP (Gu and Yakel, 2011), and genetic depletion of α7 nAChRs in an early-stage AD mouse model exacerbated cognitive deficits and septohippocampal pathology (Hernandez et al., 2010). Interestingly, higher concentrations or chronic exposure to AβO appears to corrupt α7nAChR function, which can be prevented by intervening small molecules (Wang et al., 2009, 2012) or by genetic depletion of α7nAChR (Dziewczapolski et al., 2009). It remains to be determined whether biasing signaling via PrPC/α7nAChR by further increasing STI1 levels could be used to prevent the toxic actions of AβOs in vivo.
Conclusion
Our studies suggest the possibility that STI1 may influence toxic responses to Aβ oligomers in AD. Increased levels of STI1 observed in AD brain may exert a protective role, although this obviously cannot prevent toxicity in advanced disease. It is possible that higher levels of STI1 are part of a compensatory response that may mitigate toxicity. Future experiments using tissue-specific elimination of STI1 may help to clarify this issue.
STI1 is a cochaperone known to interact with Hsp90 and Hsp70 to facilitate client transfer (Southworth and Agard, 2011). Our experiments in neurons support the importance of extracellular STI1 in protection against AβO toxicity; however, we cannot completely exclude that intracellular STI1 may also participate in cellular resilience in this condition (Beraldo et al., 2013). Increased chaperone activity may also play a role in protection against prolonged AβO exposure (Resenberger et al., 2012), which induces oxidative stress (De Felice et al., 2007) and mitochondrial damage in neurons (Paula-Lima et al., 2011), leading to increased load of misfolded proteins (Li et al., 2009). Interestingly, we showed recently that, after 9 months of age, APP/Ps1dE9 mice seem to present increased oxidative stress, revealed by increased PrPC β processing (Ostapchenko et al., 2013). Our present findings describe a novel neuroprotective role for the PrPC ligand STI1, which added to recent systems biology reports (Ambegaokar and Jackson, 2011; Zhang et al., 2013), implicates STI1 in distinct aspects of AD.
Footnotes
This work was supported by the Alzheimer's Association, PrioNet-Canada, Canadian Institutes of Health Research Grant MOP 93651, São Paulo State Foundation (FAPESP) Grants 2009/14027-2 (V.R.M.) and 2012/04370-4 (G.N.M.H.), National Institute for Translational Neuroscience, Canadian Foundation for Innovation, and Ontario Research Fund. A.H.M. and A.M. were recipients of Master's Program Ontario Graduate Scholarship. B.L.T. received an FAPESP fellowship. We thank PrP5, a PrioNet-Canada facility, and David Wishart for the gift of PrP 112-231.
The authors declare no competing interests.
References
- Ambegaokar SS, Jackson GR. Functional genomic screen and network analysis reveal novel modifiers of tauopathy dissociated from tau phosphorylation. Hum Mol Genet. 2011;20:4947–4977. doi: 10.1093/hmg/ddr432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, Chiesa R, Gobbi M, Salmona M, Forloni G. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A. 2010;107:2295–2300. doi: 10.1073/pnas.0911829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, Scott M, Walsh DM, Rowan MJ. Alzheimer's disease brain-derived amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci. 2011;31:7259–7263. doi: 10.1523/JNEUROSCI.6500-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bate C, Williams A. Amyloid-beta-induced synapse damage is mediated via cross-linkage of cellular prion proteins. J Biol Chem. 2011;286:37955–37963. doi: 10.1074/jbc.M111.248724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova I, De Strooper B. Prion protein in Alzheimer's pathogenesis: a hot and controversial issue. EMBO Mol Med. 2010;2:289–290. doi: 10.1002/emmm.201000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraldo FH, Soares IN, Goncalves DF, Fan J, Thomas AA, Santos TG, Mohammad AH, Roffé M, Calder MD, Nikolova S, Hajj GN, Guimaraes AL, Massensini AR, Welch I, Betts DH, Gros R, Drangova M, Watson AJ, Bartha R, Prado VF, et al. Stress-inducible phosphoprotein 1 has unique cochaperone activity during development and regulates cellular response to ischemia via the prion protein. FASEB J. 2013;27:3594–3607. doi: 10.1096/fj.13-232280. [DOI] [PubMed] [Google Scholar]
- Beraldo FH, Arantes CP, Santos TG, Queiroz NG, Young K, Rylett RJ, Markus RP, Prado MA, Martins VR. Role of alpha7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J Biol Chem. 2010;285:36542–36550. doi: 10.1074/jbc.M110.157263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraldo FH, Arantes CP, Santos TG, Machado CF, Roffe M, Hajj GN, Lee KS, Magalhães AC, Caetano FA, Mancini GL, Lopes MH, Américo TA, Magdesian MH, Ferguson SS, Linden R, Prado MA, Martins VR. Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin gamma1 chain. FASEB J. 2011;25:265–279. doi: 10.1096/fj.10-161653. [DOI] [PubMed] [Google Scholar]
- Brinker A, Scheufler C, Von Der Mulbe F, Fleckenstein B, Herrmann C, Jung G, Moarefi I, Hartl FU. Ligand discrimination by TPR domains. Relevance and selectivity of EEVD-recognition in Hsp70 x Hop x Hsp90 complexes. J Biol Chem. 2002;277:19265–19275. doi: 10.1074/jbc.M109002200. [DOI] [PubMed] [Google Scholar]
- Caetano FA, Lopes MH, Hajj GN, Machado CF, Pinto Arantes C, Magalhães AC, Vieira Mde P, Américo TA, Massensini AR, Priola SA, Vorberg I, Gomez MV, Linden R, Prado VF, Martins VR, Prado MA. Endocytosis of prion protein is required for ERK1/2 signaling induced by stress-inducible protein 1. J Neurosci. 2008;28:6691–6702. doi: 10.1523/JNEUROSCI.1701-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caetano FA, Beraldo FH, Hajj GN, Guimaraes AL, Jürgensen S, Wasilewska-Sampaio AP, Hirata PH, Souza I, Machado CF, Wong DY, De Felice FG, Ferreira ST, Prado VF, Rylett RJ, Martins VR, Prado MA. Amyloid-beta oligomers increase the localization of prion protein at the cell surface. J Neurochem. 2011;117:538–553. doi: 10.1111/j.1471-4159.2011.07225.x. [DOI] [PubMed] [Google Scholar]
- Chen S, Yadav SP, Surewicz WK. Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role OF N-terminal residues. J Biol Chem. 2010;285:26377–26383. doi: 10.1074/jbc.M110.145516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarini LB, Freitas AR, Zanata SM, Brentani RR, Martins VR, Linden R. Cellular prion protein transduces neuroprotective signals. EMBO J. 2002;21:3317–3326. doi: 10.1093/emboj/cdf324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, Strittmatter SM, Wisniewski T. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer's disease model mouse. BMC Neurosci. 2010;11:130. doi: 10.1186/1471-2202-11-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker H, Jürgensen S, Adrover MF, Brito-Moreira J, Bomfim TR, Klein WL, Epstein AL, De Felice FG, Jerusalinsky D, Ferreira ST. N-methyl-D-aspartate receptors are required for synaptic targeting of Alzheimer's toxic amyloid-beta peptide oligomers. J Neurochem. 2010;115:1520–1529. doi: 10.1111/j.1471-4159.2010.07058.x. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, Viola KL, Zhao WQ, Ferreira ST, Klein WL. Protection of synapses against Alzheimer's-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc Natl Acad Sci U S A. 2009;106:1971–1976. doi: 10.1073/pnas.0809158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. β-Amyloid activates the mitogen-activated protein kinase cascade via hippocampal α7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziewczapolski G, Glogowski CM, Masliah E, Heinemann SF. Deletion of the α7 nicotinic acetylcholine receptor gene improves cognitive deficits and synaptic pathology in a mouse model of Alzheimer's disease. J Neurosci. 2009;29:8805–8815. doi: 10.1523/JNEUROSCI.6159-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira ST, Klein WL. The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer's disease. Neurobiol Learn Mem. 2011;96:529–543. doi: 10.1016/j.nlm.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo CP, Clarke JR, Ledo JH, Ribeiro FC, Costa CV, Melo HM, Mota-Sales AP, Saraiva LM, Klein WL, Sebollela A, De Felice FG, Ferreira ST. Memantine rescues transient cognitive impairment caused by high-molecular-weight abeta oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J Neurosci. 2013;33:9626–9634. doi: 10.1523/JNEUROSCI.0482-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer MJ. Amine coupling through EDC/NHS: a practical approach. Methods Mol Biol. 2010;627:55–73. doi: 10.1007/978-1-60761-670-2_3. [DOI] [PubMed] [Google Scholar]
- Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR, Clarke AR, Rowan MJ, Walsh DM, Collinge J. Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat Commun. 2011;2:336. doi: 10.1038/ncomms1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Laurén J, Gimbel ZA, Strittmatter SM. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci. 2010;30:6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Yakel JL. Timing-dependent septal cholinergic induction of dynamic hippocampal synaptic plasticity. Neuron. 2011;71:155–165. doi: 10.1016/j.neuron.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajj GN, Arantes CP, Dias MV, Roffé M, Costa-Silva B, Lopes MH, Porto-Carreiro I, Rabachini T, Lima FR, Beraldo FH, Prado MM, Linden R, Martins VR. The unconventional secretion of stress-inducible protein 1 by a heterogeneous population of extracellular vesicles. Cell Mol Life Sci. 2013;70:3211–3227. doi: 10.1007/s00018-013-1328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez CM, Dineley KT. α7 nicotinic acetylcholine receptors in Alzheimer's disease: neuroprotective, neurotrophic or both? Curr Drug Targets. 2012;13:613–622. doi: 10.2174/138945012800398973. [DOI] [PubMed] [Google Scholar]
- Hernandez CM, Kayed R, Zheng H, Sweatt JD, Dineley KT. Loss of α7 nicotinic receptors enhances β-amyloid oligomer accumulation, exacerbating early-stage cognitive decline and septohippocampal pathology in a mouse model of Alzheimer's disease. J Neurosci. 2010;30:2442–2453. doi: 10.1523/JNEUROSCI.5038-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung LW, Ciccotosto GD, Giannakis E, Tew DJ, Perez K, Masters CL, Cappai R, Wade JD, Barnham KJ. Amyloid-β peptide (Aβ) neurotoxicity is modulated by the rate of peptide aggregation: Aβ dimers and trimers correlate with neurotoxicity. J Neurosci. 2008;28:11950–11958. doi: 10.1523/JNEUROSCI.3916-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/S1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Kessels HW, Nguyen LN, Nabavi S, Malinow R. The prion protein as a receptor for amyloid-beta. Nature. 2010;466:E3–E4. doi: 10.1038/nature09217. discussion E4–E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachaturian ZS. Diagnosis of Alzheimer's disease. Arch Neurol. 1985;42:1097–1105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- Kudo W, Lee HP, Zou WQ, Wang X, Perry G, Zhu X, Smith MA, Petersen RB, Lee HG. Cellular prion protein is essential for oligomeric amyloid-beta-induced neuronal cell death. Hum Mol Genet. 2012;21:1138–1144. doi: 10.1093/hmg/ddr542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson M, Sherman MA, Amar F, Nuvolone M, Schneider JA, Bennett DA, Aguzzi A, Lesné SE. The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer's disease. J Neurosci. 2012;32:16857–16871a. doi: 10.1523/JNEUROSCI.1858-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Magalhães AC, Zanata SM, Brentani RR, Martins VR, Prado MA. Internalization of mammalian fluorescent cellular prion protein and N-terminal deletion mutants in living cells. J Neurochem. 2001;79:79–87. doi: 10.1046/j.1471-4159.2001.00529.x. [DOI] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima FR, Arantes CP, Muras AG, Nomizo R, Brentani RR, Martins VR. Cellular prion protein expression in astrocytes modulates neuronal survival and differentiation. J Neurochem. 2007;103:2164–2176. doi: 10.1111/j.1471-4159.2007.04904.x. [DOI] [PubMed] [Google Scholar]
- Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev. 2008;88:673–728. doi: 10.1152/physrev.00007.2007. [DOI] [PubMed] [Google Scholar]
- Lopes MH, Hajj GN, Muras AG, Mancini GL, Castro RM, Ribeiro KC, Brentani RR, Linden R, Martins VR. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J Neurosci. 2005;25:11330–11339. doi: 10.1523/JNEUROSCI.2313-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdesian MH, Nery AA, Martins AH, Juliano MA, Juliano L, Ulrich H, Ferreira ST. Peptide blockers of the inhibition of neuronal nicotinic acetylcholine receptors by amyloid beta. J Biol Chem. 2005;280:31085–31090. doi: 10.1074/jbc.M502406200. [DOI] [PubMed] [Google Scholar]
- Martins VR, Beraldo FH, Hajj GN, Lopes MH, Lee KS, Prado MA, Linden R. Prion protein: orchestrating neurotrophic activities. Curr Issues Mol Biol. 2010;12:63–86. [PubMed] [Google Scholar]
- Martyn AC, De Jaeger X, Magalhães AC, Kesarwani R, Gonçalves DF, Raulic S, Guzman MS, Jackson MF, Izquierdo I, Macdonald JF, Prado MA, Prado VF. Elimination of the vesicular acetylcholine transporter in the forebrain causes hyperactivity and deficits in spatial memory and long-term potentiation. Proc Natl Acad Sci U S A. 2012;109:17651–17656. doi: 10.1073/pnas.1215381109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/WNL.34.7.939. [DOI] [PubMed] [Google Scholar]
- Michalski B, Fahnestock M. Pro-brain-derived neurotrophic factor is decreased in parietal cortex in Alzheimer's disease. Brain Res Mol Brain Res. 2003;111:148–154. doi: 10.1016/S0169-328X(03)00003-2. [DOI] [PubMed] [Google Scholar]
- Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Mucke L, Selkoe DJ. Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012;2:a006338. doi: 10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Urtreger A, Göldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M, Dani JA, Patrick JW, Beaudet AL. Mice deficient in the α7 neuronal nicotinic acetylcholine receptor lack α-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci. 1997;17:9165–9171. doi: 10.1523/JNEUROSCI.17-23-09165.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostapchenko VG, Makarava N, Savtchenko R, Baskakov IV. The polybasic N-terminal region of the prion protein controls the physical properties of both the cellular and fibrillar forms of PrP. J Mol Biol. 2008;383:1210–1224. doi: 10.1016/j.jmb.2008.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostapchenko VG, Beraldo FH, Guimaraes AL, Mishra S, Guzman M, Fan J, Martins VR, Prado VF, Prado MA. Increased prion protein processing and expression of metabotropic glutamate receptor 1 in a mouse model of Alzheimer's disease. J Neurochem. 2013 doi: 10.1111/jnc.12296. doi: 10.1111/jnc.12296. Advance online publication. Retrieved September 12, 2013. [DOI] [PubMed] [Google Scholar]
- Paula-Lima AC, Adasme T, SanMartín C, Sebollela A, Hetz C, Carrasco MA, Ferreira ST, Hidalgo C. Amyloid beta-peptide oligomers stimulate RyR-mediated Ca2+ release inducing mitochondrial fragmentation in hippocampal neurons and prevent RyR-mediated dendritic spine remodeling produced by BDNF. Antioxid Redox Signal. 2011;14:1209–1223. doi: 10.1089/ars.2010.3287. [DOI] [PubMed] [Google Scholar]
- Paula-Lima AC, Brito-Moreira J, Ferreira ST. Deregulation of excitatory neurotransmission underlying synapse failure in Alzheimer's disease. J Neurochem. 2013;126:191–202. doi: 10.1111/jnc.12304. [DOI] [PubMed] [Google Scholar]
- Prado VF, Martins-Silva C, de Castro BM, Lima RF, Barros DM, Amaral E, Ramsey AJ, Sotnikova TD, Ramirez MR, Kim HG, Rossato JI, Koenen J, Quan H, Cota VR, Moraes MF, Gomez MV, Guatimosim C, Wetsel WC, Kushmerick C, Pereira GS, Gainetdinov RR, Izquierdo I, Caron MG, Prado MA. Mice deficient for the vesicular acetylcholine transporter are myasthenic and have deficits in object and social recognition. Neuron. 2006;51:601–612. doi: 10.1016/j.neuron.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Müller V, Krishnan R, Vabulas RM, Kretzschmar HA, Lindquist S, Hartl FU, Multhaup G, Winklhofer KF, Tatzelt J. The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J. 2011;30:2057–2070. doi: 10.1038/emboj.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resenberger UK, Müller V, Munter LM, Baier M, Multhaup G, Wilson MR, Winklhofer KF, Tatzelt J. The heat shock response is modulated by and interferes with toxic effects of scrapie prion protein and amyloid beta. J Biol Chem. 2012;287:43765–43776. doi: 10.1074/jbc.M112.389007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roffé M, Beraldo FH, Bester R, Nunziante M, Bach C, Mancini G, Gilch S, Vorberg I, Castilho BA, Martins VR, Hajj GN. Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc Natl Acad Sci U S A. 2010;107:13147–13152. doi: 10.1073/pnas.1000784107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roffé M, Hajj GN, Azevedo HF, Alves VS, Castilho BA. IMPACT is a developmentally regulated protein in neurons that opposes the eukaryotic initiation factor 2alpha kinase GCN2 in the modulation of neurite outgrowth. J Biol Chem. 2013;288:10860–10869. doi: 10.1074/jbc.M113.461970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano SA, Cordeiro Y, Lima LM, Lopes MH, Silva JL, Foguel D, Linden R. Reciprocal remodeling upon binding of the prion protein to its signaling partner hop/STI1. FASEB J. 2009;23:4308–4316. doi: 10.1096/fj.09-138974. [DOI] [PubMed] [Google Scholar]
- Santos TG, Beraldo FH, Hajj GN, Lopes MH, Roffe M, Lupinacci FC, Ostapchenko VG, Prado VF, Prado MA, Martins VR. Laminin-gamma1 chain and stress inducible protein 1 synergistically mediate PrPC-dependent axonal growth via Ca2+ mobilization in dorsal root ganglia neurons. J Neurochem. 2013;124:210–223. doi: 10.1111/jnc.12091. [DOI] [PubMed] [Google Scholar]
- Sebollela A, Freitas-Correa L, Oliveira FF, Paula-Lima AC, Saraiva LM, Martins SM, Mota LD, Torres C, Alves-Leon S, de Souza JM, Carraro DM, Brentani H, De Felice FG, Ferreira ST. Amyloid-beta oligomers induce differential gene expression in adult human brain slices. J Biol Chem. 2012;287:7436–7445. doi: 10.1074/jbc.M111.298471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Kihara T, Hongo H, Wu X, Kem WR, Shimohama S, Akaike A, Niidome T, Sugimoto H. Neuroprotection by donepezil against glutamate excitotoxicity involves stimulation of alpha7 nicotinic receptors and internalization of NMDA receptors. Br J Pharmacol. 2010;161:127–139. doi: 10.1111/j.1476-5381.2010.00894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Solforosi L, Criado JR, McGavern DB, Wirz S, Sánchez-Alavez M, Sugama S, DeGiorgio LA, Volpe BT, Wiseman E, Abalos G, Masliah E, Gilden D, Oldstone MB, Conti B, Williamson RA. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science. 2004;303:1514–1516. doi: 10.1126/science.1094273. [DOI] [PubMed] [Google Scholar]
- Southworth DR, Agard DA. Client-loading conformation of the Hsp90 molecular chaperone revealed in the cryo-EM structure of the human Hsp90:Hop complex. Mol Cell. 2011;42:771–781. doi: 10.1016/j.molcel.2011.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui S, Hahn JN, Johnson TA, Ali Z, Jirik FR. Absence of the cellular prion protein exacerbates and prolongs neuroinflammation in experimental autoimmune encephalomyelitis. Am J Pathol. 2008;173:1029–1041. doi: 10.2353/ajpath.2008.071062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci. 2012;15:1227–1235. doi: 10.1038/nn.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB. beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem. 2000;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- Wang HY, Stucky A, Liu J, Shen C, Trocme-Thibierge C, Morain P. Dissociating β-amyloid from α7 nicotinic acetylcholine receptor by a novel therapeutic agent, S 24795, normalizes α7 nicotinic acetylcholine and NMDA receptor function in Alzheimer's disease brain. J Neurosci. 2009;29:10961–10973. doi: 10.1523/JNEUROSCI.6088-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Bakshi K, Frankfurt M, Stucky A, Goberdhan M, Shah SM, Burns LH. Reducing amyloid-related Alzheimer's disease pathogenesis by a small molecule targeting filamin A. J Neurosci. 2012;32:9773–9784. doi: 10.1523/JNEUROSCI.0354-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Helmerhorst E, Taddei K, Plewright B, Van Bronswijk W, Martins R. Alzheimer's β-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci. 2002;22:RC221. doi: 10.1523/JNEUROSCI.22-10-j0001.2002. (1–5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- You H, Tsutsui S, Hameed S, Kannanayakal TJ, Chen L, Xia P, Engbers JD, Lipton SA, Stys PK, Zamponi GW. Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-d-aspartate receptors. Proc Natl Acad Sci U S A. 2012;109:1737–1742. doi: 10.1073/pnas.1110789109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanata SM, Lopes MH, Mercadante AF, Hajj GN, Chiarini LB, Nomizo R, Freitas AR, Cabral AL, Lee KS, Juliano MA, de Oliveira E, Jachieri SG, Burlingame A, Huang L, Linden R, Brentani RR, Martins VR. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002;21:3307–3316. doi: 10.1093/emboj/cdf325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, Fluder E, Clurman B, Melquist S, Narayanan M, Suver C, Shah H, Mahajan M, Gillis T, Mysore J, MacDonald ME, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013;153:707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]