

Abstract
The spike-timing-dependent plasticity (STDP), a synaptic learning rule for encoding learning and memory, relies on relative timing of neuronal activity on either side of the synapse. GABAergic signaling has been shown to control neuronal excitability and consequently the spike timing, but whether GABAergic circuits rule the STDP remained unknown. Here we show that GABAergic signaling governs the polarity of STDP, because blockade of GABAA receptors was able to completely reverse the temporal order of plasticity at corticostriatal synapses in rats and mice. GABA controls the polarity of STDP in both striatopallidal and striatonigral output neurons. Biophysical simulations and experimental investigations suggest that GABA controls STDP polarity through depolarizing effects at distal dendrites of striatal output neurons by modifying the balance of two calcium sources, NMDARs and voltage-sensitive calcium channels. These findings establish a central role for GABAergic circuits in shaping STDP and suggest that GABA could operate as a Hebbian/anti-Hebbian switch.
Introduction
Synaptic plasticity is the main neuronal substrate for learning and memory (Martin and Morris, 2002). The synaptic strength between neurons can be modified by the relative timing of their neuronal firing on either side of the synapse, a phenomenon called spike-timing-dependent plasticity (STDP; Markram et al., 1997; Caporale and Dan, 2008; Sjöström et al., 2008). GABAergic circuits, via feedforward inhibition, control the input-output gain function of principal neurons by efficiently modulating their spike timing (Pouille and Scanziani, 2001; Wehr and Zador, 2003; Higley and Contreras, 2006). Indeed, feedforward GABAergic signaling may restrict the time window in which synaptic inputs can summate and eventually reach the spiking threshold. Such control of the spike timing could lead to a major modulation of the induction of long-term synaptic plasticity such as STDP, in which occurrence of long-term potentiation (LTP) or long-term depression (LTD) relies on the precise relative timing (in milliseconds) of presynaptic and postsynaptic action potentials. In addition, STDP is dependent on the spatial location of dendritic inputs and is modulated by factors affecting dendritic voltage-gated channel properties (Froemke et al., 2010). Despite the tight control of GABAergic circuits onto output neurons, their contribution to activity-dependent plasticity remained unknown.
We previously characterized a potent and reliable STDP at corticostriatal synapses in the striatal output neurons, the medium-sized spiny neurons (MSNs; Fino et al., 2005; Fino and Venance, 2010). MSN activity is tightly controlled by different local GABAergic circuits. Striatum is mainly composed of GABAergic neurons (interneurons and MSNs), which form feedback and feedforward inhibitory circuits. Feedback inhibitory circuits are formed by MSN collaterals and feedforward inhibition involves different subtypes of GABAergic interneurons (Koós and Tepper, 1999; Tepper et al., 2008; Planert et al., 2010) that are efficiently recruited by cortical activity with shorter latencies than MSNs (Mallet et al., 2005; Fino et al., 2008). Functionally, feedforward inhibition mediates the most powerful inhibition in striatum and provides strong perisomatic synapses onto hundreds of surrounding MSNs (Tepper et al., 2008).
We investigated here the impact of striatal inhibitory circuits on STDP-timing rules in MSNs. We observed that the blockade of GABAA receptors reversed the polarity of STDP in the striatum in juvenile and adult animals. Using a combination of modeling and experimental approaches, we predict that this reversion is due to a depolarizing effect of GABA within the dendritic tree, which would modulate two different sources of calcium, NMDARs and voltage-sensitive calcium channels (VSCCs), and control the orientation of the plasticity.
Materials and Methods
Electrophysiological recordings
All experiments were performed in accordance with European Union guidelines (directive 86/609/EEC). Connections between the somatosensory cortex (layer 5) and the striatum are preserved in a horizontal plane (Fino et al., 2005, 2008, 2010). We used horizontal brain slices (330 μm) from OFA rats (P15-P90) and 300 μm slices from parvalbumin-GFP mice (P20-P25) and D1-eGFP mice (P18-P32) of either sex. We used parvalbumin-GFP mice to target fast-spiking (FS) interneurons and D1-eGFP to target the two MSN subpopulations, D1-expressing (GFP-positive) or D2-expressing (GFP-negative) MSNs. Patch-clamp recordings (at 34°C) of MSNs or FS cells were performed as described previously (Fino et al., 2005, 2008, 2010). Briefly, borosilicate glass pipettes (5–8 MΩ) contained the following (in mm): 105 K-gluconate, 30 KCl (or 127 K-gluconate and 13 KCl for experiments with a chloride reversal potential ECl− = −60 mV), 10 HEPES, 10 phosphocreatine, 4 ATP-Mg, 0.3 GTP-Na, and 0.3 EGTA adjusted to pH 7.35 with KOH. The composition of the extracellular solution was as follows (in mm): 125 NaCl, 2.5 KCl, 25 glucose, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, and 10 μm pyruvic acid bubbled with 95% O2 and 5% CO2. Signals were amplified using EPC10–2 amplifiers (HEKA Elektronik). Current-clamp recordings were filtered at 2.5 kHz and sampled at 5 kHz and voltage-clamp recordings were filtered at 5 kHz and sampled at 10 kHz using the program Patchmaster v2x32 (HEKA Elektronik). The series resistance was compensated at 75–80%.
STDP induction protocols
Electrical stimulations were performed with a bipolar electrode (Phymep) placed in layer 5 of the somatosensory cortex. Electrical stimulations were monophasic at constant current (Iso-Flex AMPI; Science Products). Repetitive control stimuli were applied at 0.1 Hz and currents were adjusted to evoke ∼50–200 pA EPSCs. MSNs were held at their physiological membrane potential, on average −72.3 ± 0.6 mV (n = 92) and there was no statistical difference in the holding membrane potentials between the different experimental conditions. STDP protocols consisted of pairings of presynaptic and postsynaptic stimulations (100–150 times at 1 Hz) with a time shifting (Δt) of several milliseconds. We varied the Δt between −25 and +30 ms to fully explore the time window of plasticity induction. We observed reliable plasticity for −25<Δt<0 ms for post-pre pairings and 0<Δt<+30 ms for pre-post pairings and a lack of induction of LTP or LTD for longer Δt (Fino et al., 2005, 2010). Presynaptic stimulations correspond to cortical stimulations and the postsynaptic stimulation to an action potential evoked by a depolarizing current step (30 ms duration) in the MSN. Neurons were recorded for 10 min during baseline and for at least 60 min after the STDP protocol; long-term synaptic efficacy changes were measured after 60 min. Variation of series resistance >20% led to the rejection of the experiment.
Chemicals
We used the following chemicals at the indicated concentrations: DL-2-amino-5-phosphono-pentanoic acid (D-AP5, 40 μm; Tocris Bioscience), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μm; Tocris Bioscience), bicuculline methiodide (20 μm; Sigma), SR 95531 hydrobromide (gabazine 10 μm; Tocris Bioscience), NMDA (10 mm; Tocris Bioscience), glycine (10 mm; Tocris Bioscience), strychnine (1 mm; Tocris Bioscience), isoguvacine (5 mm; Tocris Bioscience), GABA (5 mm; Tocris Bioscience), tetraethylammonium chloride (TEA-Cl, 20 mm; Tocris Bioscience), and 4-aminopyridine (4-AP, 5 mm; Tocris Bioscience) were dissolved in the extracellular solution. N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3carboxamide (AM251, 3 μm; Tocris Bioscience) and picrotoxin (PTX, 50 μm extracellular and i-PTX, 500 μm-1 mm intracellular; Sigma) were also used. PTX was dissolved in ethanol and then added in the external solution at a final ethanol concentration of of 0.01–0.02%.
Electrophysiological data analysis
Off-line analysis was performed using Igor-Pro 6.0.3 (Wavemetrics), Fitmaster (HEKA Elektronik), and MiniAnalysis 6.0.7 (Synaptosoft) software. Statistical analysis was performed using Prism 5.0 (GraphPad). All results are expressed as mean ± SEM and statistical significance was assessed using the Student's t test, paired test, or the nonparametric Wilcoxon signed-rank test when appropriate at the significance levels (p) indicated.
Reversal potential of the GABAA-mediated current
Cell-attached recordings (voltage-clamp mode with injecting current sets to 0 pA) were performed to determine the reversal potential of GABAAR-mediated currents. The value of the driving force of chloride ions through GABAARs is based on resting membrane potential (RMP) and the reversal potential of GABAAR (EGABA), both measured by cell-attached recordings of single-channel iGABA and iNMDA (Dehorter et al., 2009). We estimated RMP from iNMDA known to reverse at a membrane potential close to 0 mV, and EGABA was determined based on the relationship between iGABA and the extracellular potential to the patch of the membrane as follows: EGABA = DFGABA + RMP. Two intrapipette solutions were used and contained the following (in mm): for iNMDA, 140 NaCl, 3.5 KCl, 1.8 CaCl2, 10 HEPES pH = 7.3, 10 μm NMDA, 10 μm glycine, and 1 μm strychnine; for iGABA, 120 NaCl, 20 TEA-Chloride, 5 KCl, 5 4-aminopyridine, 0.1 CaCl2, 10 MgCl2, 10 glucose, 10 HEPES buffered to pH = 7.3, GABA 5 μm, isoguvacine 5 μm, and CsCl 3 μm.
MSN biophysical model
A biophysically detailed model of a MSN was developed based on a previously published model for ventral striatum (Wolf et al., 2005), but further modified to match our experimental conditions and more recent studies on striatal MSNs. In another published study, we presented previously a “sister model” for MSNs in dorsal striatum (Evans et al., 2012).
Our present model shows significant inward rectification when clamped at the potentials negative to resting level (Fig. 5A), consistent with classic studies on MSN electrophysiology properties in dorsal striatum. The current-frequency behavior of the model matches our experimental conditions (Fig. 5B). Furthermore, it has TTX-dependent backpropagating action potentials (bAPs) and an improved calcium dynamics (Fig. 5C,D).
Figure 5.

Biophysical model predicts a depolarizing effect of GABA. A–D, Model validation. A, Simulation of voltage trace in the soma during current step injections. Inward-rectification is significant with hyperpolarizing inputs. B, Current-firing frequency behavior for a real and the model MSN; experimental data were from MSNs recorded in this study. C, Calcium transients recorded in the proximal dendrites (40–60 μm from the soma) after a depolarizing pulse (20 mV, 1 ms duration) in the soma. Simulated blockade of sodium currents reduces the dendritic calcium (red curve), consistent with the observations on TTX-dependent bAP. D, Simulation of calcium transients in the proximal dendrites (50–60 μm from the soma) after a spike train (theta burst) delivered to the MSN. The simulated calcium transient trace (red curve) matches the original experimental curve (black curve). E, Simulation of voltage sample traces during post-pre and pre-post STDP pairings. GABAergic inputs depolarize the membrane locally during post-pre and pre-post pairings (black arrows). This enhances and prolongs the depolarization during the decaying phase of the bAP for post-pre pairings; a bimodal voltage fluctuation can sometimes be seen locally at the distal dendrites/spines. For pre-post pairings, an early depolarization before the bAP is obvious locally. The simulated spine is located ∼130 μm from the soma.
Morphology and passive properties.
The morphology of our model was identical to one described previously (Evans et al., 2012). In brief, the model has four primary dendrites (20 μm), eight secondary dendrites (24 μm), and 16 tertiary dendritic branches (each tertiary branch consisting of 11 compartments of 36 μm). These morphology parameters were used to compensate for the additional spine membrane area as in previous models (Wolf et al., 2005). To match the present experimental conditions, the input resistance of the model was tuned to be 210 MΩ and the RMP was adjusted to be −80 mV. The other passive parameters used in the simulations were specific axial resistance at 4 Ω · m in the soma/proximal/secondary dendrites and 2 Ω · m2 in the distal dendrites, specific membrane resistance at 8 Ω · m2, reversal potential of leak channels at −60 mV, and membrane capacitance at 0.01 F/m2.
Calcium channels and buffers.
The calcium channels in our model were all taken from previous published models (Wolf et al., 2005): Q-, R-, T-, N-, and L-type (both CaV1.2 and CaV1.3) calcium channels. The details of their kinetics can be found in Evans et al. (2012). We used a classic calcium buffer model, which was used in experimental studies of calcium dynamics in both hippocampal neurons (Sabatini et al., 2002; Yasuda et al., 2004) and MSNs (Carter and Sabatini, 2004). Using calcium buffer parameters obtained from MSN experimental data (Carter and Sabatini, 2004), our model can account for the effects of different calcium dyes (Sabatini et al., 2002) and therefore reproduce dendritic calcium behavior in MSNs (Fig. 5D, modified from Day et al., 2008). Table 1 shows the maximal conductance of noncalcium channels in the model; Table 2 shows the maximal permeability of calcium channels in the model.
Table 1.
Maximal conductance of noncalcium channels in the model (S/m2)
| Channel name | Soma | Proximal dendrites | Distal dendrites |
|---|---|---|---|
| Naf | 15,000 | 1560 | 195 |
| NaP | 0.4 | 1.38e-3 | 1.38e-3 |
| KAf | 2250 | 630 | 210 |
| KAs | 166.4 | 15.216 | 15.216 |
| Kir | 4.34 | 4.34 | 4.34 |
| Kdr | 28 | 28 | 28 |
| SK | 0.5 | 0.5 | 0.5 |
| BK | 10 | 10 | 10 |
Naf indicates fast sodium channel; NaP, persistent sodium channel; KAf, fast A-type potassium channel; KAs, slow A-type potassium channels; Kir, inward-rectifier potassium channel (Kir); Kdr, delayed rectifier potassium channels; SK, small-conductance calcium-dependent[1] potassium channel; BK, large-conductance calcium-dependent potassium channel.
Table 2.
Maximal permeability of calcium channels in the model (m/s)
| Channel name | Soma | Proximal dendrites | Distal dendrites |
|---|---|---|---|
| CaQ | 3e-7 | ||
| CaN | 1.05e-7 | ||
| CaR | 13e-7 | 13e-7 | 13e-7 |
| CaV1.2 | 2.8475e-8 | 2.8475e-8 | 2.8475e-8 |
| CaV1.3 | 2.125e-7 | 2.125e-7 | 2.125e-7 |
| CaT | 11.75e-9 | 11.75e-9 | 11.75e-9 |
Other ion channels.
The model also contains most of the other ion channels introduced in the previous model (Wolf et al., 2005): fast and persistent sodium channels, fast and slow A-type potassium channels, inward-rectifier potassium channels, small-conductance calcium-dependent potassium channels, and large-conductance calcium-dependent potassium channels. The fast A-type potassium channel adopts a conventional form “m4h” instead of “m2h” described previously (Wolf et al., 2005). The kinetics of inward-rectifier potassium channels, fast A-type potassium channels, delayed rectifier potassium channels, and small-conductance calcium-dependent potassium channels were modified in accordance with available data described previously (Evans et al., 2012). The ion channel distributions in the model are described in Table 1.
Spines.
Another salient feature in our model was to include some spines explicitly. The spine consists of two cylindrical compartments: the spine-head and the spine-neck. Because the morphology parameters were already set to compensate for the additional spine membrane area (Wolf et al., 2005), we only inserted 10–15 spines onto the dendritic locations of interest. As for ion channels in spines, we included experimentally verified calcium channels (Carter and Sabatini, 2004), R-, T-, and L-type calcium channels, onto the spine-head. NMDA/AMPA synapses were also added to the spines. The kinetic parameters of NMDA/AMPA channels were taken from dorsal striatum (Chapman et al., 2003). As in the previous model (Wolf et al., 2005), we introduced calcium pools coupled to different calcium sources to keep track of the calcium: CaV1.3/CaV1.2 (the “L-type calcium pool”) and NMDA (the “NMDA calcium pool”). Maximal permeability of calcium channels in the spines (m/s): CaT, 2.35e-8; CaR, 1.3e-6; CaV1.2, 5.695e-8; and CaV1.3, 4.25e-7.
GABAergic input.
Tonic background GABAergic inputs were explicitly modeled based on our experimental data and other published experimental studies (Ade et al., 2008; Santhakumar et al., 2010). The GABA synapse was modeled with the conductance (1500 pS) set to fit our experimental observations of an amplitude ∼13–15 pA when measured in the soma. In our experiments, FS cell spike latency after cortical stimulation was 3.3 ± 0.5 ms (n = 6), whereas IPSC latency in MSN after FS cell spikes was 1.3 ± 0.2 ms (n = 5). Therefore, we assumed that the latency of GABAergic input after NMDA/AMPA inputs was ∼3–5 ms. In our model, when GABAergic input was included, 3 GABAergic synapses were inserted onto the dendritic compartment neighboring to the spine (∼20–30 μm) and activated 5 ms after NMDA/AMPA inputs. The feedforward GABAergic activations were given asynchronously at up to 200 Hz to resemble GABAergic inputs clustered temporally.
Simulation environment.
GENESIS v2.3 (Bower and Beeman, 2007) was the main simulator running in the Linux environment (Fedora 11/12/13, X86_64; CPU, Q6600; RAM, 4GB). The simulation time step was set to be 5 μs.
The model will be deposited in ModelDB (http://senselab.med.yale.edu/modeldb/).
Results
Inhibition of GABAARs reverses the polarity of the corticostriatal STDP
In control conditions (i.e., without any pharmacological treatment), consistent with previous results (Fino et al., 2005, 2010), we observed a robust bidirectional STDP in MSNs: post-pre pairings induced LTP (mean value of the EPSC amplitude: 161.2 ± 14.1% recorded 60 min after STDP protocol induction, for −30<Δt<0 ms, p < 0.001, n = 21) and pre-post pairings induced LTD (72.3 ± 6.9%, for 0<Δt<+30 ms, p < 0.01, n = 14) (Fig. 1A–C). The impact of GABAergic signaling on STDP was assessed by inhibiting GABAARs with bicuculline methiodide (20 μm, n = 12), PTX (50 μm, n = 11), or gabazine (10 μm, n = 10). PTX acts as a blocker of GABAARs, whereas bicuculline and gabazine are GABAAR-competitive antagonists. Remarkably, the STDP-timing rule was reversed when GABAARs were blocked. Indeed, when bicuculline, PTX, or gabazine was applied, post-pre pairings induced LTD instead of LTP observed in control conditions, and vice versa for pre-post pairings (i.e., LTD in control became LTP when GABAARs were inhibited; Fig. 1A–C). For example, with PTX, post-pre pairings induced LTD (85.2 ± 29.0%, p < 0.01, n = 5) and, conversely, pre-post pairings with PTX induced LTP (220.3 ± 45.4%, p < 0.05, n = 6). On average, when bicuculline, PTX, and gabazine treatments were considered altogether, post-pre pairings induced LTD (84.6 ± 3.0%, p < 0.001, n = 17) and pre-post pairings induced LTP (197.3 ± 21.0%, p < 0.001, n = 16) (Fig. 1B–D). No significant difference was observed between the effects of the three different GABA blockers/antagonists. Last, we investigated whether the effects of GABA on STDP-timing rules exists up to adulthood (i.e., in P60-P90 rats) and we indeed observed that GABA tightly controlled STDP in adult animals (Fig. 1D). We conclude that GABAAR blockade induced a reversion of polarity of STDP in MSNs of juvenile and adult animals.
Figure 1.

GABAAR inhibition reverses the STDP. (A) Representative STDP experiments illustrating the time courses of plasticity induced by post-pre and pre-post pairings in control conditions and with PTX (50 μm). In control conditions (black traces), post-pre and pre-post pairings induced LTP and LTD, respectively, as illustrated by EPSCs (averages of 10 raw traces) evoked before (gray traces) or after (black and red traces) the STDP protocol. With PTX, post-pre pairings induced LTD and pre-post pairings induced LTP (red traces). Low traces show monitoring of input resistance (filled circles) and holding current (diamonds) during the entire experiment in control (black) and picrotoxin (red) conditions. This illustrates the stability of the recordings along time for the different experimental conditions. B, Summary graphs showing that GABAAR blockade reversed STDP polarity (control: black circles; GABAAR inhibition [PTX, bicuculline, and gabazine]: red circles). Each empty symbol represents one neuron and filled symbols are the averages. C, Summary bar graphs showing the inversion of plasticity after GABAAR inhibition with bicucculline (20 μm), gabazine (10 μm), or PTX (50 μm). Post-pre pairings (left) induced LTP in control and LTD after GABAAR inhibition by any of the three treatments; conversely, pre-post pairings (right) induced LTD in control and LTP with GABAAR inhibition. D, The reversion of STDP polarity by PTX is also observed in adult animals (P60-P90) as illustrated by the summary bar graph. In control conditions, post-pre and pre-post pairings induced LTP (121.0 ± 6.0%, p < 0.05, n = 9) and LTD (69.4 ± 7.1, p < 0.01, n = 7), respectively. When GABAARs were blocked with PTX (50 μm), post-pre pairings induced LTD (56.4 ± 9.0%, p < 0.05, n = 3) and pre-post pairings induced LTP (125.0 ± 7.0, p < 0.05, n = 5). The black arrows indicate the STDP protocol induction. Data: mean ± SEM. *p < 0.05.
GABA controls STDP in both striatopallidal and striatonigral MSNs
Corticostriatal projections contact two MSN populations belonging to either the direct (striatonigral) or indirect (striato-pallido-subthalamo-nigral) striatal output pathways (Gerfen and Surmeier, 2011). We investigated whether GABA could act differently depending on these anatomo-functional MSN subpopulations. The two functional MSN subtypes express different dopaminergic receptors, D1-like for the direct pathway and D2-like for the indirect pathway, allowing us to target them specifically with transgenic D1-eGFP mice and to investigate STDP in D1+ and non-D1+ MSNs (Fig. 2). We tested the STDP in both MSN subpopulations in control conditions and in GABAAR blockade conditions (PTX, 50 μm). In control conditions, we observed that pre-post pairings induced LTD in both D1+ (p < 0.05, n = 4) and non-D1+ (p < 0.05, n = 4) MSNs, whereas post-pre pairings induced LTP in non-D1+ MSNs (p < 0.05, n = 7). These observations are similar to those described above for rats (Fig. 1B). We did not observe any significant plasticity for post-pre pairings in D1+ MSNs (n = 5), consistent with a previous study performed with these D1-eGFP mice (Shen et al., 2008). In GABAAR inhibition, PTX reversed the polarity of the plasticity, because post-pre pairings induced LTD in non-D1+ cells (p < 0.05, n = 7); an absence of plasticity was still observed in D1+ MSNs (n = 3). In addition, PTX precluded the LTD observed in control conditions after pre-post pairings in D1+ (n = 6) and non-D1+ (n = 7) MSNs (Fig. 2A,B). This indicates that GABA exerts a similar effect on STDP for both striatopallidal and striatonigral MSNs.
Figure 2.
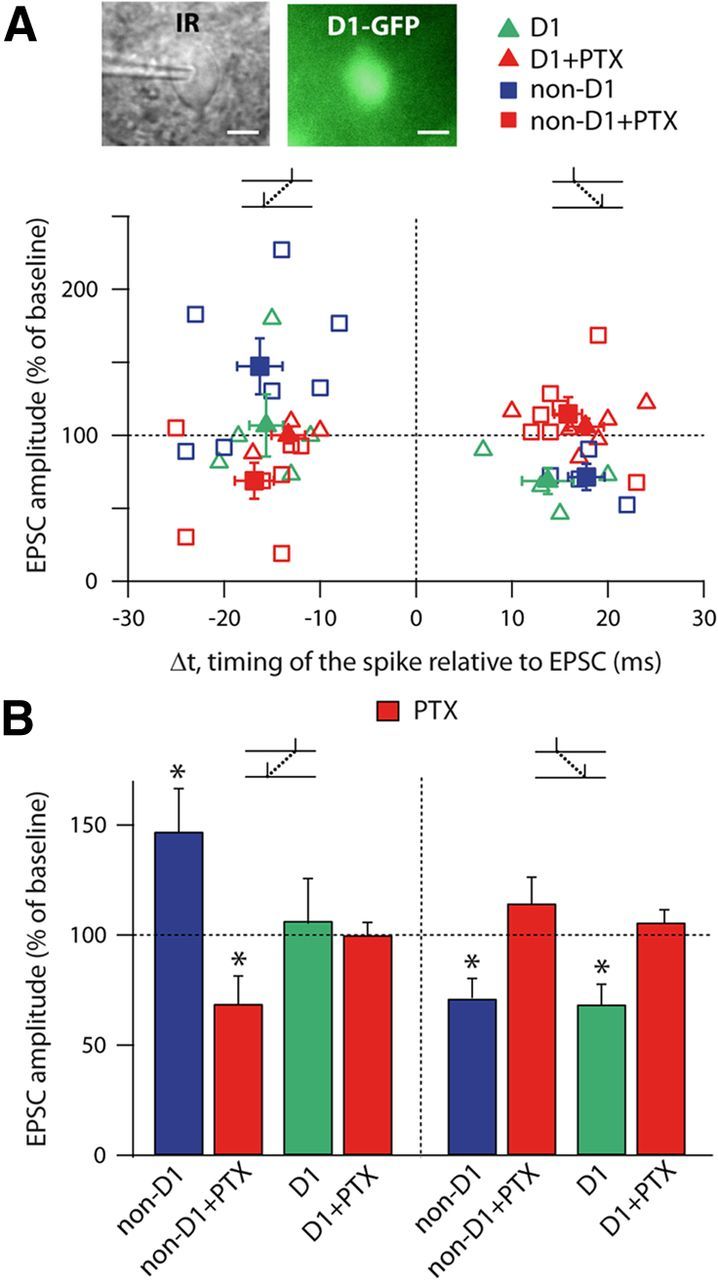
Similar effect of GABA on striato-pallidal and striato-nigral MSNs. A, Recording of D1-eGFP positive MSN (scale bars, 10 μm). Summary graphs showing STDP occurrence depending on the D1+ (triangles) or non-D1+ (squares) MSN recordings, in control conditions (green and blue symbols) or with PTX (red symbols). Each empty symbol represents one neuron and filled symbols are the averages. B, Summary bar graph of the effect of PTX on STDP in D1+ (green) and non-D1+ (blue) MSNs.
GABAARs located postsynaptically in MSNs are sufficient to control STDP polarity
To investigate the locus of GABA effects, one should distinguish a direct effect of GABA onto the postsynaptic MSN from broader network effects that could be induced by bath-applied GABA blockers. We therefore investigated whether GABAARs located on the sole postsynaptic MSN were able to control the STDP-timing rule. To address this question, we took advantage of a potent effect of PTX on GABAARs when applied intracellularly (Yazaki-Sugiyama et al., 2009). We recorded pairs of neighboring MSNs (<50 μm away, n = 11 pairs), one MSN filled with PTX added into the intracellular solution (i-PTX, 1 mm) through the patch-clamp pipette and a neighboring control MSN recorded with regular internal solution. We first ensured that i-PTX efficiently blocked IPSCs by recording spontaneous IPSCs in i-PTX conditions or when PTX was bath-applied (e-PTX; Fig. 3A). We observed no significant difference, because both the amplitude and frequency of IPSCs were similarly abolished. In MSN pairs (n = 11), the STDP-timing rule was reversed exclusively in the MSN recorded with i-PTX, but unaffected in the control neighboring MSN (Fig. 3B). Indeed, MSNs recorded with i-PTX displayed LTD after post-pre pairings (75.2 ± 6.4%, p < 0.05, n = 5), whereas control neighbor MSNs exhibited LTP (187.5 ± 17.5%, p < 0.05, n = 5). Conversely, after pre-post pairings, MSNs recorded with i-PTX displayed LTP (175.2 ± 32.6%, p < 0.05, n = 6), whereas control neighbor MSNs exhibited LTD (77.4 ± 7.9%, p < 0.05, n = 6). Therefore, blockade of GABAARs restricted to the postsynaptic recorded MSN was sufficient to reverse the STDP-timing rule.
Figure 3.
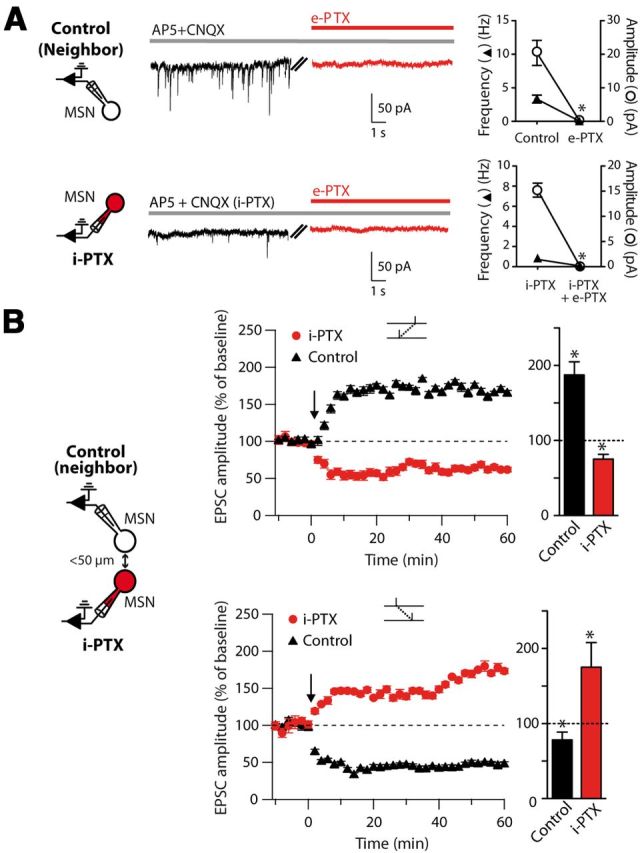
Inhibition of GABAARs in the postsynaptic neuron only is sufficient to reverse STDP. A, Raw traces of 50 s recordings in the presence or absence of e-PTX (in D-AP5/CNQX) in a pair of MSNs recorded simultaneously, one with regular internal solution (neighboring MSN) and one with PTX inside the internal solution (i-PTX, 1 mm). Right panels, frequency and amplitude of IPSCs in the presence or absence of e-PTX and i-PTX (in D-AP5/CNQX). This shows that i-PTX blocks IPSCs as efficiently as e-PTX in MSNs. B, MSN paired recordings with one MSN recorded with control intracellular solution and a neighboring MSN recorded with i-PTX (1 mm), the two neurons being <50 μm away. Post-pre pairings induced LTD in i-PTX recorded MSNs, whereas LTP was observed in neighboring control MSNs (top graphs). Conversely, after pre-post pairings, LTP was observed in i-PTX MSN, whereas LTD was observed in control MSNs (bottom graphs).
GABA has a depolarizing effect on MSNs at rest
The next step was to determine how GABA could have such a strong effect and reverses the plasticity polarity. Given that membrane properties modulate the STDP-timing rule, GABAergic-induced shunt could affect bAPs, a key parameter in orientating STDP preferentially toward a potentiation or depression (Froemke et al., 2010). We analyzed the effects of the GABAAR blockade by PTX or bicuculline on membrane properties. Input resistance, impedance amplitude profile, resonance, and EPSPs/EPSCs kinetics did not display significant variation between control and PTX conditions (Fig. 4). In particular, we observed no significant difference between the amplitude of EPSCs before and after PTX or bicuculline treatment (EPSC amplitude in control conditions was 163.4 ± 15.3 pA and in GABAAR blockade conditions 154.7 ± 14.9 pA, p > 0.05, n = 25). Therefore, somatic recordings did not reveal a significant GABA shunt that could have changed the dendritic electrotonic length and therefore the STDP polarity.
Figure 4.
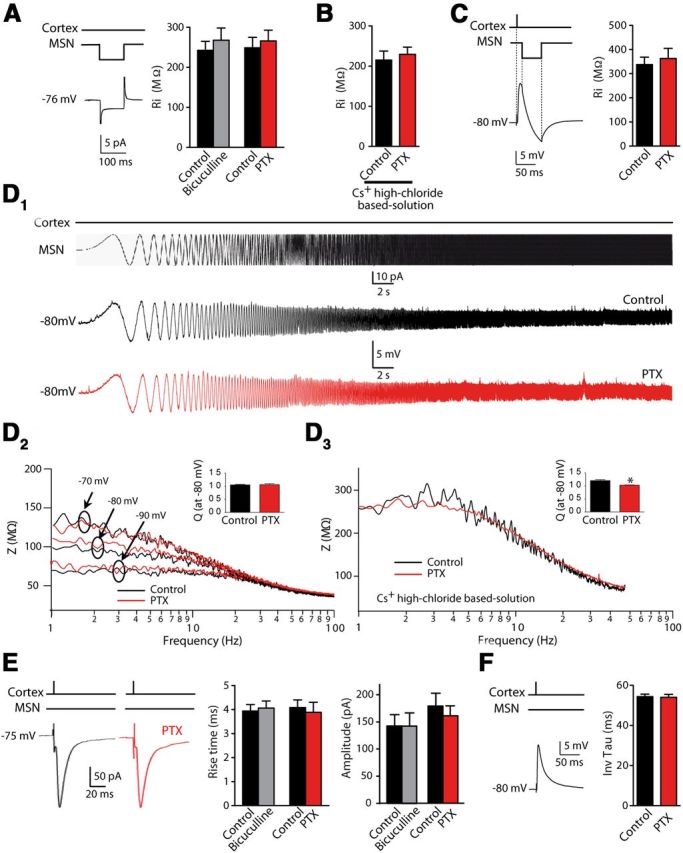
MSN membrane properties are not significantly affected after GABAAR blockade. A–C, Measurement of the input resistance (Ri) before and after bicuculline (20 μm) or PTX (50 μm). Ri was measured with a hyperpolarized pulse (5 pA, 100 ms duration) applied to MSN recorded with regular or high-chloride (136 mm) cesium (Cs+)-based intracellular solution (A,B) or during an evoked corticostriatal EPSP. A hyperpolarized pulse (5 mV during 50 ms) was applied to the MSN 5 ms after a cortical stimulation (C). Ri, reflecting the cellular conductance state, did not display significant variation in control or PTX/bicuculline conditions. D, Impedance amplitude profile (ZAP) and resonance value (Q) tested at different holding membrane potentials (−90, −80, and −70 mV) with regular solution did not show any significant variation in control or PTX. There was a slight change when recorded with high-chloride Cs+-based intracellular solution. D1, Illustrative ZAP protocol applied to one MSN in control and with PTX. This protocol consisted of a sinusoidal current of 25 pA amplitude with an increasing frequency from 1 to 100 Hz lasting for 30 s. Below, sample traces of voltage response to the protocol before (black) and after (red) PTX bath application. D2, Impedance computed as the ratio of the fast Fourier transforms of voltage and current at different membrane potentials (−70, −80, and −90 mV). Inset: Q value at a membrane potential of −80 mV before and after PTX bath application. D3, MSN impedance at −80 mV using a high chloride and Cs+-based internal solution. Inset: resonance at −80 mV before and after PTX bath application. E, F, Kinetics of EPSCs such as rise time or amplitude (E) and EPSP decay time (F) were not significantly affected by PTX or bicuculline treatment.
Although GABA shunting effects were not detected in somatic recordings, we hypothesized that dendritic GABA effects could be significant locally and attenuated before reaching the soma. To investigate this, we built a biophysically detailed model of the MSN (see Materials and Methods) to investigate the effect of GABA on dendritic signals. The model was constructed based on an MSN model (Wolf et al., 2005) capturing the majority of ion channels known to be present in MSNs. We further adjusted the model to include additional experimentally established features such as significant inward rectification, TTX-dependent bAP, dendritic calcium dynamics, and realistic input-output frequency curve (Fig. 5A–D). The model MSN was designed to be activated by glutamatergic and GABAergic inputs, and model simulations predicted that GABAergic inputs would depolarize the distal dendrites (Fig. 5E). Moreover, GABAergic inputs in the dendrites, simulated as activated mainly after the presynaptic stimulation, could hardly be seen in the soma even though they were potent locally (Fig. 5E). A depolarizing effect is likely attributed to the positive difference between the EGABA and the RMP of MSNs. Therefore, to confirm the model prediction, we experimentally estimated the EGABA with cell-attached recordings of iNMDA and iGABA(A) (Fig. 6A). We measured a driving force of chloride ions through GABAARs of 17.2 ± 7 mV from EGABA = −60.8 mV and RMP = −78.1 ± 1.1 mV (n = 4), confirming the depolarizing effect of GABA predicted by the model. Knowing the physiological chloride reversal potential in MSNs, we then verified that our observations of reversion of plasticity with GABAAR blockade (recorded with ECl = −40 mV; Fig. 1) were still valid for ECl = −60 mV; that is, with chloride having a weaker driving force but still depolarizing due to the hyperpolarized MSN RMP (−72.3 ± 0.6 mV, n = 92). We observed a similar effect of the GABA on STDP polarity, with ECl = −60 mV (Fig. 6B). Indeed, in control conditions, post-pre and pre-post pairings induced LTP (158.8 ± 15.5%, p < 0.01, n = 10) and LTD (55.1 ± 8.8%, p < 0.01, n = 6), respectively. With PTX, such plasticity polarity was reversed: post-pre pairings induced LTD (70.7 ± 8.2%, p < 0.05, n = 6) and pre-post pairings induced LTP (144.2 ± 17.3%, p < 0.05, n = 7). Therefore, we show that GABA exerts a strong control of STDP at the physiological ECl measured in MSNs.
Figure 6.

GABA controls plasticity polarity at physiological ECl. A, Cell-attached recordings of unitary NMDA currents at various holding potentials (top traces) and iNMDA-V relationship. RMP is determined at the value indicated by the arrow on the graph. Cell-attached recordings of unitary GABAA currents at various holding potentials (bottom traces) and iGABA-V relationship. The driving force of chloride ions (DFGABA) through GABAARs is determined at the value indicated by the arrow. To extract EGABA, we used the relationship: EGABA = DFGABA + RMP. B, Left: Summary graphs showing that GABAARs blockade at the physiological ECl = −60 mV reversed STDP polarity (control: black circles; PTX: red circles). Each empty symbol represents one neuron and filled symbols are the averages. Right: Summary bar graphs showing the inversion of plasticity after PTX with ECl = −60 mV. GABAAR blockade has a similar effect (reversion of STDP polarity) at the physiological ECl = −60 mV than with a more depolarized chloride reversal (−40 mV).
Because we did not detect a significant effect of GABA on MSN membrane properties or on cortically evoked EPSC/EPSP (Fig. 4), we explored the effect of direct GABAergic transmission onto MSNs. We performed paired recordings to monitor the transmission between one single GABAergic interneuron and one MSN. The sources of GABA are diverse in striatum, but the most powerful inhibition is mediated by FS interneurons (Tepper et al., 2008), which receive monosynaptic afferents from cortex as MSNs (Fino et al., 2005, 2008; Mallet et al., 2005). FS interneurons express parvalbumin (PV), allowing us to record them in PV-GFP mice (Fig. 7A). First, using paired cell-attached recordings of FS-MSN pairs (n = 8), we observed that FS fired action potentials in response to lower intensity of cortical stimulation than MSNs, as illustrated by their relative input/output gain functions (Fig. 7B). This indicates a greater responsiveness of FS cells leading to their activation before MSNs. Of 8 FS-MSN pairs, we found that 6 pairs (75%) were connected, which is in agreement with the high connectivity described for these neurons (Planert et al., 2010). We observed that FS cells induced a depolarizing IPSPs in MSNs when they were maintained at their RMP (averaged MSN RMP was −78.7 ± 2.8 mV and averaged amplitude of unitary IPSP was 2.05 ± 0.62 mV, n = 6 pairs; Fig. 7C). These results confirm the model prediction of a depolarizing effect of GABA on MSNs. In addition, previous studies described an inhibitory weight of FS cells on MSNs (Koós and Tepper, 1999; Tepper et al., 2008). We tested the effect of FS cells on depolarized MSNs and indeed observed a strong inhibitory effect (Fig. 7D,E). We observed that FS cell firing was able to delay an action potential in MSNs, the delay ranging from 3–4 ms to 40 ms (n = 5), depending on the number of action potentials evoked in the FS cell (1–2 spikes vs 3–4 spikes; Fig. 7D). In addition, FS firing decreased the firing rate of MSNs (22.5% averaged decrease of frequency rate, p < 0.05, n = 5; Fig. 7E). In conclusion, although GABA displayed inhibitory effects on firing MSN, we demonstrated that GABA induced a depolarizing effect on MSN at rest, which thus validated a prediction of our biophysical model.
Figure 7.

GABA has a dual effect in MSNs. A, FS-MSN paired recordings (<50 μm away). Infrared (top) and fluorescent (bottom) microphotograph of a corticostriatal slice from PV-GFP mice (scale bars, 10 μm). Right, Electrophysiological characterization of the two cells: MSN (black) and FS cell (blue) raw traces show voltage responses to series of 500 ms current pulses from −100 pA with 40 pA steps (black and blue traces) and to +50 pA above AP threshold (red traces). B, Input/output gain function of FS cell and MSN in response to cortical stimulations. The cortical stimulation intensity is normalized to the FS spike threshold. C, Illustration of a FS-MSN connected pair. When the MSN is at its RMP, spikes evoked in the FS triggered depolarizing IPSPs in the MSN. D, E, When MSN is depolarized above AP threshold, spikes evoked in FS leads to an inhibitory effect. FS firing delays an action potential in MSN (orange trace compared with the control without FS firing in black; D). FS firing induces a decrease of the frequency rate of MSN (E). In this example, the initial firing rate of the MSN is 13.8 Hz and significantly decreases to 9.7 Hz (30% decrease) during the 500 ms of FS cell stimulation (in blue) and comes back to the initial frequency after the end of the stimuli.
GABAAR activation boosts both L-type VSCC- and NMDAR-dependent calcium rise
Finally, we used the model predictions to understand which mechanisms could explain the effect of GABA on STDP rules. For this purpose, we first implemented the model by exploring the signaling pathways involved in the reversed STDP in GABAAR blockade conditions. It has been shown previously that for corticostriatal STDP, LTP is dependent on NMDAR activation and LTD relies on type-1 cannabinoid receptor (CB1R) activation (Adermark and Lovinger, 2007; Shen et al., 2008; Fino et al., 2010). Interestingly, the signaling pathways required for STDP induction were similar in control and GABAAR blockade conditions (Fig. 8). LTP induced by post-pre pairings in control or pre-post pairings with PTX (50 μm) were NMDAR activation dependent, because they were blocked by D-AP5, an NMDAR blocker (50 μm, n = 5 for post-pre pairings in control and n = 6 for pre-post pairings with PTX; Fig. 8A). Indeed, when D-AP5 was applied together with PTX, pre-post pairings did not induce LTP anymore (111.3 ± 7.7%, p > 0.05, n = 6). LTD induced by pre-post pairings in control or post-pre pairings with PTX were CB1R activation dependent, because they were blocked by AM251, a CB1R antagonist (3 μm, n = 5 for control pre-post and n = 5 for PTX post-pre pairings). Indeed, bath application of AM251 prevented the induction of post-pre LTD (113.4 ± 8.9%, p > 0.05, n = 5; Fig. 8B). Therefore, NMDAR-mediated calcium elevations are necessary for LTP formation, whereas LTD induction is mediated by endocannabinoid signaling via L-type VSCC activation (Adermark and Lovinger, 2007; Shen et al., 2008; Fino et al., 2010), in particular, CaV1.3, a low-voltage-activated L-type calcium channel (Olson et al., 2005). Accordingly, we used our biophysical model to investigate these two calcium sources to predict how NMDAR and L-type VSCC calcium-mediated currents in dendrites were affected by GABA during STDP induction protocols. Simulations predicted that, during both post-pre and pre-post pairings, GABAergic inputs would increase both the NMDAR and L-type VSCC-dependent calcium elevations (Fig. 9A,B). Although the interactions between the NMDAR and L-type VSCC calcium influxes are expected to be nonlinear (Kotaleski and Blackwell, 2010), an altered balance between the calcium sources could likely affect the probability for LTP or LTD induction. We investigated how successively increasing GABAergic inputs influenced the balance between NMDAR- and L-type VSCC-dependent calcium influxes. Simulations predicted that GABA would push the ratio between peak amplitudes of NMDAR- and L-type VSCC-mediated calcium toward NMDAR-mediated calcium with post-pre pairings and toward L-type VSCC-mediated calcium during pre-post pairings. Increasing twofold GABAergic inputs strengthened these trends consistently (Fig. 9C,D).
Figure 8.

Pharmacology of STDP in control and GABA blockade conditions. A, B, Summary bar graphs of pharmacological experiments for post-pre sequences (A) or pre-post sequences (B). LTP induced in control with post-pre sequences or with post-pre pairings in GABAAR inhibition was NMDAR dependent, because it was abolished with D-AP5 (50 μm). When D-AP5 was applied together with PTX, pre-post pairings no longer induced LTP. LTD observed in control after pre-post pairings or after post-pre pairings in GABAAR inhibition was CB1R dependent, because it was prevented by AM251 (3 μm). Indeed, bath application of AM251, a CB1R selective antagonist, prevented the induction of post-pre LTD.
Figure 9.

Biophysical simulation of GABA effects on L-type VSCC- and NMDAR-dependent calcium influxes. A, B, Distributions of spine NMDAR-dependent (A) and L-type VSCC-dependent (B) calcium levels along the dendrites in simulated neurons during STDP protocols (n = 15). Results are mean values of the MSN model with 15 sets of random GABA inputs, which are the same as in next panels. Insets: Simulated calcium sample traces. Baselines aligned here. C, D, Ratio between NMDAR and L-type VSCC calcium in simulated neurons (n = 15) during post-pre (C) or pre-post (D) STDP protocols. Arrows indicate the effect on the balance between NMDA- and L-type calcium with successively increasing GABA inputs.
Why does GABA modulate NMDAR- and L-type VSCC-dependent calcium influxes differentially depending on post-pre or pre-post pairings? One plausible explanation is the distinct control of the low-voltage-activated L-type calcium channel CaV1.3 operated by the temporal patterns of bAP GABAergic inputs integration. In post-pre conditions, falling in the “tail” of the decaying phase of the bAP, GABAergic inputs were more separated from the bAP, which only induced a short depolarizing “dendritic plateau” at ∼−65/−70 mV (Fig. 5E). In contrast, in the pre-post conditions, by being tightly integrated into the rising phase of bAP, GABAergic inputs rapidly elevated the local membrane potential to −60 mV and induced a longer depolarizing “dendritic plateau” (Fig. 5E). Considering CaV1.3 channel activates at −60/−50 mV (Xu and Lipscombe, 2001), the temporal patterns of bAP-GABAergic inputs integration in pre-post conditions will greatly facilitate opening of CaV1.3 channel (Shindou et al., 2011) and thus affect the calcium balance. In conclusion, our model suggests that GABAergic inputs tightly influence the NMDAR/L-type VSCC calcium balance in distal dendrites, a key element for the induction of either LTP or LTD.
Discussion
GABAergic circuits are known to modulate strongly the spike timing of output neurons in various brain structures (Pouille and Scanziani, 2001; Wehr and Zador, 2003; Higley and Contreras, 2006), including the striatum (Tepper et al., 2008), but how they could control plasticity was unclear. We demonstrate here that GABA tightly controls STDP-timing rules at corticostriatal synapses. Previous studies investigating different forms of long-term plasticity in MSNs were performed with blocked GABAAR-mediated transmission and we report here that this condition has a potent control over the polarity of the plasticity. Our results reconcile the different reported polarity of the corticostriatal STDP as depending on the use (Hebbian STDP; Pawlak and Kerr, 2008; Shen et al., 2008) or not (anti-Hebbian STDP; Fino et al., 2005, 2010; Schulz et al., 2010) of GABAAR antagonists. Therefore, GABA appears to act as a Hebbian/anti-Hebbian switch. Using D1-GFP mice, we found that GABA displays a significant effect on STDP for MSNs of both direct and indirect pathways. This is also consistent with previous observations reporting that FS interneurons contact both subpopulations of MSNs and exert a strong inhibitory weight on both (Bennett and Bolam, 1994; Planert et al., 2010). The control of STDP by GABA relies on postsynaptic GABAARs, as evidenced by the fact that inhibition of GABAARs only in the postsynaptic neuron was sufficient to reverse STDP. We investigated the effect GABA has in the single postsynaptic MSN and observed in silico that the effect of GABA during STDP paradigms affected both L-type-VSCC and NMDAR activation through a depolarizing effect in the MSN distal dendrites.
What are the effects of GABA during STDP?
Paired FS-MSN recordings showed a depolarizing effect of GABA when MSNs were at rest. Consistent with this, previous studies (in vitro as well as in vivo) have reported that GABA enhances MSN excitability (Mercuri et al., 1991; Blackwell et al., 2003; Bracci and Panzeri, 2006; Dehorter et al., 2009). Nevertheless, such a depolarizing effect of GABA on MSN dendrites does not always imply an excitatory effect. Indeed, we observed that FS cells delayed an evoked action potential and slowed down the frequency firing rate in the postsynaptic MSN, as described previously (Koós and Tepper, 1999). In addition, blocking GABAAR by local PTX injection in vivo enhances the firing activity in MSNs (Mallet et al., 2005). Interestingly, our observations are consistent with in vivo experiments showing that corticostriatal STDP with intact physiological GABAergic transmission displays a similar anti-Hebbian polarity (Schulz et al., 2010). Therefore, the effects of GABA might depend on: (1) the state of activation of MSNs and thus the membrane potential relative to EGABA, and (2) the axo-dendritic compartment (dendrites, spines, soma) considered.
The feedforward inhibition mediated by FS interneurons is efficiently recruited by cortical activity and exerts the strongest inhibition on MSNs (Koós and Tepper, 1999; Tepper et al., 2008). Nevertheless, in addition to FS interneurons, which have been more extensively documented, striatal GABAergic circuits also comprise at least two other types of GABAergic interneurons, NO-synthase- and calretinin-containing interneurons and collaterals from MSNs (Tepper et al., 2008). Even though their inhibitory weight seems to be smaller than FS interneurons, they could be involved in GABAergic control of MSN STDP. Therefore, the effect of GABA could rely on the different subtypes of striatal interneurons and/or MSN collaterals and their associated signaling. In addition, we consider here the GABAergic inputs in a specific static state, but it has been shown that GABAergic synapses are also subject to activity-dependent plasticity (Kullmann and Lamsa, 2011). Indeed, coincident presynaptic and postsynaptic spiking alters the activity of KCC2, a K–Cl cotransporter, resulting in changes in the reversal potential of GABAergic synaptic currents (Woodin et al., 2003). Therefore, STDP itself could modulate the strength of GABAergic inhibition, which in turn controls the polarity of the plasticity. It will therefore be determinant to take into account the interplay of both excitatory and inhibitory synapses plasticity to understand how neuronal circuits encode learning and memory.
Detection of cortical patterns and action selection
In control conditions, we observed anti-Hebbian learning rules at corticostriatal synapses (Fino and Venance, 2010). Our present observations show that GABAergic circuits would be determinant to maintain corticostriatal plasticity in such anti-Hebbian orientation. This is consistent with in vivo experiments with intact GABAergic microcircuits reporting an anti-Hebbian learning rule at corticostriatal synapses (Schulz et al., 2010). Computational studies proposed that anti-Hebbian learning allows decorrelating the association between frequent patterns and thus increases its detection for infrequent patterns (Földiák, 1990; Roberts and Leen, 2010). In striatum, this mechanism might be essential for novelty detection, which favors selection between existed sensory-motor association and the newly arising patterns. During sensory control of motor cortical activation, a “hotspot” in sensory cortex can give rise to broad and persistent sensory and motor cortical activations (Matyas et al., 2010), both types of cortical inputs being further processed in striatum. A new incoming sensory input, likely presented as another hotspot in sensory cortex, would be drowned out by massive and persistent cortical activities. At corticostriatal synapses, thanks to the anti-Hebbian learning rule, the old associated sensory signals will be greatly reduced, whereas the novel sensory signals, disconnected from the previous patterns, would be easily detected. Therefore, the anti-Hebbian learning rule in striatum might allow picking up new inputs and in turn help the cortex learn how to select between the old and new patterns via the corticobasal ganglia loop. GABAergic circuits, by promoting this anti-Hebbian learning rule at corticostriatal synapses, would thus play a central role in the function of novelty detection operated by the striatum.
In addition, network stability, a key point in theories of correlation-based learning, faces an unsolved issue: plain Hebbian learning without additional mechanisms leads to the divergence of synaptic weights because even random correlation leads to weight growth (Wörgötter and Porr, 2005). Therefore, GABAergic tone as an add-on mechanism could help to reconcile the network stability with Hebbian-correlation-based learning.
Implications for diseases
Finally, the present findings should facilitate the investigation of the dysfunctions of synaptic plasticity in central disorders affecting GABAergic circuits. Several pathologies, such as epilepsy, autism, dystonia, schizophrenia, or Tourette syndrome, affect GABAergic microcircuits (Rubenstein and Merzenich, 2003; Kalanithi et al., 2005; Chao et al., 2010), and are thus expected to switch STDP polarity and impair proper learning and memory. This is also particularly exemplified by dystonia-like behaviors observed after an inhibition of FS interneuron activity (by selective inhibition of excitatory inputs to FS cells; Gittis et al., 2011). Interestingly, in a mouse model of dystonia associated with reduced striatal GABA levels, an impairment of bidirectional synaptic plasticity in the striatum was reported (Martella et al., 2009). More generally, subtle perturbation of GABAergic neuronal function contributes to neuropsychiatric phenotypes (Chao et al., 2010) and likely to changes in synaptic plasticity properties or rules.
Footnotes
This work was supported by INSERM, College de France, the FP7 Marie Curie International Outgoing Fellowship program, the Swedish Research Council, and the Stockholm Brain Institute. We thank J.M. Deniau, O.J. Manzoni, and M. Vandecasteele for critical comments and J.C. Poncer for kindly providing parvalbumin-GFP mice.
The authors declare no competing financial interests.
References
- Ade KK, Janssen MJ, Ortinski PI, Vicini S. Differential tonic GABA conductances in striatal medium spiny neurons. J Neurosci. 2008;28:1185–1197. doi: 10.1523/JNEUROSCI.3908-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adermark L, Lovinger DM. Combined activation of L-type Ca2+ channels and synaptic transmission is sufficient to induce striatal long-term depression. J Neurosci. 2007;27:6781–6787. doi: 10.1523/JNEUROSCI.0280-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BD, Bolam JP. Synaptic input and output of parvalbumin-immunoreactive neurons in the neostriatum of the rat. Neuroscience. 1994;62:707–719. doi: 10.1016/0306-4522(94)90471-5. [DOI] [PubMed] [Google Scholar]
- Blackwell KT, Czubayko U, Plenz D. Quantitative estimate of synaptic inputs to striatal neurons during up and down states in vitro. J Neurosci. 2003;23:9123–9132. doi: 10.1523/JNEUROSCI.23-27-09123.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower JM, Beeman D. Constructing realistic neural simulations with GENESIS. Methods Mol Biol. 2007;401:103–125. doi: 10.1007/978-1-59745-520-6_7. [DOI] [PubMed] [Google Scholar]
- Bracci E, Panzeri S. Excitatory GABAergic effects in striatal projection neurons. J Neurophysiol. 2006;95:1285–1290. doi: 10.1152/jn.00598.2005. [DOI] [PubMed] [Google Scholar]
- Caporale N, Dan Y. Spike timing-dependent plasticity: a Hebbian learning rule. Annu Rev Neurosci. 2008;31:25–46. doi: 10.1146/annurev.neuro.31.060407.125639. [DOI] [PubMed] [Google Scholar]
- Carter AG, Sabatini BL. State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron. 2004;44:483–493. doi: 10.1016/j.neuron.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JL, Noebels JL, Rosenmund C, Zoghbi HY. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman DE, Keefe KA, Wilcox KS. Evidence for functionally distinct synaptic NMDA receptors in ventromedial versus dorsolateral striatum. J Neurophysiol. 2003;89:69–80. doi: 10.1152/jn.00342.2002. [DOI] [PubMed] [Google Scholar]
- Day M, Wokosin D, Plotkin JL, Tian X, Surmeier DJ. Differential excitability and modulation of striatal medium spiny neuron dendrites. J Neurosci. 2008;28:11603–11614. doi: 10.1523/JNEUROSCI.1840-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehorter N, Guigoni C, Lopez C, Hirsch J, Eusebio A, Ben-Ari Y, Hammond C. Dopamine-deprived striatal GABAergic interneurons burst and generate repetitive gigantic IPSCs in medium spiny neurons. J Neurosci. 2009;29:7776–7787. doi: 10.1523/JNEUROSCI.1527-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RC, Morera-Herreras T, Cui Y, Du K, Sheehan T, Kotaleski JH, Venance L, Blackwell KT. The effects of NMDA subunit composition on calcium influx and spike timing-dependent plasticity in striatal medium spiny neurons. PLoS Comput Biol. 2012;8:e1002493. doi: 10.1371/journal.pcbi.1002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Venance L. Spike-timing dependent plasticity in the striatum. Front Synaptic Neurosci. 2010;2:6. doi: 10.3389/fnsyn.2010.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Glowinski J, Venance L. Bidirectional activity-dependent plasticity at corticostriatal synapses. J Neurosci. 2005;25:11279–11287. doi: 10.1523/JNEUROSCI.4476-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Deniau JM, Venance L. Cell-specific spike-timing-dependent plasticity in GABAergic and cholinergic interneurons in corticostriatal rat brain slices. J Physiol. 2008;586:265–282. doi: 10.1113/jphysiol.2007.144501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Paille V, Cui Y, Morera-Herreras T, Deniau JM, Venance L. Distinct coincidence detectors govern the corticostriatal spike timing-dependent plasticity. J Physiol. 2010;588:3045–3062. doi: 10.1113/jphysiol.2010.188466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Földiák P. Forming sparse representations by local anti-Hebbian learning. Biol Cybern. 1990;64:165–170. doi: 10.1007/BF02331346. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Letzkus JJ, Kampa BM, Hang GB, Stuart GJ. Dendritic synapse location and neocortical spike-timing-dependent plasticity. Front Synaptic Neurosci. 2010;2:29. doi: 10.3389/fnsyn.2010.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Leventhal DK, Fensterheim BA, Pettibone JR, Berke JD, Kreitzer AC. Selective inhibition of striatal fast-spiking interneurons causes dyskinesias. J Neurosci. 2011;31:15727–15731. doi: 10.1523/JNEUROSCI.3875-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Contreras D. Balanced excitation and inhibition determine spike timing during frequency adaptation. J Neurosci. 2006;26:448–457. doi: 10.1523/JNEUROSCI.3506-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalanithi PS, Zheng W, Kataoka Y, DiFiglia M, Grantz H, Saper CB, Schwartz ML, Leckman JF, Vaccarino FM. Altered parvalbumin-positive neuron distribution in basal ganglia of individuals with Tourette syndrome. Proc Natl Acad Sci U S A. 2005;102:13307–13312. doi: 10.1073/pnas.0502624102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koós T, Tepper JM. Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat Neurosci. 1999;2:467–472. doi: 10.1038/8138. [DOI] [PubMed] [Google Scholar]
- Kotaleski JH, Blackwell KT. Modelling the molecular mechanisms of synaptic plasticity using systems biology approaches. Nat Rev Neurosci. 2010;11:239–251. doi: 10.1038/nrn2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Lamsa KP. LTP and LTD in cortical GABAergic interneurons: emerging rules and roles. Neuropharmacology. 2011;60:712–719. doi: 10.1016/j.neuropharm.2010.12.020. [DOI] [PubMed] [Google Scholar]
- Mallet N, Le Moine C, Charpier S, Gonon F. Feedforward inhibition of projection neurons by fast-spiking GABA interneurons in the rat striatum in vivo. J Neurosci. 2005;25:3857–3869. doi: 10.1523/JNEUROSCI.5027-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Martella G, Tassone A, Sciamanna G, Platania P, Cuomo D, Viscomi MT, Bonsi P, Cacci E, Biagioni S, Usiello A, Bernardi G, Sharma N, Standaert DG, Pisani A. Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: role of endogenous acetylcholine. Brain. 2009;132:2336–2349. doi: 10.1093/brain/awp194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, Morris RG. New life in an old idea: the synaptic plasticity and memory hypothesis revisited. Hippocampus. 2002;12:609–636. doi: 10.1002/hipo.10107. [DOI] [PubMed] [Google Scholar]
- Matyas F, Sreenivasan V, Marbach F, Wacongne C, Barsy B, Mateo C, Aronoff R, Petersen CC. Motor control by sensory cortex. Science. 2010;330:1240–1243. doi: 10.1126/science.1195797. [DOI] [PubMed] [Google Scholar]
- Mercuri NB, Calabresi P, Stefani A, Stratta F, Bernardi G. GABA depolarizes neurons in the rat striatum: an in vivo study. Synapse. 1991;8:38–40. doi: 10.1002/syn.890080106. [DOI] [PubMed] [Google Scholar]
- Olson PA, Tkatch T, Hernandez-Lopez S, Ulrich S, Ilijic E, Mugnaini E, Zhang H, Bezprozvanny I, Surmeier DJ. G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J Neurosci. 2005;25:1050–1062. doi: 10.1523/JNEUROSCI.3327-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak V, Kerr JN. Dopamine receptor activation is required for corticostriatal spike-timing-dependent plasticity. J Neurosci. 2008;28:2435–2446. doi: 10.1523/JNEUROSCI.4402-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planert H, Szydlowski SN, Hjorth JJ, Grillner S, Silberberg G. Dynamics of synaptic transmission between fast-spiking interneurons and striatal projection neurons of the direct and indirect pathways. J Neurosci. 2010;30:3499–3507. doi: 10.1523/JNEUROSCI.5139-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- Roberts PD, Leen TK. Anti-hebbian spike-timing-dependent plasticity and adaptive sensory processing. Front Comput Neurosci. 2010;4:156. doi: 10.3389/fncom.2010.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183X.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG, Svoboda K. The life cycle of Ca(2+) ions in dendritic spines. Neuron. 2002;33:439–452. doi: 10.1016/S0896-6273(02)00573-1. [DOI] [PubMed] [Google Scholar]
- Santhakumar V, Jones RT, Mody I. Developmental regulation and neuroprotective effects of striatal tonic GABAA currents. Neuroscience. 2010;167:644–655. doi: 10.1016/j.neuroscience.2010.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JM, Redgrave P, Reynolds JN. Cortico-striatal spike-timing dependent plasticity after activation of subcortical pathways. Front Synaptic Neurosci. 2010;2:23. doi: 10.3389/fnsyn.2010.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindou T, Ochi-Shindou M, Wickens JR. A Ca(2+) threshold for induction of spike-timing-dependent depression in the mouse striatum. J Neurosci. 2011;31:13015–13022. doi: 10.1523/JNEUROSCI.3206-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström PJ, Rancz EA, Roth A, Hausser M. Dendritic excitability and synaptic plasticity. Physiol Rev. 2008;88:769–840. doi: 10.1152/physrev.00016.2007. [DOI] [PubMed] [Google Scholar]
- Tepper JM, Wilson CJ, Koós T. Feedforward and feedback inhibition in neostriatal GABAergic spiny neurons. Brain Res Rev. 2008;58:272–281. doi: 10.1016/j.brainresrev.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehr M, Zador AM. Balanced inhibition underlies tuning and sharpens spike timing in auditory cortex. Nature. 2003;426:442–446. doi: 10.1038/nature02116. [DOI] [PubMed] [Google Scholar]
- Wolf JA, Moyer JT, Lazarewicz MT, Contreras D, Benoit-Marand M, O'Donnell P, Finkel LH. NMDA/AMPA ratio impacts state transitions and entrainment to oscillations in a computational model of the nucleus accumbens medium spiny projection neuron. J Neurosci. 2005;25:9080–9095. doi: 10.1523/JNEUROSCI.2220-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodin MA, Ganguly K, Poo MM. Coincident pre- and postsynaptic activity modifies GABAergic synapses by postsynaptic changes in Cl- transporter activity. Neuron. 2003;39:807–820. doi: 10.1016/S0896-6273(03)00507-5. [DOI] [PubMed] [Google Scholar]
- Wörgötter F, Porr B. Temporal sequence learning, prediction, and control: a review of different models and their relation to biological mechanisms. Neural Comput. 2005;17:245–319. doi: 10.1162/0899766053011555. [DOI] [PubMed] [Google Scholar]
- Xu W, Lipscombe D. Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci. 2001;21:5944–5951. doi: 10.1523/JNEUROSCI.21-16-05944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda R, Nimchinsky EA, Scheuss V, Pologruto TA, Oertner TG, Sabatini BL, Svoboda K. Imaging calcium concentration dynamics in small neuronal compartments. Sci STKE. 2004;2004:pl5. doi: 10.1126/stke.2192004pl5. [DOI] [PubMed] [Google Scholar]
- Yazaki-Sugiyama Y, Kang S, Câteau H, Fukai T, Hensch TK. Bidirectional plasticity in fast-spiking GABA circuits by visual experience. Nature. 2009;462:218–221. doi: 10.1038/nature08485. [DOI] [PubMed] [Google Scholar]