

Abstract
Presynaptic long-term depression (LTD) of synapse efficacy generally requires coordinated activity between presynaptic and postsynaptic neurons and a retrograde signal synthesized by the postsynaptic cell in an activity-dependent manner. In this study, we examined LTD in the rat nucleus tractus solitarii (NTS), a brainstem nucleus that relays homeostatic information from the internal body to the brain. We found that coactivation of N-methyl-D-aspartate receptors (NMDARs) and type 1 cannabinoid receptors (CB1Rs) induces LTD at the first central excitatory synapse between visceral fibers and NTS neurons. This LTD is presynaptically expressed. However, neither postsynaptic activation of NMDARs nor postsynaptic calcium influx are required for its induction. Direct activation of NMDARs triggers cannabinoid-dependent LTD. In addition, LTD is unaffected by blocking 2-arachidonyl-glycerol synthesis, but its induction threshold is lowered by preventing fatty acid degradation. Altogether, our data suggest that LTD in NTS neurons may be entirely expressed at the presynaptic level by local anandamide synthesis.
Introduction
Recent advances have led to the notion that the induction and expression of long-term depression (LTD) is attributable to diverse mechanisms (Collingridge et al., 2010). Although these various forms of LTD may appear similar because they reduce the average synaptic strength of a given synapse, they are likely to subserve different functions across the mammalian brain. Whereas postsynaptic mechanisms have been mainly proposed to account for reduced synapse efficacy, presynaptic mechanisms, such as a reduction in release probability, have increasingly received support over the past decade (Collingridge et al., 2010). Presynaptic or postsynaptic LTD generally requires coordinated activity between presynaptic and postsynaptic neurons for their induction. For presynaptic LTD, a retrograde signal synthesized by the postsynaptic cell in an activity-dependent manner is thus required. Among them, endocannabinoids (ECs) have emerged as the best-characterized retrograde messengers of LTS of synaptic plasticity (Gerdeman et al., 2002; Chevaleyre and Castillo, 2003). However, a recent study indicates that presynaptic LTD can be induced without the requirement of postsynaptic signaling (Rodríguez-Moreno et al., 2013).
Although most studies on synaptic plasticity have been conducted on brain structures involved in memory or motor control, relatively few studies are available on brain regions devoted to autonomic functions such as the brainstem. Visceral afferent fibers carry a large volume of information about the physiological status of the internal body directly to brainstem circuits regulating autonomic functions (Baude et al., 2009; Browning and Travagli, 2011; Andresen et al., 2012). The cell bodies for these afferent sensory fibers are contained in the nodose ganglion. These neurons connect directly onto nucleus tractus solitarii (NTS) neurons located in the brainstem via monosynaptic glutamatergic synapses. An important question is to know whether visceral information processing at the NTS level goes beyond a simple relaying and distribution role of visceral signals or is subjected to plastic changes (Browning and Travagli, 2011; Andresen et al., 2012). Previous studies in immature preweaning rats have described a form of postsynaptic LTD at excitatory synapses between visceral afferent fibers and NTS neurons (Zhou et al., 1997; Poon et al., 2000). Here we report that, in postweaning juvenile rats, low-frequency stimulation (LFS) of visceral afferent fibers induces a presynaptic form of LTD. This LTD induction requires the activation of both type 1 cannabinoid (CB1Rs) and NMDA (NMDARs) receptors but requires neither the activity of the postsynaptic neuron nor the release of 2-arachidonyl glycerol (2-AG). Our results suggest that LTD may be entirely expressed at the presynaptic level and involve anandamide (AEA) synthesis.
Materials and Methods
Animals.
Postweaning juvenile male Wistar rats (25–32 d) were anesthetized first with halothane before slice preparation. All procedures were in agreement with the European Communities Council directive (86/609/EEC).
Slice preparation.
Preparation of medullary slice was performed as described previously (Lachamp et al., 2003; Balland et al., 2006). For each experiment, two rats were decapitated, and the brainstem was quickly removed, cooled to 4°C, and cut on a vibratome (VT1000S; Leica Microsystems), into 300-μm-thick medullary slices, in oxygenated (95% O2, 5% CO2, pH 7.4) saline containing the following (in mm): 130 NaCl, 3 KCl, 0.5 CaCl2, 4 MgCl2, 26 NaHCO3, 1.25 KH2PO4,10 glucose, 0.5 ascorbate, 2 pyruvate, and 3 myoinositol. Slices were then allowed to recover for 1 h in a similar saline containing 2 mm CaCl2 and 2 mm MgCl2 at room temperature.
Electrophysiology.
For recordings, slices were perfused in a chamber at 3 ml/min with a physiological solution containing 2 mm CaCl2, 2 mm MgCl2, and a mixture of GABAA receptor blockers (20 μm bicuculine and 50 μm picrotoxin) plus d-serine (30 μm) at 32–34°C. Because the NTS is functionally divided into different subnuclei in the adult rat, we restricted our study to the medial NTS region on subpostremal slices. The dorsal motor nucleus of the vagus nerve and the tractus solitarius (TS; a fiber bundle formed by visceral afferent fibers within the brainstem; Andresen et al., 2001) were taken as anatomical landmarks to reproducibly target the recording region. Therefore, only two slices were selected per animal. Patch electrodes (2.5–3 MΩ) contained the following (in mm): 120 cesium methane sulfonate, 10 NaCl, 1 MgCl2, 0.1 CaCl2, 1 EGTA, 2 ATP, 0.3 GTP, 10 glucose, and 10 HEPES, pH 7.4. In some experiments, EGTA was replaced with BAPTA (10–20 mm) and the osmolarity was adjusted by reduction of cesium methane sulfonate. The junction potential was calculated to be 10 mV and was corrected for after the recordings. Stimulation of the TS (1–10 V; 100 μs duration) was applied using a tungsten bipolar electrode at low frequency (0.05 Hz) unless otherwise stated. EPSCs were deemed monosynaptic by virtue of their short latencies and small jitter (SD <200 μs) from stimulus onset (Doyle and Andresen, 2001; Lachamp et al., 2003; Balland et al., 2006). Miniature EPSCs (mEPSCs) were recorded in the presence of tetrodotoxin (TTX; 1 μm).
Drugs.
NMDA, d-aminophosphonovalerate (APV), arachidonyl-2′-chloroethylamide (ACEA), N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251), N-formyl-l-leucine (1S)-1-[[(2S,3S)-3-hexyl-4-oxo-2-oxetanyl]methyl]dodecyl ester (THL), N-phenyl-4-(3-phenyl-1,2,4-thiadiazol-5-yl)-1-piperazinecarboxamide [JNJ 1661010 (JNJ)], and 5,7-dichloro-4-hydroxyquinoline-2-carboxylic acid (DCK) were purchased from Tocris Cookson. N-ω-Nitro-l-arginine (LNNA) was purchased from Sigma-Aldrich. Lipophilic compounds were first prepared as stock solution in either dimethylsulfoxide (AM251, THL) or ethanol (ACEA, JNJ) at a concentration of 10–100 mm. The final concentration of the solvents were <0.01% for the highest concentration of the drugs. When testing lipophilic compounds, control experiments were routinely done by perfusing solution containing the solvent only. Other chemicals were obtained from Sigma. For all experiments, only one neuron per slice was recorded, and, when using lipophilic compounds, tubing was rinsed with ethanol between each application.
LTD induction protocol.
Two TS stimuli separated by 50 ms were delivered every 20 s during the control period, and a 2 mV, 20 ms negative step was delivered at the end of the acquisition window to measure input and access resistances (see below). After 10 min of stable synaptic responses at a potential of −80 mV (see below), neurons were held at −40 mV, and afferent fibers were stimulated at 4 Hz during 4 min to induce LTD. Then the holding potential was reverted to −80 mV, and the control stimulation was resumed. LTD was monitored for at least 20 min after its induction. All drugs were applied via the general perfusion system, allowing for a rapid wash-in of the slice. Antagonists were applied at least 15 min before LTD induction. For the inhibitor of nitric oxide (NO) synthase (200 μm, LNNA), the inhibitor of fatty acid synthase (THL, 10 μm) or the inhibitor of fatty acid hydrolase (JNJ, 2 μm) slices were preincubated 1–2 h before recording with the drug and during the whole recording session.
Depolarization-induced suppression of EPSC experiments.
For studying depolarization-induced suppression of EPSC (DSE), EPSCs were evoked at a rate of 0.2 Hz. After a control period (20–30 EPSCs), the neuron was depolarized to 0 mV for 5 or 25 s. The control mean amplitude was calculated and used for normalization of all EPSCs. DSE was calculated as the ratio between the mean peak amplitude of three consecutive EPSCs after the pulse normalized to the mean peak amplitude of control EPSCs.
Data acquisition.
Whole-cell patch-clamp recordings of NTS neurons were made with an Axopatch 200B (Molecular Devices), filtered at 5 kHz, and digitized at 20 kHz. Series resistance (Rs < 15 MΩ) and input resistance were continuously monitored throughout the experiment by delivering a voltage step command at the end of each trace recorded. Neurons in which Rs or input resistance were unstable were discarded. Neurons with an inward current greater than −120 pA at a holding potential of −80 mV were also discarded.
Analyses.
EPSC amplitudes were plotted against the stimulus number, and linear fit was applied to the data. Only experiments in which this linear regression did not significantly deviate from zero slope during the control period (before LTD induction) were selected for additional analysis. Input resistance was measured from the steady-state current induced by a 20 ms test pulse from −80 to −82 mV. Access resistance was estimated as follows: the peak current induced by the test pulse was extrapolated to the start of the pulse with an exponential fit to obtain its amplitude; then access resistance was calculated by dividing 2 mV by the peak current amplitude. LTD strength was estimated by measuring EPSC amplitude on 20–25 successive responses before LFS and 20 min after LTD induction unless otherwise noted. Paired-pulse ratio (PPR) was measured as follows: a series (20–25 trials) of two electrical pulses separated by a 50 ms interval were applied to the TS, and the ratio of the mean amplitude of the second response to the mean amplitude of the first one was calculated. The coefficient of variation (CV) was calculated for at least 25 consecutive trials as follows. The variance of EPSC peak was obtained from 25 EPSCs. Then, the variance of the background noise was measured from 25 interleaved traces in which no stimuli were delivered and subtracted from the EPSC variance (Balland et al., 2008). Then the square root of this corrected variance was divided by the mean EPSC amplitude. mEPSC analysis was performed with the Clampfit module of the Axon software (Molecular Devices) by using the template search method. Ten minutes after break-in, 150 contiguous events per cell were analyzed and averaged to yield mean mEPSC amplitude for each cell for different periods of time (before drug and during the last minutes of drug application). The mean frequency of AMPA mEPSC was determined for each neuron as the number of events per second binned every 60 s.
Statistical analyses.
Significance of the difference between means of two samples was computed using unpaired or paired nonparametric tests (Mann–Whitney and Wilcoxon's tests, respectively). When three samples or more were compared, the variance of the whole population was first analyzed by a one-way nonparametric ANOVA (Kruskal–Wallis test). For comparing LTD parameters at different time points, repeated-measures ANOVA (RM ANOVA) was used. If the null hypothesis was rejected, the post hoc Dunnett's test was then used to compare groups. Statistical data are given as mean ± SEM. The differences were considered significant at p < 0.05. Statistical tests were computed by using the GraphPad Software Instat software. For mEPSC analysis, differences in amplitude or event interval distribution were assessed by the Kolgomorov–Smirnov test included in the Clampfit software.
Results
Characterization of EPSC LTD in NTS neurons
At a holding potential of −80 mV, stimulation of the TS evoked a rapid EPSC with a mean latency of 3 ± 0.2 ms. TS-evoked EPSCs (n = 13; Fig. 1A) had invariant latencies (jitter <200 μs), indicating that they corresponded to monosynaptic connections (Doyle and Andresen, 2001). In addition, NTS synapses were characterized by a consistent paired-pulse depression when two TS shocks separated by 50 ms were applied (PPR, 0.54 ± 0.06, n = 23; Fig. 1B,E).
Figure 1.
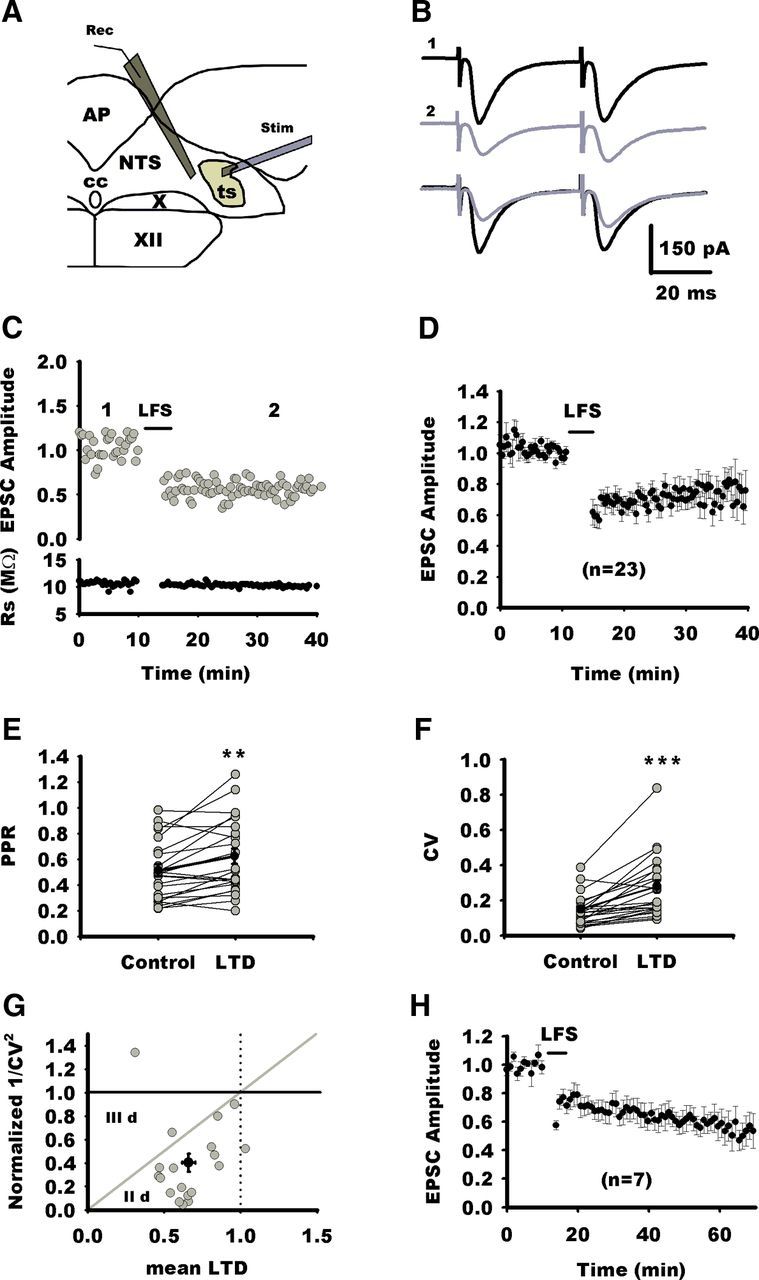
Characterization of LTD at NTS excitatory synapses. A, Schematic view showing the location of stimulating electrode (Stim) and the recording pipette (Rec). The stimulating electrode is placed on the TS, and neurons were recorded from the medial part of the NTS at the level of the area postrema (AP). cc, Central canal; X, dorsal nucleus of the vagus nerve; XII, hypoglossal nucleus. B, Average synaptic responses to TS stimulation before (1) and after (2) LTD of induction of an NTS neuron. Superimposed traces are shown below. C, Normalized individual response amplitudes to stimulation of visceral afferent fibers (TS stimulation; gray circles) recorded before and after LTD induction by LFS as a function of time for the same neuron as in B. Rs is also plotted against time. D, Normalized EPSC amplitudes for all neurons of the control group before and after induction of LTD by LFS. E, Changes in PPR before and after LFS in NTS neurons (gray circles). On average, PPR is significantly increased during LTD (black circles). F, Significant increase in EPSC CV before and after LFS in NTS neurons. G, Normalized plot of 1/CV2 versus mean LTD after LFS yield data points (gray circles) to gather below the diagonal in the IId region (Faber and Korn, 1991), suggesting that LTD is expressed at the presynaptic level (black circle indicates mean changes in 1/CV2 vs mean changes in EPSC amplitude ± SEM). H, Normalized EPSC amplitudes for seven neurons before and 1 h after induction of LTD by LFS (for clarity, each data point represents the amplitude of the average of 3 consecutive EPSCs). For all figures, *p < 0.05; **p < 0.01; ***p < 0.001).
LFS of TS (4 Hz, 4 min) at a holding potential of −40 mV induced LTD at all the excitatory synaptic connections but one, which was maintained for at least 25 min after LFS (Fig. 1C,D). For 23 NTS neurons recorded, mean EPSC amplitudes after LFS was 68 ± 4.5% (p = 0.0012) of the baseline values (Fig. 1D). To test whether LTD induction involves a presynaptic or postsynaptic mechanism, we first analyzed changes in PPR before and after LFS. As illustrated in Figure 1E, LTD was associated with an increase in PPR (129 ± 6%, n = 23, p = 0.0083). We also analyzed changes in CV of the ESPC before and after LFS. We found an increase in CV after LTD (210 ± 19%, n = 23, p = 0.0001; Fig. 1F). Finally, we represented changes in 1/CV2 (Fig. 1G) as a function of changes in mean EPSC amplitude (Faber and Korn, 1991). According to this model, neurons in which LTD is attributable to a reduction in presynaptic release give rise to experimental data points below the diagonal in the IId region. As illustrated in Figure 1G, most experimental data points lay below the diagonal. Altogether, changes in PPR and CV support the presynaptic expression of LTD at excitatory synapses in NTS neurons. Finally, we extended the recording period after LTD induction to better characterize the time course of LTD. For seven neurons, recordings could be maintained up to 60 min after LFS (Fig. 1H). Mean EPSC amplitude was 69 ± 7% (p = 0.04), 60 ± 7% (p = 0.03), and 55 ± 9% (p = 0.02) of the baseline values 20, 40, and 50 min after LTD induction, respectively (RM ANOVA; p = 0.008). LTD strength was larger 50 min after induction (p = 0.006, compared with strength at 20 min) than earlier, which indicated that, once induced, LTD developed for at least 1 h. In addition, PPR increased at 20 and 50 min (152 ± 35%, p = 0.04; 165 ± 50%, p = 0.02; RM ANOVA; p = 0.019).
LTD induced by LFS requires NMDARs not located in the postsynaptic compartment
Previous experiment in immature preweaning rats (aged 3–21 postnatal days; Zhou et al., 1997) have demonstrated that LTD required activation of NMDARs. As shown in Figure 2A, 10 min application of APV (50 μm) altered neither EPSC amplitude nor PPR, arguing against an endogenous tonic modulation of neurotransmission by NMDARs [EPCS amplitude (last 5 min), 97 ± 5% of 10 min baseline, n = 10, p = 0.11; PPR, 105 ± 4.5% of baseline, p = 0.5]. In contrast, APV reduced LTD compared with the control group (91 ± 4% of control, n = 11; APV vs control group; p = 0.02; Fig. 2). Consistently, neither PPR nor CV exhibited changes after LFS (110 ± 22%, p = 0.89; 120 ± 11%; p = 0.7, respectively; Fig. 2E,F). However, the residual depression observed after blockade of NMDARs was significant when compared with baseline amplitudes before LTD induction (p = 0.01). Therefore, NMDAR activation is required for LTD induction by LFS, but other mechanisms may also be involved in the residual depression observed after NMDAR blockade (see Discussion). Although, the PPR and CV changes demonstrate that the LTD is expressed presynaptically, NMDARs could be located in the presynaptic or postsynaptic compartment. Monosynaptic responses elicited by solitary tract stimulation often lack an NMDA component, thereby suggesting that at least some NTS glutamatergic synapses are devoid of NMDARs (Doyle and Andresen, 2001; Jin et al., 2003). To determine whether NMDARs were located presynaptically or postsynaptically, we used a specific postsynaptic blockade of NMDARs. Figure 2H illustrates the effect of intracellular perfusion of MK-801 (iMK-801; 1 mm), a pore blocker of NMDARs. We first recorded from NTS neurons with control intracellular solution (Fig. 2H). At a holding potential of +30 mV, most neurons (five of six) displayed outward synaptically evoked current with fast and slow components, corresponding to the activation of AMPARs and NMDARs, respectively (Balland et al., 2006). The amplitude of the slow component, measured 30 ms after the peak, amounted to ∼44 ± 9% of the amplitude of the peak current (Fig. 2H, n = 6). In another series, we then recorded NTS neurons with iMK-801. Interestingly, for all these neurons (n = 8), the synaptically evoked current exhibited no or a very weak slow component (4 ± 0.9% of the peak current, control vs MK-801, p = 0.008; Fig. 2H). After 10 min of recording at +30 mV and continuous TS stimulation, we did not notice an increase in the blockade of the slow component (4.8 ± 0.1% of the peak current; Fig. 2H), indicating that MK-801 had blocked the NMDARs very rapidly. We then tested the effect of iMK-801 on the LFS LTD induction. As depicted in Figure 3A1, LFS of the TS was still able to induce an LTD comparable in magnitude with control (71 ± 5%, n = 8; iMK-801 vs control group, p = 0.8; Fig. 3E). Accordingly, LTD was associated with significant changes in PPR and CV (128 ± 5%, p = 0.015; 197 ± 20%, p = 0.007, respectively; Fig. 3A2). We then further investigated whether postsynaptic rise in intracellular calcium was required for the induction of LTD by LFS. For doing so, we recorded NTS neurons with an intracellular medium containing BAPTA (10–20 mm), a fast calcium chelator. As illustrated in Figure 3B1, LFS still induced LTD of EPSC comparable with control (67 ± 8%; BAPTA vs control group, p = 0.8; Fig. 3E). Here again, LTD was associated with significant changes in PPR and CV (125 ± 8%, p = 0.01; 226 ± 28%, p = 0.009, respectively; Fig. 3B2). Finally, we tried to induce LTD while maintaining NTS neurons at a holding potential of −80 mV to prevent NMDAR activation and voltage-dependent channel activation. As depicted in Figure 3C1, EPSCs were still depressed after LFS. The LTD was similar to control (69.5 ± 5%; −80 mV vs control group, p = 0.6; Fig. 3E) and associated with significant changes in PPR and CV (150 ± 10%, p = 0.0039; 210 ± 30%, p = 0.004, respectively; Fig. 3C2). We also represented changes in 1/CV2 (Fig. 3D) as a function of changes in mean EPSC amplitude for all experimental conditions. All experimental points gained from groups expressing LTD gather below the diagonal in the area corresponding to presynaptic LTD. Although postsynaptic calcium influx via NMDARs appear not to be required for LTD induction in NTS neurons, a recent study has shown that a metabotropic action of NMDARs can induce LTD in hippocampal neurons without raising postsynaptic calcium (Nabavi et al., 2013). This novel form of LTD is prevented by APV but not by antagonists acting at the glycine-binding site on the NR1 subunit of NMDARs, such as DCK. We thus recorded from NTS neurons in the presence of DCK (50 μm). In this condition, the blockade of LFS LTD was similar to what we observed with APV (89.5 ± 4% of baseline values before LTD induction, not different from APV, n = 5, p = 0.87; Fig. 2G). Likewise, PPR was unchanged after LFS (101 ± 6%, p = 0.8). Therefore, LFS LTD in NTS neurons is not mediated via a metabotropic action of the NMDARs.
Figure 2.
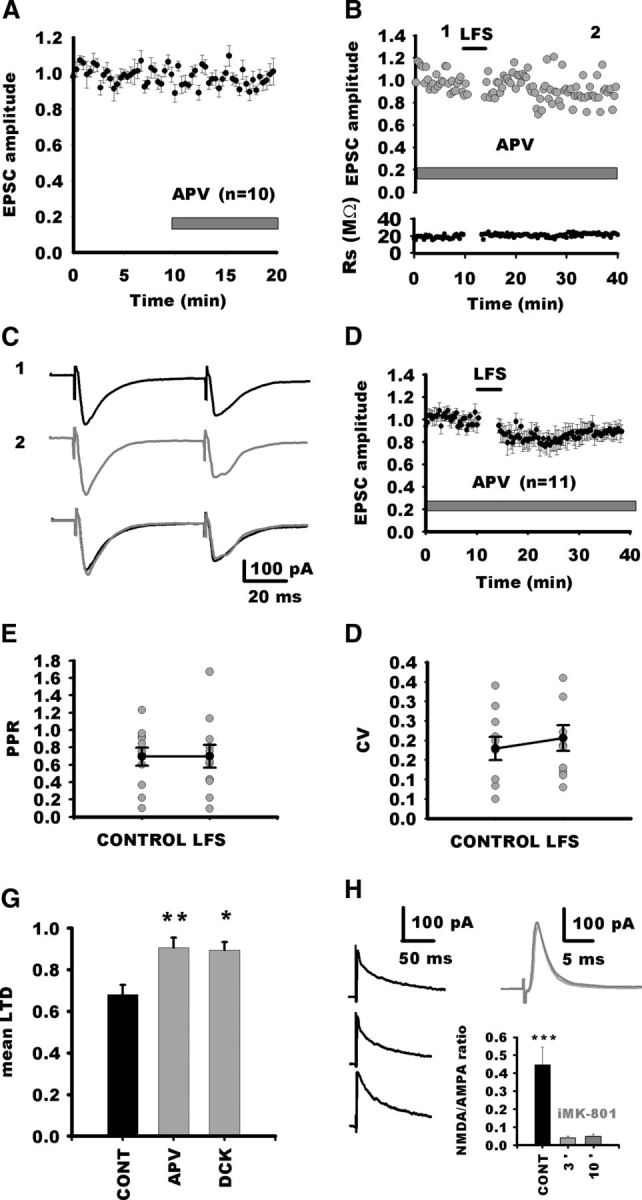
Role of NMDARs in LTD induction. A, Normalized response amplitudes to stimulation of visceral afferent fibers recorded before and during application of APV. NMDAR blockade does not alter EPSC amplitude. B, Normalized individual response amplitudes to stimulation of visceral afferent fibers (TS stimulation; gray circles) recorded before and after LFS of the TS in the presence of APV as a function of time for an NTS neuron. Rs is also plotted against time. In this condition, LFS of the TS does not induce LTD. C, Average synaptic responses to TS stimulation before (1) and after (2) LTD of induction of the same neuron in A. Superimposed traces are shown below. D, Normalized EPSC amplitudes for all neurons recorded in the presence of APV before and after LFS. Note that, after a transient depression, EPSC amplitude reverted to nearly control amplitude. E, Absence of changes in PPR before and after LFS in NTS neurons in the presence of APV. F, Absence of changes in EPSC CV before and after LFS in NTS neurons in the presence of APV. G, Mean LTD for the control group (CONT) and for groups after NMDAR blockade by APV or DCK. H, Blockade of NMDARs with iMK-801. Left black traces, Typical EPSCs recorded at a holding potential of +30 mV with control intracellular saline in three NTS neurons. The synaptic responses consisted of a fast AMPA current that peaked in <2 ms and a slow NMDA current that lasted longer. Right traces, Example of a synaptic responses at +30 mV for an NTS neurons recorded with iMK-801 recorded 3 min (black) and 10 min (gray) after seal opening. Note the absence of the slow NMDA current. The average NMDA/AMPA ratio of EPSCs for neurons recorded with control or iMK-801 (3 and 10 min after seal opening) saline is shown of the graph. All traces are the average of five consecutive EPCSs.
Figure 3.
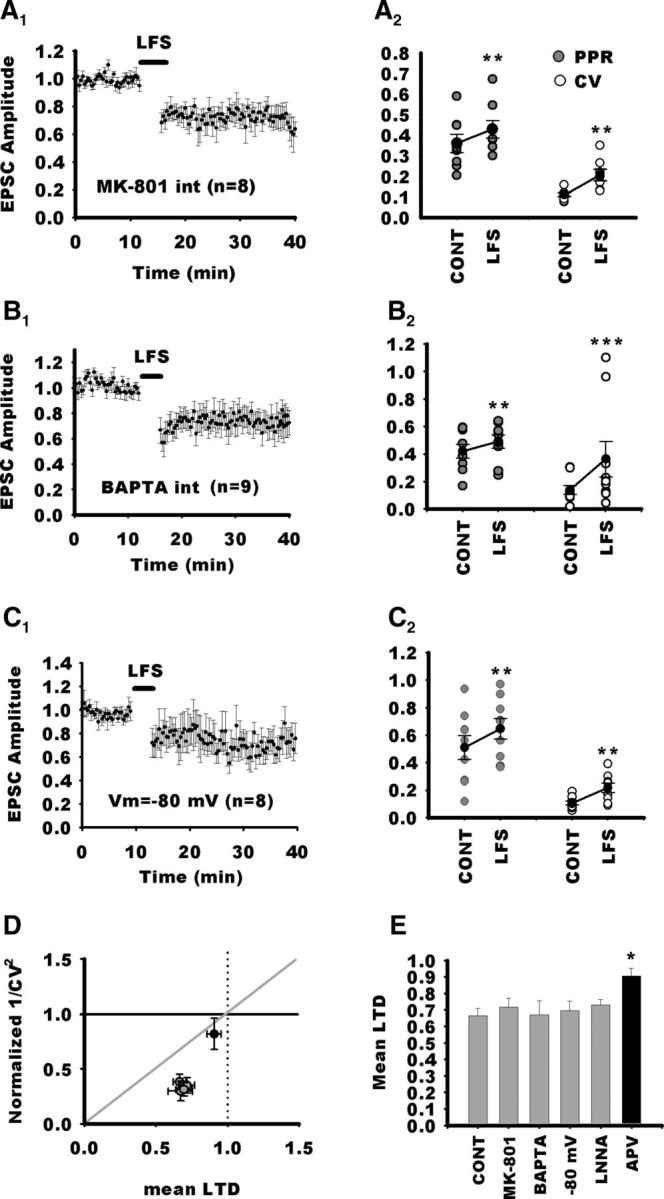
Postsynaptic NMDARs are not required for LTD in NTS neurons. A1, Normalized EPSC amplitudes for NTS neurons recorded with an intracellular saline containing the pore blocker of NMDARs, MK-801. Despite postsynaptic blockade of NMDARs, LFS still induces a prominent LTD of the EPSC. A2, Accordingly, PPR and CV were significantly increased during LTD. B1, Normalized EPSC amplitudes for NTS neurons recorded with an intracellular saline containing the fast calcium chelator BAPTA. In this condition, LFS still induces a prominent LTD of the EPSC. B2, Accordingly, PPR and CV were significantly increased during LTD. C1, Normalized EPSC amplitudes for NTS neurons maintained at a holding potential of −80 mV throughout the experiment to prevent NMDARs to open during LFS. Despite postsynaptic blockade of NMDARs, LTD of the EPSC is not blocked. C2, Accordingly, PPR and CV were significantly increased during LTD. D, Normalized plot of mean 1/CV2 versus mean changes in EPSC after LFS for the above experiments (gray circles) and after blockade of NMDARs by APV (black circles). Note that experimental points gained from groups expressing LTD (control, MK-801, BAPTA, −80 mV; gray circles) gather below the diagonal nearly at the same coordinates. In contrast, for the APV group (black circles), the mean data point is close to the identity point. E, Mean LTD of the different groups. Loading the postsynaptic neuron with MK-801 does not change LTD magnitude compared with control. Consistent with this, maintaining at a negative potential does not block LTD. Likewise, chelating intracellular calcium of the postsynaptic cell with BAPTA does not interfere with LTD. In addition, blockage of the NO synthase by LNNA does not suppress LTD. The results argue that the NMDAR dependence of LTD induced by LFS does not arise postsynaptically and does not depend on NO production. CONT, Control.
LTD induction via presynaptic NMDAR activation has been reported in several systems (Casado et al., 2002; Sjöström et al., 2003; Rodríguez-Moreno et al., 2011). For downstream NMDAR activation, several second messengers have been described to alter presynaptic release of transmitter. Among them, NO is involved in cerebellar presynaptic LTD (Casado et al., 2002). Therefore, we tested the role of NO in LTD induced by LFS. Addition of the NO inhibitor (LNNA, 200 μm) did not block LTD (72 ± 3.5%, n = 11, LNNA vs control, p = 0.57; Fig. 3E). Consistently, LTD was accompanied by changes in PPR and CV (130 ± 11%, p = 0.007; 151 ± 19%, p = 0.006, respectively).
Activation of CB1Rs by AEA is required for LTD induction by LFS
ECs have been identified as messengers that mediate short- and long-term modulation of synaptic transmission (Heifets and Castillo, 2009). In the NTS, EC receptors are expressed in terminal-like structures (Van Sickle et al., 2003; Ray et al., 2009). Therefore, we tested the possibility that activation of NMDARs during LFS leads to EC synthesis and triggers LTD. As shown in Figure 4A, 10 min application of AM251 (5 μm) altered neither EPSC amplitude nor PPR, arguing against an endogenous tonic modulation of neurotransmission by NMDARs [EPCS amplitude (last 5 min), 98 ± 7% of 10 min baseline, n = 6, p = 0.097; PPR, 105 ± 6% of baseline, p = 0.84]. As shown in Figure 4, B and C, blocking CB1Rs by the selective antagonist AM251 (5 μm) reduced LTD compared with the control group (90 ± 2% of control, n = 16; AM251 vs control group, p = 0.003). In addition, neither PPR nor CV exhibited significant changes after LFS (100 ± 8%, p = 0.6; 107 ± 7%, p = 0.7, respectively; Fig. 4D). However, as observed with APV, the residual depression was significantly different from baseline values before LTD induction (p = 0.001). To rule out a synergistic effect of NMDARs and CB1R activation, we also tested the effect of blocking both receptors on LTD induction. Coapplication of APV and AM251 did not increase the blockade of LTD compared with APV or AM251 alone (90.5 ± 5%, n = 4, p = 0.9; Fig. 4E). CB1R LTD is induced by 2-AG release in several systems (Chevaleyre and Castillo, 2003; Heifets and Castillo, 2009). We tested the effect of preincubating and perfusing slices with THL (10 μm), an inhibitor of fatty acid synthesis (Fig. 4F). In these conditions, LTD was not suppressed in NTS neurons compared with control slices (62 ± 6 vs 60 ± 10% for control and THL, respectively; n = 5 in each condition, p = 0.88). As expected, CV and PPR were similarly affected in treated and control neurons (238 ± 36 vs 208 ± 20% for CV in control and THL, respectively; 120 ± 5 vs 137 ± 12% for PPR in control and THL, respectively). Therefore, LFS LTD in NTS neurons is not mediated by release of 2-AG. We then wondered whether AEA was involved in LTD instead. Therefore, we tried to reduce its degradation by inhibiting fatty acid amide hydrolase (FAAH) as described previously (Puente et al., 2011). We checked whether FAAH inhibition lowered the threshold for LFS LTD induction. In control experiments, LFS of afferent fibers for 0.5 min led to a nonsignificant reduction in synaptic strength (92 ± 8%, p = 0.4, n = 8; Fig. 4G). Blockade of AEA hydrolysis by JNJ pretreatment (2 μm) resulted in a strong LTD induced by the same subthreshold protocol (55 ± 12%, control vs JNJ, p = 0.02, n = 6; Fig. 4G). Then we analyzed the effect of FAAH inhibition on LTD strength induced by control LFS protocol. As shown in Figure 4H, LTD developed more rapidly than in control conditions, but 30 and 50 min after induction, LTD strength converged to similar values for both groups (60 ± 7 vs 43 ± 12%, p = 0.09; 55 ± 9 vs 41 ± 11%, p = 0.5 for control and JNJ, respectively, at 30 and 50 min after LFS; n = 7 for control and n = 5 for JNJ). This suggests that control LFS induced a saturating LTD that was more rapidly reached by reducing fatty acid degradation. Finally, because most visceral afferent terminals bear presynaptic vallinoid receptors (TRPV1R; Jin et al., 2004; Peters et al., 2010) that may be activated by AEA and contribute to LTD as described in other systems (Grueter et al., 2010; Puente et al., 2011), we tested the potential role of TRPV1R in LTD induction. Blockade of TRPV1R by capsazepine (CPZ; 10 μm) did not prevent LFS LTD in NTS neurons (78 ± 9% of control; n = 7; CPZ vs control group, p = 0.4; data not shown). Together, these data suggest that LFS LTD in the NTS requires the activation of CB1Rs and the release of AEA.
Figure 4.
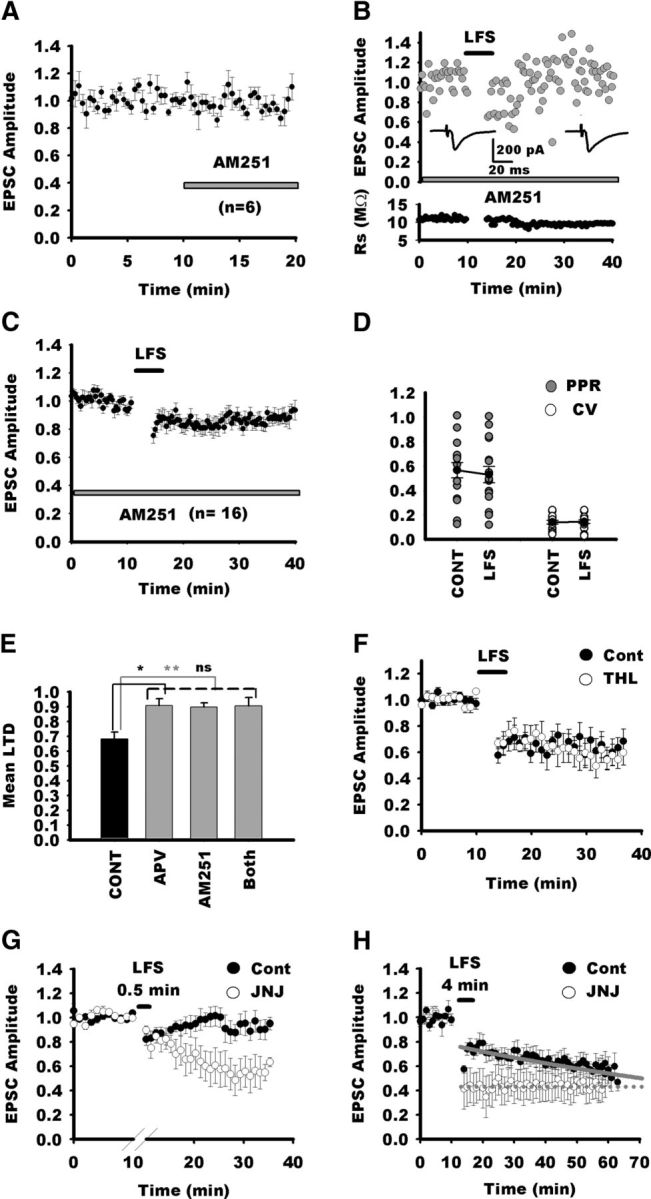
LTD in NTS neurons requires activation of CB1Rs. A, Blockade of CB1Rs with AM251 does not alter EPSC amplitude during baseline recordings. Normalized EPSC amplitudes for all neurons recorded before and after AM251 application. B, Normalized individual response amplitudes to stimulation of visceral afferent fibers (TS stimulation; gray circles) recorded before and after LTD induction by LFS during blockade of CB1Rs. In this condition, LFS does not induce LTD in this neuron. C, Normalized EPSC amplitudes for all neurons recorded in the presence of AM251 before and after LFS. Note that, after a transient depression, EPSC amplitude reverted to nearly control amplitude. D, Absence of changes in PPR an CV before (CONT) and after LFS in NTS neurons in the presence of AM251. E, Comparison of LTD amplitude in the control group in the presence of APV, AM251, or both. Note that blockade of both NMDARs and CB1Rs do not better suppress LTD that APV or AM251 alone, arguing against an additive effect of NMDARs and CB1Rs on LTD induction. F, Blockade of fatty acid synthesis does not suppress LFS LTD. Normalized EPSC amplitudes for NTS neurons recorded in control condition (black circles, n = 5) or after pretreatment and perfusion with THL (white circles, n = 5; for clarity, each data point represents the amplitude of the average of 3 consecutive EPSC). In both groups, LFS induced a similar LTD. G, Blockade of FAAH with JNJ lowers the threshold for LFS LTD induction. Normalized EPSC amplitudes for NTS neurons recorded in before and after 0.5 min LFS (black circles, n = 8) or after pretreatment and perfusion with JNJ (white circles, n = 6; for clarity, each data point represents the amplitude of the average of 3 consecutive EPSCs). Note that, in control conditions, 0.5 min LFS of visceral afferent fibers did not induce LTD, whereas a prominent depression was observed after JNJ treatment. H, Same experiment as in G but with control LFS (4 min). Note that, in the untreated control group (n = 7), LTD reached gradually a maximum value within 50 min after LFS, whereas in the JNJ group (n = 5), maximum LTD is already reached after LFS. Gray lines, Exponential fits for LTD time course after LFS in both groups. Exponential equation is f(t) = a × exp(−b × t). a and b are equal to 0.77 and 0.05 for the control group and to 0.44 and 0 for the JNJ group, respectively.
Activation of CB1Rs occurs downstream of NMDAR activation
Because our previous experiments suggested that NMDARs and CB1Rs were not activated in parallel, we tested the effects of their sequential activation. Direct activation of CB1Rs by applying the specific agonist (ACEA, 3 μm; Fig. 5A) produced a long-lasting depression [chemical LTD (cLTD)] that was similar in magnitude to LFS-induced LTD (71 ± 7% of control, n = 8). During ACEA application, PPR increased (128 ± 10%, p = 0.02) and CV was significantly augmented (160 ± 22%, p = 0.01), confirming a direct activation of presynaptic CB1Rs (Fig. 5B). Several studies have reported that CB1R activation does not trigger cLTD in the absence of a coincident presynaptic activity (Sjöström et al., 2003; Singla et al., 2007; Heifets and Castillo, 2009). Therefore, we performed additional experiments during which TS stimulation was discontinued during the first 10 min of ACEA application. In these conditions, ACEA did not produce cLTD at NTS synapses (Fig. 5A; 100 ± 3%, n = 5; p = 0.0043 compared with ACEA alone with stimulation 71 ± 7%). However, when stimulation of afferent fibers was resumed after 10 min ACEA application, the same synapses depressed gradually and reached a similar level to that induced during continuous presynaptic stimulation (p = 0.4). We then verified that ACEA cLTD and LFS LTD shared similar biochemical pathways by conducting occlusion experiments. In six neurons, application of ACEA (15 min; Fig. 5C) induced a cLTD that stabilized at 67 ± 6%. Visceral afferent were then stimulated to induce LFS LTD, and amplitude of EPSC was measured 20 min after LFS. EPSC amplitude was 58 ± 10% of control values (p = 0.01) but not significantly different from values obtained during ACEA application (p = 0.23; RM ANOVA, p = 0.0032). Therefore, ACEA cLTD and LFS LTD are likely to share similar biochemical pathways.
Figure 5.

A, Direct activation of CB1Rs (ACEA, n = 6, black circles; each data point represents the amplitude of the average of 3 consecutive EPSC) induces a long-lasting EPSC depression. ACEA-induced depression requires presynaptic activity because application of ACEA produces an EPSC depression only after onset of presynaptic stimulation (gray circles; 10 min delay between NMDA application onset and stimulation onset). In addition, blockade of NMDARs (white circles, n = 7) does not suppress ACEA-induced depression of EPSC. B, In both conditions (ACEA or ACEA + APV), drug-induced EPSC depression is accompanied by a significant increase in PPR and CV. C, Occlusion experiments: after a stable depression induced by ACEA, LFS induces a transient EPSC depression that nearly reverts back to values before LFS. D, Direct activation of NMDARs by application of NMDA (black circles) induces a long-lasting EPSC depression that does not revert after washout. However, this NMDA cLTD is prevented by blockade of CB1Rs (white circles). E, NMDA cLTD is accompanied by changes in PPR and CV, whereas blockade of CB1Rs with AM251 prevents increase in PPR and reduces the increase in CV. F, NMDA cLTD requires presynaptic activity because application of NMDA produces an EPSC depression only after onset of presynaptic stimulation (black circles, continuous stimulation; gray circles, 10 min delay between NMDA application onset and stimulation onset). CONT, Control.
We then examined the interplay between NMDARs and CB1Rs during LTD. We first tested whether NMDAR activation was necessary to activate CB1Rs. As shown in Figure 5A, blockade of NMDARs by APV did not prevent ACEA to induce a long-lasting depression of EPSC (73 ± 5% of control, n = 7; APV vs ACEA alone, p = 0.8). In addition, both PPR and CV were significantly increased (130 ± 8%, p = 0.03; 170 ± 15%, p = 0.031; Fig. 5B).
Data suggest that activation of CB1Rs may occur downstream of activation of NMDARs. To test this hypothesis, we recorded NTS neurons with iMK-801. Application of NMDA (25 μm) induced a long-lasting depression of EPSCs that took place in ∼5 min and did not reverse after 15–20 min washout (71 ± 5%, n = 7; Fig. 5D). Here again, both PPR and CV were significantly increased (126 ± 6%, p = 0.030; 181 ± 20%, p = 0.031; Fig. 5E). However, blocking CB1Rs completely suppressed the NMDA cLTD (Fig. 5D; 101 ± 3%, n = 5; AM251 vs NMDA alone, p = 0.005). PPR remained unchanged (Fig. 5E; 97 ± 3%; p = 0.6), but CV was still slightly increased (120 ± 7%; p = 0.045). We then wondered whether presynaptic stimulation was also necessary for the NMDA cLTD. In the absence of presynaptic activity, NMDA application did not produce cLTD at NTS synapses (Fig. 5F; 94 ± 4%, n = 7; p = 0.01 compared with NMDA with stimulation, 72 ± 3%). However, when stimulation of afferent fibers was resumed after 10 min NMDA application, the same synapses depressed gradually and reached a similar level to that induced by NMDA with continuous presynaptic stimulation (p = 0.4). Finally, we recorded mEPSCs in NTS neurons with iMK-801. On average, NMDA application did not decreased the frequency of mEPSCs (measured 10–15 min after NMDA application onset; 94.7 ± 5%, p = 0.6; range, 79–115%; n = 7; Fig. 6A,B). Accordingly, mEPSC amplitude did not change significantly (22.4 ± 2.8 vs 22.5 ± 3 pA; ratio range, 91–116%; p = 0.4; Fig. 6C). Data indicate that NMDA does not alter mEPSC frequency and amplitude.
Figure 6.

A, Normalized frequency of mEPSC (TTX, 1 μm; n = 7 neurons, 1 min bin) before and during NMDA application. No significant change in frequency was observed. B, Cumulative distribution of mEPSC amplitude for all neurons before and during NMDA application. Inset, Example of an averaged mEPSC before and during NMDA application for an NTS neurons. C, Cumulative distribution of mEPSC interevent intervals for all neurons before and during NMDA application. Inset, Example of mEPSC recordings before and during NMDA application for the same NTS neurons as in C. D, Absence of DSE in NTS neurons. Normalized EPSC amplitudes for seven neurons in response to stimulation of visceral afferent fibers (0.2 Hz; black circles) recorded before and after postsynaptic depolarizations (0 mV, 5s). Note that EPSC amplitude remained unaffected by the postsynaptic depolarizations. Traces in black, dark gray, and gray represent the average EPSCs (3 consecutive responses) for a representative NTS neuron before and after the conditional test pulses. E, Schematic diagram of LTD mechanisms. Glutamate released by visceral afferent fibers activates NMDARs that may be located in the presynaptic terminal or in a neighboring neuron. NMDAR activation leads to calcium influx and EC synthesis. ECs acting on CB1Rs would reduce release probability of synaptic vesicles and therefore induce LTD.
In addition to LTD, ECs may be released by the postsynaptic neuron in response to depolarization and transiently inhibit DSE or inhibitory synaptic transmission (DSI; Hashimotodani et al., 2008; Kano et al., 2009) by acting on presynaptic CB1Rs. EPSCs (0.2 Hz) were evoked in NTS neurons during the control period, and then NTS neurons were depolarized to 0 mV for 5 s during the interstimulation interval. After additional EPSCs, this protocol was repeated (Fig. 6D). No significant changes of the EPSC amplitude were observed at the first response after the conditioning pulses (101 ± 9% of control value for the first assay, p = 0.7; 95 ± 8% for the second, p = 0.3; n = 7). To confirm that the absence of DSE was not attributable to insufficient depolarization, pulses were increased to 25 s for another set of neurons. DSE was not induced in these conditions (94 ± 4% of control value, p = 0.22; range, 85–106%; n = 6).
Discussion
In this study, we showed that NTS neurons expressed a presynaptic form of LTD. This LTD relies on a reduction of vesicular release triggered by activation of presynaptic CB1Rs. AEA appears to be synthetized and released during NMDAR activation. However, neither postsynaptic NMDARs nor coincident activity in the postsynaptic neuron is mandatory for LTD induction. Therefore, this form of LTD may be entirely expressed at the presynaptic level and requires AEA release (Fig. 6E).
Several observations indicate that LFS LTD expression in NTS neurons is presynaptic. LFS LTD was associated with a decrease in short-term depression of EPSCs, measured by the PPR, and with an increase in the EPSC variability, measured by the CV. Changes in these parameters have been widely used to distinguish presynaptic from postsynaptic effects, and a recent study in NTS neurons confirmed that changes in release probability alter both PPR and CV, whereas reduction in postsynaptic AMPAR currents leaves them unchanged (Almado et al., 2012).
The induction of LFS LTD also requires activation of CB1Rs but not NO synthesis. In situ hybridization studies have demonstrated that the cell bodies of visceral afferent fibers expressed CB1Rs (Burdyga et al., 2010). Terminal-like staining for CB1Rs has been observed in the NTS (Partosoedarso et al., 2003; Derbenev et al., 2004). In addition, activation of CB1Rs decreases calcium influx in vagal afferent terminals (Rogers and Hermann, 2012). Blockade of CB1Rs not only reduces LTD but prevents the associated changes in PPR and CV, supporting the presynaptic hypothesis. Accordingly, direct activation of CB1Rs induces cLTD of EPSCs with similar changes in quantal parameters. This ACEA cLTD is not observed if presynaptic terminals are left unstimulated. Coincident presynaptic activity has been shown to be mandatory to trigger EC LTD in several systems, even when LFS is used (Ronesi et al., 2004; Singla et al., 2007; Heifets and Castillo, 2009). Presynaptic calcium entry during stimulation appears to be necessary to complement CB1R activation (Singla et al., 2007; Heifets et al., 2008). The requirement of both CB1R activation and presynaptic activity for LTD may confer to the synapse a relative specificity because it may limit the action of ECs on presynaptic active synapses only.
As described previously in neonate rodents, NMDAR activation was necessary for LFS LTD in NTS neurons (Zhou et al., 1997; Poon et al., 2000). However, in this previous study, only half of the NTS expressed LTD and a postsynaptic mechanism was proposed to explain it, but changes in PPR and CV had not been measured. Because this previous work had been done in immature rats, aged 3–21 postnatal days, the differences we observed here may reflect developmental changes in LTD mechanisms. In our study, although bath-applied NMDAR antagonists reduces LTD, loading NTS neurons with iMK-801 does not prevent it. In agreement with the experiment in which we used intracellular BAPTA, it indicates that postsynaptic NMDARs and calcium influx in the recorded neurons are not necessary for LTD induction. The dual requirement of NMDARs and CB1Rs for LTD induction is reminiscent of previous studies in neocortical layer 5 pyramidal neurons (Sjöström et al., 2003). In this model, blockade of NMDARs primarily suppresses ACEA cLTD, suggesting that presynaptic NMDARs interacts with CB1Rs. However, in NTS neurons, ACEA cLTD is not blocked by pretreatment with NMDAR antagonist. In addition, NMDA cLTD is prevented by blockade of CB1Rs, and both NMDA and ACEA cLTD require presynaptic activity to develop. This suggests that NMDAR activation occurs upstream of CB1R activation and may be responsible for EC release. EC release can be generally triggered by activation of voltage-dependent calcium channels during postsynaptic spiking or by activation of postsynaptic NMDARs (Heifets and Castillo, 2009; Castillo et al., 2012). In other systems, activation of group I mGluRs coupled to Gαq/11-protein activates phospholipases and eventually leads to presynaptic EC LTD (Heifets and Castillo, 2009). This seems unlikely for NTS neurons because their direct activation by (S)-3,5-dihydroxyphenylglycine does not affect excitatory transmission, even at saturating concentration (3 mm; Chen et al., 2002; Fernandes et al., 2011). In addition, NMDA cLTD is completely prevented by CB1R antagonists, suggesting that NMDAR and CB1R activation are sufficient to induce LFS LTD in NTS neurons. Therefore, NMDAR activation located outside the recorded neurons appear to be required to trigger AEA release. Accordingly, NMDAR subunit NR1 is abundantly expressed on vagal terminals within the NTS and by their cell bodies in the nodose ganglia (Shigemoto et al., 1992; Aicher et al., 1999). Therefore, one explanation may be that NMDARs involved in LFS LTD are located at the presynaptic level and that their activation leads to AEA release in the same compartment. Altogether, these results suggest that, in NTS neurons, LTD induction is independent of postsynaptic NMDARs, a mechanism that departs from the classical Hebbian rule based on detection of coincident presynaptic and postsynaptic activity by postsynaptic NMDARs. In this view, LFS LTD in the NTS shares similarities with the phenomenon of self-depression described recently in the mouse barrel cortex in response to sensory stimulation (Rodríguez-Moreno et al., 2013). The NTS being a sensory nucleus for visceral afferent, it may be a general mechanism devoted to filter uncorrelated incoming activity.
Although CB1 LTD is induced by 2-AG in several systems (Chevaleyre and Castillo, 2004; Heifets and Castillo, 2009), AEA is likely to be involved in presynaptic LTD in the NTS as reported in other systems (Ade and Lovinger, 2007; Puente et al., 2011; Lerner and Kreitzer, 2012). However, we cannot rule out that other endogenous ECs or related fatty acid-derived ligands may participate in LTD because some of them share with AEA the same degradation pathway (Walker et al., 2002). Interestingly, although NTS neurons do not express DSE, arguing against 2-AG expression at excitatory synapses, DSI has been reported previously (Chen et al., 2010). In addition 2-AG synthesizing enzymes are mainly expressed in the postsynaptic dendritic compartment, whereas enzymes for AEA synthesis may be located in presynaptic terminals (Nyilas et al., 2008; Puente et al., 2011; Pickel et al., 2012) Precise location of EC enzymes in the NTS at the subcellular level at excitatory and inhibitory synapses requires additional investigations.
Another explanation for our results may be that NMDAR activation occurs in neighboring cells. Activation of NMDARs in glial cells seems unlikely because calcium rises in glial cells induced by visceral fibers stimulation are insensitive to NMDAR blockade (McDougal et al., 2011). Although part of excitatory synapses between visceral fibers and NTS neurons do not express synaptic NMDARs (Doyle and Andresen, 2001; Jin et al., 2003), they are present extrasynaptically. In addition, EC may diffuse over micrometric distances (Chevaleyre and Castillo, 2004). Synaptic density in the NTS is rather low (Lachamp et al., 2002), but some excitatory contacts gather in multi-synaptic arrangements (Chounlamountry and Kessler, 2011). Therefore, NMDAR activation on neighboring neurons may trigger EC release. In this view, EC diffusion would induce hetero-neuronal LTD in the recorded neuron whether or not its synapses bear NMDARs. This holds true if AEA is produced in presynaptic terminals. This hetero-neuronal plasticity may be important for adjusting synaptic gain in neighboring active synapses on different NTS neurons. However, LFS LTD is not completely blocked by NMDARs and/or CB1Rs antagonists, suggesting that additional mechanisms may participate in its expression. Because this residual depression is not accompanied by changes in CV or PPR, one may speculate that LFS LTD involves presynaptic and postsynaptic independent mechanisms as observed in other systems (Grueter et al., 2010; Puente et al., 2011; Izumi and Zorumski, 2012).
In conclusion, we found that AEA mediates presynaptic LTD of excitatory transmission at the first central synapse for visceral afferent fibers. NTS excitatory synapses are characterized by a very high release probability, making them highly reliable for transmitting relatively low frequency signals (Andresen et al., 2004, 2013; Kline, 2008). Presynaptic LTD would not only reduce information inflow from the periphery but also alter synapse dynamics attributable to redistribution of synapse efficacy that may affect temporal dynamics of neural circuits (Abbott and Nelson, 2000; Silberberg et al., 2004a,b). In addition, visceral afferent fibers directly connect onto both excitatory and inhibitory NTS neurons (Bailey et al., 2008). Therefore, LTD would differentially affect information processing in NTS neural circuits depending on the balance of the depression between excitatory and inhibitory neurons (McBain and Kauer, 2009).
The NTS is the first site of visceral information processing and plays a key role in the control of autonomic functions (Andresen et al., 2004; Travagli et al., 2006; Kline, 2008; Baude et al., 2009). Interestingly, several studies have shown that ECs likely act as signaling molecules at the NTS level to regulate autonomic reflex functions (for review, see Andresen et al., 2012). In addition, several studies have shown that, after long-term exposition of rats to various chronic environmental conditions (hypoxia, ozone; Chen et al., 2003; Kline et al., 2007; Almado et al., 2012), synaptic transmission is depressed for hours in NTS neurons. The mechanism of this depression may involve either a reduction in vesicular release or synapse elimination (Kline et al., 2007; Almado et al., 2012). Whether this depression shares similar mechanisms with EC LTD described in our study remains an open question. Nevertheless, these data open new perspectives to understand how visceral information is processed within the NTS.
Footnotes
This study was supported by National Center of Scientific Research and the Aix-Marseille University. We thank Dominique Debanne, Jean-Marc Goaillard, and Oliver Manzoni for critical reading of this manuscript.
The authors declare no competing financial interests.
References
- Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nat Neurosci. 2000;3:1178–1183. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- Ade KK, Lovinger DM. Anandamide regulates postnatal development of long-term synaptic plasticity in the rat dorsolateral striatum. J Neurosci. 2007;27:2403–2409. doi: 10.1523/JNEUROSCI.2916-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aicher SA, Sharma S, Pickel VM. N-methyl-d-aspartate receptors are present in vagal afferents and their dendritic targets in the nucleus tractus solitarius. Neuroscience. 1999;91:119–132. doi: 10.1016/S0306-4522(98)00530-2. [DOI] [PubMed] [Google Scholar]
- Almado CE, Machado BH, Leão RM. Chronic intermittent hypoxia depresses afferent neurotransmission in NTS neurons by a reduction in the number of active synapses. J Neurosci. 2012;32:16736–16746. doi: 10.1523/JNEUROSCI.2654-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andresen MC, Doyle MW, Jin YH, Bailey TW. Cellular mechanisms of baroreceptor integration at the nucleus tractus solitarius. Ann N Y Acad Sci. 2001;940:132–141. doi: 10.1111/j.1749-6632.2001.tb03672.x. [DOI] [PubMed] [Google Scholar]
- Andresen MC, Doyle MM, Bailey TW, Jin YH. Differentiation of autonomic reflex control begins with cellular mechanisms at the first synapse within the nucleus tractus solitarius. Braz J Med Biol Res. 2004;37:549–558. doi: 10.1590/S0100-879X2004000400012. [DOI] [PubMed] [Google Scholar]
- Andresen MC, Fawley JA, Hofmann ME. Peptide and lipid modulation of glutamatergic afferent synaptic transmission in the solitary tract nucleus. Front Neurosci. 2012;6:191. doi: 10.3389/fnins.2012.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TW, Appleyard SM, Jin YH, Andresen MC. Organization and properties of GABAergic neurons in solitary tract nucleus (NTS) J Neurophysiol. 2008;99:1712–1722. doi: 10.1152/jn.00038.2008. [DOI] [PubMed] [Google Scholar]
- Balland B, Lachamp P, Strube C, Kessler JP, Tell F. Glutamatergic synapses in the rat nucleus tractus solitarii develop by direct insertion of calcium-impermeable AMPA receptors and without activation of NMDA receptors. J Physiol. 2006;574:245–261. doi: 10.1113/jphysiol.2006.108738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balland B, Lachamp P, Kessler JP, Tell F. Silent synapses in developing rat nucleus tractus solitarii have AMPA receptors. J Neurosci. 2008;28:4624–4634. doi: 10.1523/JNEUROSCI.5355-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baude A, Strube C, Tell F, Kessler JP. Glutamatergic neurotransmission in the nucleus tractus solitarii: structural and functional characteristics. J Chem Neuroanat. 2009;38:145–153. doi: 10.1016/j.jchemneu.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Browning KN, Travagli RA. Plasticity of vagal brainstem circuits in the control of gastrointestinal function. Auton Neurosci. 2011;161:6–13. doi: 10.1016/j.autneu.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdyga G, Varro A, Dimaline R, Thompson DG, Dockray GJ. Expression of cannabinoid CB1 receptors by vagal afferent neurons: kinetics and role in influencing neurochemical phenotype. Am J Physiol Gastrointest Liver Physiol. 2010;299:G63–G69. doi: 10.1152/ajpgi.00059.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado M, Isope P, Ascher P. Involvement of presynaptic N-methyl-d-aspartate receptors in cerebellar long-term depression. Neuron. 2002;33:123–130. doi: 10.1016/S0896-6273(01)00568-2. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Bonham AC, Plopper CG, Joad JP. Plasticity in respiratory motor control: selected contribution: neuroplasticity in nucleus tractus solitarius neurons after episodic ozone exposure in infant primates. J Appl Physiol. 2003;94:819–827. doi: 10.1152/japplphysiol.00552.2002. [DOI] [PubMed] [Google Scholar]
- Chen CY, Bonham AC, Dean C, Hopp FA, Hillard CJ, Seagard JL. Retrograde release of endocannabinoids inhibits presynaptic GABA release to second-order baroreceptive neurons in NTS. Auton Neurosci. 2010;158:44–50. doi: 10.1016/j.autneu.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Ling EH, Horowitz JM, Bonham AC. Synaptic transmission in nucleus tractus solitarius is depressed by Group II and III but not Group I presynaptic metabotropic glutamate receptors in rats. J Physiol. 2002;538:773–786. doi: 10.1113/jphysiol.2001.012948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/S0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron. 2004;43:871–881. doi: 10.1016/j.neuron.2004.08.036. [DOI] [PubMed] [Google Scholar]
- Chounlamountry K, Kessler JP. The ultrastructure of perisynaptic glia in the nucleus tractus solitarii of the adult rat: comparison between single synapses and multisynaptic arrangements. Glia. 2011;59:655–663. doi: 10.1002/glia.21135. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci. 2010;11:459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- Derbenev AV, Stuart TC, Smith BN. Cannabinoids suppress synaptic input to neurones of the rat dorsal motor nucleus of the vagus nerve. J Physiol. 2004;559:923–938. doi: 10.1113/jphysiol.2004.067470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle MW, Andresen MC. Reliability of monosynaptic sensory transmission in brain stem neurons in vitro. J Neurophysiol. 2001;85:2213–2223. doi: 10.1152/jn.2001.85.5.2213. [DOI] [PubMed] [Google Scholar]
- Faber DS, Korn H. Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophys J. 1991;60:1288–1294. doi: 10.1016/S0006-3495(91)82162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes LG, Jin YH, Andresen MC. Heterosynaptic crosstalk: GABA-glutamate metabotropic receptors interactively control glutamate release in solitary tract nucleus. Neuroscience. 2011;174:1–9. doi: 10.1016/j.neuroscience.2010.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Grueter BA, Brasnjo G, Malenka RC. Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci. 2010;13:1519–1525. doi: 10.1038/nn.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Maejima T, Fukami K, Kano M. Pharmacological evidence for the involvement of diacylglycerol lipase in depolarization-induced endocanabinoid release. Neuropharmacology. 2008;54:58–67. doi: 10.1016/j.neuropharm.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Heifets BD, Castillo PE. Endocannabinoid signaling and long-term synaptic plasticity. Annu Rev Physiol. 2009;71:283–306. doi: 10.1146/annurev.physiol.010908.163149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heifets BD, Chevaleyre V, Castillo PE. Interneuron activity controls endocannabinoid-mediated presynaptic plasticity through calcineurin. Proc Natl Acad Sci U S A. 2008;105:10250–10255. doi: 10.1073/pnas.0711880105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Zorumski CF. NMDA receptors, mGluR5, and endocannabinoids are involved in a cascade leading to hippocampal long-term depression. Neuropsychopharmacology. 2012;37:609–617. doi: 10.1038/npp.2011.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YH, Bailey TW, Li BY, Schild JH, Andresen MC. Purinergic and vanilloid receptor activation releases glutamate from separate cranial afferent terminals in nucleus tractus solitarius. J Neurosci. 2004;24:4709–4717. doi: 10.1523/JNEUROSCI.0753-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YH, Bailey TW, Doyle MW, Li BY, Chang KS, Schild JH, Mendelowitz D, Andresen MC. Ketamine differentially blocks sensory afferent synaptic transmission in medial nucleus tractus solitarius (mNTS) Anesthesiology. 2003;98:121–132. doi: 10.1097/00000542-200301000-00021. [DOI] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Kline DD. Plasticity in glutamatergic NTS neurotransmission. Respir Physiol Neurobiol. 2008;164:105–111. doi: 10.1016/j.resp.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD, Ramirez-Navarro A, Kunze DL. Adaptive depression in synaptic transmission in the nucleus of the solitary tract after in vivo chronic intermittent hypoxia: evidence for homeostatic plasticity. J Neurosci. 2007;27:4663–4673. doi: 10.1523/JNEUROSCI.4946-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachamp P, Tell F, Kessler JP. Successive episodes of synapses production in the developing rat nucleus tractus solitarii. J Neurobiol. 2002;52:336–342. doi: 10.1002/neu.10091. [DOI] [PubMed] [Google Scholar]
- Lachamp P, Balland B, Tell F, Crest M, Kessler JP. Synaptic localization of the glutamate receptor subunit GluR2 in the rat nucleus tractus solitarii. Eur J Neurosci. 2003;17:892–896. doi: 10.1046/j.1460-9568.2003.02494.x. [DOI] [PubMed] [Google Scholar]
- Lerner TN, Kreitzer AC. RGS4 is required for dopaminergic control of striatal LTD and susceptibility to parkinsonian motor deficits. Neuron. 2012;73:347–359. doi: 10.1016/j.neuron.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBain CJ, Kauer JA. Presynaptic plasticity: targeted control of inhibitory networks. Curr Opin Neurobiol. 2009;19:254–262. doi: 10.1016/j.conb.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougal DH, Hermann GE, Rogers RC. Vagal afferent stimulation activates astrocytes in the nucleus of the solitary tract via AMPA receptors: evidence of an atypical neural–glial interaction in the brainstem. J Neurosci. 2011;31:14037–14045. doi: 10.1523/JNEUROSCI.2855-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabavi S, Kessels HW, Alfonso S, Aow J, Fox R, Malinow R. Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc Natl Acad Sci U S A. 2013;110:4027–4032. doi: 10.1073/pnas.1219454110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyilas R, Dudok B, Urbán GM, Mackie K, Watanabe M, Cravatt BF, Freund TF, Katona I. Enzymatic machinery for endocannabinoid biosynthesis associated with calcium stores in glutamatergic axon terminals. J Neurosci. 2008;28:1058–1063. doi: 10.1523/JNEUROSCI.5102-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partosoedarso ER, Abrahams TP, Scullion RT, Moerschbaecher JM, Hornby PJ. Cannabinoid1 receptor in the dorsal vagal complex modulates lower oesophageal sphincter relaxation in ferrets. J Physiol. 2003;550:149–158. doi: 10.1113/jphysiol.2003.042242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JH, McDougall SJ, Fawley JA, Smith SM, Andresen MC. Primary afferent activation of thermosensitive TRPV1 triggers asynchronous glutamate release at central neurons. Neuron. 2010;65:657–669. doi: 10.1016/j.neuron.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickel VM, Shobin ET, Lane DA, Mackie K. Cannabinoid-1 receptors in the mouse ventral pallidum are targeted to axonal profiles expressing functionally opposed opioid peptides and contacting N-acylphosphatidylethanolamine-hydrolyzing phospholipase D terminals. Neuroscience. 2012;227:10–21. doi: 10.1016/j.neuroscience.2012.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon CS, Zhou Z, Champagnat J. NMDA receptor activity in utero averts respiratory depression and anomalous long-term depression in newborn mice. J Neurosci. 2000;20:RC73. doi: 10.1523/JNEUROSCI.20-09-j0003.2000. (1–6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente N, Cui Y, Lassalle O, Lafourcade M, Georges F, Venance L, Grandes P, Manzoni OJ. Polymodal activation of the endocannabinoid system in the extended amygdala. Nat Neurosci. 2011;14:1542–1547. doi: 10.1038/nn.2974. [DOI] [PubMed] [Google Scholar]
- Ray AP, Griggs L, Darmani NA. Δ9-Tetrahydrocannabinol suppresses vomiting behavior and Fos expression in both acute and delayed phases of cisplatin-induced emesis in the least shrew. Behav Brain Res. 2009;196:30–36. doi: 10.1016/j.bbr.2008.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, Kohl MM, Reeve JE, Eaton TR, Collins HA, Anderson HL, Paulsen O. Presynaptic induction and expression of timing-dependent long-term depression demonstrated by compartment-specific photorelease of a use-dependent NMDA receptor antagonist. J Neurosci. 2011;31:8564–8569. doi: 10.1523/JNEUROSCI.0274-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, González-Rueda A, Banerjee A, Upton AL, Craig MT, Paulsen O. Presynaptic self-depression at developing neocortical synapses. Neuron. 2013;77:35–42. doi: 10.1016/j.neuron.2012.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RC, Hermann GE. Tumor necrosis factor activation of vagal afferent terminal calcium is blocked by cannabinoids. J Neurosci. 2012;32:5237–5241. doi: 10.1523/JNEUROSCI.6220-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi J, Gerdeman GL, Lovinger DM. Disruption of endocannabinoid release and striatal long-term depression by postsynaptic blockade of endocannabinoid membrane transport. J Neurosci. 2004;24:1673–1679. doi: 10.1523/JNEUROSCI.5214-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto R, Ohishi H, Nakanishi S, Mizuno N. Expression of the mRNA for the rat NMDA receptor (NMDAR1) in the sensory and autonomic ganglion neurons. Neurosci Lett. 1992;144:229–232. doi: 10.1016/0304-3940(92)90756-W. [DOI] [PubMed] [Google Scholar]
- Silberberg G, Bethge M, Markram H, Pawelzik K, Tsodyks M. Dynamics of population rate codes in ensembles of neocortical neurons. J Neurophysiol. 2004a;91:704–709. doi: 10.1152/jn.00415.2003. [DOI] [PubMed] [Google Scholar]
- Silberberg G, Wu C, Markram H. Synaptic dynamics control the timing of neuronal excitation in the activated neocortical microcircuit. J Physiol. 2004b;556:19–27. doi: 10.1113/jphysiol.2004.060962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla S, Kreitzer AC, Malenka RC. Mechanisms for synapse specificity during striatal long-term depression. J Neurosci. 2007;27:5260–5264. doi: 10.1523/JNEUROSCI.0018-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/S0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Travagli RA, Hermann GE, Browning KN, Rogers RC. Brainstem circuits regulating gastric function. Annu Rev Physiol. 2006;68:279–305. doi: 10.1146/annurev.physiol.68.040504.094635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Sickle MD, Oland LD, Mackie K, Davison JS, Sharkey KA. Δ9-Tetrahydrocannabinol selectively acts on CB1 receptors in specific regions of dorsal vagal complex to inhibit emesis in ferrets. Am J Physiol Gastrointest Liver Physiol. 2003;285:G566–G576. doi: 10.1152/ajpgi.00113.2003. [DOI] [PubMed] [Google Scholar]
- Walker JM, Krey JF, Chu CJ, Huang SM. Endocannabinoids and related fatty acid derivatives in pain modulation. Chem Phys Lipids. 2002;121:159–172. doi: 10.1016/S0009-3084(02)00152-4. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Champagnat J, Poon CS. Phasic and long-term depression in brainstem nucleus tractus solitarius neurons: differing roles of AMPA receptor desensitization. J Neurosci. 1997;17:5349–5356. doi: 10.1523/JNEUROSCI.17-14-05349.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]